
Anti-Cancer Effects of Auranofin in Human Lung Cancer Cells by Increasing Intracellular ROS Levels and Depleting GSH Levels


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Reagents
2.3. Cell Proliferation Assay
2.4. Determination of Intracellular ROS and O2•− Levels
2.5. Detection of Intracellular GSH
2.6. Detection of Apoptosis
2.7. Measurement of Mitochondrial Membrane Potential (MMP; ΔΨm)
2.8. Western Blotting
2.9. TrxR Activity Assay
2.10. Statistical Analysis
3. Results
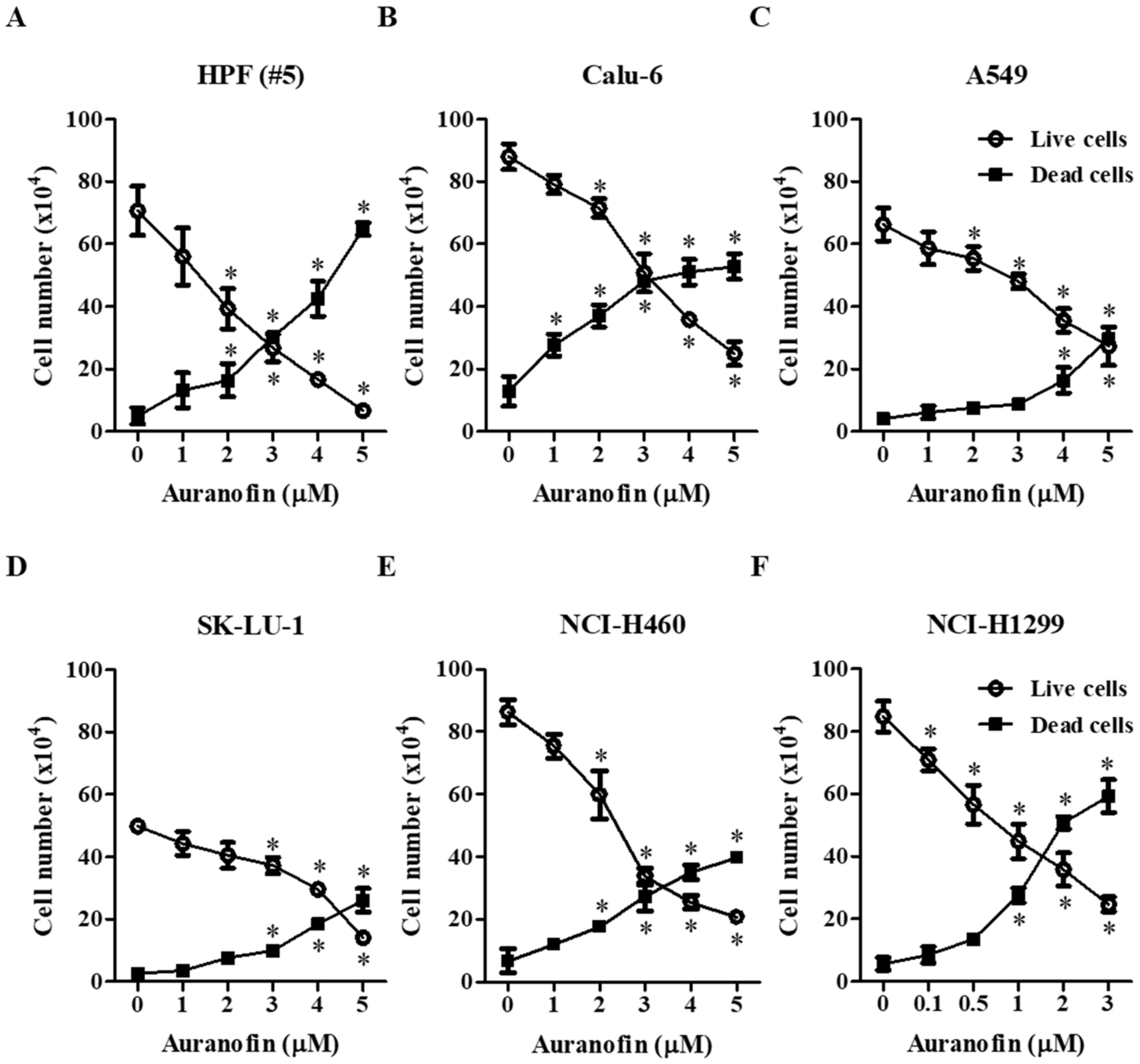
3.1. Effects of Auranofin on Cell Growth and Death in Lung Cancer Cells
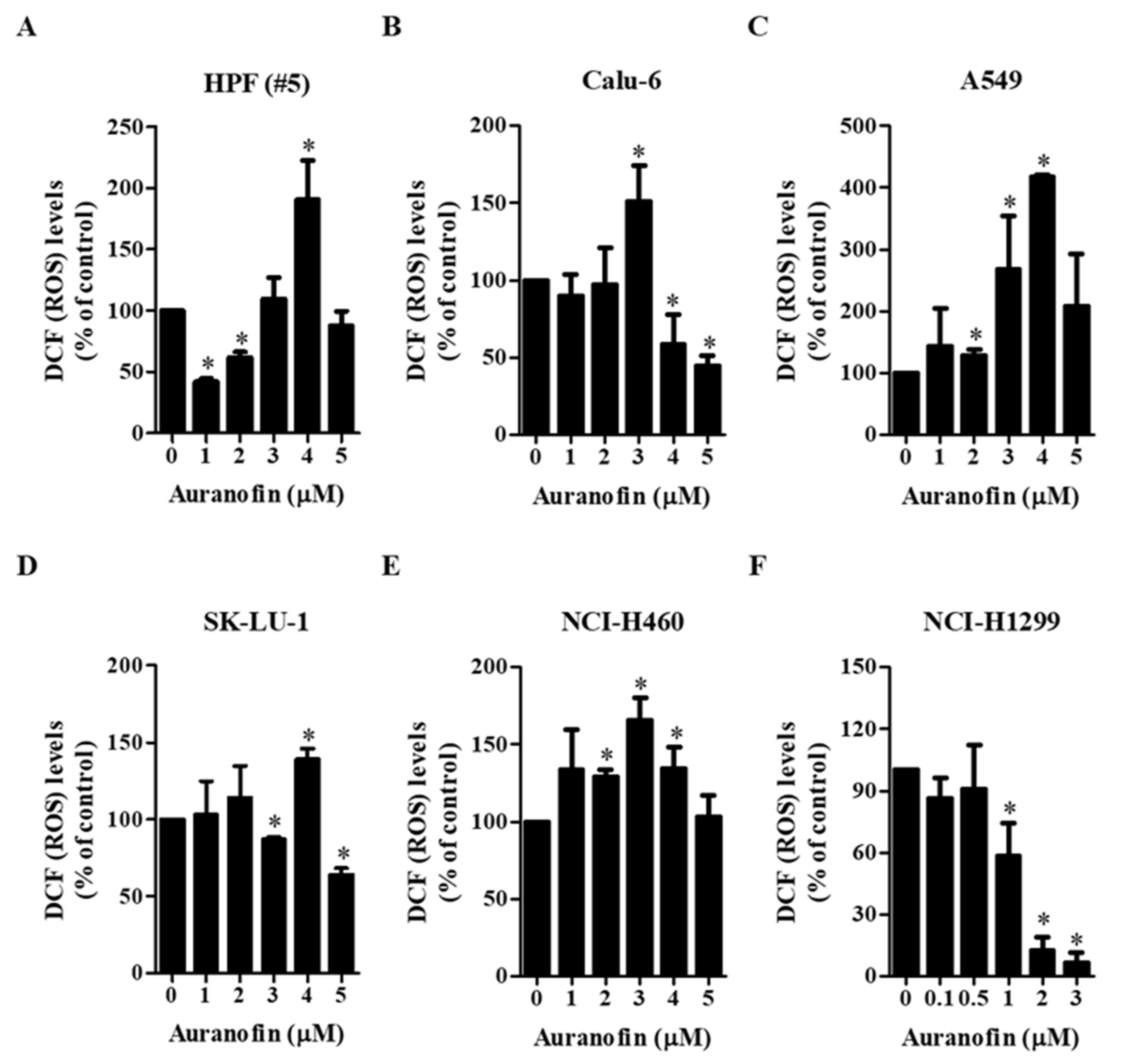
3.2. Effects of Auranofin on Intracellular ROS Levels in Lung Cancer Cells
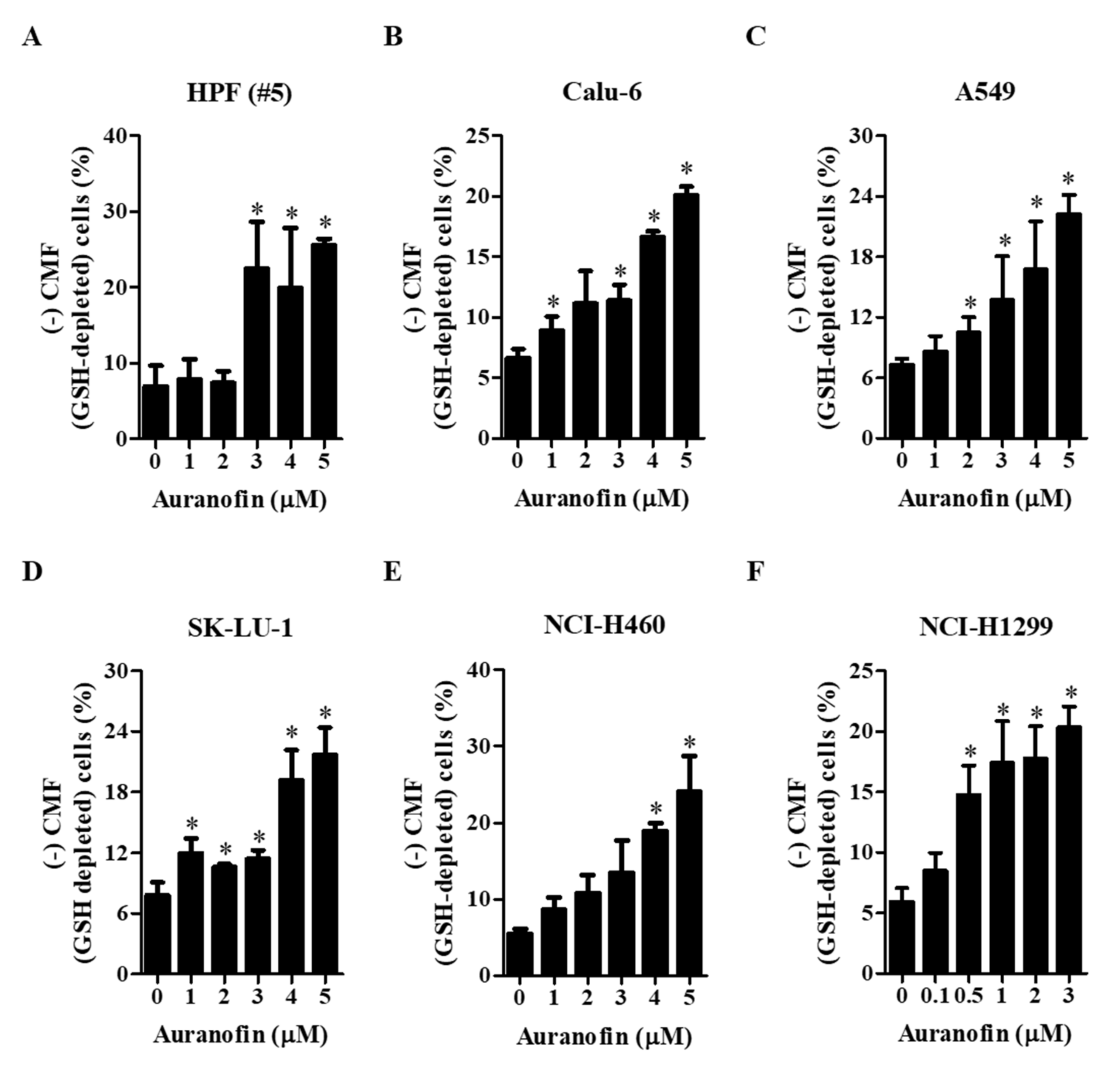
3.3. Effects of Auranofin on Intracellular GSH Depletion in Lung Cancer Cells
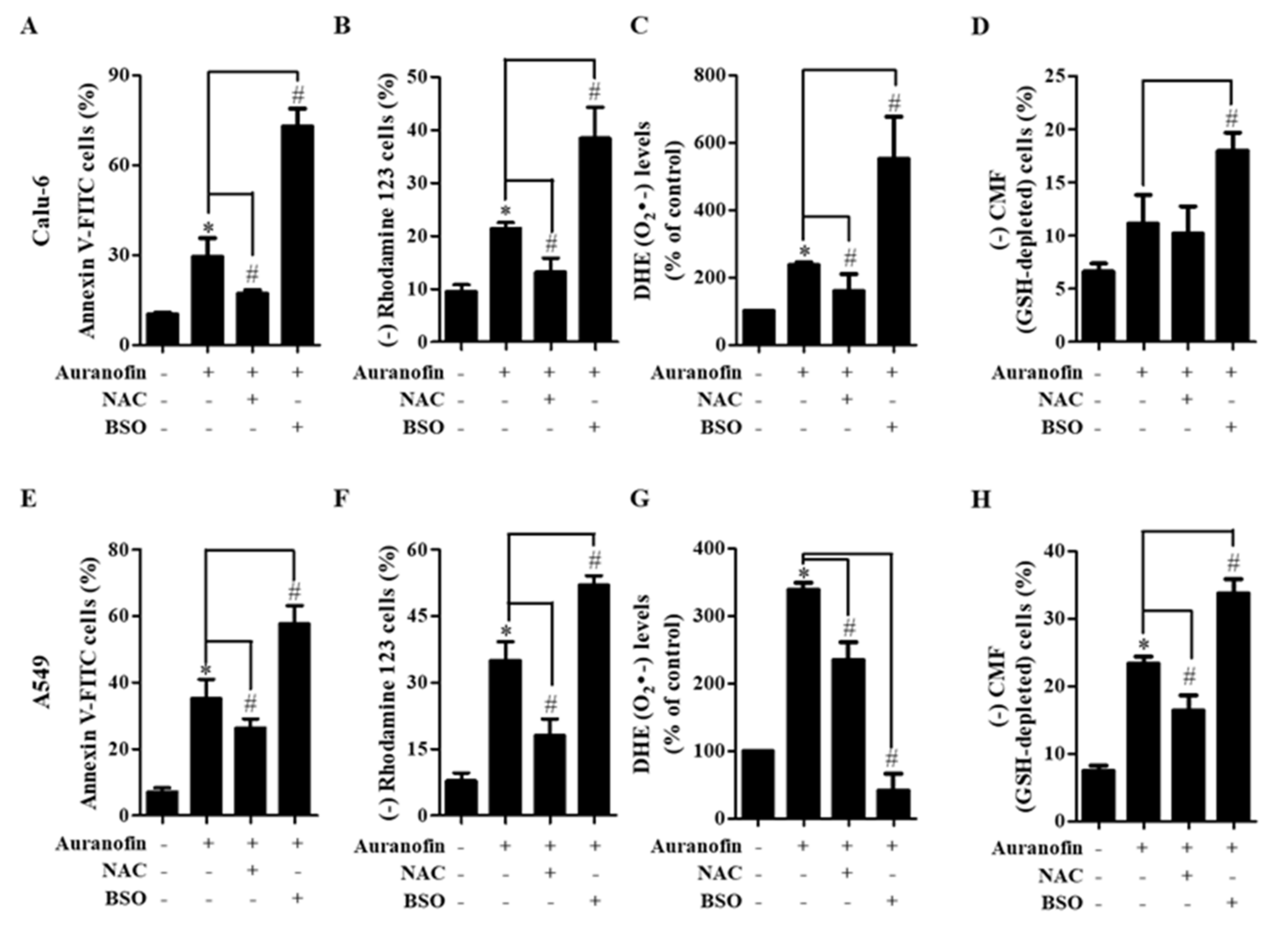
3.4. Effects of NAC and BSO on Cell Death, MMP (ΔΨm), ROS Level, and GSH Depletion in Auranofin-Treated Calu-6 and A549 Cells
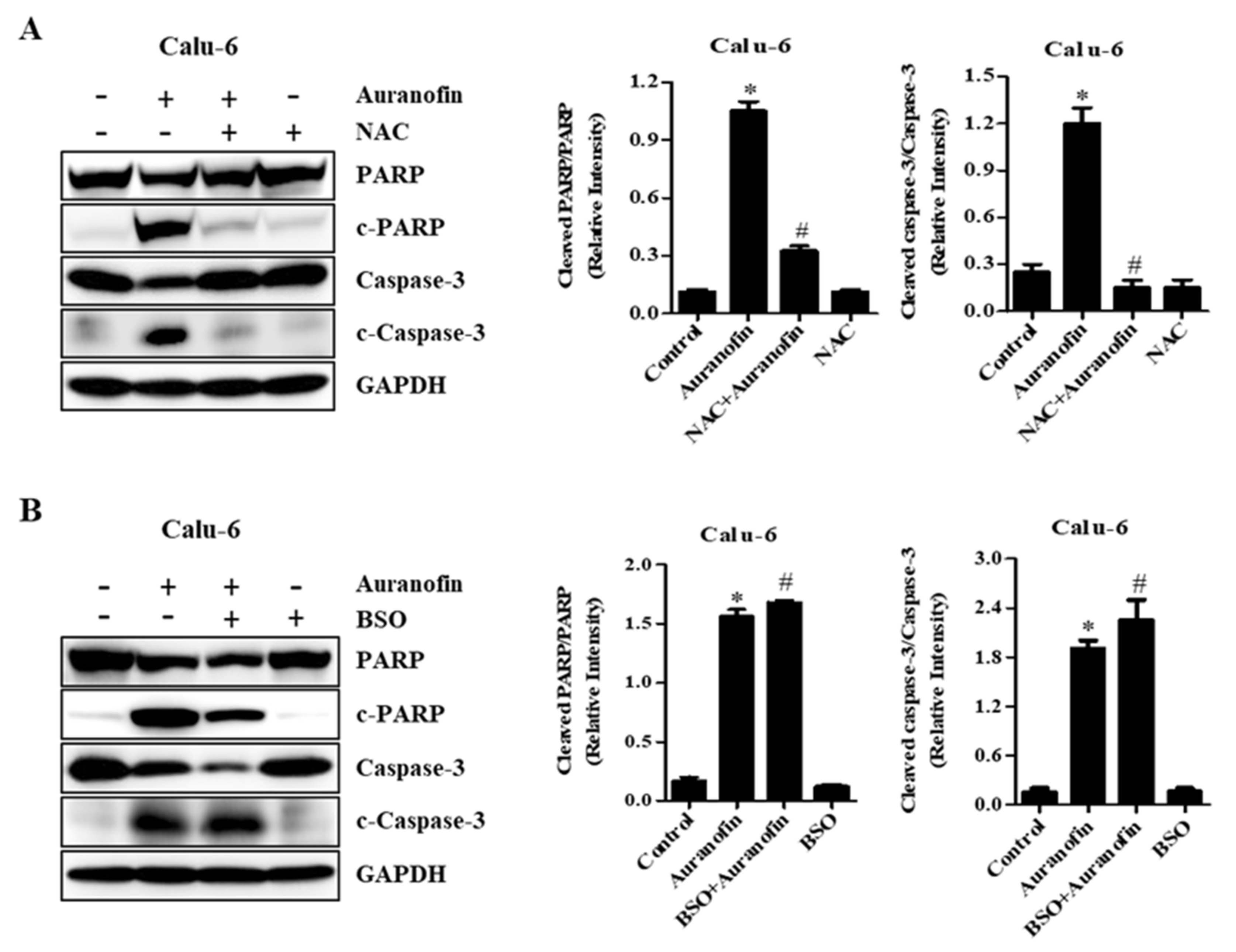
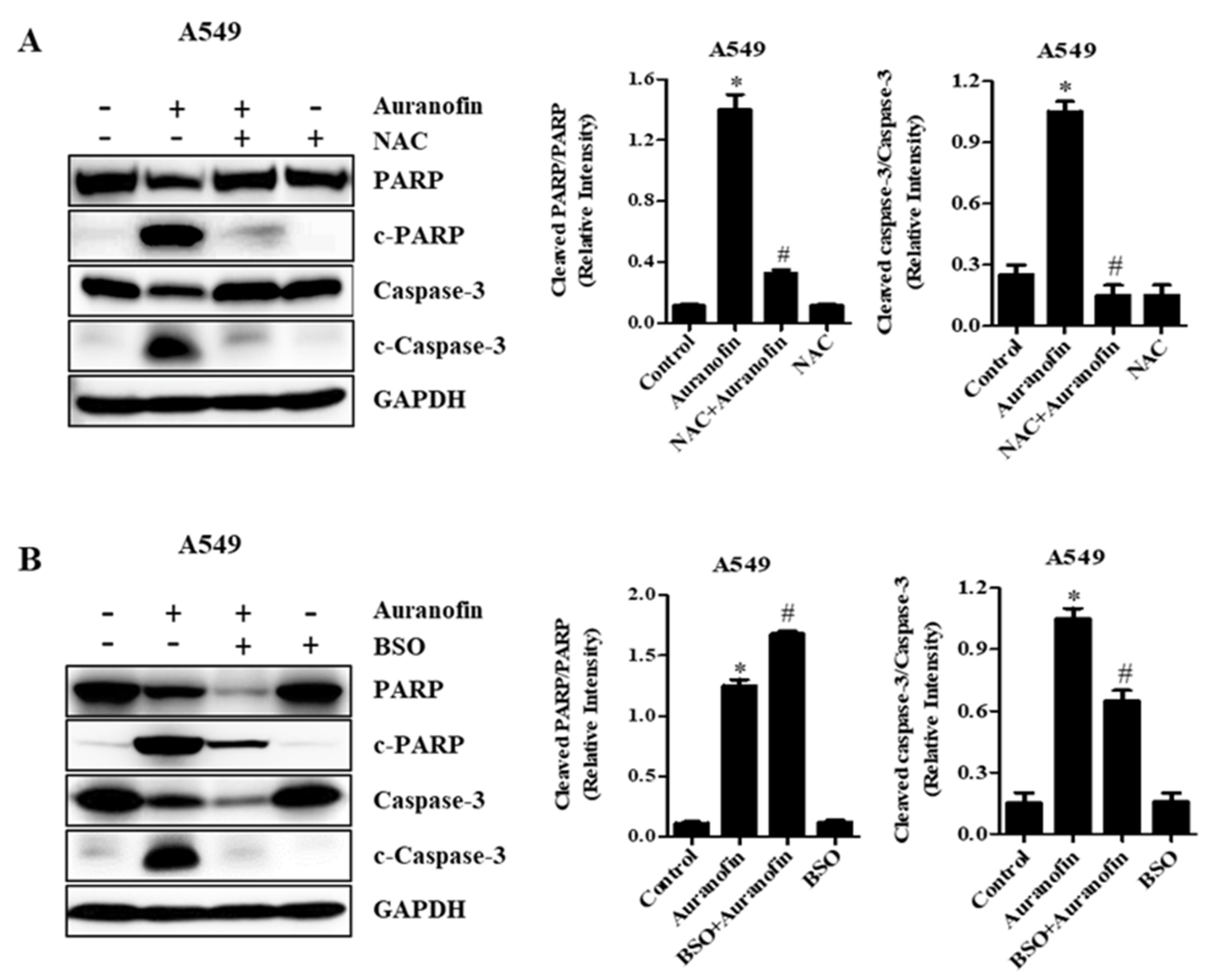
3.5. Effects of NAC and BSO on Apoptosis-Related Protein Levels in Calu-6 and A549 Cells
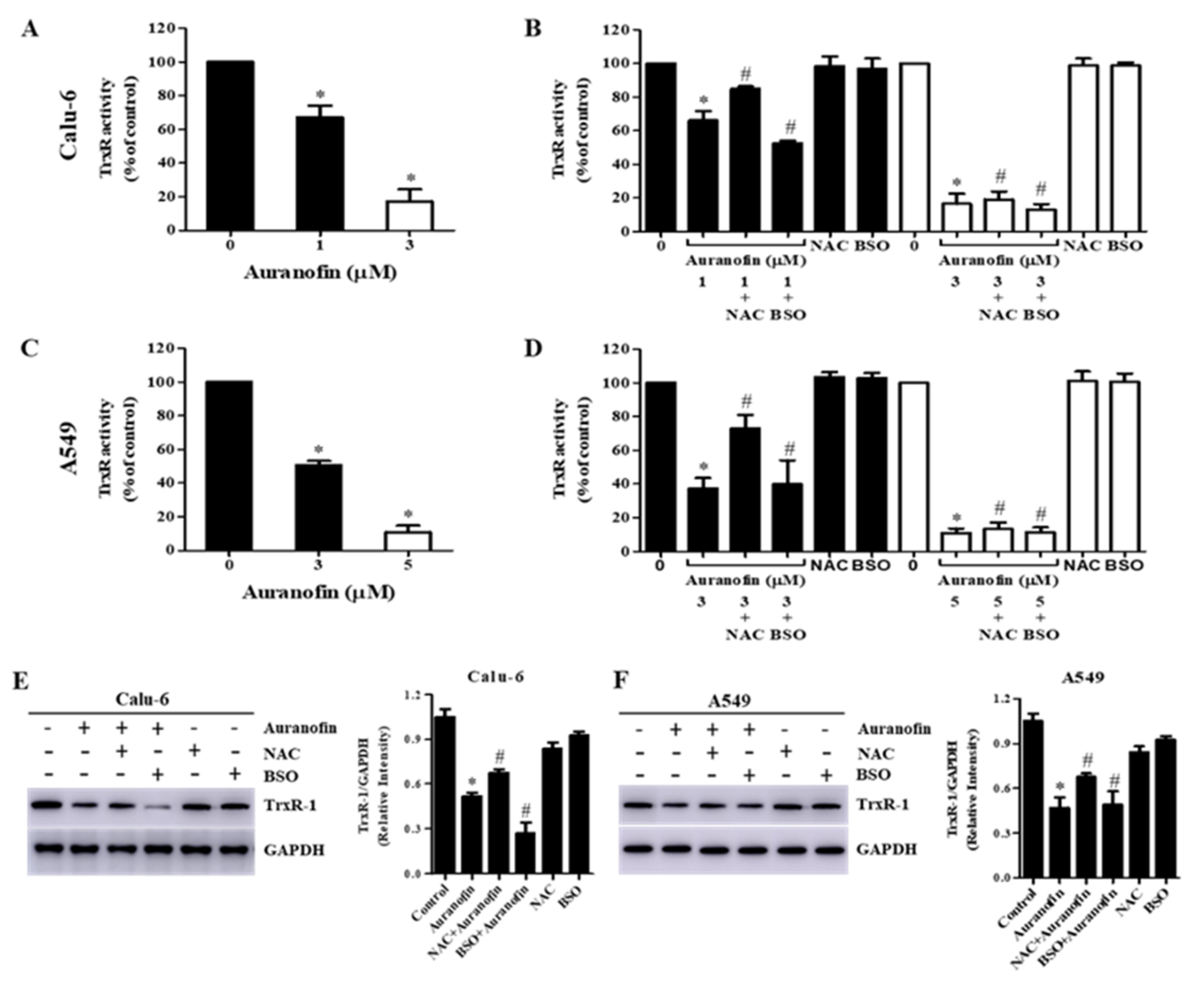
3.6. Effects of NAC and BSO on TrxR Activity in Auranofin-Treated Calu-6 and A549 Cells
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| BSO | l-buthionine sulfoximine |
| CMFDA | 5-chloromethylfluorescein diacetate |
| DHE | dihydroethidium |
| DMSO | dimethyl sulfoxide |
| DTNB | 5,5′-dithiobis (2-nitrobenzoic acid) |
| Ex/Em | Excitation/Emission |
| FBS | fetal bovine serum |
| FDA | Food and Drug Administration |
| FITC | fluorescein isothiocyanate |
| GSH | glutathione |
| H2DCFDA | 2′,7′-dichlorodihydrofluorescein diacetate |
| H2O2 | hydrogen peroxide |
| HPF | human pulmonary fibroblast |
| LUAD | lung adenocarcinoma |
| LULC | lung large-cell carcinoma |
| LUSC | lung squamous cell carcinoma |
| MMP | mitochondrial membrane potential (∆Ψm) |
| NAC | N-acetyl cysteine |
| NADPH | nicotinamide adenine dinucleotide phosphate |
| NSCLC | non-small-cell lung cancer |
| ONOO• | peroxynitrite |
| PARP | poly (ADP ribose) polymerase |
| PBS | phosphate-buffered saline |
| PVDF | poly-vinylidene difluoride |
| redox | reduction-oxidation |
| RIPA | radio immunoprecipitation assay |
| ROS | reactive oxygen species |
| SCLC | small-cell lung cancer |
| SDS-PAGE | sodium dodecyl sulfate-polyacrylamide gel electrophoresis |
| Trx | thioredoxin |
| TrxR | thiore-doxin reductase |
