
Targeting Microglia in Alzheimer’s Disease: From Molecular Mechanisms to Potential Therapeutic Targets for Small Molecules


Abstract
1. Introduction

{kind=link}
{kind=link}
{kind=link}
| Drug | Approved Indication | Mode of Action | Dose | Titration Scheme | References |
|---|---|---|---|---|---|
| Memantine | Moderate-to-severe Alzheimer’s disease (AD) | Non-competitively antagonize N-methyl-D-aspartic acid receptor | 5–20 mg/day | Initially 5 mg/day, subsequently increase 5 mg at weekly intervals to a maximum dose of 20 mg/day | [2,38,39,40,41,42,43] |
| Galantamine | Mild-to-moderate AD | Selectively, reversibly, and competitively suppress AChE | 16–24 mg/day | Initially 8 mg once per day for four weeks, subsequently increase to 16 mg once per day for minimum four weeks; maintenance therapy is 16–24 mg once per day | [38,39,42,43,44] |
| Rivastigmine | Mild-to-moderate AD | Pseudo-selectively and irreversibly suppress butyrylcholinesterase and AChE | 1.5–6 mg/day | Initially 1.5 mg two times per day and the dose can be increased up to 1.5 mg two times per day at intervals of minimum two weeks as per the tolerance; the maximum dose is 6 mg two times per day | [38,39,42,43,45] |
| Donepezil | All stages of AD | Selectively, non-competitively, and reversibly suppress AChE | 5–10 mg/day | Initially 5 mg/day; if necessary, the dose can be increased up to 10 mg after 1 month | [38,39,42,43,46,47] |
2. Molecular Pathogenesis of Alzheimer’s Disease
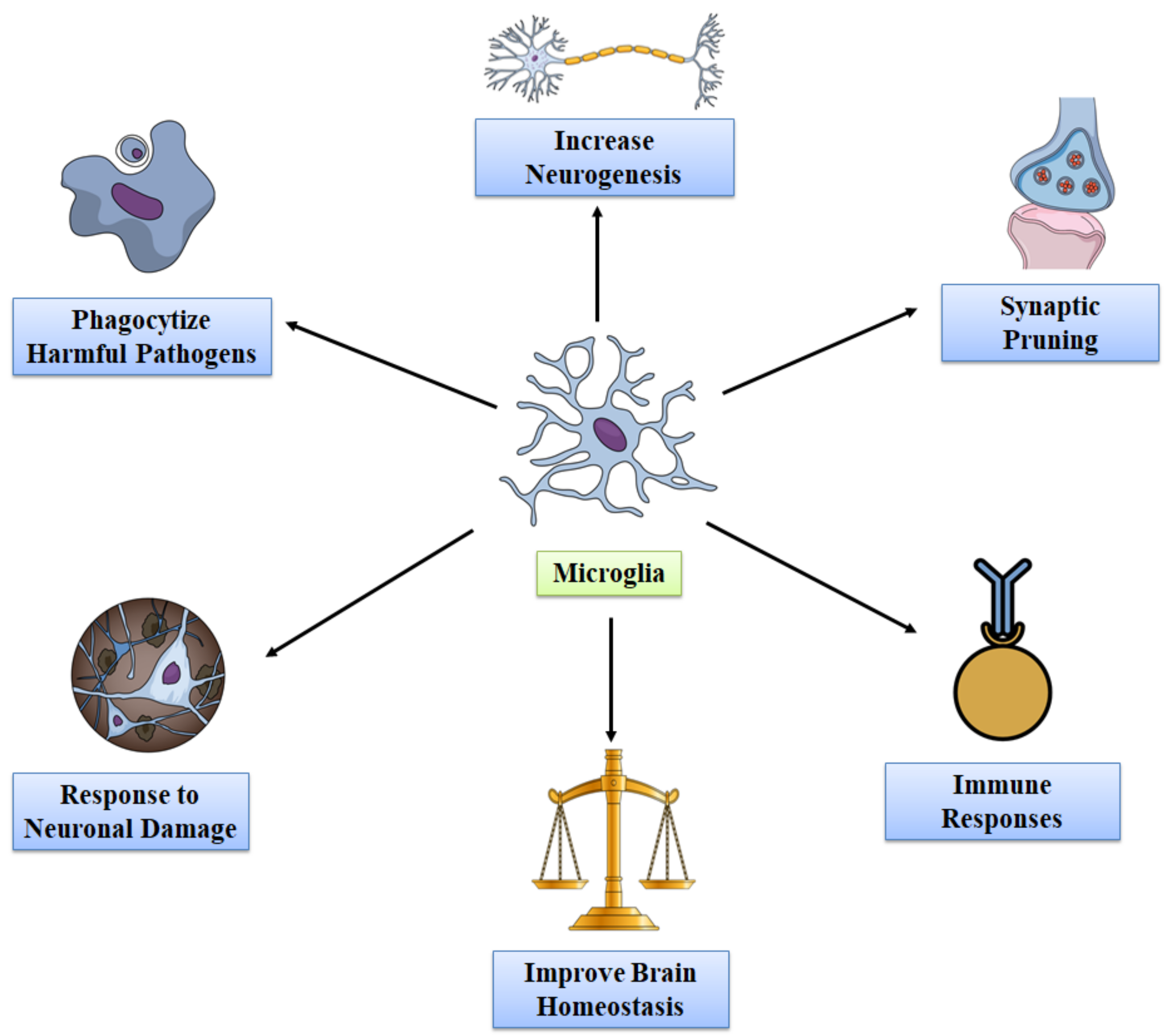
3. Functions of Microglia in Healthy Adult Brain
4. Effect of Microglia on Aging Brain
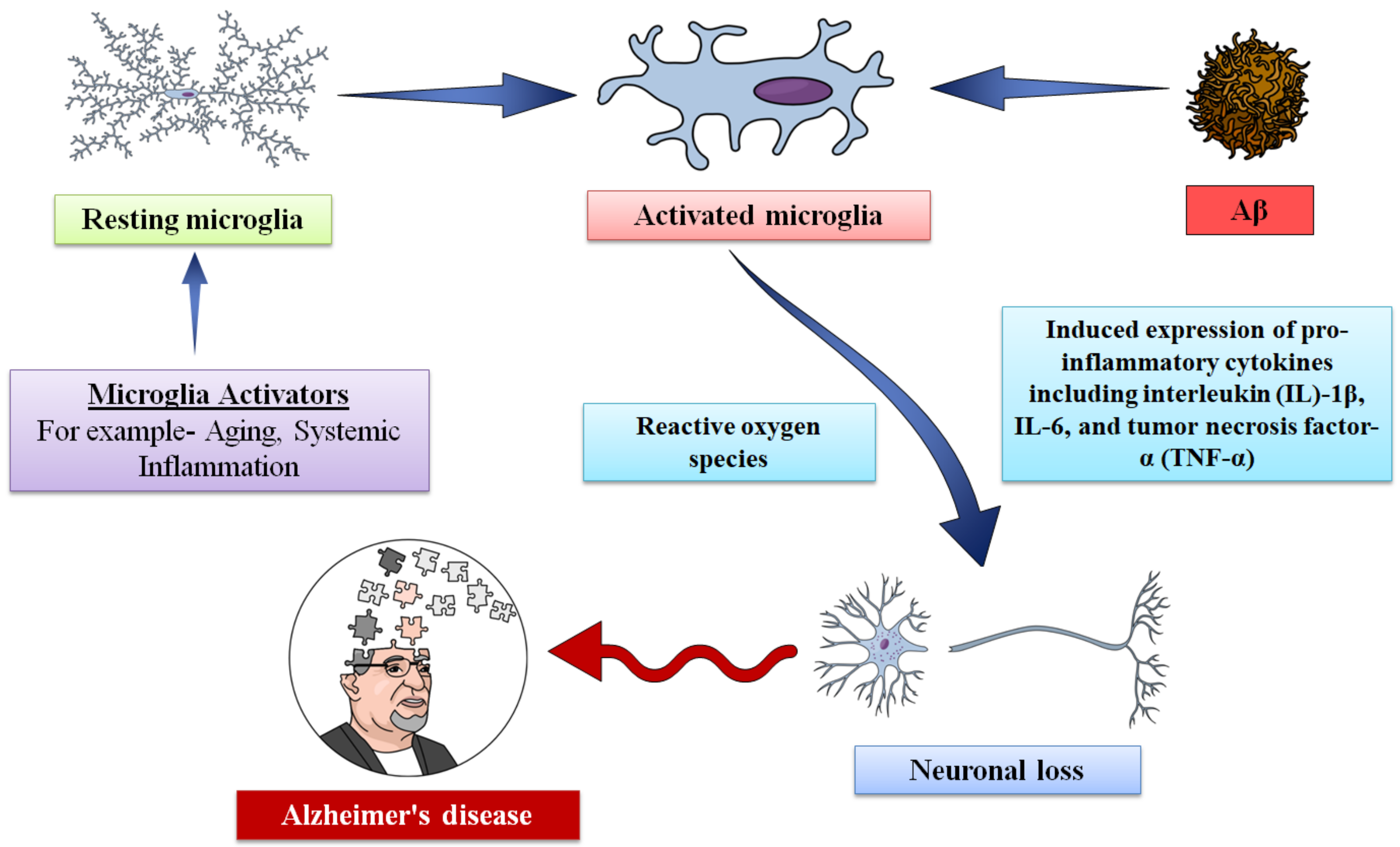
5. Role of Microglia in Alzheimer’s Disease
5.1. Microglial Mitophagy
5.2. Role of Microglia in Amyloid Beta
5.3. Effects of Microglia in Tau Protein
5.4. Effect of Microglia in Neuroinflammation
5.5. Detrimental Activities of Microglia in AD
6. Targeting Microglia for the Treatment of Alzheimer’s Disease
6.1. Therapeutics to Modify Microglia
6.1.1. CSF1R Inhibitors
6.1.2. Stem Cell Therapy
6.2. Targeting Microglial Immunoreceptors
6.2.1. Targeting TREM2 Gene
6.2.2. Targeting CD33 Gene
6.3. Targeting Inflammatory Response Mediated by Microglia
6.3.1. Non-Steroidal Anti-Inflammatory Drugs
6.3.2. NLRP3 Inflammasome Inhibitors
