
A Novel ERK2 Degrader Z734 Induces Apoptosis of MCF–7 Cells via the HERC3/p53 Signaling Pathway

 ,
,
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
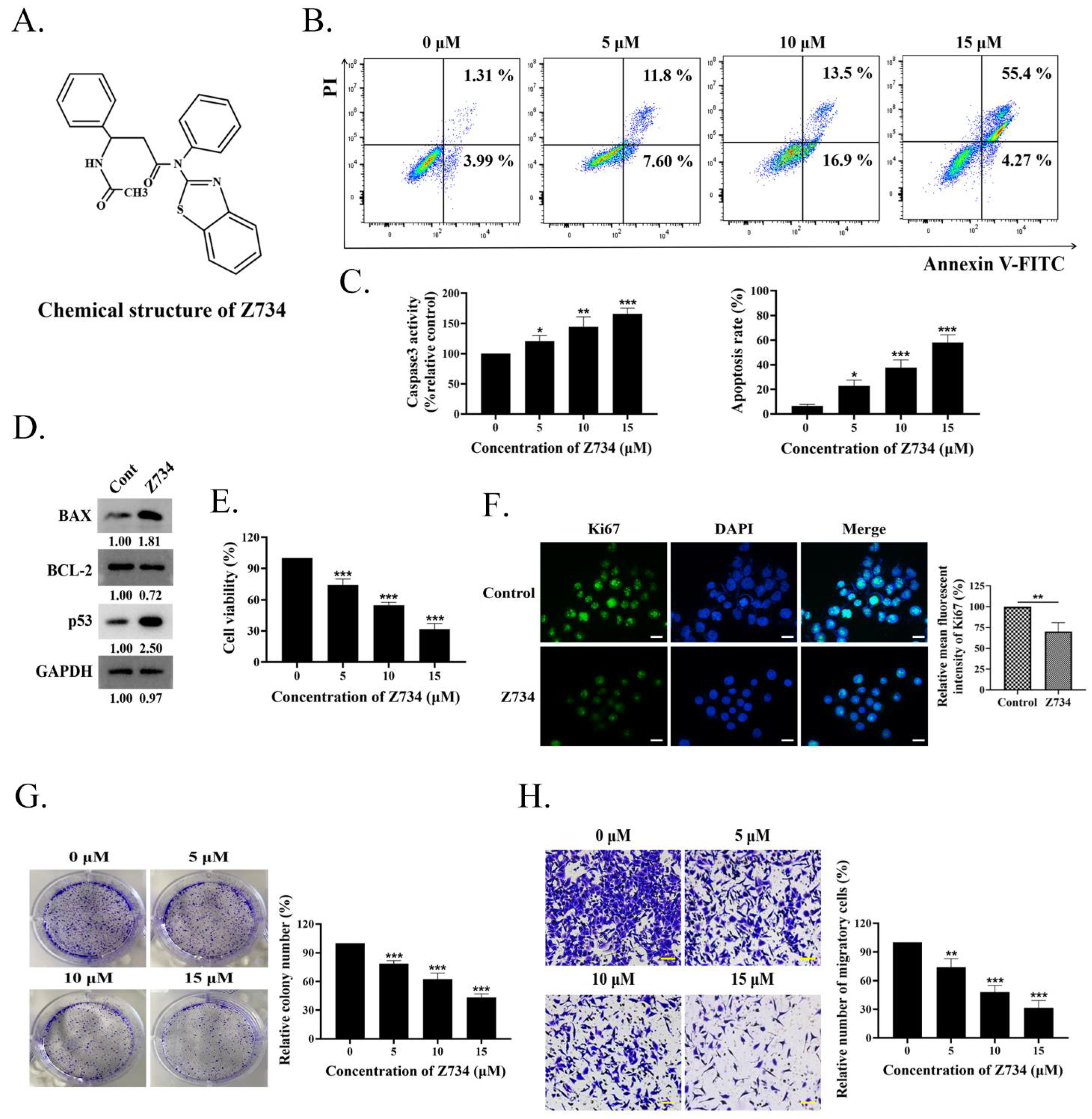
2.1. Z734 Causes Apoptosis and Inhibits MCF–7 Cells Proliferation and Migration
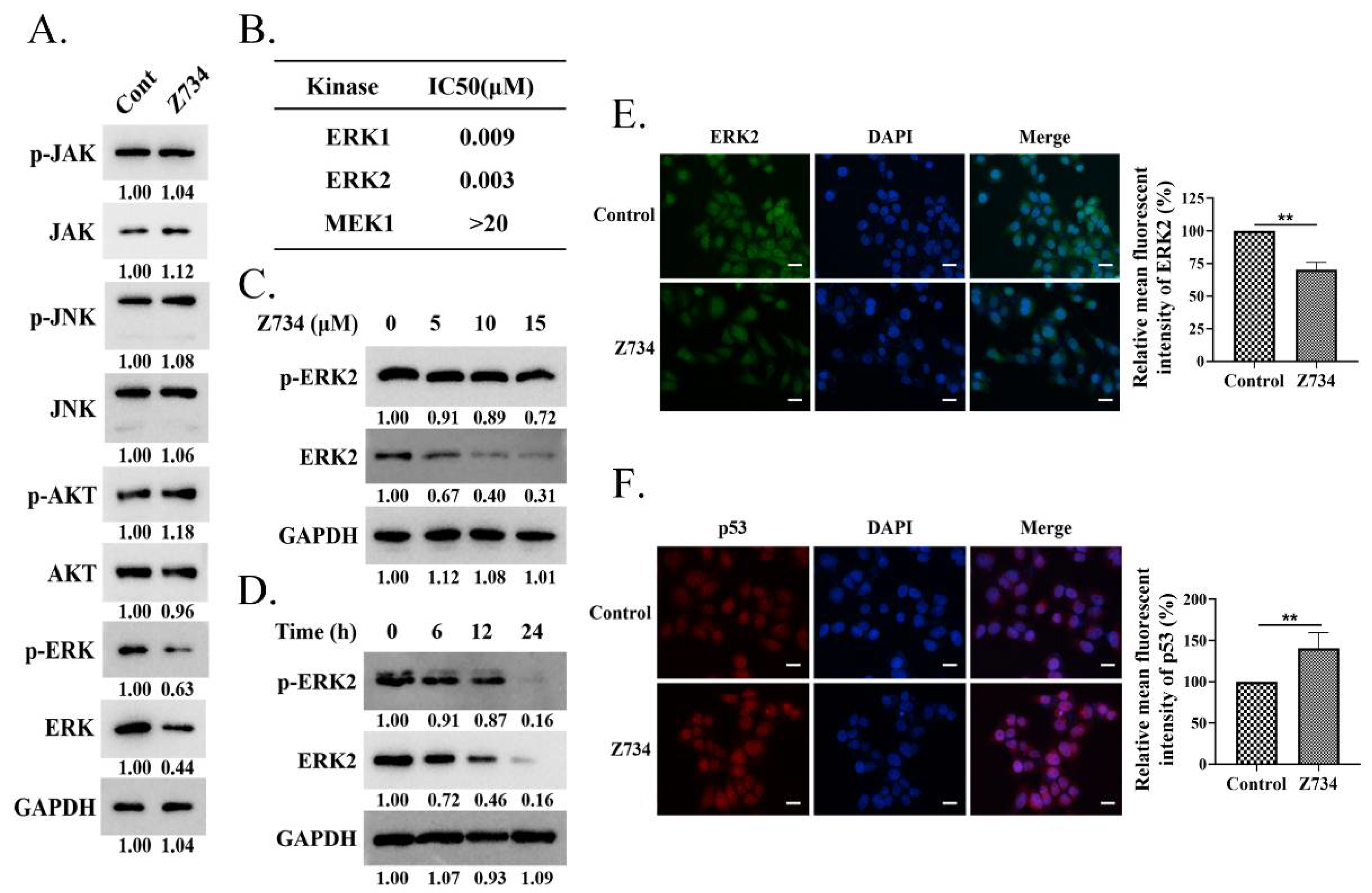
2.2. Z734 Downregulates the Expression of ERK2 in MCF–7 Cells
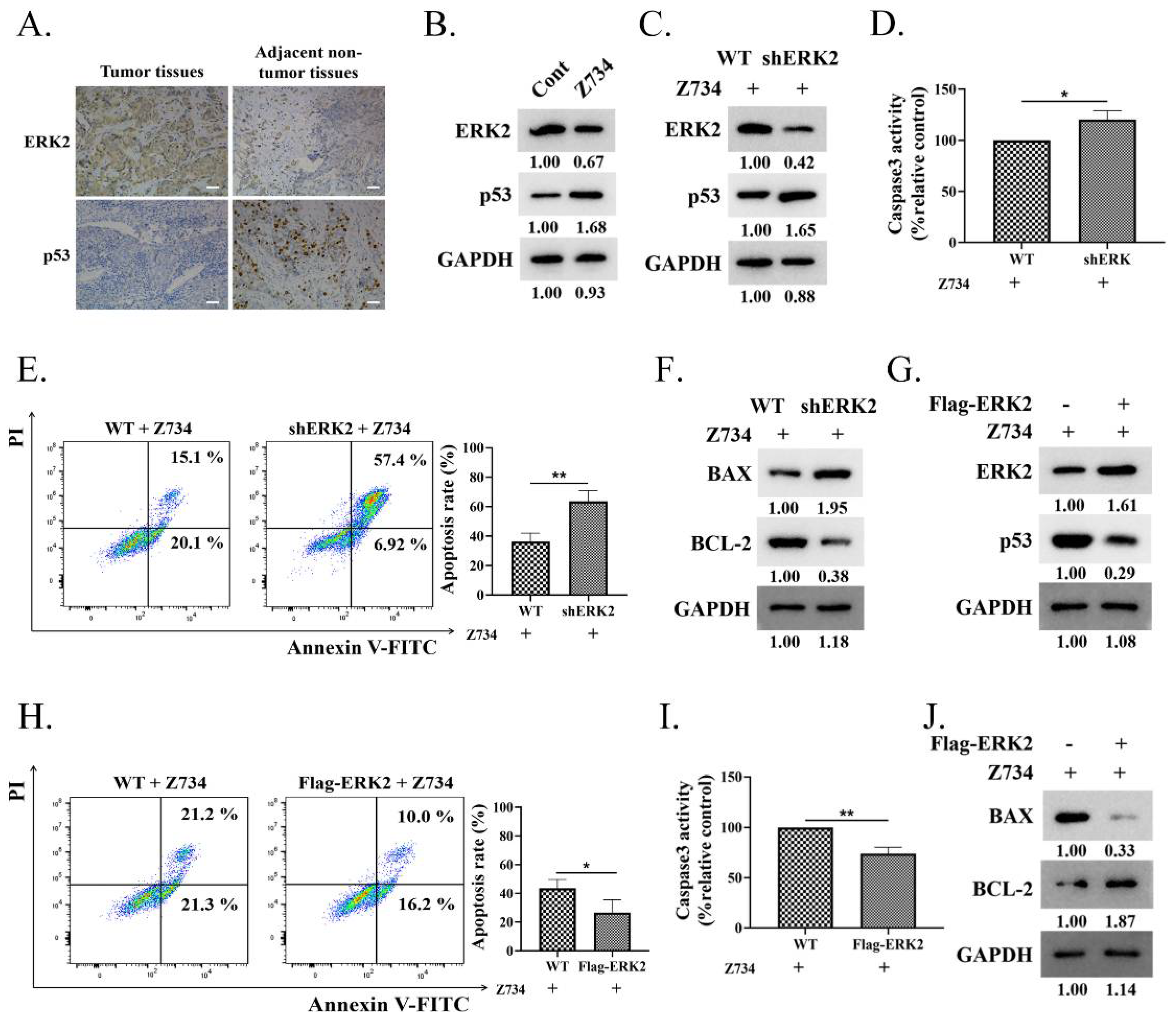
2.3. ERK2 Inhibition Promotes Z734–Mediated Apoptosis in MCF–7 Cells
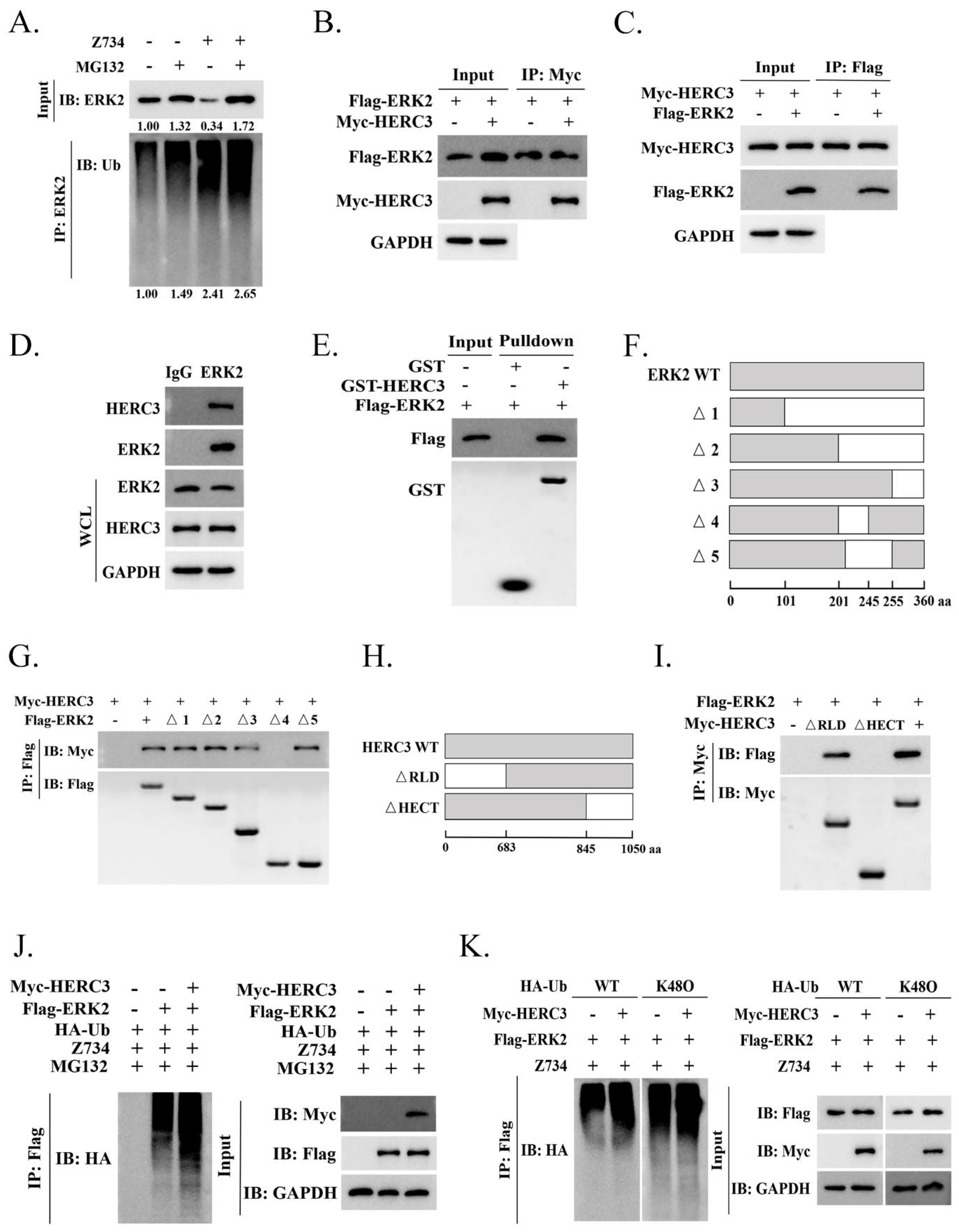
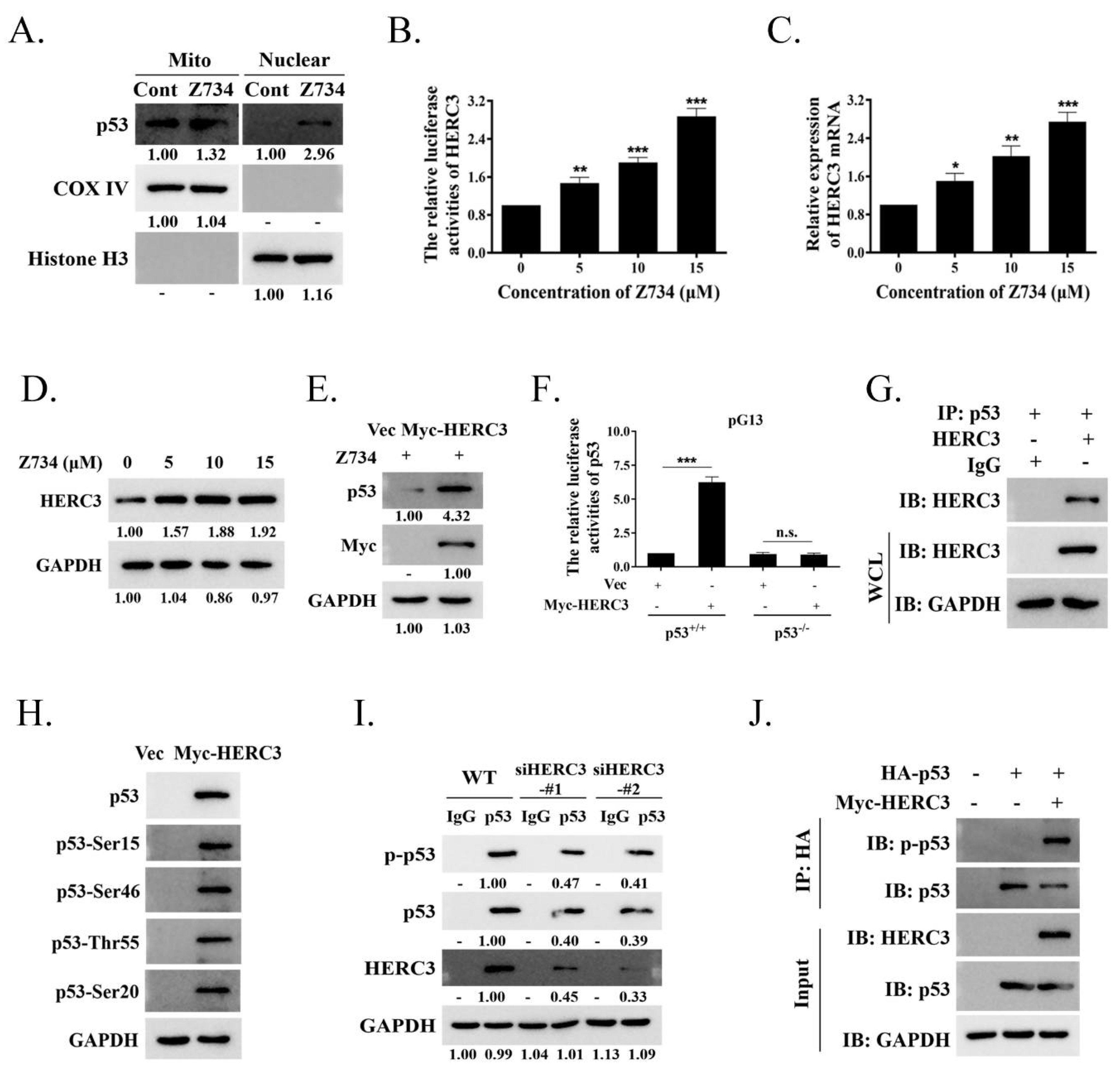
2.4. HERC3 Interacts with ERK2 and Promotes in the Z734–Mediated Ubiquitination of ERK2
2.5. Z734–Mediated HERC3 Increasing Promotes p53 Level and p53 Phosphorylation
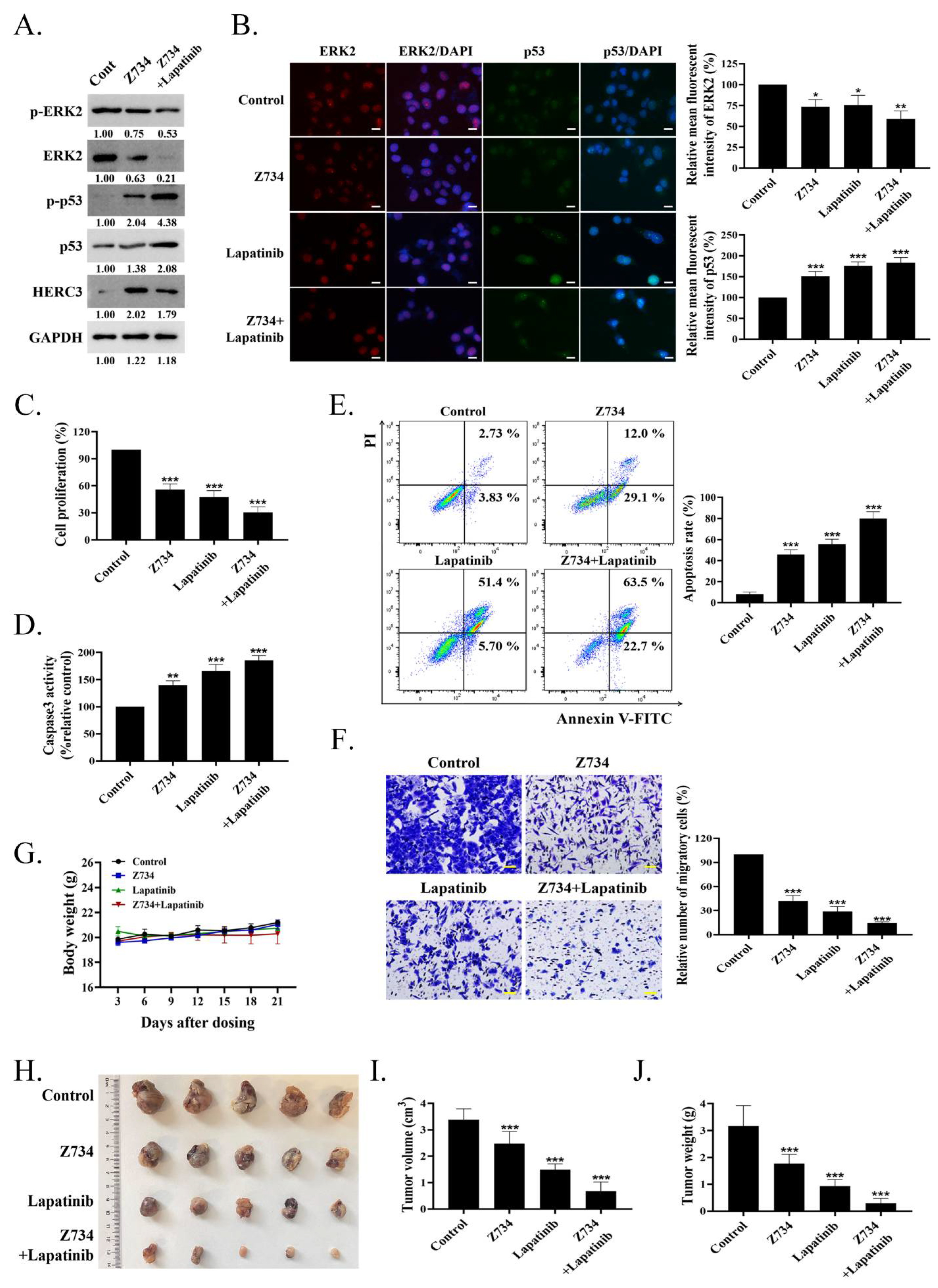
2.6. A Combination of Z734 and Lapatinib Inhibits the Growth of Breast Cancer
3. Discussion
4. Materials and Methods
4.1. Human Specimens
4.2. Cell Lines and Transfection
4.3. Construction of Plasmids
4.4. Real–Time Quantitative PCR (qRT–PCR)
4.5. Generation of CRISPR–Cas9–Based Knockout Cells
4.6. Cell Viability Assays
4.7. Luciferase Reporter Assay
4.8. Western Blot Analysis (WB)
4.9. Immunoprecipitation (Co–IP)
4.10. Immunofluorescence (IF)
4.11. Flow Cytometric Assays
4.12. IMAP Enzymatic Assay
4.13. Caspase3 Activity Assays
4.14. ELISAs
4.15. Colony Formation Assays
4.16. Migration Analysis
4.17. Animal Studies
4.18. Histopathology
4.19. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Mikalsen, T.; Gerits, N.; Moens, U. Inhibitors of signal transduction protein kinases as targets for cancer therapy. Biotechnol. Annu. Rev. 2006, 12, 153–223. [Google Scholar] [PubMed]
- Zhang, W.; Liu, H.T. MAPK signal pathways in the regulation of cell proliferation in mammalian cells. Cell Res. 2002, 12, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Chapman, P.B.; Hauschild, A.; Robert, C.; Haanen, J.B.; Ascierto, P.; Larkin, J.; Dummer, R.; Garbe, C.; Testori, A.; Maio, M.; et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N. Engl. J. Med. 2011, 364, 2507–2516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hauschild, A.; Grob, J.J.; Demidov, L.V.; Jouary, T.; Gutzmer, R.; Millward, M.; Rutkowski, P.; Blank, C.U.; Miller, W.H., Jr.; Kaempgen, E.; et al. Dabrafenib in BRAF-mutated metastatic melanoma: A multicentre, open-label, phase 3 randomised controlled trial. Lancet 2012, 380, 358–365. [Google Scholar] [CrossRef]
