
Determination of 241Am in Environmental Samples: A Review


Abstract
1. Introduction
1.1. Nuclear Physical and Chemical Properties of 241Am
1.2. Sources of 241Am in the Environment
1.3. Distribution and Transfer of 241Am in the Environment
2. Sample Pre-Treatment and Pre-Concentration
2.1. Sample Pre-Treatment
2.2. Pre-Concentration
3. Chemical Separation and Purification Procedures
3.1. Solvent Extraction
3.2. Ion-Exchange Chromatography
3.3. Extraction Chromatography
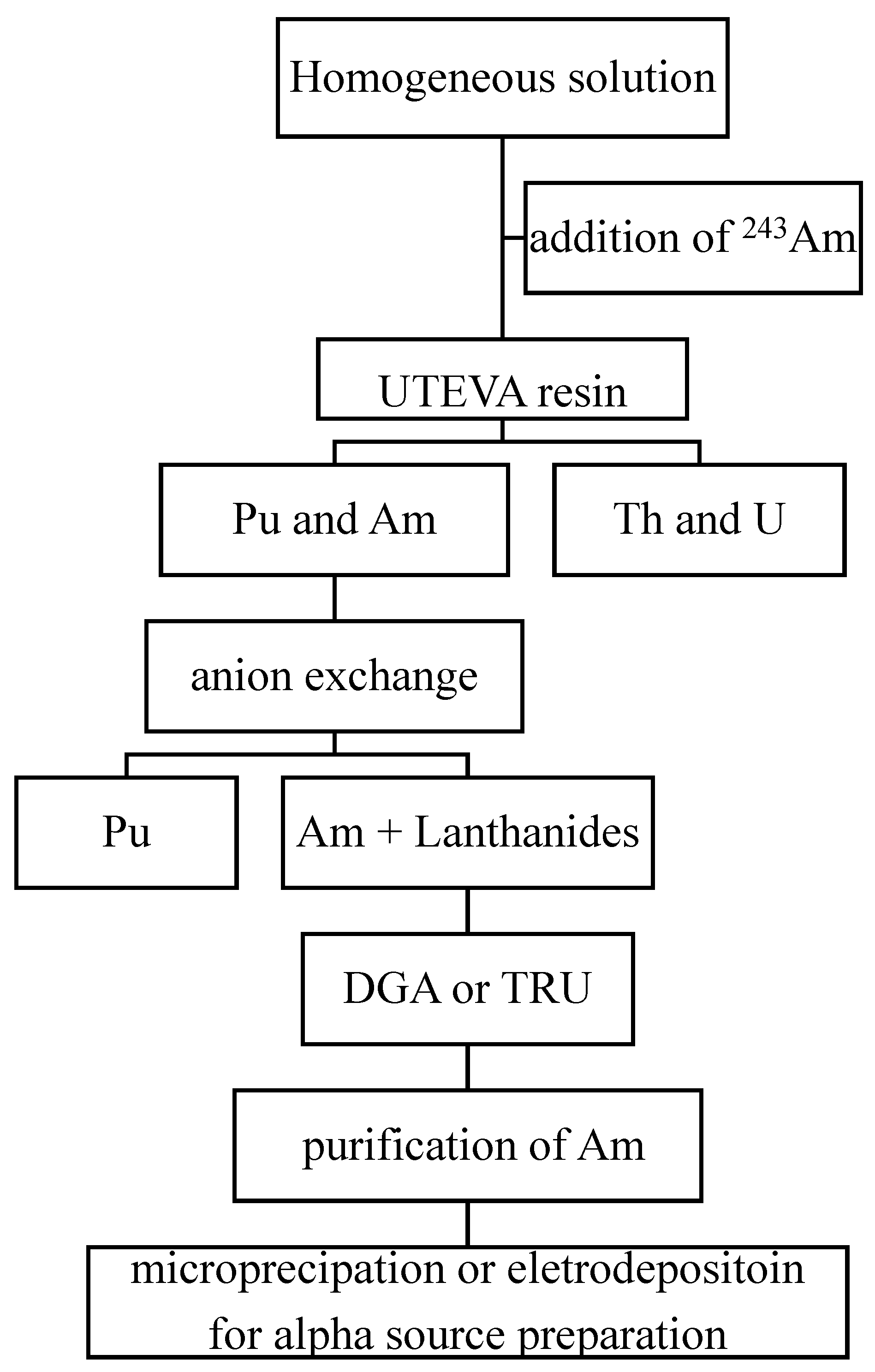
3.4. Combined Procedures for 241Am Determination
3.5. Automated Systems for the Separation of Am
4. Source Preparation
4.1. Source Preparation for Alpha Spectrometry
4.2. Source Preparation for Mass Spectrometry
5. Alpha Spectrometry for 241Am Measurement
6. Mass Spectrometry for 241Am Measurement
6.1. Inductively Coupled Plasma Mass Spectrometry (ICP-MS)
6.2. Thermal Ionization Mass Spectrometry (TIMS)
6.3. Accelerator Mass Spectrometry (AMS)
6.4. Quality Control and Uncertainty for 241Am Determination
7. Speciation Analyses of 241Am in Environmental Samples
7.1. Soluble Species of 241Am in Natural Water
7.2. Particle- and Colloid-Associated 241Am in Natural Water
7.3. Fractionation Analyses of 241Am in Soil and Sediment
8. Tracer Applications of 241Am in the Environment
8.1. 241Am as a Time-Marker for Sediment Dating
8.2. 241Am Signatures in Nuclear Forensics
9. Conclusions and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| AMS | Accelerator-based mass spectrometry |
| BTBP | 2,6-bis(5,6-dialkyl-1,2,4-triazinyl-3-yl)dipyridine |
| BTP | 2,6-bis(5,6-dialkyl-1,2,4-triazinyl-3-yl)pyridine |
| CA-BTP | bis-2,6-(5,6,7,8-tetrahydro-5,9,9-trimethyl-5,8-methano-1,2,4-benzotriazin-3-yl) pyridine |
| CMPO | octyl, phenyl-N, N-di-iso-butyl carbamoylmethyl phosphine oxide |
| CRM | certified reference material |
| CyMe4BTBP | 6,6′-bis(5,5,8,8-tetramethyl-5,6,7,8-tetrahydrobenzo [1,2,4]triazin-3-yl)-2,2′-bipyridine |
| CyMe4BTPhen | 2,9-bis(5,5,8,8-tetramethyl-5,6,7,8-tetrahydro-benzo [1,2,4]triazin-3-yl)-1,10-phenanthroline |
| DAm | weight distribution ration |
| DDCP | dibutyl-N,N′-diethylcarbamylphosphonate |
| DF | decontamination factor |
| DGA | di glycol amide |
| DIPEX | a commercial resin consisting of bis(2-ethylhexyl)methanediphosphonic acid |
| DIPHONIX | a commercial resin containing sulfonic and gem-diphosphonic acid groups |
| DTPA | 2-hydorxy-1,3-diaminopropane tetra-acetic acid |
| EC | extraction chromatography |
| FI | flow injection |
| FPs | fission products |
| FWHW | full width at half maximum |
| HDEHP | di-2-ethylhexylphosphoric acid |
| IAEA | international atomic energy agency |
| ICP-MS | inductively coupled plasma mass spectrometry |
| ID | isotope dilution |
| LOD | limit of detection |
| LOQ | limit of quantification |
| MC | multiple-ion collector |
| MCFI | multi-commuted flow injection |
| MNPs | magnetic nanoparticles |
| MOB-BTP | 3,3′-dimethoxy-phenyl-bis-1,2,4-triazinyl-2,6-pyridine |
| MPFS | multi-pumping flow system |
| MSFI | multi-syringe flow injection |
| PI | pressurized injection |
| PIPS | passivated ion-implanted planar silicon |
| PMBP | 1-benzyl-3-methyl-4-benzoyl acetyl acetone |
| Q-ICP-MS | quadrupole ICP-MS |
| RE | rare-earth |
| SAMMS | self-assembled monolayer on mesoporous supports |
| SF | sector field |
| STS | Semipalatinsk Test Site |
| SI | sequential injection |
| TBP | tributyl phosphate |
| TE | total evaporation |
| TEVA | tetravalent actinide |
| TIMS | thermal ionization mass spectrometry |
| TIOA | tri-iso-octylamine |
| TOA | trioctylamine |
| TONA | tri-n-octylaime |
| TOPO | trioctyl phosphine oxide |
| TRU | trans-uranium |
| TTA | triethylene tetramine |
| UTEVA | uranium and tetravalent actinides |
| WHO | World Health Organization |
| XRD | X-ray diffraction |
References
- Warwick, P.; Croudace, I.; Carpenter, R. Review of analytical techniques for the determination of Americium-241 in soils and sediments. Appl. Radiat. Isot. 1996, 47, 627–642. [Google Scholar] [CrossRef]
- Hou, X.; Roos, P. Critical comparison of radiometric and mass spectrometric methods for the determination of radionuclides in environmental, biological and nuclear waste samples. Anal. Chim. Acta 2008, 608, 105–139. [Google Scholar] [CrossRef] [PubMed]
- Vajda, N.; Kim, C. Determination of Transuranium Isotopes (Pu, Np, Am) by Radiometric Techniques: A Review of Analytical Methodology. Anal. Chem. 2011, 83, 4688–4719. [Google Scholar] [CrossRef]
- Vajda, N.; Kim, C. Determination of Am-241 isotope: A review of analytical methodology. J. Radioanal. Nucl. Chem. 2010, 284, 341–366. [Google Scholar] [CrossRef]
- Aggarwal, S.K. A review on the mass spectrometric studies of americium: Present status and future perspective. Mass Spectrom. Rev. 2018, 37, 43–56. [Google Scholar] [CrossRef] [PubMed]
- Tavcar, P.; Jakopic, R.; Benedik, L. Sequential determination of 241Am, 237Np, Pu radioisotopes and 90Sr in soil and sediment samples. Acta Chim. Slov. 2005, 52, 60–66. [Google Scholar]
- Osváth, S.; Vajda, N.; Molnár, Z. Development of a complex method for the determination of actinoides. J. Radioanal. Nucl. Chem. 2009, 281, 461–465. [Google Scholar] [CrossRef]
- Burns, J.; Shehee, T.; Clearfield, A.; Hobbs, D. Separation of Americium from Curium by Oxidation and Ion Exchange. Anal. Chem. 2012, 84, 6930–6932. [Google Scholar] [CrossRef]
- Thakur, P.; Mulholland, G.P. Determination of 237Np in environmental and nuclear samples: A review of the analytical method. Appl. Radiat. Isot. 2012, 70, 1747–1778. [Google Scholar] [CrossRef]
- UNSCEAR. Sources and Effects of Ionizing Radiation[R]; United Nations Scientific Committee on the Effects of Atomic Radiation: Vienna, Austria, 2000; ISBN 92-1-142238-8. [Google Scholar]
- Pazukhin, E.M.; Drozd, I.P.; Tokarevskii, V.V. Chernobyl accident and the problem of 241Am. Radiochemistry 1995, 36, 590–597. [Google Scholar]
- Muravitsky, A.V.; Razbudey, V.F.; Tokarevsky, V.V.; Vorona, P.N. Time-dependent 241Am activity in the environment from decay of 241Pu released in the Chernobyl accident. Appl. Radiat. Isot. 2005, 63, 487–492. [Google Scholar] [CrossRef] [PubMed]
- Hunt, J.; Leonard, K.; Hughes, L. Artificial radionuclides in the Irish Sea from Sellafield: Remobilisation revisited. J. Radiol. Prot. 2013, 33, 261–279. [Google Scholar] [CrossRef] [PubMed]
- Cundy, A.B.; Croudace, I.W.; Warwick, P.E.; Oh, J.; Haslett, S.K. Accumulation of COGEMA-La Hague-derived Reprocessing Wastes in French Salt Marsh Sediments. Environ. Sci. Technol. 2002, 36, 4990–4997. [Google Scholar] [CrossRef] [PubMed]
- Mackenzie, A.B.; Scott, R.D. Sellafield waste radionuclides in Irish sea intertidal and salt marsh sediments. Environ. Geochem. Hlth. 1993, 15, 173–184. [Google Scholar] [CrossRef]
- Ikaheimonen, T.; Ilus, E.; Klemola, S.; Dahlgaard, H.; Ryan, T.; Eriksson, M. Plutonium and americium in the sediments off the Thule air base, Greenland. J. Radioanal. Nucl. Chem. 2002, 252, 339–344. [Google Scholar] [CrossRef]
- Aarkrog, A.; Boelskifte, S.; Dahlgaard, H.; Duniec, S.; Holm, E.; Smith, J.N. Studies of transuranics in an arctic marine environment. J. Radioanal. Nucl. Chem. 1987, 115, 39–50. [Google Scholar] [CrossRef]
- Aarkrog, A. Inventory of Nuclear Releases in the World. In Radioecology and the Restoration of Radioactive-Contaminated Sites; Luykx, F.F., Frissel, M.J., Eds.; NATO ASI Series; Springer: Dordrecht, The Netherlands, 1996; Volume 13, pp. 31–43. ISBN 978-94-009-0301-2. [Google Scholar]
- Ahn, J.; Carson, C.; Jensen, M.; Juraku, K.; Nagasaki, S.; Tanaka, S. Reflections on the Fukushima Daiichi Nuclear Accident: Toward Social-Scientific Literacy and Engineering Resilience; Springer Nature: Berlin/Heidelberg, Germany, 2015. [Google Scholar]
- Duffa, C.; Renaud, P.; Goutelard, F. Activities and transfers of Pu and Am in rice samples from Camargue, France. J. Radioanal. Nucl. Chem. 2002, 252, 247–248. [Google Scholar] [CrossRef]