6.3.3. P2X7R Inhibitors
7. Future Directions
8. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- De Ture, M.A.; Dickson, D.W. The neuropathological diagnosis of Alzheimer’s disease. Mol. Neurodegener. 2019, 14, 32. [Google Scholar] [CrossRef] [PubMed]
- Karthika, C.; Appu, A.P.; Akter, R.; Rahman, H.; Tagde, P.; Ashraf, G.M.; Abdel-Daim, M.M.; Hassan, S.S.U.; Abid, A.; Bungau, S. Potential innovation against Alzheimer’s disorder: A tricomponent combination of natural antioxidants (vitamin E, quercetin, and basil oil) and the development of its intranasal delivery. Environ. Sci. Pollut. Res. 2022, 29, 10950–10965. [Google Scholar] [CrossRef] [PubMed]
- Glabe, C.C. Amyloid Accumulation and Pathogensis of Alzheimer’s Disease: Significance of Monomeric, Oligomeric and Fibrillar Aβ. Alzheimer’s Dis. 2005, 38, 167–177. [Google Scholar] [CrossRef]
- Sastre, M.; Klockgether, T.; Heneka, M.T. Contribution of inflammatory processes to Alzheimer’s disease: Molecular mechanisms. Int. J. Dev. Neurosci. 2006, 24, 167–176. [Google Scholar] [CrossRef]
- Hardy, J.; Selkoe, D.J. The Amyloid Hypothesis of Alzheimer’s Disease: Progress and Problems on the Road to Therapeutics. Science 2002, 297, 353–356. [Google Scholar] [CrossRef]
- Shie, F.-S.; LeBoeur, R.C.; Jin, L.-W. Early intraneuronal Aβ deposition in the hippocampus of APP transgenic mice. NeuroReport 2003, 14, 123–129. [Google Scholar] [CrossRef]
- Takahashi, R.H.; Capetillo-Zarate, E.; Lin, M.T.; Milner, T.A.; Gouras, G.K. Co-occurrence of Alzheimer’s disease β-amyloid and tau pathologies at synapses. Neurobiol. Aging 2010, 31, 1145–1152. [Google Scholar] [CrossRef]
- Maragakis, N.J.; Rothstein, J.D. Mechanisms of Disease: Astrocytes in neurodegenerative disease. Nat. Clin. Pract. Cardiovasc. Med. 2006, 2, 679–689. [Google Scholar] [CrossRef]
- Ransohoff, R.M.; Perry, V.H. Microglial Physiology: Unique Stimuli, Specialized Responses. Annu. Rev. Immunol. 2009, 27, 119–145. [Google Scholar] [CrossRef]
- Glass, C.K.; Saijo, K.; Winner, B.; Marchetto, M.C.; Gage, F.H. Mechanisms Underlying Inflammation in Neurodegeneration. Cell 2010, 140, 918–934. [Google Scholar] [CrossRef]
- Ginhoux, F.; Lim, S.; Hoeffel, G.; Low, D.; Huber, T. Origin and differentiation of microglia. Front. Cell Neurosci. 2013, 7, 45. [Google Scholar] [CrossRef]