| •OH | hydroxyl radical |
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed]
- Blandin Knight, S.; Crosbie, P.A.; Balata, H.; Chudziak, J.; Hussell, T.; Dive, C. Progress and prospects of early detection in lung cancer. Open Biol. 2017, 7, 170070. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, T.; Miura, T.; Degawa, Y.; Fujita, Y.; Inoue, M.; Kawaguchi, M.; Furihata, C. Comparison of lung cancer cell lines representing four histopathological subtypes with gene expression profiling using quantitative real-time PCR. Cancer Cell Int. 2010, 10, 2. [Google Scholar] [CrossRef] [PubMed]
- Xu, Q.; Wang, Y.; Liu, H.; Meng, S.; Zhou, S.; Xu, J.; Schmid-Bindert, G.; Zhou, C. Treatment outcome for patients with primary NSCLC and synchronous solitary metastasis. Clin. Transl. Oncol. 2013, 15, 802–809. [Google Scholar] [CrossRef]
- De Silva, M.; Itchins, M.; Pavlakis, N. Breakthrough 5-year survival with pembrolizumab in Keynote-001 study: Horizon shifting in advanced non-small cell lung cancer with immune check point inhibition. Ann. Transl. Med. 2020, 8, 555. [Google Scholar] [CrossRef]
- Noreldeen, H.A.A.; Liu, X.; Xu, G. Metabolomics of lung cancer: Analytical platforms and their applications. J. Sep. Sci. 2020, 43, 120–133. [Google Scholar] [CrossRef]
- Sharma, P.; Mehta, M.; Dhanjal, D.S.; Kaur, S.; Gupta, G.; Singh, H.; Thangavelu, L.; Rajeshkumar, S.; Tambuwala, M.; Bakshi, H.A.; et al. Emerging trends in the novel drug delivery approaches for the treatment of lung cancer. Chem. Biol. Interact. 2019, 309, 108720. [Google Scholar] [CrossRef]
- Nagasaka, M.; Gadgeel, S.M. Role of chemotherapy and targeted therapy in early-stage non-small cell lung cancer. Expert Rev. Anticancer 2018, 18, 63–70. [Google Scholar] [CrossRef]
- Wang, L.; Li, X.; Ren, Y.; Geng, H.; Zhang, Q.; Cao, L.; Meng, Z.; Wu, X.; Xu, M.; Xu, K. Cancer-associated fibroblasts contribute to cisplatin resistance by modulating ANXA3 in lung cancer cells. Cancer Sci. 2019, 110, 1609–1620. [Google Scholar] [CrossRef]
- Becker, A.; van Wijk, A.; Smit, E.F.; Postmus, P.E. Side-effects of long-term administration of erlotinib in patients with non-small cell lung cancer. J. Thorac. Oncol. 2010, 5, 1477–1480. [Google Scholar] [CrossRef]
- Davalli, P.; Mitic, T.; Caporali, A.; Lauriola, A.; D’Arca, D. ROS, Cell Senescence, and Novel Molecular Mechanisms in Aging and Age-Related Diseases. Oxid. Med. Cell Longev. 2016, 2016, 3565127. [Google Scholar] [CrossRef]