- Yamamoto, M.; Sakaguchi, A.; Ochiai, S.; Takada, T.; Hamataka, K.; Murakami, T.; Nagao, S. Isotopic Pu, Am and Cm signatures in environmental samples contaminated by the Fukushima Dai-ichi Nuclear Power Plant accident. J. Environ. Radioact. 2014, 132, 31–46. [Google Scholar] [CrossRef]
- Lujanienė, G.; Valiulis, D.; Byčenkienė, S.; Aakalys, J.; Povinec, P.P. Plutonium isotopes and 241Am in the atmosphere of Lithuania: A comparison of different source terms. Atmos. Environ. 2012, 61, 419–427. [Google Scholar] [CrossRef]
- Thakur, P.; Mulholland, G.P. Determination of Pu, Am, U and Cs in large soil samples in the vicinity of the USDOE Waste Isolation Pilot Plant. J. Radioanal. Nucl. Chem. 2011, 288, 499–506. [Google Scholar] [CrossRef]
- Popov, L.; Mihailova, G.; Naidenov, I. Determination of activity ratios of 238,239+240,241Pu, 241Am, 134,137Cs, and 90Sr in Bulgarian soils. J. Radioanal. Nucl. Chem. 2010, 285, 223–237. [Google Scholar] [CrossRef]
- Ni, Y.; Wang, Z.; Guo, Q.; Zheng, J.; Li, S.; Lin, J.; Tan, Z.; Huang, W. Distinctive distributions and migrations of 239+240Pu and 241Am in Chinese forest, grassland and desert soils. Chemosphere 2018, 212, 1002–1009. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Zhang, H.; Fang, S.; Hou, X.; Zhang, L.; Dang, H.; Yi, X.; Zhai, S.; Wang, W.; Xu, J. Determination of ultra-low level 241Am in soil and sediment using chemical separation and triple quadrupole inductively coupled plasma mass spectrometry measurement with He-NH3 as collision-reaction gas. Spectrochim. Acta Part B At. Spectrosc. 2021, 178, 106113. [Google Scholar] [CrossRef]
- Holgye, Z.; Schlesingerova, E.; Tecl, J.; Filgas, R. 238Pu, 239,240Pu, 241Am, 90Sr and 137Cs in soils around nuclear research centre Rez, near Prague. J. Environ. Radioact. 2004, 71, 115–125. [Google Scholar] [CrossRef]
- Cwanek, A.; Mietelski, J.W.; Bokas, E.; Olech, M.A.; Anczkiewicz, R.; Misiak, R. Sources and variation of isotopic ratio of airborne radionuclides in Western Arctic lichens and mosses. Chemosphere 2020, 239, 124783. [Google Scholar] [CrossRef] [PubMed]
- Jia, G.; Desideri, D.; Guerra, F.; Meli, M.A.; Testa, C. Concentration and vertical distribution of plutonium and americium in Italian mosses and lichens. J. Radioanal. Nucl. Chem. 1997, 222, 3–9. [Google Scholar] [CrossRef]
- Wan Mahmood, Z.U.Y.; Shahar, H.; Ahmad, Z.; Wo, Y.M.; Abu Bakar, A.S. Behavior and distribution of 239+240Pu and 241Am in the east coast of Peninsular Malaysia marine environment. J. Radioanal. Nucl. Chem. 2010, 286, 265–272. [Google Scholar] [CrossRef]
- Mulsow, S.; Coquery, M.; Dovlete, C.; Gastaud, J.; Ikeuchi, Y.; Pham, M.K.; Povinec, P.P. Radionuclide concentrations in underground waters of Mururoa and Fangataufa Atolls. Sci. Total Environ. 1999, 237–238, 287–300. [Google Scholar] [CrossRef]
- Lee, S.; Povinec, P.P.; Wyse, E.; Pham, M.K.; Hong, G.; Chung, C.; Kim, S.; Lee, H. Distribution and inventories of 90Sr, 137Cs, 241Am and Pu isotopes in sediments of the Northwest Pacific Ocean. Mar. Geol. 2005, 216, 249–263. [Google Scholar] [CrossRef]
- Ugur, A.; Yener, G. Plutonium isotopes, 241Am and 137Cs activity concentrations in marine sediments of Gokova Bay, Aegean Turkish Coast. J. Radioanal. Nucl. Chem. 2002, 252, 47–51. [Google Scholar] [CrossRef]
- Mihai, S.; Hurtgen, C. Plutonium and americium in sediment samples along the Romanian sector of the Danube river and the Black Sea coast. J. Radioanal. Nucl. Chem. 1997, 222, 275–278. [Google Scholar] [CrossRef]
- Kim, C.K.; Morita, S.; Seki, R.; Takaku, Y.; Ikeda, N.; Assinder, D.J. Distribution and behaviour of 99Tc, 237Np, 239,240Pu, and 241Am in the coastal and estuarine sediments of the Irish Sea. J. Radioanal. Nucl. Chem. 1992, 156, 201–213. [Google Scholar] [CrossRef]
- Desideri, D.; Giuliani, S.; Meli, M.A.; Testa, C.; Triulzi, C.; Vaghi, M. Presence of 137Cs, Pu isotopes and 241Am in ligurian and Tyrrhenian Seas sediments. J. Radioanal. Nucl. Chem. 2004, 260, 9–12. [Google Scholar] [CrossRef]
- Caroli, S.; Forte, M.; Nuccetelli, C.; Rusconi, R.; Risica, S. A short review on radioactivity in drinking water as assessed by radiometric and Inductively Coupled Plasma-Mass Spectrometry techniques. Microchem. J. 2013, 107, 95–100. [Google Scholar] [CrossRef]
- Liu, N.; Liao, J.; Yang, Y.; Luo, S.; Luo, Q.; An, Z.; Duan, Y.; Liu, M.; Zhao, P. Biosorption of 241Am by Saccharomyces cerevisiae: Preliminary investigation on mechanism. J. Radioanal. Nucl. Chem. 2008, 275, 173–180. [Google Scholar] [CrossRef]
- Baigazinov, Z.; Lukashenko, S.N.; Panitsky, V.; Kadyrova, N.Z.; Karatayev, S.S.; Mamyrbayeva, S.; Baigazy, S.; Bazarbaeva; Kabdyrakova, A.B.; Kunduzbaeva, E.; et al. The transfer of 239 + 240Pu, 241Am, 137Cs and 90Sr to the tissues of horses. J. Environ. Radioact. 2020, 222, 106322. [Google Scholar] [CrossRef]
- Mamyrbayeva, A.S.; Baigazinov, Z.A.; Lukashenko, S.N.; Panitskiy, A.V.; Karatayev, S.S.; Baigazy, S.A.; Bazarbayeva, A.B.; Zhadyranova, A.A.; Kenzhina, L.B.; Mukhamediyarov, N.; et al. The excretion of 241Am and 137Cs from the broilers organs after long-term application. J. Environ. Radioact. 2021, 229–230, 106543. [Google Scholar] [CrossRef]
- Bolotov, B.M.; Gaitinov, A.C.; Polyakov, A.I.; Chuburkov, Y.T.; Perelygin, V.P.; Drobina, T.P.; Kravets, L.I.; Timokhin, S.N.; Mietelski, J.M.; Szeglowski, Z.; et al. On the determination of and content in human tissues. Radiat. Meas. 2003, 36, 541–545. [Google Scholar] [CrossRef]
- Sokolik, G.A.; Ovsiannikova, S.V.; Ivanova, T.G.; Leinova, S.L. Soil-plant transfer of plutonium and americium in contaminated regions of Belarus after the Chernobyl catastrophe. Environ. Int. 2004, 30, 939–947. [Google Scholar] [CrossRef]
- Lehto, J.; Vaaramaa, K.; Leskinen, A. 137Cs, 239,240Pu and 241Am in boreal forest soil and their transfer into wild mushrooms and berries. J. Environ. Radioact. 2013, 116, 124–132. [Google Scholar] [CrossRef]
- Wang, Z.T.; Zheng, J.; Tagami, K.; Uchida, S. Newly derived transfer factors for Th, Am, Pu, and Cl since publication of IAEA TRS No. 472: A review. J. Radioanal. Nucl. Chem. 2015, 306, 11–20. [Google Scholar] [CrossRef]
- Wang, Z.; Yang, G.; Zheng, J.; Cao, L.; Yu, H.; Zhu, Y.; Tagami, K.; Uchida, S. Effect of Ashing Temperature on Accurate Determination of Plutonium in Soil Samples. Anal. Chem. 2015, 87, 5511–5515. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, S.J.; Hensley, C.A.; Armenta, C.E.; Peters, R.J. Environmental and Human Monitoring of Americium-241 Utilizing Extraction Chromatography and α-Spectrometry. Anal. Chem. 1997, 69, 809–812. [Google Scholar] [CrossRef] [PubMed]
- Garcia, R.; Kahn, B. Total dissolution of environmental and biological samples by closed-vessel microwave digestion for radiometric analysis. J. Radioanal. Nucl. Chem. 2001, 250, 85–91. [Google Scholar] [CrossRef]
- Mietelski, J.W.; Kubica, B.; Gaca, P.; Tomankiewicz, E.; Błażej, S.; Tuteja-Krysa, M.; Stobiński, M. 238Pu, 239 + 240Pu, 241Am, 90Sr and 137Cs in mountain soil samples from the Tatra National Park (Poland). J. Radioanal. Nucl. Chem. 2008, 275, 523–533. [Google Scholar] [CrossRef]
- Luisier, F.; Alvarado, J.; Steinmann, P.; Krachler, M.; Froidevaux, P. A new method for the determination of plutonium and americium using high pressure microwave digestion and alpha-spectrometry or ICP-SMS. J. Radioanal. Nucl. Chem. 2009, 281, 425–432. [Google Scholar] [CrossRef]
- Maxwell, S. Rapid method for determination of plutonium, americium and curium in large soil samples. J. Radioanal. Nucl. Chem. 2008, 275, 395–402. [Google Scholar] [CrossRef]
- Maxwell, S.L.; Culligan, B.; Hutchison, J.B.; Mcalister, D.R. Rapid fusion method for the determination of Pu, Np, and Am in large soil samples. J. Radioanal. Nucl. Chem. 2015, 305, 599–608. [Google Scholar] [CrossRef]
- Maxwell, S.; Culligan, B.; Kelsey-Wall, A.; Shaw, P. Rapid radiochemical method for determination of actinides in emergency concrete and brick samples. Anal. Chim. Acta 2011, 701, 112–118. [Google Scholar] [CrossRef]
- Reis, A.; Temba, E.; Kastner, G.; Monteiro, R. Combined procedure using radiochemical separation of plutonium, americium and uranium radionuclides for alpha-spectrometry. J. Radioanal. Nucl. Chem. 2011, 287, 567–572. [Google Scholar] [CrossRef]