- Flaherty, K.T.; Robert, C.; Hersey, P.; Nathan, P.; Garbe, C.; Milhem, M.; Demidov, L.V.; Hassel, J.C.; Rutkowski, P.; Mohr, P.; et al. Improved survival with MEK inhibition in BRAF-mutated melanoma. N. Engl. J. Med. 2012, 367, 107–114. [Google Scholar] [CrossRef] [Green Version]
- Diamond, E.L.; Durham, B.H.; Ulaner, G.A.; Drill, E.; Buthorn, J.; Ki, M.; Bitner, L.; Cho, H.; Young, R.J.; Francis, J.H.; et al. Efficacy of MEK inhibition in patients with histiocytic neoplasms. Nature 2019, 567, 521–524. [Google Scholar] [CrossRef]
- Roskoski, R., Jr. Targeting ERK1/2 protein-serine/threonine kinases in human cancers. Pharmacol. Res. 2019, 142, 151–168. [Google Scholar] [CrossRef]
- Shi, H.; Moriceau, G.; Kong, X.; Lee, M.K.; Lee, H.; Koya, R.C.; Ng, C.; Chodon, T.; Scolyer, R.A.; Dahlman, K.B.; et al. Melanoma whole-exome sequencing identifies V600EB-RAF amplification-mediated acquired B-RAF inhibitor resistance. Nat. Commun. 2012, 3, 724. [Google Scholar] [CrossRef] [Green Version]
- Kidger, A.M.; Sipthorp, J.; Cook, S.J. ERK1/2 inhibitors: New weapons to inhibit the RAS-regulated RAF-MEK1/2-ERK1/2 pathway. Pharmacol. Ther. 2018, 187, 45–60. [Google Scholar] [CrossRef]
- Liu, F.; Yang, X.; Geng, M.; Huang, M. Targeting ERK, an Achilles’ Heel of the MAPK pathway, in cancer therapy. Acta Pharm. Sin. B 2018, 8, 552–562. [Google Scholar] [CrossRef] [PubMed]
- Peh, J.; Boudreau, M.W.; Smith, H.M.; Hergenrother, P.J. Overcoming Resistance to Targeted Anticancer Therapies through Small-Molecule-Mediated MEK Degradation. Cell Chem. Biol. 2018, 25, 996–1005.e4. [Google Scholar] [CrossRef] [PubMed]