- Sena, L.A.; Chandel, N.S. Physiological roles of mitochondrial reactive oxygen species. Mol. Cell. 2012, 48, 158–167. [Google Scholar] [CrossRef]
- Brieger, K.; Schiavone, S.; Miller, F.J., Jr.; Krause, K.H. Reactive oxygen species: From health to disease. Swiss Med. Wkly. 2012, 142, w13659. [Google Scholar] [CrossRef]
- Trachootham, D.; Alexandre, J.; Huang, P. Targeting cancer cells by ROS-mediated mechanisms: A radical therapeutic approach? Nat. Rev. Drug Discov. 2009, 8, 579–591. [Google Scholar] [CrossRef]
- Nogueira, V.; Hay, N. Molecular pathways: Reactive oxygen species homeostasis in cancer cells and implications for cancer therapy. Clin. Cancer Res. 2013, 19, 4309–4314. [Google Scholar] [CrossRef]
- Perillo, B.; Di Donato, M.; Pezone, A.; Di Zazzo, E.; Giovannelli, P.; Galasso, G.; Castoria, G.; Migliaccio, A. ROS in cancer therapy: The bright side of the moon. Exp. Mol. Med. 2020, 52, 192–203. [Google Scholar] [CrossRef]
- Hua, P.; Sun, M.; Zhang, G.; Zhang, Y.; Song, G.; Liu, Z.; Li, X.; Zhang, X.; Li, B. Costunolide Induces Apoptosis through Generation of ROS and Activation of P53 in Human Esophageal Cancer Eca-109 Cells. J. Biochem. Mol. Toxicol. 2016, 30, 462–469. [Google Scholar] [CrossRef]
- Park, W.H. Gallic acid induces HeLa cell death via increasing GSH depletion rather than ROS levels. Oncol. Rep. 2017, 37, 1277–1283. [Google Scholar] [CrossRef]
- Gao, C.; Sun, X.; Wu, Z.; Yuan, H.; Han, H.; Huang, H.; Shu, Y.; Xu, M.; Gao, R.; Li, S.; et al. A Novel Benzofuran Derivative Moracin N Induces Autophagy and Apoptosis Through ROS Generation in Lung Cancer. Front Pharm. 2020, 11, 391. [Google Scholar] [CrossRef]
- Lu, J.; Holmgren, A. The thioredoxin antioxidant system. Free Radic. Biol. Med. 2014, 66, 75–87. [Google Scholar] [CrossRef]
- Jia, J.J.; Geng, W.S.; Wang, Z.Q.; Chen, L.; Zeng, X.S. The role of thioredoxin system in cancer: Strategy for cancer therapy. Cancer Chemother. Pharm. 2019, 84, 453–470. [Google Scholar] [CrossRef]
- Arner, E.S.; Holmgren, A. Physiological functions of thioredoxin and thioredoxin reductase. Eur. J. Biochem. 2000, 267, 6102–6109. [Google Scholar] [CrossRef]
- Selenius, M.; Rundlof, A.K.; Olm, E.; Fernandes, A.P.; Bjornstedt, M. Selenium and the selenoprotein thioredoxin reductase in the prevention, treatment and diagnostics of cancer. Antioxid. Redox. Signal 2010, 12, 867–880. [Google Scholar] [CrossRef]
- Karlenius, T.C.; Tonissen, K.F. Thioredoxin and Cancer: A Role for Thioredoxin in all States of Tumor Oxygenation. Cancers 2010, 2, 209–232. [Google Scholar] [CrossRef]