- Sidhu, R. A robust procedure for the determination of plutonium and americium in seawater. J. Radioanal. Nucl. Chem. 2003, 256, 501–504. [Google Scholar] [CrossRef]
- Joshi, S.R. Lanthanum fluoride coprecipitation technique for the preparation of actinides for alpha-particle spectrometry. J. Radioanal. Nucl. Chem. 1985, 90, 409–414. [Google Scholar] [CrossRef]
- Rao, R.R.; Cooper, E.L. Separation of low levels of actinides by selective oxidation/reduction and co-precipitation with neodymium fluoride. J. Radioanal. Nucl. Chem. 1995, 197, 133–148. [Google Scholar] [CrossRef]
- Kimura, T. Simultaneous determination of neptunium, plutonium, americium and curium using coprecipitation with bismuth phosphate. J. Radioanal. Nucl. Chem. 1990, 139, 297–305. [Google Scholar] [CrossRef]
- Sajeniouk, A.D. Routine radiochemical method for the determination of 90Sr, 238Pu, 239 + 240Pu, 241Am and 244Cm in environmental samples. J. Radioanal. Nucl. Chem. 2005, 264, 337–342. [Google Scholar] [CrossRef]
- Crespo, M.T.; Gascón, J.L.; Aceña, M.L. Techniques and analytical methods in the determination of uranium, thorium, plutonium, americium and radium by adsorption on manganese dioxide. Sci. Total Environ. 1993, 130–131, 383–391. [Google Scholar] [CrossRef]
- Suarez-Navarro, J.; Pujol, L.; de Pablo, M. Rapid determination of gross alpha-activity in sea water by coprecipitation. J. Radioanal. Nucl. Chem. 2002, 253, 47–52. [Google Scholar] [CrossRef]
- Montaña, M.; Camacho, A.; Vallés, I.; Serrano, I. Experimental analysis of the mass efficiency curve for gross alpha activity and morphological study of the residue obtained by the co-precipitation method. Appl. Radiat. Isot. 2012, 70, 1541–1548. [Google Scholar] [CrossRef]
- Singhal, R.K.; Basu, H.; Reddy, A.V.R. Removal of environmental level of 239 + 240Pu and 241Am from groundwater by using humic coated colloidal suspension of goethite (α-FeO(OH)). J. Radioanal. Nucl. Chem. 2013, 295, 1345–1351. [Google Scholar] [CrossRef]
- Kikunaga, H.; Kasamatsu, Y.; Takamiya, K.; Ohtsuki, T.; Yuki, H.; Yokoyama, A.; Nakanishi, T.; Mitsugashira, T. Development of a rapid source preparation method for high-resolution α-particle spectrometry. Appl. Radiat. Isot. 2009, 67, 539–543. [Google Scholar] [CrossRef]
- Fryxell, G.E.; Lin, Y.; Fiskum, S.; Birnbaum, J.C.; Wu, H.; Kemner, K.; Kelly, S. Actinide Sequestration Using Self-Assembled Monolayers on Mesoporous Supports. Environ. Sci. Technol. 2005, 39, 1324–1331. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Fiskum, S.K.; Yantasee, W.; Wu, H.; Mattigod, S.V.; Vorpagel, E.; Fryxell, G.E.; Raymond, K.N.; Xu, J. Incorporation of Hydroxypyridinone Ligands into Self-Assembled Monolayers on Mesoporous Supports for Selective Actinide Sequestration. Environ. Sci. Technol. 2005, 39, 1332–1337. [Google Scholar] [CrossRef] [PubMed]
- Yantasee, W.; Sangvanich, T.; Creim, J.A.; Pattamakomsan, K.; Wiacek, R.J.; Fryxell, G.E.; Addleman, R.S.; Timchalk, C. Functional Sorbents for Selective Capture of Plutonium, Americium, Uranium, and Thorium in Blood. Health Phys. 2010, 99, 413–419. [Google Scholar] [CrossRef] [PubMed]
- Kaur, M.; Zhang, H.; Martin, L.; Todd, T.; Qiang, Y. Conjugates of Magnetic Nanoparticle—Actinide Specific Chelator for Radioactive Waste Separation. Environ. Sci. Technol. 2013, 47, 11942–11959. [Google Scholar] [CrossRef]
- Testa, C.; Desideri, D.; Guerra, F.; Meli, M.A.; Roselli, C.; Jia, G. The importance of separation chemistry for the determination of radionuclides in environmental samples. J. Radioanal. Nucl. Chem. 1998, 229, 23–32. [Google Scholar] [CrossRef]
- Seike, M.; Eguchi, M.; Shinohara, A.; Yoshimura, T. Solvent extraction behaviors of Americium(III) and Europium(III) using 2-hydroxy-2-methyloctanoic acid and 2-hydroxy-2-trifluoromethyloctanoic acid. J. Radioanal. Nucl. Chem. 2015, 303, 1413–1416. [Google Scholar] [CrossRef]
- Kolarik, Z.; Müllich, U.; Gassner, F. Selective extraction of Am(III)over Eu(III) by 2,6-ditriazolyl- and 2,6-ditriazinylpyridine. Solvent Extr. Ion Exch. 1999, 17, 23–32. [Google Scholar] [CrossRef]
- Panak, P.J.; Geist, A. Complexation and Extraction of Trivalent Actinides and Lanthanides by Triazinylpyridine N-Donor Ligands. Chem. Rev. 2013, 113, 1199–1236. [Google Scholar] [CrossRef]
- Xu, Y.; Kim, S.; Ito, T.; Hitomi, K.; Kuraoka, E.; Usuda, S.; Ishii, K. Adsorption behavior of trivalent americium and rare earth ions onto a macroporous silica-based isobutyl-BTP/SiO2-P adsorbent in nitric acid solution. J. Radioanal. Nucl. Chem. 2014, 299, 149–155. [Google Scholar] [CrossRef]
- Liu, R.; Wei, Y.; Xu, Y.; Usuda, S.; Kim, S.; Yamazaki, H.; Ishii, K. Evaluation study on properties of isohexyl-BTP/SiO2-P resin for direct separation of trivalent minor actinides from HLLW. J. Radioanal. Nucl. Chem. 2012, 292, 537–544. [Google Scholar] [CrossRef]
- Usuda, S.; Wei, Y.; Liu, R.; Li, Z.; Xu, Y.; Wu, Y.; Kim, S. Challenges to develop single-column MA(III) separation from HLLW using R-BTP type adsorbents. Sci. China Chem. 2012, 55, 1732–1738. [Google Scholar] [CrossRef]
- Usuda, S.; Wei, Y.; Xu, Y.; Li, Z.; Liu, R.; Kim, S.; Wakui, Y.; Hayashi, H.; Yamazaki, H. Development of a simplified separation process of trivalent minor actinides from fission products using novel R-BTP/SiO2-P adsorbents. J. Nucl. Sci. Technol. 2012, 49, 334–342. [Google Scholar] [CrossRef]
- Tevepaugh, K.N.; Coonce, J.; Tai, S.; Delmau, L.H.; Carrick, J.D.; Ensor, D.D. Chromatographic separation of americium from europium using bis-2,6-(5,6,7,8-tetrahydro-5,9,9-trimethyl-5,8-methano-1,2,4-benzotriazin-3-yl) pyridine. J. Radioanal. Nucl. Chem. 2017, 314, 371–376. [Google Scholar] [CrossRef]
- Tevepaugh, K.N.; Carrick, J.D.; Tai, S.; Coonce, J.G.; Delmau, L.H.; Ensor, D.D. Separation of Americium from Europium using Camphor-BisTriazinyl Pyridine: A Fundamental Study. Solvent Extr. Ion Exch. 2016, 34, 13–25. [Google Scholar] [CrossRef]
- Hill, T.G.; Chin, A.L.; Tai, S.; Carrick, J.D.; Ensor, D.D.; Delmau, L.H. Separation of americium from europium using 3,3′-dimethoxy-phenyl-bis-1,2,4-triazinyl-2,6-pyridine. Sep. Sci. Technol. 2018, 53, 1848–1855. [Google Scholar] [CrossRef]
- Mahmoud, J.; Higginson, M.; Thompson, P.; Gilligan, C.; Livens, F.; Heath, S.L. Rapid separation of americium from complex matrices using solvent impregnated triazine extraction chromatography resins. J. Chromatogr. A 2022, 1669, 462950. [Google Scholar] [CrossRef]
- Kiliari, T.; Pashalidis, I. Americium and samarium determination in aqueous solutions after separation by cation-exchange. J. Radioanal. Nucl. Chem. 2014, 299, 721–724. [Google Scholar] [CrossRef]
- Odintsov, A.A.; Pazukhin, E.M.; Khan, V.E. Procedure for Simultaneous Determination of Uranium and Transuranium Elements in Groundwater and Liquid Radioactive Wastes from the Shelter. Radiochemistry 2005, 47, 510–515. [Google Scholar] [CrossRef]
- Jia, G.; Desideri, D.; Guerra, F.; Meli, M.A.; Testa, C. Determination of plutonium and americium in moss and lichen samples. J. Radioanal. Nucl. Chem. 1997, 220, 15–19. [Google Scholar] [CrossRef]
- Desideri, D.; Meli, M.A.; Roselli, C.; Testa, C.; Boulyga, S.F.; Becker, J.S. Determination of 236U and transuranium elements in depleted uranium ammunition by α-spectrometry and ICP-MS. Anal. Bioanal. Chem. 2002, 374, 1091–1095. [Google Scholar] [CrossRef]
- Desideri, D.; Feduzi, L.; Meli, M.; Roselli, C. Sequential determination of Am, Cm, Pu, Np and U by extraction chromatography. Microchem. J. 2011, 97, 264–268. [Google Scholar] [CrossRef]
- Mohandas, J.; Srinivasa Rao, V.; Vijayakumar, N.; Kumar, T.; Velmurugan, S.; Narasimhan, S.V. Uptake of actinides by sulphonated phosphinic acid resin from acid medium. J. Radioanal. Nucl. Chem. 2014, 302, 1185–1188. [Google Scholar] [CrossRef]
- Horwitz, E.P.; Chiarizia, R.; Dietz, M.L.; Diamond, H.; Nelson, D.M. Separation and preconcentration of actinides from acidic media by extraction chromatography. Anal. Chim. Acta 1993, 281, 361–372. [Google Scholar] [CrossRef]