- Vollmer, S.; Cunoosamy, D.; Lv, H.F.; Feng, H.X.; Li, X.; Nan, Z.Y.; Yang, W.Z.; Perry, M.W.D. Design, Synthesis, and Biological Evaluation of MEK PROTACs. J. Med. Chem. 2020, 63, 157–162. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Wang, H.; Zheng, C.X.; Liu, Z.M.; Gao, X.Z.; Xu, F.R.; Niu, Y.; Zhang, L.R.; Xu, P. Research progress of MEK1/2 inhibitors and degraders in the treatment of cancer. Eur. J. Med. Chem. 2021, 218, 113386. [Google Scholar] [CrossRef]
- Zhang, Z.; He, G.; Lv, Y.; Liu, Y.; Niu, Z.; Feng, Q.; Hu, R.; Xu, J. HERC3 regulates epithelial-mesenchymal transition by directly ubiquitination degradation EIF5A2 and inhibits metastasis of colorectal cancer. Cell Death Dis. 2022, 13, 74. [Google Scholar] [CrossRef]
- Cruz, C.; Ventura, F.; Bartrons, R.; Rosa, J.L. HERC3 binding to and regulation by ubiquitin. FEBS Lett. 2001, 488, 74–80. [Google Scholar] [CrossRef]
- Garcia-Gonzalo, F.R.; Rosa, J.L. The HERC proteins: Functional and evolutionary insights. Cell. Mol. Life Sci. 2005, 62, 1826–1838. [Google Scholar] [CrossRef]
- Li, H.; Li, J.; Chen, L.; Qi, S.; Yu, S.; Weng, Z.; Hu, Z.; Zhou, Q.; Xin, Z.; Shi, L.; et al. HERC3-Mediated SMAD7 Ubiquitination Degradation Promotes Autophagy-Induced EMT and Chemoresistance in Glioblastoma. Clin. Cancer Res. 2019, 25, 3602–3616. [Google Scholar] [CrossRef] [Green Version]
- Zhu, D.; Yang, D.; Li, X.; Feng, F. Heterogeneous expression and biological function of SOX18 in osteosaroma. J. Cell. Biochem. 2018, 119, 4184–4192. [Google Scholar] [CrossRef]
- Lane, D.P. Cancer. p53, guardian of the genome. Nature 1992, 358, 15–16. [Google Scholar] [CrossRef]
- Zilfou, J.T.; Lowe, S.W. Tumor suppressive functions of p53. Cold Spring Harb. Perspect. Biol. 2009, 1, a001883. [Google Scholar] [CrossRef] [PubMed]
- Aubrey, B.J.; Kelly, G.L.; Janic, A.; Herold, M.J.; Strasser, A. How does p53 induce apoptosis and how does this relate to p53–mediated tumour suppression? Cell Death Differ. 2018, 25, 104–113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vogelstein, B.; Lane, D.; Levine, A.J. Surfing the p53 network. Nature 2000, 408, 307–310. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Wang, Y.; Yang, J.; Zhang, W.; Meng, K.; Sun, Y.; Li, Y.; He, Q.Y. RNF128 Promotes Invasion and Metastasis Via the EGFR/MAPK/MMP-2 Pathway in Esophageal Squamous Cell Carcinoma. Cancers 2019, 11, 840. [Google Scholar] [CrossRef] [Green Version]