- Mohammadi, F.; Soltani, A.; Ghahremanloo, A.; Javid, H.; Hashemy, S.I. The thioredoxin system and cancer therapy: A review. Cancer Chemother. Pharmacol. 2019, 84, 925–935. [Google Scholar] [CrossRef]
- Zheng, X.; Ma, W.; Sun, R.; Yin, H.; Lin, F.; Liu, Y.; Xu, W.; Zeng, H. Butaselen prevents hepatocarcinogenesis and progression through inhibiting thioredoxin reductase activity. Redox Biol. 2018, 14, 237–249. [Google Scholar] [CrossRef]
- Bhatia, M.; McGrath, K.L.; Di Trapani, G.; Charoentong, P.; Shah, F.; King, M.M.; Clarke, F.M.; Tonissen, K.F. The thioredoxin system in breast cancer cell invasion and migration. Redox Biol. 2016, 8, 68–78. [Google Scholar] [CrossRef]
- Lim, J.Y.; Yoon, S.O.; Hong, S.W.; Kim, J.W.; Choi, S.H.; Cho, J.Y. Thioredoxin and thioredoxin-interacting protein as prognostic markers for gastric cancer recurrence. World J. Gastroenterol. 2012, 18, 5581–5588. [Google Scholar] [CrossRef]
- Uzawa, K.; Iwasawa, S.; Yamano, Y.; Takiguchi, Y.; Tanzawa, H.; Tatsumi, K. Upregulation of thioredoxin reductase 1 in human oral squamous cell carcinoma. Oncol. Rep. 2011, 25, 637–644. [Google Scholar] [CrossRef]
- Fernandes, A.P.; Capitanio, A.; Selenius, M.; Brodin, O.; Rundlöf, A.-K.; Björnstedt, M. Expression profiles of thioredoxin family proteins in human lung cancer tissue: Correlation with proliferation and differentiation. Histopathology 2009, 55, 313–320. [Google Scholar] [CrossRef]
- Onodera, T.; Momose, I.; Kawada, M. Potential Anticancer Activity of Auranofin. Chem. Pharm. Bull. 2019, 67, 186–191. [Google Scholar] [CrossRef] [PubMed]
- Chaffman, M.; Brogden, R.; Heel, R.; Speight, T.; Avery, G.J.D. Auranofin. A preliminary review of its pharmacological properties and therapeutic use in rheumatoid arthritis. Drugs 1984, 27, 378–424. [Google Scholar] [CrossRef] [PubMed]
- Madeira, J.M.; Gibson, D.L.; Kean, W.F.; Klegeris, A. The biological activity of auranofin: Implications for novel treatment of diseases. Inflammopharmacology 2012, 20, 297–306. [Google Scholar] [CrossRef] [PubMed]
- Roder, C.; Thomson, M.J. Auranofin: Repurposing an Old Drug for a Golden New Age. Drugs R&D 2015, 15, 13–20. [Google Scholar] [CrossRef]
- Rigobello, M.P.; Messori, L.; Marcon, G.; Cinellu, M.A.; Bragadin, M.; Folda, A.; Scutari, G.; Bindoli, A. Gold complexes inhibit mitochondrial thioredoxin reductase: Consequences on mitochondrial functions. J. Inorg. Biochem. 2004, 98, 1634–1641. [Google Scholar] [CrossRef]