- Horwitz, E.P.; Dietz, M.L.; Chiarizia, R.; Diamond, H.; Maxwell, S.L.; Nelson, M.R. Separation and preconcentration of actinides by extraction chromatography using a supported liquid anion exchanger: Application to the characterization of high-level nuclear waste solutions. Anal. Chim. Acta 1995, 310, 63–78. [Google Scholar] [CrossRef]
- Horwitz, E.P.; Chiarizia, R.; Dietz, M.L. DIPEX: A new extraction chromatographic material for the separation and preconcentration of actinides from aqueous solution. React. Funct. Polym. 1997, 33, 25–36. [Google Scholar] [CrossRef]
- Macsik, Z.; Groska, J.; Vajda, N.; Vogt, S.; Kis-Benedek, G.; Kim, C.S.; Maddison, A.; Donohue, D. Improved radioanalytical method for the simultaneous determination of Th, U, Np, Pu and Am(Cm) on a single TRU column by alpha spectrometry and ICP-MS. Radiochim. Acta 2013, 101, 241–252. [Google Scholar] [CrossRef]
- Yi, X.W.; Li, D.M.; Dang, H.J.; Li, M.; Zhang, H.T. Separation of Transuranium Elements With UTEVA Extraction Chromatography. J. Nucl. Radiochem. 2010, 32, 22–26. [Google Scholar]
- Kazi, Z.H.; Cornett, J.R.; Zhao, X.; Kieser, L. Americium and plutonium separation by extraction chromatography for determination by accelerator mass spectrometry. Anal. Chim. Acta 2014, 829, 75–80. [Google Scholar] [CrossRef]
- Kazi, Z.H.; Cornett, R.J.; Zhao, X.; Kieser, W.E. Comparison of the measurement of Pu and Am isotopes by AMS using fluoride and oxide anion beams. J. Anal. Atom. Spectrom. 2015, 3, 2224–2235. [Google Scholar] [CrossRef]
- Xiao, G.; Saunders, D.; Jones, R.L.; Caldwell, K.L. Determination of 241Am in urine using sector field inductively coupled plasma mass spectrometry (SF-ICP-MS). J. Radioanal. Nucl. Chem. 2014, 301, 285–291. [Google Scholar] [CrossRef][Green Version]
- Chiariza, R.; Horwitz, E.P.; Alexandrators, S.D.; Gula, M.J. Diphonix® Resin: A Review of Its Properties and Applications. Sep. Sci. Technol. 1997, 32, 1–35. [Google Scholar] [CrossRef]
- Croudace, I.W.; Warwick, P.E.; Greenwood, R.C. A novel approach for the rapid decomposition of ActinideTM resin and its application to measurement of uranium and plutonium in natural waters. Anal. Chim. Acta 2006, 577, 111–118. [Google Scholar] [CrossRef] [PubMed]
- Dulanská, S.; Antalík, I.; Labaška, M.; Remenec, B.; Mátel, A. Rapid determination of 239,240Pu, 238Pu, 241Am and 90Sr in high contaminated samples waste using combined SPE sorbents AnaLig® Pu-02, AnaLig® Sr-01 and DGA® Resin. J. Radioanal. Nucl. Chem. 2013, 295, 1635–1639. [Google Scholar] [CrossRef]
- Dulanská, S.; Bilohuščin, J.; Remenec, B.; Galanda, D.; Mátel, L.U. Determination of 239Pu, 241Am and 90Sr in urine using pre-filter material and combined sorbents AnaLig® Pu-02, AnaLig® Sr-01, DGA® Resin. J. Radioanal. Nucl. Chem. 2015, 304, 127–132. [Google Scholar] [CrossRef]
- Maxwell, S.L.; Culligan, B.K. New column separation method for emergency urine samples. J. Radioanal. Nucl. Chem. 2009, 279, 105–111. [Google Scholar] [CrossRef][Green Version]
- Lee, M.H.; Jeon, Y.S.; Song, K. Determination of activity concentrations and activity ratios of plutonium, americium and curium isotopes in radioactive waste samples. J. Radioanal. Nucl. Chem. 2009, 280, 457–465. [Google Scholar] [CrossRef]
- Michel, H.; Levent, D.; Barci, V.; Barci-Funel, G.; Hurel, C. Soil and sediment sample analysis for the sequential determination of natural and anthropogenic radionuclides. Talanta 2008, 74, 1527–1533. [Google Scholar] [CrossRef]
- Luo, M.; Xing, S.; Yang, Y.; Song, L.; Ma, Y.; Wang, Y.; Dai, X.; Happel, S. Sequential analyses of actinides in large-size soil and sediment samples with total sample dissolution. J. Environ. Radioact. 2018, 187, 73–80. [Google Scholar] [CrossRef]
- Luo, M.; Liu, D.; Yang, Y.; Dai, X.; Wu, Y.; Shi, K. Simultaneous determination of actinides and 90Sr in large-size soil and sediment samples. J. Environ. Radioact. 2022, 247, 106854. [Google Scholar] [CrossRef]
- Grate, J.W.; Egorov, O.B.; Fiskum, S.K. Automated extraction chromatographic separations of actinides using separation-optimized sequential injection techniques. Analyst 1999, 124, 1143–1150. [Google Scholar] [CrossRef]
- Charlton, J.J.; Sepaniak, M.J.; Sides, A.K.; Schaaff, T.G.; Mann, D.K.; Bradshaw, J.A. The automation and optimization of solid phase extraction inductively coupled plasma mass spectrometry analysis for the high throughput determination of aqueous levels of U, Th, Np, Pu, and Am. J. Anal. Atom. Spectrom. 2013, 28, 711–718. [Google Scholar] [CrossRef]
- Guérin, N.; Nadeau, K.; Potvin, S.; Hardy, J.; Larivière, D. Automated pressurized injection system for the separation of actinides by extraction chromatography. J. Radioanal. Nucl. Chem. 2013, 295, 1803–1811. [Google Scholar] [CrossRef]
- Qiao, J. Dynamic Flow Approaches for Automated Radiochemical Analysis in Environmental, Nuclear and Medical Applications. Molecules 2020, 25, 1462. [Google Scholar] [CrossRef] [PubMed]
- Truscott, J.B.; Jones, P.; Fairman, B.E.; Evans, E.H. Determination of actinides in environmental and biological samples using high-performance chelation ion chromatography coupled to sector-field inductively coupled plasma mass spectrometry. J. Chromatogr. A 2001, 928, 91–98. [Google Scholar] [CrossRef]
- Peterson, D.S.; Montoya, V.M. Separation of Actinides Using Capillary Extraction Chromatography-Inductively Coupled Plasma Mass Spectrometry. J. Chromatogr. Sci. 2009, 47, 545–548. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kim, H.; Chung, K.H.; Jung, Y.; Jang, M.; Kang, M.J.; Choi, G. A rapid and efficient automated method for the sequential separation of plutonium and radiostrontium in seawater. J. Radioanal. Nucl. Chem. 2015, 304, 321–327. [Google Scholar] [CrossRef]
- Wang, W.; Evans, R.D.; Evans, H.E. A rapid, automated system for the separation, preconcentration and measurement of 90Sr, and U, Am and Pu isotopes. Talanta 2021, 233, 122507. [Google Scholar] [CrossRef]
- Higginson, M.; Palmer, K.; King, J.; Dawkins, B.; Huggins, T.; Ingman, L.; Taylor, F.; Xu, N.; Kaye, P. Development of automated separations for actinides analysis. J. Radioanal. Nucl. Chem. 2019, 320, 689–698. [Google Scholar] [CrossRef]
- Egorov, O.; O’Hara, M.; Grate, J.; Ruzicka, J. Sequential injection renewable separation column instrument for automated sorbent extraction separations of radionuclides. Anal. Chem. 1999, 71, 345–352. [Google Scholar] [CrossRef]
- Grate, J.W.; Egorov, O.B. Investigation and Optimization of On-Column Redox Reactions in the Sorbent Extraction Separation of Americium and Plutonium Using Flow Injection Analysis. Anal. Chem. 1998, 70, 3920–3929. [Google Scholar] [CrossRef]
- Egorov, O.B.; Grate, J.W.; O’Hara, M.J.; Farmer, O.T., III. Extraction chromatographic separations and analysis of actinides using sequential injection techniques with on-line inductively coupled plasma mass spectrometry (ICP MS) detection. Analyst 2001, 126, 1594–1601. [Google Scholar] [CrossRef]
- Mirashi, N.N.; Aggarwal, S.K. Studies for simultaneous quantitative electrodeposition of plutonium and americium for alpha-spectrometry. J. Radioanal. Nucl. Chem. 2009, 279, 777–781. [Google Scholar] [CrossRef]
- Irlweck, K. Effects of Fe(III) ions on the electrodeposition of trace amounts of plutonium and americium. J. Radioanal. Nucl. Chem. 1996, 214, 429–437. [Google Scholar] [CrossRef]
- Bajo, S.; Eikenberg, J. Electrodeposition of actinides for alpha-spectrometry. J. Radioanal. Nucl. Chem. 1999, 242, 745–751. [Google Scholar] [CrossRef]
- Tran, Q.; Pierre, S.; de Sanoit, J.; Pomorski, M.; Bergonzo, P. Electro-Precipitation of Actinides on Boron-Doped Diamond Thin Films for Solid Sources Preparation for High-Resolution Alpha-Particle Spectrometry. Appl. Sci. 2019, 9, 1473. [Google Scholar] [CrossRef]
- Glover, S.E.; Filby, R.H.; Clark, S.B.; Grytdal, S.P. Optimization and characterization of a sulfate based electrodeposition method for alpha-spectroscopy of actinide elements using chemometric analysis. J. Radioanal. Nucl. Chem. 1998, 234, 213–220. [Google Scholar] [CrossRef]