- Wu, Q.; Yuan, X.; Li, B.; Han, R.; Zhang, H.; Xiu, R. Salvianolic Acid Alleviated Blood-Brain Barrier Permeability in Spontaneously Hypertensive Rats by Inhibiting Apoptosis in Pericytes via P53 and the Ras/Raf/MEK/ERK Pathway. Drug Des. Dev. Ther. 2020, 14, 1523–1534. [Google Scholar] [CrossRef] [Green Version]
- Hafner, A.; Bulyk, M.L.; Jambhekar, A.; Lahav, G. The multiple mechanisms that regulate p53 activity and cell fate. Nat. Rev. Mol. Cell. Biol. 2019, 20, 199–210. [Google Scholar] [CrossRef]
- Wawryk-Gawda, E.; Chylinska-Wrzos, P.; Lis-Sochocka, M.; Chlapek, K.; Bulak, K.; Jedrych, M.; Jodlowska-Jedrych, B. P53 protein in proliferation, repair and apoptosis of cells. Protoplasma 2014, 251, 525–533. [Google Scholar] [CrossRef] [Green Version]
- Kruse, J.P.; Gu, W. Modes of p53 regulation. Cell 2009, 137, 609–622. [Google Scholar] [CrossRef] [Green Version]
- Chen, J. The Cell-Cycle Arrest and Apoptotic Functions of p53 in Tumor Initiation and Progression. Cold Spring Harb. Perspect. Med. 2016, 6, a026104. [Google Scholar] [CrossRef]
- Mustachio, L.M.; Chelariu-Raicu, A.; Szekvolgyi, L.; Roszik, J. Targeting KRAS in Cancer: Promising Therapeutic Strategies. Cancers 2021, 13, 1204. [Google Scholar] [CrossRef]
- Roberts, P.J.; Der, C.J. Targeting the Raf-MEK-ERK mitogen-activated protein kinase cascade for the treatment of cancer. Oncogene 2007, 26, 3291–3310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vousden, K.H.; Lu, X. Live or let die: The cell’s response to p53. Nat. Rev. Cancer 2002, 2, 594–604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sugiura, R.; Satoh, R.; Takasaki, T. ERK: A Double-Edged Sword in Cancer. ERK-Dependent Apoptosis as a Potential Therapeutic Strategy for Cancer. Cells 2021, 10, 2509. [Google Scholar] [CrossRef] [PubMed]
- Komander, D.; Rape, M. The ubiquitin code. Annu. Rev. Biochem. 2012, 81, 203–229. [Google Scholar] [CrossRef] [Green Version]