- Hansen, D.V.; Hanson, J.E.; Sheng, M. Microglia in Alzheimer’s disease. J. Cell Biol. 2017, 217, 459–472. [Google Scholar] [CrossRef]
- Ginhoux, F.; Greter, M.; Leboeuf, M.; Nandi, S.; See, P.; Gokhan, S.; Mehler, M.F.; Conway, S.J.; Ng, L.G.; Stanley, E.R.; et al. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science 2010, 330, 841–845. [Google Scholar] [CrossRef]
- Butovsky, O.; Jedrychowski, M.P.; Moore, C.S.; Cialic, R.; Lanser, A.J.; Gabriely, G.; Koeglsperger, T.; Dake, B.; Wu, P.M.; E Doykan, C.; et al. Identification of a unique TGF-β–dependent molecular and functional signature in microglia. Nat. Neurosci. 2013, 17, 131–143. [Google Scholar] [CrossRef]
- Elmore, M.R.P.; Najafi, A.R.; Koike, M.A.; Dagher, N.N.; Spangenberg, E.E.; Rice, R.A.; Kitazawa, M.; Matusow, B.; Nguyen, H.; West, B.L.; et al. CSF1 receptor signaling is necessary for microglia viability, which unmasks a cell that rapidly repopulates the microglia-depleted adult brain. Neuron 2014, 82, 380–397. [Google Scholar] [CrossRef]
- Nayak, D.; Roth, T.L.; McGavern, D.B. Microglia Development and Function. Annu. Rev. Immunol. 2014, 32, 367–402. [Google Scholar] [CrossRef]
- Colonna, M.; Butovsky, O. Microglia Function in the Central Nervous System during Health and Neurodegeneration. Annu. Rev. Immunol. 2017, 35, 441–468. [Google Scholar] [CrossRef]
- Schafer, D.P.; Lehrman, E.K.; Kautzman, A.G.; Koyama, R.; Mardinly, A.R.; Yamasaki, R.; Ransohoff, R.M.; Greenberg, M.E.; Barres, B.A.; Stevens, B. Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron 2012, 74, 691–705. [Google Scholar] [CrossRef]
- Frost, J.L.; Schafer, D.P. Microglia: Architects of the Developing Nervous System. Trends Cell Biol. 2016, 26, 587–597. [Google Scholar] [CrossRef]
- Nimmerjahn, A.; Kirchhoff, F.; Helmchen, F. Resting Microglial Cells Are Highly Dynamic Surveillants of Brain Parenchyma in Vivo. Science 2005, 308, 1314–1318. [Google Scholar] [CrossRef]
- Au, N.P.B.; Ma, C.H.E. Recent Advances in the Study of Bipolar/Rod-Shaped Microglia and their Roles in Neurodegeneration. Front. Aging Neurosci. 2017, 9, 128. [Google Scholar] [CrossRef]
- Spittau, B. Aging Microglia—Phenotypes, Functions and Implications for Age-Related Neurodegenerative Diseases. Front. Aging Neurosci. 2017, 9, 194. [Google Scholar] [CrossRef]