- Janda, J.; Sládek, P.; Sas, D. Electrodeposition of selected alpha-emitting radionuclides from oxalate-ammonium sulfate electrolyte and measured by means of solid-state alpha spectrometry. J. Radioanal. Nucl. Chem. 2010, 286, 687–691. [Google Scholar] [CrossRef]
- Lee, K.B.; Man Lee, J.; Soon Park, T.; Oh, P. Preparation and activity measurement of electrodeposited alpha-emitting sources. Appl. Radiat. Isot. 2006, 64, 1260–1264. [Google Scholar] [CrossRef]
- Oh, J.; Warwick, P.E.; Croudace, I.W.; Lee, S. Evaluation of three electrodeposition procedures for uranium, plutonium and americium. Appl. Radiat. Isot. 2014, 87, 233–237. [Google Scholar] [CrossRef]
- Hindman, F.D. Actinide separations for α spectrometry using neodymium fluoride coprecipitation. Anal. Chem. 1986, 58, 1238–1241. [Google Scholar] [CrossRef]
- Sill, C.W. Precipitation of actinides as fluorides or hydroxides for high-resolution alpha spectrometry. Nucl. Chem. Waste Manag. 1987, 7, 201–215. [Google Scholar] [CrossRef]
- Lozano, J.C.; Fernandez, F.; Gomez, J.M.G. Preparation of Alpha-spectrometric sources by coprecipitation with Fe(OH)3: Application to actinides. Appl. Radiat. Isot. 1997, 48, 383–389. [Google Scholar] [CrossRef]
- Dion, M.P.; Liezers, M.; Farmer, O.T.; Miller, B.W.; Morley, S.; Barinaga, C.; Eiden, G. Improving alpha spectrometry energy resolution by ion implantation with ICP-MS. J. Radioanal. Nucl. Chem. 2015, 303, 877–884. [Google Scholar] [CrossRef]
- Quinto, F.; Blechschmidt, I.; Garcia Perez, C.; Geckeis, H.; Geyer, F.; Golser, R.; Huber, F.; Lagos, M.; Lanyon, B.; Plaschke, M.; et al. Multiactinide Analysis with Accelerator Mass Spectrometry for Ultratrace Determination in Small Samples: Application to an in Situ Radionuclide Tracer Test within the Colloid Formation and Migration Experiment at the Grimsel Test Site (Switzerland). Anal. Chem. 2017, 89, 7182–7189. [Google Scholar] [CrossRef]
- Dai, X.; Christl, M.; Sheila, K.; Synal, H. Determination of Atto- to Femtogram Levels of Americium and Curium Isotopes in Large-Volume Urine Samples by Compact Accelerator Mass Spectrometry. Anal. Chem. 2016, 88, 2832–2837. [Google Scholar] [CrossRef]
- Aggarwal, S.K.; Duggal, R.K.; Jain, H.C. Alpha spectrum evaluation method for the simultaneous determination of plutonium, americium and curium. J. Radioanal. Nucl. Chem. 1986, 107, 263–277. [Google Scholar] [CrossRef]
- Aggarwal, S.K.; Alamelu, D.; Shah, P.M.; Mirashi, N.N. Determination of 241Am/243Am ratios. J. Radioanal. Nucl. Chem. 2007, 273, 771–774. [Google Scholar] [CrossRef]
- Bortels, G.; Collaers, P. Analytical function for fitting peaks in alpha-particle spectra from Si detectors. Int. J. Radiat. Appl. Instrum. Part A Appl. Radiat. Isot. 1987, 38, 831–837. [Google Scholar] [CrossRef]
- Devol, T.; Ringberg, A.; Dewberry, R. Isotopic analysis of plutonium using a combination of alpha and internal conversion electron spectroscopy. J. Radioanal. Nucl. Chem. 2002, 254, 71–79. [Google Scholar] [CrossRef]
- Afonin, M.; Simonoff, M.; Donard, O.; Michel, H.; Ardisson, G. Pu and Am determination in the environment—Method development. Czechoslov. J. Phys. 2003, 53, A15–A23. [Google Scholar] [CrossRef]
- Varga, Z.; Surányi, G.; Vajda, N.; Stefánka, Z. Determination of plutonium and americium in environmental samples by inductively coupled plasma sector field mass spectrometry and alpha spectrometry. Microchem. J. 2007, 85, 39–45. [Google Scholar] [CrossRef]
- Becker, J.S. Inductively coupled plasma mass spectrometry (ICP-MS) and laser ablation ICP-MS for isotope analysis of long-lived radionuclides. Int. J. Mass Spectrom. 2005, 242, 183–195. [Google Scholar] [CrossRef]
- Chartier, F.; Aubert, M.; Pilier, M. Determination of Am and Cm in spent nuclear fuels by isotope dilution inductively coupled plasma mass spectrometry and isotope dilution thermal ionization mass spectrometry after separation by high-performance liquid chromatography. Fresenius’ J. Anal. Chem. 1999, 364, 320–327. [Google Scholar] [CrossRef]
- Wang, Z.; Zheng, J.; Cao, L.; Tagami, K.; Uchida, S. Method for Ultratrace Level 241Am Determination in Large Soil Samples by Sector Field-Inductively Coupled Plasma Mass Spectrometry: With Emphasis on the Removal of Spectral Interferences and Matrix Effect. Anal. Chem. 2016, 88, 7387–7394. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; Yamada, M. Isotope Dilution Sector-Field Inductively Coupled Plasma Mass Spectrometry Combined with Extraction Chromatography for Rapid Determination of 241Am in Marine Sediment Samples: A Case Study in Sagami Bay, Japan. J. Oceanogr. 2008, 64, 541–550. [Google Scholar] [CrossRef]
- Zhang, W.; Lin, J.; Zhang, H.; Fang, S.; Li, C.; Yi, X.; Dang, H.; Xu, Y.; Wang, W.; Xu, J. Determination of ultra-trace level 241Am in soil by triple-quadrupole inductively coupled plasma-mass spectrometry with mass-shift mode combined with chemical separation. J. Anal. Atom. Spectrom. 2022, 37, 1044–1052. [Google Scholar] [CrossRef]
- Goldstein, S.J.; Hinrichs, K.A.; Nunn, A.J.; Gurganus, D.W.; Amato, R.S.; Oldham, W.J. Sequential chemical separations and multiple ion counting ICP-MS for 241Pu—241Am—237Np dating of environmental collections on a single aliquot. J. Radioanal. Nucl. Chem. 2018, 318, 695–701. [Google Scholar] [CrossRef]
- Hang, W.; Zhu, L.; Zhong, W.; Mahan, C. Separation of actinides at ultra-trace level from urine matrix using extraction chromatography-inductively coupled plasma mass spectrometry. J. Anal. Atom. Spectrom. 2004, 19, 966–972. [Google Scholar] [CrossRef]
- Epov, V.N. Comparative study of three sample preparation approaches for the fast determination of americium in urine by flow injection ICP-MS. Can. J. Anal. Sci. Spectrosc. 2005, 50, 14–22. [Google Scholar]
- Guerin, N.; Calmette, R.; Johnson, T.; Lariviere, D. Multi-dimensional extraction chromatography of actinides for alpha and mass spectrometry. Anal. Methods-UK 2011, 3, 1560–1567. [Google Scholar] [CrossRef]
- Agarande, M.; Benzoubir, S.; Bouisset, P.; Calmet, D. Determination of 241Am in sediments by isotope dilution high resolution inductively coupled plasma mass spectrometry (ID HR ICP-MS). Appl. Radiat. Isot. 2001, 55, 161–165. [Google Scholar] [CrossRef]
- Varga, Z. Application of inductively coupled plasma sector field mass spectrometry for low-level environmental americium-241 analysis. Anal. Chim. Acta 2007, 587, 165–169. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Xie, Y.; Lin, J.; Li, Z.; Tan, Z. Rapid method for sequential determination of Pu and Am in soil and sediment samples by sector-field inductively coupled plasma mass spectrometry. J. Radioanal. Nucl. Chem. 2021, 328, 137–147. [Google Scholar] [CrossRef]
- Habibi, A.; Vivien, C.; Boulet, B.; Cossonnet, C.; Gurriaran, R.; Gleizes, M.; Cote, G.; Larivière, D. A rapid sequential separation of actinides and radiostrontium coupled to ICP-MS and gas proportional counting. J. Radioanal. Nucl. Chem. 2016, 310, 217–227. [Google Scholar] [CrossRef]
- Boulyga, S.F. Mass spectrometric analysis of long-lived radionuclides in bio-assays. Int. J. Mass Spectrom. 2011, 307, 200–210. [Google Scholar] [CrossRef]
- Mathew, K.J.; Ottenfeld, C.F.; Keller, R.C.; Kuhn, K.J.; Fulwyler, J.B. Preparation of 241Am/243Am gravimetric mixtures and development of Am isotopic and assay measurement techniques using thermal ionization mass spectrometry. Int. J. Mass Spectrom. 2020, 458, 116430. [Google Scholar] [CrossRef]
- Quemet, A.; Ruas, A.; Dalier, V.; Rivier, C. Americium isotope analysis by Thermal Ionization Mass Spectrometry using the total evaporation method. Int. J. Mass Spectrom. 2018, 431, 8–14. [Google Scholar] [CrossRef]
- Quemet, A.; Angenieux, M.; Ruas, A. Nd, Am and Cm isotopic measurements after simultaneous separation in transmutation irradiated samples. J. Anal. Atom. Spectrom. 2021, 36, 1758–1767. [Google Scholar] [CrossRef]
- Bürger, S.; Riciputi, L.R.; Bostick, D.A.; Turgeon, S.; Mcbay, E.H.; Lavelle, M. Isotope ratio analysis of actinides, fission products, and geolocators by high-efficiency multi-collector thermal ionization mass spectrometry. Int. J. Mass Spectrom. 2009, 286, 70–82. [Google Scholar] [CrossRef]