- Ping, Z.; Siegal, G.P.; Harada, S.; Eltoum, I.E.; Youssef, M.; Shen, T.; He, J.; Huang, Y.; Chen, D.; Li, Y.; et al. ERBB2 mutation is associated with a worse prognosis in patients with CDH1 altered invasive lobular cancer of the breast. Oncotarget 2016, 7, 80655–80663. [Google Scholar] [CrossRef] [Green Version]
- Rossi, F.A.; Calvo Roitberg, E.H.; Enrique Steinberg, J.H.; Joshi, M.U.; Espinosa, J.M.; Rossi, M. HERC1 Regulates Breast Cancer Cells Migration and Invasion. Cancers 2021, 13, 1309. [Google Scholar] [CrossRef]
- Zhou, H.; Shi, R.; Wei, M.; Zheng, W.L.; Zhou, J.Y.; Ma, W.L. The expression and clinical significance of HERC4 in breast cancer. Cancer Cell Int. 2013, 13, 113. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.; Ji, K.; Wu, M.; Hao, B.; Yao, K.T.; Xu, Y. A miRNA-HERC4 pathway promotes breast tumorigenesis by inactivating tumor suppressor LATS1. Protein Cell 2019, 10, 595–605. [Google Scholar] [CrossRef] [Green Version]
- Tang, J.; Yang, Q.; Cui, Q.; Zhang, D.; Kong, D.; Liao, X.; Ren, J.; Gong, Y.; Wu, G. Weighted gene correlation network analysis identifies RSAD2, HERC5, and CCL8 as prognostic candidates for breast cancer. J. Cell. Physiol. 2020, 235, 394–407. [Google Scholar] [CrossRef]
- Wu, W.; Sato, K.; Koike, A.; Nishikawa, H.; Koizumi, H.; Venkitaraman, A.R.; Ohta, T. HERC2 is an E3 ligase that targets BRCA1 for degradation. Cancer Res. 2010, 70, 6384–6392. [Google Scholar] [CrossRef] [Green Version]
- Peng, Y.; Dai, H.; Wang, E.; Lin, C.C.; Mo, W.; Peng, G.; Lin, S.Y. TUSC4 functions as a tumor suppressor by regulating BRCA1 stability. Cancer Res. 2015, 75, 378–386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Li, Y.; Peng, Y.; Zheng, X.; Fan, S.; Yi, Y.; Zeng, P.; Chen, H.; Kang, H.; Zhang, Y.; et al. ΔNp63α down-regulates c-Myc modulator MM1 via E3 ligase HERC3 in the regulation of cell senescence. Cell Death Differ. 2018, 25, 2118–2129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galluzzi, L.; Morselli, E.; Kepp, O.; Tajeddine, N.; Kroemer, G. Targeting p53 to mitochondria for cancer therapy. Cell Cycle 2008, 7, 1949–1955. [Google Scholar] [CrossRef] [PubMed]