- Davies, D.S.; Ma, J.; Jegathees, T.; Goldsbury, A.C. Microglia show altered morphology and reduced arborization in human brain during aging and Alzheimer’s disease. Brain Pathol. 2016, 27, 795–808. [Google Scholar] [CrossRef]
- Bisht, K.; Sharma, K.P.; Lecours, C.; Sánchez, M.G.; El Hajj, H.; Milior, G.; Olmos-Alonso, A.; Gómez-Nicola, D.; Luheshi, G.; Vallières, L.; et al. Dark microglia: A new phenotype predominantly associated with pathological states. Glia 2016, 64, 826–839. [Google Scholar] [CrossRef]
- Plescher, M.; Seifert, G.; Hansen, J.N.; Bedner, P.; Steinhäuser, C.; Halle, A. Plaque-dependent morphological and electrophysiological heterogeneity of microglia in an Alzheimer’s disease mouse model. Glia 2018, 66, 1464–1480. [Google Scholar] [CrossRef]
- Sanchez-Mejias, E.; Navarro, V.; Jimenez, S.; Micó, M.V.S.; Sanchez-Varo, R.; Nuñez-Diaz, C.; Trujillo-Estrada, L.; Davila, J.C.; Vizuete, M.; Gutierrez, A.; et al. Soluble phospho-tau from Alzheimer’s disease hippocampus drives microglial degeneration. Acta Neuropathol. 2016, 132, 897–916. [Google Scholar] [CrossRef]
- Navarro, V.; Sanchez-Mejias, E.; Jimenez, S.; Muñoz-Castro, C.; Sanchez-Varo, R.; Davila, J.C.; Vizuete, M.; Gutierrez, A.; Vitorica, J. Microglia in Alzheimer’s Disease: Activated, Dysfunctional or Degenerative. Front. Aging Neurosci. 2018, 10, 140. [Google Scholar] [CrossRef]
- Doorn, K.J.; Goudriaan, A.; Blits-Huizinga, C.; Bol, J.G.; Rozemuller, A.J.; Hoogland, P.V.; Lucassen, P.J.; Drukarch, B.; Van de Berg, W.; Van Dam, A.-M. Increased Amoeboid Microglial Density in the Olfactory Bulb of Parkinson’s and Alzheimer’s Patients. Brain Pathol. 2013, 24, 152–165. [Google Scholar] [CrossRef] [PubMed]
- Tischer, J.; Krueger, M.; Mueller, W.; Staszewski, O.; Prinz, M.; Streit, W.J.; Bechmann, I. Inhomogeneous distribution of Iba-1 characterizes microglial pathology in Alzheimer’s disease. Glia 2016, 64, 1562–1572. [Google Scholar] [CrossRef] [PubMed]
- Streit, W.J.; Braak, H.; Xue, Q.-S.; Bechmann, I. Dystrophic (senescent) rather than activated microglial cells are associated with tau pathology and likely precede neurodegeneration in Alzheimer’s disease. Acta Neuropathol. 2009, 118, 475–485. [Google Scholar] [CrossRef] [PubMed]