- Quinto, F.; Golser, R.; Lagos, M.; Plaschke, M.; Schäfer, T.; Steier, P.; Geckeis, H. Accelerator Mass Spectrometry of Actinides in Ground—and Seawater: An Innovative Method Allowing for the Simultaneous Analysis of U, Np, Pu, Am, and Cm Isotopes below ppq Levels. Anal. Chem. 2015, 87, 5766–5773. [Google Scholar] [CrossRef]
- Cornett, R.J.; Kazi, Z.H.; Zhao, X.L.; Chartrand, M.G.; Charles, R.J.; Kieser, W.E. Actinide measurements by AMS using fluoride matrices. Nucl. Instrum. Methods Phys. Res. Sect. B Beam Interact. Mater. At. 2015, 361, 317–321. [Google Scholar] [CrossRef]
- Christl, M.; Dai, X.; Lachner, J.; Kramer-Tremblay, S.; Synal, H. Low energy AMS of americium and curium. Nucl. Instrum. Methods Phys. Res. Sect. B Beam Interact. Mater. At. 2014, 331, 225–232. [Google Scholar] [CrossRef]
- Srncik, M.; Hrnecek, E.; Steier, P.; Wallner, G. Determination of U, Pu and Am isotopes in Irish Sea sediment by a combination of AMS and radiometric methods. J. Environ. Radioact. 2011, 102, 331–335. [Google Scholar] [CrossRef]
- Agapkina, G.I.; Tikhomirov, F.A.; Shcheglov, A.I.; Kracke, W.; Bunzl, K. Association of Chernobyl-derived 239+240Pu, 241Am, 90Sr and 137Cs with organic matter in the soil solution. J. Environ. Radioact. 1995, 29, 257–269. [Google Scholar] [CrossRef]
- Novikov, A.P. Migration and concentration of artificial radionuclides in environmental objects. Geochem. Int. 2010, 48, 1263–1387. [Google Scholar] [CrossRef]
- Novikov, A.P.; Tkachev, V.V.; Myasoedov, B.F. Speciation methods of actinides in trace concentrations. C. R. Chim. 2004, 7, 1219–1225. [Google Scholar] [CrossRef]
- Altmaier, M.; Gaona, X.; Fanghänel, T. Recent Advances in Aqueous Actinide Chemistry and Thermodynamics. Chem. Rev. 2013, 113, 901–943. [Google Scholar] [CrossRef]
- Guo, H.; Kang, M.L.; Chen, W.L.; Long, J.C.; Zhao, Z. Speciation and solubility of americium in Beishan groundwater. Radiat. Prot. 2016, 36, 40–46. [Google Scholar]
- Ikeda-Ohno, A.; Harrison, J.J.; Thiruvoth, S.; Wilsher, K.; Wong, H.K.Y.; Johansen, M.P.; Waite, T.D.; Payne, T.E. Solution Speciation of Plutonium and Americium at an Australian Legacy Radioactive Waste Disposal Site. Environ. Sci. Technol. 2014, 48, 10045–10053. [Google Scholar] [CrossRef]
- Salbu, B. Speciation of radionuclides—Analytical challenges within environmental impact and risk assessments. J. Environ. Radioact. 2007, 96, 47–53. [Google Scholar] [CrossRef]
- León Vintró, L.; Mitchell, P.I.; Omarova, A.; Burkitbayev, M.; Jiménez Nápoles, H.; Priest, N.D. Americium, plutonium and uranium contamination and speciation in well waters, streams and atomic lakes in the Sarzhal region of the Semipalatinsk Nuclear Test Site, Kazakhstan. J. Environ. Radioact. 2009, 100, 308–314. [Google Scholar] [CrossRef] [PubMed]
- Vangenechten, J.H.D.; Chughtai, N.A.; Bierkens, J.; Vanderborght, O.L.J. Similarity of 241Am and 59Fe specification in selected freshwaters and of their adsorption on crayfish exoskeleton. J. Environ. Radioact. 1987, 5, 275–286. [Google Scholar] [CrossRef]
- Molero, J.; Sanchez-Cabeza, J.A.; Merino, J.; Batlle, J.V.; Mitchell, P.I.; Vidal-Quadras, A. Particulate distribution of plutonium and americium in surface waters from the Spanish Mediterranean coast. J. Environ. Radioact. 1995, 28, 271–283. [Google Scholar] [CrossRef]
- Tessier, A.; Campbell, P.G.C.; Bisson, M. Sequential extraction procedure for the speciation of particulate trace metals. Anal. Chem. 1979, 51, 844–851. [Google Scholar] [CrossRef]
- Szabó, G.; Guczi, J.; Nisbet, A. Investigation of the solid phase speciation of 90Sr, 137Cs, 239Pu and 241Am in soils determined by extraction and ultra-filtration methods. J. Radioanal. Nucl. Chem. 1997, 226, 255–259. [Google Scholar] [CrossRef]
- Bolsunovsky, A.; Bondareva, L. Actinides and other radionuclides in sediments and submerged plants of the Yenisei River. J. Alloys Compd. 2007, 444, 495–499. [Google Scholar] [CrossRef]
- Alberts, J.J.; Wahlgren, M.A.; Orlandini, K.A.; Durbahn, C.A. The distributions of 239,240Pu, 238Pu, 241Am and 137Cs among chemically-defined components of sediments, settling particulates and net plankton of Lake Michigan. J. Environ. Radioact. 1989, 9, 89–103. [Google Scholar] [CrossRef]
- Ibrahim, S.A.; Salazar, W.R. Physicochemical Speciation of Americium in Soils from Rocky Flats, Colorado, USA. J. Radioanal. Nucl. Chem. 2000, 243, 347–351. [Google Scholar] [CrossRef]
- álvarez, A.; Gascó, C.; Navarro, N.; Antón, M.; Sancho, C. Determination of actinides in samples obtained during dismantling activities. J. Radioanal. Nucl. Chem. 2005, 265, 383–387. [Google Scholar] [CrossRef]
- Lujanienė, G.; Aninkevičius, V.; Lujanas, V. Artificial radionuclides in the atmosphere over Lithuania. J. Environ. Radioact. 2009, 100, 108–119. [Google Scholar] [CrossRef]
- Shi, Y.M.; Wang, X.H.; Zhou, G.Q.; Zhang, H.T.; Xie, J.C.; Li, M. Existing Form of 241Am in Sandy Soil. At. Energy Sci. Technol. 2012, 46, 263–267. [Google Scholar]
- Desideri, D.; Meli, M.; Roselli, C.; Testa, C.; Degetto, S. Speciation of natural and antropogenic radionuclides in different sea sediment samples. J. Radioanal. Nucl. Chem. 2001, 248, 727–733. [Google Scholar] [CrossRef]
- Lujanienė, G.; Beneš, P.; Atamberg, K.; Jokšas, K.; Kulakauskaitė, I. Pu and Am sorption to the Baltic Sea bottom sediments. J. Radioanal. Nucl. Chem. 2013, 295, 1957–1967. [Google Scholar] [CrossRef]
- Ketterer, M.E.; Hafer, K.M.; Jones, V.J.; Appleby, P.G. Rapid dating of recent sediments in Loch Ness: Inductively coupled plasma mass spectrometric measurements of global fallout plutonium. Sci. Total Environ. 2004, 322, 221–229. [Google Scholar] [CrossRef] [PubMed]
- Arnaud, F.; Magand, O.; Chapron, E.; Bertrand, S.; Boës, X.; Charlet, F.; Mélières, M.A. Radionuclide dating (210Pb, 137Cs, 241Am) of recent lake sediments in a highly active geodynamic setting (Lakes Puyehue and Icalma—Chilean Lake District). Sci. Total Environ. 2006, 366, 837–850. [Google Scholar] [CrossRef]
- Appleby, P.G. Radiometric dating of sediment records in European mountain lakes. J. Limnol. 2000, 59, 1–14. [Google Scholar] [CrossRef]
- Kuzmenkova, N.V.; Ivanov, M.M.; Alexandrin, M.Y.; Grachev, A.M.; Rozhkova, A.K.; Zhizhin, K.D.; Grabenko, E.A.; Golosov, V.N. Use of natural and artificial radionuclides to determine the sedimentation rates in two North Caucasus lakes. Environ. Pollut. 2020, 262, 114269. [Google Scholar] [CrossRef]
- Appleby, P.G.; Richardson, N.; Nolan, P.J. 241Am dating of lake sediments. Hydrobiologia 1991, 214, 35–42. [Google Scholar] [CrossRef]
- Mayer, K.; Wallenius, M.; Varga, Z. Nuclear Forensic Science: Correlating Measurable Material Parameters to the History of Nuclear Material. Chem. Rev. 2013, 113, 884–900. [Google Scholar] [CrossRef]
- Keegan, R.P.; Gehrke, R.J. A method to determine the time since last purification of weapons grade plutonium. Appl. Radiat. Isot. 2003, 59, 137–143. [Google Scholar] [CrossRef]
- Archer, D.E.; Luke, S.J.; Parker, W. Pu300: A Tool for Measurement of Plutonium Age for Arms Control Transparency via Gamma-Ray Spectroscopy[R]; UCRL-JC-136626; Lawrence Livermore National Lab. (LLNL): Livermore, CA, USA, 2000. [Google Scholar]
- Wallenius, M.; Mayer, K. Age determination of plutonium material in nuclear forensics by thermal ionisation mass spectrometry. Fresenius’ J. Anal. Chem. 2000, 366, 234–238. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Zhu, F.R.; Xu, J.; Dai, Y.H.; Li, D.M.; Yi, X.W.; Zhang, L.X.; Zhao, Y.G. Age determination of plutonium material by alpha spectrometry and thermal ionization mass spectrometry. Radiochim. Acta 2008, 96, 327–331. [Google Scholar] [CrossRef]
- Chen, Y.; Chang, Z.; Zhao, Y.; Zhang, J.; Li, J.; Shu, F. Studies on the age determination of trace plutonium. J. Radioanal. Nucl. Chem. 2009, 281, 675–678. [Google Scholar] [CrossRef]