- Zhang, X.; Selvaraju, K.; Saei, A.A.; D’Arcy, P.; Zubarev, R.A.; Arnér, E.S.; Linder, S. Repurposing of auranofin: Thioredoxin reductase remains a primary target of the drug. Biochimie 2019, 162, 46–54. [Google Scholar] [CrossRef]
- Fiskus, W.; Saba, N.; Shen, M.; Ghias, M.; Liu, J.; Das Gupta, S.; Chauhan, L.; Rao, R.; Gunewardena, S.; Schorno, K.; et al. Auranofin Induces Lethal Oxidative and Endoplasmic Reticulum Stress and Exerts Potent Preclinical Activity against Chronic Lymphocytic Leukemia. Cancer Res. 2014, 74, 2520–2532. [Google Scholar] [CrossRef]
- Zou, P.; Chen, M.; Ji, J.; Chen, W.; Chen, X.; Ying, S.; Zhang, J.; Zhang, Z.; Liu, Z.; Yang, S.; et al. Auranofin induces apoptosis by ROS-mediated ER stress and mitochondrial dysfunction and displayed synergistic lethality with piperlongumine in gastric cancer. Oncotarget 2015, 6, 36505–36521. [Google Scholar] [CrossRef]
- Cui, X.Y.; Park, S.H.; Park, W.H. Auranofin inhibits the proliferation of lung cancer cells via necrosis and caspase-dependent apoptosis. Oncol. Rep. 2020, 44, 2715–2724. [Google Scholar] [CrossRef]
- Ott, I. On the medicinal chemistry of gold complexes as anticancer drugs. Co-ord. Chem. Rev. 2009, 253, 1670–1681. [Google Scholar] [CrossRef]
- Boullosa, L.F.; Van Loenhout, J.; Flieswasser, T.; De Waele, J.; Hermans, C.; Lambrechts, H.; Cuypers, B.; Laukens, K.; Bartholomeus, E.; Siozopoulou, V.; et al. Auranofin reveals therapeutic anticancer potential by triggering distinct molecular cell death mechanisms and innate immunity in mutant p53 non-small cell lung cancer. Redox Biol. 2021, 42, 101949. [Google Scholar] [CrossRef]
- Dunigan-Russell, K.; Li, Q.; Li, R.; Locy, M.L.; Wall, S.B.; Tipple, T.E. The thioredoxin reductase inhibitor auranofin induces heme oxygenase-1 in lung epithelial cells via Nrf2-dependent mechanisms. Am. J. Physiol. Cell. Mol. Physiol. 2018, 315, L545–L552. [Google Scholar] [CrossRef]
- Li, H.; Hu, J.; Wu, S.; Wang, L.; Cao, X.; Zhang, X.; Dai, B.; Cao, M.; Shao, R.; Zhang, R.; et al. Auranofin-mediated inhibition of PI3K/AKT/mTOR axis and anticancer activity in non-small cell lung cancer cells. Oncotarget 2015, 7, 3548–3558. [Google Scholar] [CrossRef]
- Gandin, V.; Fernandes, A.P.; Rigobello, M.P.; Dani, B.; Sorrentino, F.; Tisato, F.; Björnstedt, M.; Bindoli, A.; Sturaro, A.; Rella, R.; et al. Cancer cell death induced by phosphine gold(I) compounds targeting thioredoxin reductase. Biochem. Pharmacol. 2010, 79, 90–101. [Google Scholar] [CrossRef]