- Yin, Z.; Raj, D.; Saiepour, N.; Van Dam, D.; Brouwer, N.; Holtman, I.R.; Eggen, B.J.; Möller, T.; Tamm, J.A.; Abdourahman, A.; et al. Immune hyperreactivity of Aβ plaque-associated microglia in Alzheimer’s disease. Neurobiol. Aging 2017, 55, 115–122. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, H.M.; Grössinger, E.M.; Horiuchi, M.; Davis, K.W.; Jin, L.-W.; Maezawa, I.; Wulff, H. Differential Kv1.3, KCa3.1, and Kir2.1 expression in “classically” and “alternatively” activated microglia. Glia 2016, 65, 106–121. [Google Scholar] [CrossRef] [PubMed]
- Frank-Cannon, T.C.; Alto, L.T.; McAlpine, F.E.; Tansey, M.G. Does neuroinflammation fan the flame in neurodegenerative diseases? Mol. Neurodegener. 2009, 4, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Jha, M.K.; Lee, W.-H.; Suk, K. Functional polarization of neuroglia: Implications in neuroinflammation and neurological disorders. Biochem. Pharmacol. 2016, 103, 1–16. [Google Scholar] [CrossRef]
- Tang, Y.; Le, W. Differential Roles of M1 and M2 Microglia in Neurodegenerative Diseases. Mol. Neurobiol. 2016, 53, 1181–1194. [Google Scholar] [CrossRef]
- Song, G.J.; Nam, Y.; Jo, M.; Jung, M.; Koo, J.Y.; Cho, W.; Koh, M.; Park, S.B.; Suk, K. A novel small-molecule agonist of PPAR-γ potentiates an anti-inflammatory M2 glial phenotype. Neuropharmacology 2016, 109, 159–169. [Google Scholar] [CrossRef]
- Wang, Y.; Ulland, T.K.; Ulrich, J.D.; Song, W.; Tzaferis, J.A.; Hole, J.T.; Yuan, P.; Mahan, T.E.; Shi, Y.; Gilfillan, S.; et al. TREM2-mediated early microglial response limits diffusion and toxicity of amyloid plaques. J. Exp. Med. 2016, 213, 667–675. [Google Scholar] [CrossRef]
- Bondi, M.W.; Edmonds, E.C.; Salmon, D.P. Alzheimer’s disease: Past, present, and future. J. Int. Neuropsychol. Soc. 2017, 23, 818–831. [Google Scholar] [CrossRef]
- Dos Santos Picanco, L.C.; Ozela, P.F.; De Fatima de Brito Brito, M.; Pinheiro, A.A.; Padilha, E.C.; Braga, F.S.; De Paula da Silva, C.H.T.; Dos Santos, C.B.R.; Rosa, J.M.C.; Da Silva Hage-Melim, L.I. Alzheimer’s Disease: A Review from the Pathophysiology to Diagnosis, New Perspectives for Pharmacological Treatment. Curr. Med. Chem. 2018, 25, 3141–3159. [Google Scholar] [CrossRef]