- Eriksson, M.; Lindahl, P.; Roos, P.; Dahlgaard, H.; Holm, E. U, Pu, and Am Nuclear Signatures of the Thule Hydrogen Bomb Debris. Environ. Sci. Technol. 2008, 42, 4717–4722. [Google Scholar] [CrossRef]
- Aarkrog, A.; Dahlgaard, H.; Nilsson, K.; Holm, E. Further studies of plutonium and americium at Thule, Greenland. Health Phys. 1984, 46, 29–44. [Google Scholar] [CrossRef]
- Varga, Z.; Nicholl, A.; Zsigrai, J.; Wallenius, M.; Mayer, K. Methodology for the Preparation and Validation of Plutonium Age Dating Materials. Anal. Chem. 2018, 90, 4019–4024. [Google Scholar] [CrossRef]
- Pittet, P.; Josset, M.; Boilley, D.; Bernollin, A.; Rougier, G.; Froidevaux, P. Origin and age of an ongoing radioactive contamination of soils near La hague reprocessing plant based on 239 + 240Pu/238Pu and 241Am/241Pu current ratios and 90Sr and Ln(III) soil contents. Chemosphere 2021, 270, 129332. [Google Scholar] [CrossRef]
- Bouisset, P.; Nohl, M.; Cossonnet, C.; Boulet, B.; Thomas, S.; Cariou, N.; Salaun, G. Contribution of close-in fallout from the French atmospheric tests in inventories of 137Cs, 241Am and plutonium (238, 239, 240) in Gambier Islands (French Polynesia)—Signatures of stratospheric fallout in the Southern Hemisphere. J. Environ. Radioact. 2021, 235–236, 106624. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Source | The Activity of 241Am | The Activity of 241Pu | Release Period |
|---|---|---|---|
| Atmospheric nuclear weapons testing | 13 PBq | 142 PBq [10] | 1945–1980 |
| Reprocessing operations at Sellafield | 542 TBq | 22 PBq | 1952–1992 |
| 890 TBq [15] | - | up to 1990 | |
| Reprocessing operations at La Hague | - | 12 PBq [14] | 1967–1995 |
| 310 GBq | 21.8 TBq | 1995–1999 | |
| Aircraft accident in Thule, 1968 | 0.20 TBq [16,17] | 4.6 TBq [16,17] | 2002 |
| Aircraft accident in Palomares, 1966 | 0.1 TBq [18] | - | 1966 |
| Nuclear power plant accident in Chernobyl, 1986 | 0.99 MBq | 6 PBq [10] | 1986 |
| Nuclear power plant accident in Fukushima, 2011 | 89 MBq [19] | 14 GBq [19] | 2013 |
| Location | Sample Type | Concentration of 241Am | Reference |
|---|---|---|---|
| France | cultivated soil (0–20 cm) | (45 ± 7) × 10−3 Bq/kg | [20] |
| Fukushima Dai-ichi NPP, Japan | surface soil (0–2 cm) | 0.01–2.44 Bq/kg | [21] |
| litter | 0.012–1.64 Bq/kg | ||
| Vilnius, Lithuania | aerosol | 1–24.9 nBq/m3 | [22] |
| New Mexico, USA | soil (0–2 cm) in the vicinity of the USA Waste Isolation Pilot Plant | 0.003–0.067 Bq/kg | [23] |
| Bulgaria | surface soil (0–5 cm) | 0.019–0.302 Bq/kg | [24] |
| China | forest, grassland, and desert soil cores | 0.13–0.37 Bq/kg | [25] |
| Seven locations, China | soil (0–5 cm) collected from Hebei, Henan, Shandong, Inner Mongolia, Xinjiang, Sichuan, and Guangdong | 0.041–0.221 Bq/kg | [26] |
| Prague, Czech | soils (0–5 cm) around nuclear research center | 0.12 Bq/kg | [27] |
| Canadian Arctic and Alaskan tundra | lichens and mosses | 0.50 Bq/kg | [28] |
| Italy | mosses | 0.180–0.770 Bq/kg | [29] |
| lichens | 0.200–1.93 Bq/kg | ||
| Peninsular Malaysia, east coast | surface seawater | 0.5–1.9 mBq/m3 | [30] |
| Mururoa and Fangataufa Atolls, French Polynesia | groundwater | ≤0.008 Bq/L | [31] |
| Northwest Pacific Ocean | bottom sediments | 0.44–10 Bq/kg | [32] |
| Aegean Turkish coast | marine sediment | 0.003–0.33 Bq/kg | [33] |
| Black Sea coast | sediment | 0.043–0.187 Bq/kg | [34] |
| Irish Sea | sediment | 2.61–1894 Bq/kg | [35] |
| Ligurian Sea | sediment | 0.09–0.14 Bq/kg | [36] |
| Species | Transfer Factors /m2·g−1 | Remarks |
|---|---|---|
| Rice | 2.5 × 10−3 | In France [20] |
| Cereal grains | 1.5 × 10−7 to 7.7 × 10−1 | IAEA-recommended |
| Cowberry, stems and leaves | 5 × 10−4 | In Finland [43] |
| Billberry, stems and leaves | 2 × 10−4 | |
| Billberry, berries | 9 × 10−5 | |
| Lingonberry, stems and leaves | 4 × 10−4 | |
| Lingonberry, berries | 1 × 10−4 | |
| Elytrigiarepens | 1.4 × 10−7 | Contaminated regions in Belarus after Chernobyl accident [42] |
| Gramineae | 1.0 × 10−6 | |
| Carex | 2.9 × 10−6 | |
| Conium | 6.0 × 10−7 | |
| Rhinansus | 3.4 × 10−7 | |
| Moss | 4.0 × 10−6 | |
| Circiumarvens | 1.9 × 10−6 | |
| Poapratensis | 1.0 × 10−6 | |
| Leafy vegetables | 3.6 × 10−6–3.5 × 10−5 | [44] |
| Edible part of non-leafy vegetables | 9.0 × 10−5–1.0 × 10−4 | |
| Tubers | 8.4 × 10−6–1.3 × 10−5 | |
| Root crops | 6.9 × 10−6–4.0 × 10−5 |
| Reagent | Recovery/% | Function | Reference |
|---|---|---|---|
| Ferric hydroxide | >92 | Pre-concentration | [54] |
| Ferrous hydroxides | >93 | Pre-concentration | [7] |
| Lanthanide fluorides (NdF3 and CeF3) | >95 | Pre-concentration, α source preparation | [50,55,56] |
| Calcium phosphate | >95 | Pre-concentration | [50] |
| Bismuth phosphate | >95 | Pre-concentration | [57] |
| Lanthanide hydroxide | >95 | Pre-concentration, α source preparation | [58] |
| Manganese dioxide | >95 | Pre-concentration | [54,59] |
| Mix of ferric hydroxides and barium sulfate | >95 | Pre-concentration | [60,61] |
| Goethite (α-FeO(OH)) | >95 | Pre-concentration | [62] |
| Sm hydroxide | 92.7 | α source preparation | [63] |
| Name | Extractant | Supporter | Characteristics | Application | Remarks | Literature |
|---|---|---|---|---|---|---|
| TRU | CMPO-TBP | Amberlite XAD-7 | Am(III) retained on the resin; separated Am from tri, tetra, and hexavalent actinides | Determination of Th, U, Np, Pu, and Am(Cm) in sediment and swipe samples | Fe(III) retained on resin and competed with Am | [89] |
| TEVA | Aliquat 336 | Amberchrom CG-7ms | An analogue to anion-exchange resin; retained tetravalent actinides, but Am(III) was only slightly retained from nitric or hydrochloric solutions; Am(III) was retained effectively as Am(SCN)4− | Separation of Am(III) from lanthanides | A good choice for separating Am(III) from lanthanides | [87] |
| UTEVA | dipentyl-pentyl phosphonate | Amberlite XAD-7 | Retained tetra- and hexavalent actinides; Am(III) not retained from nitric solutions | Separation of Am-Pu fraction from U-Th fraction | Am and lanthanides flowed through the UTEVA resin, while Pu(IV) and U retained on UTEVA resin | [90] |
| DGA | N,N,N′,N′ tetraoctyldiglycolamide | Amberchrom CG-71 | DGA had very strong affinity to Am; the distribution coefficient for Am was higher than 104in HNO3(≥1M) | Separation of Pu and Am using single resin column | DGA resin could be used for quantitative separation of Am from various matrices | [91,92,93] |
| DIPEX | bis(2-ethylhexyl)methane diphosphonic acid | inert polymeric | DIPEX resin exhibited very strong affinity for actinides, including trivalent actinides | Pre-concentration of 241Am | The use of this resins was significantly limited mainly due to difficulties in recovering 241Am from the resin | [88] |
| DIPHONIX | geminally substituted diphosphonic acid ligands | styrene-based polymeric matrix | DIPHONIX resin exhibited very strong affinity for actinides | Pre-concentration of 241Am | The use of this resins was significantly limited mainly due to difficulties in recovering 241Am from the resin | [94,95] |
| No | Sample | Pre-Treatment | Pre-Concentration | Chemical Separation | Source Preparation | Chemical Yield | Reference |
|---|---|---|---|---|---|---|---|
| Matrix (Amount) | |||||||
| 1 | IAEA414 | 16 M nitric acid digestion | Iron oxide | DGA resin | NdF3 microprecipitates | [92] | |
| 2 | Soil sample (10 g) | Sodium hydroxide fusion | Iron hydroxide precipitate | TEVA and DGA resin, less than 8 h | CeF3 microprecipitates | 89.2% | [51] |
| 3 | Liquid waste | Evaporated to dryness; acid digestion | Oxlate acid | Pu: Dowex1 × 8 resin Am: TRU resin | Electrodeposition of H2SO4-(NH4)2SO4 | Not given | [53] |
| 4 | Low-level liquid radioactive waste | Evaporated to dryness; acid digestion | Coprecipitation on iron(II) hydroxide and calcium oxalate precipitate | Pu, Np, and U: UTEVA Am: TRU resin | NdF3 microprecipitates | 55% | [7] |
| 5 | Soil samples (10–15 g) | Acid total dissolution with HCl, HNO3, HF, and HClO4 | Leachate was filtered | Pu: AG1 × 8r resin U: UTEVA resin purification 241Am: TRU resin Separation of americium from lanthanides: TEVA resin | Electrodeposition | 85.5% | [23] |