- Weir, S.J.; DeGennaro, L.J.; Austin, C.P. Repurposing Approved and Abandoned Drugs for the Treatment and Prevention of Cancer through Public-Private Partnership. Cancer Res. 2012, 72, 1055–1058. [Google Scholar] [CrossRef]
- You, B.R.; Park, W.H. Auranofin induces mesothelioma cell death through oxidative stress and GSH depletion. Oncol. Rep. 2016, 35, 546–551. [Google Scholar] [CrossRef]
- Hwang-Bo, H.; Jeong, J.-W.; Han, M.H.; Park, C.; Hong, S.-H.; Kim, G.-Y.; Moon, S.-K.; Cheong, J.; Kim, W.-J.; Yoo, Y.H.; et al. Auranofin, an inhibitor of thioredoxin reductase, induces apoptosis in hepatocellular carcinoma Hep3B cells by generation of reactive oxygen species. Gen. Physiol. Biophys. 2017, 36, 117–128. [Google Scholar] [CrossRef]
- Ehrenfeld, V.; Heusel, J.R.; Fulda, S.; Van Wijk, S.J.L. ATM inhibition enhances Auranofin-induced oxidative stress and cell death in lung cell lines. PLoS ONE 2020, 15, e0244060. [Google Scholar] [CrossRef]
- Di Sarra, F.; Fresch, B.; Bini, R.; Saielli, G.; Bagno, A. Reactivity of Auranofin with Selenols and Thiols-Implications for the Anticancer Activity of Gold(I) Compounds. Eur. J. Inorg. Chem. 2013, 2013, 2718–2727. [Google Scholar] [CrossRef]
- Ferrer, M.D.; Sureda, A.; Tauler, P.; Palacín, C.; Tur, J.A.; Pons, A. Impaired lymphocyte mitochondrial antioxidant defences in variegate porphyria are accompanied by more inducible reactive oxygen species production and DNA damage. Br. J. Haematol. 2010, 149, 759–767. [Google Scholar] [CrossRef]
- Uttara, B.; Singh, A.V.; Zamboni, P.; Mahajan, R.T. Oxidative Stress and Neurodegenerative Diseases: A Review of Upstream and Downstream Antioxidant Therapeutic Options. Curr. Neuropharmacol. 2009, 7, 65–74. [Google Scholar] [CrossRef]
- Powis, G.; Mustacich, D.; Coon, A. Medicine, The role of the redox protein thioredoxin in cell growth and cancer. Free Radic. Biol. Med. 2000, 29, 312–322. [Google Scholar] [CrossRef]
- Ortega, A.L.; Mena, S.; Estrela, J.M. Glutathione in Cancer Cell Death. Cancers 2011, 3, 1285–1310. [Google Scholar] [CrossRef]
- Winterbourn, C.C. Toxicity of iron and hydrogen peroxide: The Fenton reaction. Toxicol. Lett. 1995, 82–83, 969–974. [Google Scholar] [CrossRef]
- Aldini, G.; Altomare, A.A.; Baron, G.; Vistoli, G.; Carini, M.; Borsani, L.; Sergio, F. N-Acetylcysteine as an antioxidant and disulphide breaking agent: The reasons why. Free Radic. Res. 2018, 52, 751–762. [Google Scholar] [CrossRef]