- Sabapathy, K.; Lane, D.P. Understanding p53 functions through p53 antibodies. J. Mol. Cell Biol. 2019, 11, 317–329. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.C.; Ma, F.; Shen, Y.; Hu, Y.Q.; Pan, S. Overexpression of SMAR1 Enhances Radiosensitivity in Human Breast Cancer Cell Line MCF7 via Activation of p53 Signaling Pathway. Oncol. Res. 2014, 22, 293–300. [Google Scholar] [CrossRef]
- Kong, W.; Jiang, X.; Mercer, W.E. Downregulation of Wip-1 phosphatase expression in MCF-7 breast cancer cells enhances doxorubicin-induced apoptosis through p53-mediated transcriptional activation of Bax. Cancer Biol. Ther. 2009, 8, 555–563. [Google Scholar] [CrossRef] [Green Version]
- Xia, L.; Paik, A.; Li, J.J. p53 activation in chronic radiation-treated breast cancer cells: Regulation of MDM2/p14ARF. Cancer Res. 2004, 64, 221–228. [Google Scholar] [CrossRef] [Green Version]
- Bilancia, D.; Rosati, G.; Dinota, A.; Germano, D.; Romano, R.; Manzione, L. Lapatinib in breast cancer. Ann. Oncol. 2007, 18 (Suppl. S6), vi26–vi30. [Google Scholar] [CrossRef]
- Schneider, T.; Martinez-Martinez, A.; Cubillos-Rojas, M.; Bartrons, R.; Ventura, F.; Rosa, J.L. The E3 ubiquitin ligase HERC1 controls the ERK signaling pathway targeting C-RAF for degradation. Oncotarget 2018, 9, 31531–31548. [Google Scholar] [CrossRef]
- Garcia-Cano, J.; Martinez-Martinez, A.; Sala-Gaston, J.; Pedrazza, L.; Rosa, J.L. HERCing: Structural and Functional Relevance of the Large HERC Ubiquitin Ligases. Front. Physiol. 2019, 10, 1014. [Google Scholar] [CrossRef]
- Sanchez-Tena, S.; Cubillos-Rojas, M.; Schneider, T.; Rosa, J.L. Functional and pathological relevance of HERC family proteins: A decade later. Cell. Mol. Life Sci. 2016, 73, 1955–1968. [Google Scholar] [CrossRef] [PubMed]
- Sala-Gaston, J.; Martinez-Martinez, A.; Pedrazza, L.; Lorenzo-Martin, L.F.; Caloto, R.; Bustelo, X.R.; Ventura, F.; Rosa, J.L. HERC Ubiquitin Ligases in Cancer. Cancers 2020, 12, 1653. [Google Scholar] [CrossRef] [PubMed]
- Mao, X.; Sethi, G.; Zhang, Z.; Wang, Q. The Emerging Roles of the HERC Ubiquitin Ligases in Cancer. Curr. Pharm. Des. 2018, 24, 1676–1681. [Google Scholar] [CrossRef]