| 6 | Radioactive sludge from nuclear power plant | Concentrated HNO3 | Single multi-stage column Pu: AnaLig@ Pu-02 resin Sr: AnaLig@ Sr-01 resin Am: DGA resin | NdF3 microprecipitates | >90% | [96] | |
| 8 | Urine (25–300 mL) | Added HNO3 | Loading after pre-filtering | Pu: AnaLig@ Pu-02 resin Sr: AnaLig@ Sr-01 resin Am: DGA resin | NdF3 microprecipitates | 25 mL:98% 300 mL: 41% | [97] |
| 9 | Urine | Concentrated HNO3 and 2 M Al(NO3)3 added to adjust the acidity of each sample | Calcium phosphate precipitation | Sr: Sr resin Am: TEVA and TRU resin | CeF3 microprecipitates | Nearly 100% | [98] |
| 10 | Liquid waste (10 mL) | Leaching with HNO3 | Pu: anion-exchange resin Am: TRU and anion-exchange resin | Microprecipitation and electrodeposition | 77–86% | [99] | |
| 11 | Soil and sediment | Leaching with HNO3 and HCl; total acid digestion with HNO3, HCl, and HF; microwave digestion with HNO3 and HF | Calcium oxalate precipitation | Sr: Sr-spec@ resin U: UTEVA resin Separation of Pu from Am: AG1X8 resin Am: mixed anion- and cation-exchange and TRU resin | Electrodeposition of H2SO4-NaHSO4 | 65–85% | [100] |
| 12 | Large-sized soil and sediment samples | Lithium metaborate fusion | Iron hydroxide precipitation and CeF3 coprecipitation | Pu: AGMP-1M and TEVA resin Am and Cm: DGA, AGMP-1M, and TEVA resin | CeF3 microprecipitation | 91% | [101] |
| 13 | Large-sized soil and sediment samples | Ashing; acid digestion | Iron hydroxide precipitation, CeF3 coprecipitation, and fluoride coprecipitation | Pu: AGMP-1M and TEVA resin Am and Cm: DGA, AGMP-1M, UTEVA, and GDA resin | CeF3 microprecipitation | 67.5–95.4% | [102] |
| Purpose | Radionuclides | Sample Type | Flow System Design | Chemical Separation | Measurement Technique | Performance | Reference |
|---|---|---|---|---|---|---|---|
| Environmental radioactivity monitoring | 239+240Pu and 241Am | Soil, vegetable ash leachate, urine, and blood | MSFIA-MPFS | Extraction chromatography (0.08 g TRU resin) | Low-background proportional counter | Chemical yield: <90% for both Pu and Am; LOD: 4 Bq/L; precision: 3%; turnover time (online separation): 40 min | [109] |
| 90Sr, 234U, 241Am, and 239Pu | Lake water | MSFI | Extraction chromatography (DGA-B resin) | ICP-MS | The limits of detection were 1.48 pg/L for 90Sr, 1.75 pg/L for 234U, 0.65 pg/L for 241Am, and 0.56 pg/L for 239Pu | [110] | |
| 237Np, 233U, 241Am, and 242Pu | Artificial solution | MSFI | Extraction chromatography (UTEVA and AG-1 resins) | Alpha spectrometry | Recovery yields: 94.2% for 233U, 87.2% for 237Np, 82.1% for 242Pu, and 98.7% for 241Am | [111] | |
| 232Th, 237Np, 238U, 241Am, and 242Pu | Large, spiked soil samples | Pressurized injection (PI) | Extraction chromatography (TEVA and DGA resins) | ICP-MS | Recovery yield: 97% for Th, U, Np, Pu, and Am | [105] | |
| Nuclear waste management | 90Sr, 241Am, and 99Tc | Aged nuclear waste | SI | Extraction chromatography (50 μL Sr resin, TRU resin, and TEVA resin) | Flow-through LSC | Chemical yields: 92 ± 2% for 90Sr and 99 ± 5% for 99Tc | [112] |
| 230 Th, 233U, 239Pu, and 241Am | Spiked sample solution in 2 M HNO3 | SI | Extraction chromatography (0.63 mL TRU resin, 20–50 μm) | Flow-through LSC and alpha spectrometry | Chemical yields: up to 102 ±4% for 241Am, up to 101 ± 3% for 239Pu, up to 93 ±4% for 233U, and up to 88 ± 3% for 230Th | [113] | |
| 237Np, 238Pu, 239+240Pu, and 241Am | Dissolved vitrified nuclear waste | SI | Extraction chromatography (0.63 mL TRU resin, 20–50 μm) | ICP-MS | U decontamination factor (for Pu determination): 3.0 × 105 | [114] | |
| 238 Pu, 239+240Pu, 241Am, 243+244Cm, and 242Cm | Vitrified glass waste, aged irradiated nuclear fuel, and waste from Handford site | SI | Extraction chromatography (0.63 mL TRU resin, 20–50 μm) | Flow-through LSC and alpha spectrometry | Chemical yields: 85% for Pu and 86% for Am | [103] |
| Isobaric and Polyatomic Interferences | Abundance of Interference Isotopes |
|---|---|
| 241Pu | |
| 240Pu1H | |
| 209Bi32S | 209Bi: 100% |
| 209BiO2 | 209Bi: 100% |
| 208Pb16O21H | 208Pb: 52.4% |
| 206Pb35Cl | 206Pb: 24.1% |
| 204Pb37Cl | 204Pb: 1.4% |
| 207Pb34S+ | 207Pb: 22.1% |
| 208Pb33S | 208Pb: 52.4% |
| 201Hg40Ar | 201Hg: 13.2% |
| 179Hf14N16O3 | 179Hf: 13.6% |
| 178Hf14N16O31H | 178Hf: 27.3% |
| 204Hg37Cl | 204Hg: 6.9% |
| 195Pt14N16O2 | 195Pt: 33.8% |
| 194Pt14N16O21H | 194Pt: 33.0% |
| 203Tl38Ar | 203Tl: 29.524% |
| 205Tl36Ar | 205Tl: 70.476% |
| MS Techniques Used | Matrix/Separation Method or Combined Method | Limit of Detection (LOD) or Limit of Quantitation (LOQ) | Reference |
|---|---|---|---|
| Q-ICP-MS | Water and urine/TRU resin | 40–150 fg/g (LOD) | [141] |
| Q-ICP-MS | Urine/flow injection and extraction chromatography | 0.9–8 fg/g (LOD) | [142] |
| Q-ICP-MS | Sediment/TEVA and DGA resins | 2 pg (LOD) | [143] |
| Q-ICP-MS, He-NH3 as collision–reaction gas | Soil samples IAEA-Soil-6 and IAEA-375/DGA resin | 0.094 fg/g (LOD) | [26] |
| Q-ICP-MS, O2/He-He as collision–reaction gas | Soil samples IAEA-Soil-6 and IAEA-375/UTEVA and DGA resins | 0.019 fg/g (LOD) | [139] |
| SF-ICP-MS | River sediment, human liver and lung samples/extraction with ammonium hydrogen oxalate | 1.2 fg/g (LOD) | [107] |
| SF-ICP-MS | Sediment/TRU resin | 0.9 fg/g (LOD) | [144] |
| SF-ICP-MS | Sediment IAEA-384, sediment IAEA-385, and seaweed IAEA-308/CaF2 precipitation and TRU resin | 0.86 fg/g (LOD) | [145] |
| SF-ICP-MS | Sediment IAEA-368/isotope dilution with 243Am, CaF2 precipitation, and TRU resin | 0.32 fg/g (LOD) | [138] |
| SF-ICP-MS | Large soil samples/Ca2C2O4 precipitation; TEVA and DGA-N resins | 0.094 fg/g (LOD) | [137] |
| SF-ICP-MS | Soil and sediment/Fe(OH)3 precipitation; UTEVA, DAG, and TEVA resins | 0.31 fg/g (LOD) | [146] |
| SF-ICP-MS | River water/TEVA, TRU, and Sr resins | 73 fg/g (LOD) | [147] |
| LA-SF-ICPMS | Mosses | 3.7 pg/g (LOD) | [148] |
| MC-ICP-MS | IAEA-385 sediment, and NIST-4350B sediment/oxalate coprecipitation; TEVA-ammonium thiocyanate column and acetone-HCl MP1 anion column | 1.4 fg (LOQ) | [140] |
| Valence | Complex | Species |
|---|---|---|
| Am(III) | Hydroxide | [Am(OH)2]+ |
| [Am(OH)3] | ||
| Am(III) | Halides | [AmF2+]/[AmCl2+] |
| [AmF2+/[AmCl2+] | ||
| Am(III) | Phosphates | [AmH2PO42+] |
| Am(III) | Nitrates | [AmNO3]2+ |
| Am(III) | Carbonates | [AmCO3]+ |
| [Am(CO3)2]− | ||
| [Am(CO3)3]3− | ||
| [AmHCO3]− | ||
| Am(III) | Silicate | [AmSiO(OH)3]2+ |
| Desired Geochemical Phases | Extraction Reagents | Temperature (°C) | Time (h) |
|---|---|---|---|
| 1. Water soluble, exchangeable | H2O, MgCl2 0.4M; pH 4.5 | room | 1 |
| 2. Carbonates | NH4Ac 1M, 25%Hac; pH 4 | room | 2 |
| 3. Oxides (Fe, Mn) | NH2OH.HCl 0.04M, HAc | room | 5 |
| 4. Organic matter | H2O2 30%, HNO3 0.02M; pH 2 | 85 | 5 |
| 5. Residue | HNO3, HCl, HF, HClO4 | 100 | 1 |
| Location/Sample | Fractionation of 241Am | Reference |
|---|---|---|
| Venice Lagoon (northern Adriatic Sea, Italy)/VLAS | carbonates > 90% | [175] |
| Gaeta Gulf (central Tyrrhenia Sea, Italy)/GGTS | carbonates > 60% | [175] |
| Marshall Islands (central Pacific Ocean)/IAEA-367 | carbonates 65%, organic matter 31% | [175] |
| Sellafield (Irish Sea, UK)/IAEA-135 | carbonates 65%, organic matter 25% | [175] |
| Baltic Sea/sediment | carbonates 21%, organic matter 42% | [176] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, H.; Hou, X.; Qiao, J.; Lin, J. Determination of 241Am in Environmental Samples: A Review. Molecules 2022, 27, 4536. https://doi.org/10.3390/molecules27144536
Zhang H, Hou X, Qiao J, Lin J. Determination of 241Am in Environmental Samples: A Review. Molecules. 2022; 27(14):4536. https://doi.org/10.3390/molecules27144536
Chicago/Turabian StyleZhang, Haitao, Xiaolin Hou, Jixin Qiao, and Jianfeng Lin. 2022. "Determination of 241Am in Environmental Samples: A Review" Molecules 27, no. 14: 4536. https://doi.org/10.3390/molecules27144536
APA StyleZhang, H., Hou, X., Qiao, J., & Lin, J. (2022). Determination of 241Am in Environmental Samples: A Review. Molecules, 27(14), 4536. https://doi.org/10.3390/molecules27144536