- Hayes, J.; McLellan, L.I. Glutathione and glutathione-dependent enzymes represent a co-ordinately regulated defence against oxidative stress. Free Radic. Res. 1999, 31, 273–300. [Google Scholar] [CrossRef]
- Iversen, R.; Andersen, P.A.; Jensen, K.S.; Winther, J.R.; Sigurskjold, B.W. Thiol-Disulfide Exchange between Glutaredoxin and Glutathione. Biochemistry 2010, 49, 810–820. [Google Scholar] [CrossRef]
- Kumar, C.; Igbaria, A.; D’Autreaux, B.; Planson, A.-G.; Junot, C.; Godat, E.; Bachhawat, A.K.; Delaunay-Moisan, A.; Toledano, M.B. Glutathione revisited: A vital function in iron metabolism and ancillary role in thiol-redox control. EMBO J. 2011, 30, 2044–2056. [Google Scholar] [CrossRef]
- Dunning, S.; Rehman, A.U.; Tiebosch, M.H.; Hannivoort, R.A.; Haijer, F.W.; Woudenberg, J.; Heuvel, F.A.V.D.; Buist-Homan, M.; Faber, K.N.; Moshage, H. Glutathione and antioxidant enzymes serve complementary roles in protecting activated hepatic stellate cells against hydrogen peroxide-induced cell death. Biochim. Biophys. Acta Mol. Basis Dis. 2013, 1832, 2027–2034. [Google Scholar] [CrossRef]
- Franco, R.; Cidlowski, J.A. Glutathione Efflux and Cell Death. Antioxidants Redox Signal. 2012, 17, 1694–1713. [Google Scholar] [CrossRef]
- Elmore, S. Apoptosis: A review of programmed cell death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef] [PubMed]
- Budihardjo, I.; Oliver, H.; Lutter, M.; Luo, X.; Wang, X. Biochemical Pathways of Caspase Activation During Apoptosis. Annu. Rev. Cell Dev. Biol. 1999, 15, 269–290. [Google Scholar] [CrossRef] [PubMed]
- Fan, T.-J.; Han, L.-H.; Cong, R.-S.; Liang, J. Caspase Family Proteases and Apoptosis. Acta Biochim. Biophys. Sin. 2005, 37, 719–727. [Google Scholar] [CrossRef] [PubMed]
- Cohen, G.M. Caspases: The executioners of apoptosis. Biochem. J. 1997, 326, 1–16. [Google Scholar] [CrossRef]
- Choudhary, G.S.; Al-Harbi, S.; Almasan, A. Caspase-3 activation is a critical determinant of genotoxic stress-induced apoptosis. In Apoptosis and Cancer; Springer: Berlin/Heidelberg, Germany, 2015; pp. 1–9. [Google Scholar]
- Brauns, S.C.; Dealtry, G.; Milne, P.; Naudé, R.; Van De Venter, M. Caspase-3 activation and induction of PARP cleavage by cyclic dipeptide cyclo(Phe-Pro) in HT-29 cells. Anticancer Res. 2005, 25, 4197–4202. [Google Scholar]
- Soini, Y.; Kahlos, K.; Näpänkangas, U.; Kaarteenaho-Wiik, R.; Säily, M.; Koistinen, P.; Pääakkö, P.; Holmgren, A.; Kinnula, V.L. Widespread expression of thioredoxin and thioredoxin reductase in non-small cell lung carcinoma. Clin. Cancer Res. 2001, 7, 1750–1757. [Google Scholar]