- Li, Y.Q.; Cheng, Z.C.; Liu, S.W.; Aubert, I.; Wong, C.S. P53 regulates disruption of neuronal development in the adult hippocampus after irradiation. Cell Death Discov. 2016, 2, 16072. [Google Scholar] [CrossRef] [Green Version]
- Bartesaghi, S.; Graziano, V.; Galavotti, S.; Henriquez, N.V.; Betts, J.; Saxena, J.; Minieri, V.; Deli, A.; Karlsson, A.; Martins, L.M.; et al. Inhibition of oxidative metabolism leads to p53 genetic inactivation and transformation in neural stem cells. Proc. Natl. Acad. Sci. USA 2015, 112, 1059–1064. [Google Scholar] [CrossRef] [Green Version]
- Mitsui, K.; Nakanishi, M.; Ohtsuka, S.; Norwood, T.H.; Okabayashi, K.; Miyamoto, C.; Tanaka, K.; Yoshimura, A.; Ohtsubo, M. A novel human gene encoding HECT domain and RCC1-like repeats interacts with cyclins and is potentially regulated by the tumor suppressor proteins. Biochem. Biophys. Res. Commun. 1999, 266, 115–122. [Google Scholar] [CrossRef]
- Hochrainer, K.; Mayer, H.; Baranyi, U.; Binder, B.; Lipp, J.; Kroismayr, R. The human HERC family of ubiquitin ligases: Novel members, genomic organization, expression profiling, and evolutionary aspects. Genomics 2005, 85, 153–164. [Google Scholar] [CrossRef]
- Mantovani, F.; Collavin, L.; Del Sal, G. Mutant p53 as a guardian of the cancer cell. Cell Death Differ. 2019, 26, 199–212. [Google Scholar] [CrossRef]
- Levine, A.J. p53: 800 million years of evolution and 40 years of discovery. Nat. Rev. Cancer 2020, 20, 471–480. [Google Scholar] [CrossRef]
- Xue, W.; Zender, L.; Miething, C.; Dickins, R.A.; Hernando, E.; Krizhanovsky, V.; Cordon-Cardo, C.; Lowe, S.W. Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature 2007, 445, 656–660. [Google Scholar] [CrossRef] [Green Version]
- Feldser, D.M.; Kostova, K.K.; Winslow, M.M.; Taylor, S.E.; Cashman, C.; Whittaker, C.A.; Sanchez-Rivera, F.J.; Resnick, R.; Bronson, R.; Hemann, M.T.; et al. Stage-specific sensitivity to p53 restoration during lung cancer progression. Nature 2010, 468, 572–575. [Google Scholar] [CrossRef] [Green Version]
- Harajly, M.; Zalzali, H.; Nawaz, Z.; Ghayad, S.E.; Ghamloush, F.; Basma, H.; Zainedin, S.; Rabeh, W.; Jabbour, M.; Tawil, A.; et al. p53 Restoration in Induction and Maintenance of Senescence: Differential Effects in Premalignant and Malignant Tumor Cells. Mol. Cell. Biol. 2016, 36, 438–451. [Google Scholar] [CrossRef] [Green Version]