- Noike, T.; Miwa, S.; Soeda, J.; Kobayashi, A.; Miyagawa, S.-I. Increased expression of thioredoxin-1, vascular endothelial growth factor, and redox factor-1 is associated with poor prognosis in patients with liver metastasis from colorectal cancer. Hum. Pathol. 2008, 39, 201–208. [Google Scholar] [CrossRef]
- Kim, S.J.; Miyoshi, Y.; Taguchi, T.; Tamaki, Y.; Nakamura, H.; Yodoi, J.; Kato, K.; Noguchi, S. High Thioredoxin Expression Is Associated with Resistance to Docetaxel in Primary Breast Cancer. Clin. Cancer Res. 2005, 11, 8425–8430. [Google Scholar] [CrossRef]
- Zhang, J.; Li, X.; Han, X.; Liu, R.; Fang, J. Targeting the Thioredoxin System for Cancer Therapy. Trends Pharmacol. Sci. 2017, 38, 794–808. [Google Scholar] [CrossRef]
- Yokomizo, A.; Ono, M.; Nanri, H.; Makino, Y.; Ohga, T.; Wada, M.; Okamoto, T.; Yodoi, J.; Kuwano, M.; Kohno, K. Cellular levels of thioredoxin associated with drug sensitivity to cisplatin, mitomycin C, doxorubicin, and etoposide. Cancer Res. 1995, 55, 4293–4296. [Google Scholar]
- Javvadi, P.; Hertan, L.; Kosoff, R.; Datta, T.; Kolev, J.; Mick, R.; Tuttle, S.W.; Koumenis, C. Thioredoxin Reductase-1 Mediates Curcumin-Induced Radiosensitization of Squamous Carcinoma Cells. Cancer Res. 2010, 70, 1941–1950. [Google Scholar] [CrossRef]
- Liang, Y.-W.; Zheng, J.; Li, X.; Zheng, W.; Chen, T. Selenadiazole derivatives as potent thioredoxin reductase inhibitors that enhance the radiosensitivity of cancer cells. Eur. J. Med. Chem. 2014, 84, 335–342. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cui, X.Y.; Park, S.H.; Park, W.H. Anti-Cancer Effects of Auranofin in Human Lung Cancer Cells by Increasing Intracellular ROS Levels and Depleting GSH Levels. Molecules 2022, 27, 5207. https://doi.org/10.3390/molecules27165207
Cui XY, Park SH, Park WH. Anti-Cancer Effects of Auranofin in Human Lung Cancer Cells by Increasing Intracellular ROS Levels and Depleting GSH Levels. Molecules. 2022; 27(16):5207. https://doi.org/10.3390/molecules27165207
Chicago/Turabian StyleCui, Xia Ying, Sun Hyang Park, and Woo Hyun Park. 2022. "Anti-Cancer Effects of Auranofin in Human Lung Cancer Cells by Increasing Intracellular ROS Levels and Depleting GSH Levels" Molecules 27, no. 16: 5207. https://doi.org/10.3390/molecules27165207
APA StyleCui, X. Y., Park, S. H., & Park, W. H. (2022). Anti-Cancer Effects of Auranofin in Human Lung Cancer Cells by Increasing Intracellular ROS Levels and Depleting GSH Levels. Molecules, 27(16), 5207. https://doi.org/10.3390/molecules27165207