- Rashmi, K.C.; Harsha Raj, M.; Paul, M.; Girish, K.S.; Salimath, B.P.; Aparna, H.S. A new pyrrole based small molecule from Tinospora cordifolia induces apoptosis in MDA-MB-231 breast cancer cells via ROS mediated mitochondrial damage and restoration of p53 activity. Chem. Biol. Interact. 2019, 299, 120–130. [Google Scholar] [CrossRef]
- Liu, Y.; Tavana, O.; Gu, W. p53 modifications: Exquisite decorations of the powerful guardian. J. Mol. Cell Biol. 2019, 11, 564–577. [Google Scholar] [CrossRef] [Green Version]
- Imami, K.; Sugiyama, N.; Imamura, H.; Wakabayashi, M.; Tomita, M.; Taniguchi, M.; Ueno, T.; Toi, M.; Ishihama, Y. Temporal profiling of lapatinib-suppressed phosphorylation signals in EGFR/HER2 pathways. Mol. Cell. Proteom. 2012, 11, 1741–1757. [Google Scholar] [CrossRef] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, S.; Xiong, Y.; Yao, R.; Tian, R.; Meng, Z.; Zaky, M.Y.; Fu, B.; Guo, D.; Wang, L.; Lin, F.; et al. A Novel ERK2 Degrader Z734 Induces Apoptosis of MCF–7 Cells via the HERC3/p53 Signaling Pathway. Molecules 2022, 27, 4337. https://doi.org/10.3390/molecules27144337
Xu S, Xiong Y, Yao R, Tian R, Meng Z, Zaky MY, Fu B, Guo D, Wang L, Lin F, et al. A Novel ERK2 Degrader Z734 Induces Apoptosis of MCF–7 Cells via the HERC3/p53 Signaling Pathway. Molecules. 2022; 27(14):4337. https://doi.org/10.3390/molecules27144337
Chicago/Turabian StyleXu, Shiyao, Yan Xiong, Rui Yao, Rong Tian, Zhuqing Meng, Mohamed Y. Zaky, Beibei Fu, Dong Guo, Lulu Wang, Feng Lin, and et al. 2022. "A Novel ERK2 Degrader Z734 Induces Apoptosis of MCF–7 Cells via the HERC3/p53 Signaling Pathway" Molecules 27, no. 14: 4337. https://doi.org/10.3390/molecules27144337
APA StyleXu, S., Xiong, Y., Yao, R., Tian, R., Meng, Z., Zaky, M. Y., Fu, B., Guo, D., Wang, L., Lin, F., Lin, X., & Wu, H. (2022). A Novel ERK2 Degrader Z734 Induces Apoptosis of MCF–7 Cells via the HERC3/p53 Signaling Pathway. Molecules, 27(14), 4337. https://doi.org/10.3390/molecules27144337