- Sanabria-Castro, A.; Alvarado-Echeverría, I.; Monge-Bonilla, C. Molecular Pathogenesis of Alzheimer’s Disease: An Update. Ann. Neurosci. 2017, 24, 46–54. [Google Scholar] [CrossRef]
- Vargas, D.M.; De Bastiani, M.A.; Zimmer, E.R.; Klamt, F. Alzheimer’s disease master regulators analysis: Search for potential molecular targets and drug repositioning candidates. Alzheimers Res. Ther. 2018, 10, 59. [Google Scholar] [CrossRef]
- Wang, Z.-Y.; Liu, J.-G.; Li, H.; Yang, H.-M. Pharmacological Effects of Active Components of Chinese Herbal Medicine in the Treatment of Alzheimer’s Disease: A Review. Am. J. Chin. Med. 2016, 44, 1525–1541. [Google Scholar] [CrossRef]
- Blesa, R.; Toriyama, K.; Ueda, K.; Knox, S.; Grossberg, G. Strategies for Continued Successful Treatment in Patients with Alzheimer’s Disease: An Overview of Switching Between Pharmacological Agents. Curr. Alzheimer Res. 2018, 15, 964–974. [Google Scholar] [CrossRef]
- Uddin, S.; Al Mamun, A.; Sumsuzzman, D.; Ashraf, G.; Perveen, A.; Bungau, S.G.; Mousa, S.A.; El-Seedi, H.R.; Bin-Jumah, M.N.; Abdel-Daim, M.M. Emerging Promise of Cannabinoids for the Management of Pain and Associated Neuropathological Alterations in Alzheimer’s Disease. Front. Pharmacol. 2020, 11, 1097. [Google Scholar] [CrossRef]
- Kaur, D.; Behl, T.; Sehgal, A.; Singh, S.; Sharma, N.; Chigurupati, S.; Alhowail, A.; Abdeen, A.; Ibrahim, S.F.; Vargas-De-La-Cruz, C.; et al. Decrypting the potential role of α-lipoic acid in Alzheimer’s disease. Life Sci. 2021, 284, 119899. [Google Scholar] [CrossRef]
- Sharma, S.; Behl, T.; Kumar, A.; Sehgal, A.; Singh, S.; Sharma, N.; Bhatia, S.; Al-Harrasi, A.; Bungau, S. Targeting Endothelin in Alzheimer’s Disease: A Promising Therapeutic Approach. BioMed Res. Int. 2021, 2021, 1–13. [Google Scholar] [CrossRef]
- Rong, X.; Jiang, L.; Qu, M.; Hassan, S.S.U.; Liu, Z. Enhancing Therapeutic Efficacy of Donepezil by Combined Therapy: A Comprehensive Review. Curr. Pharm. Des. 2021, 27, 332–344. [Google Scholar] [CrossRef]
- Nous, A.; Engelborghs, S.; Smolders, I. Melatonin levels in the Alzheimer’s disease continuum: A systematic review. Alzheimers Res. Ther. 2021, 13, 52. [Google Scholar] [CrossRef] [PubMed]
- Calabrò, M.; Rinaldi, C.; Santoro, G.; Crisafulli, C. The biological pathways of Alzheimer disease: A review. AIMS Neurosci. 2021, 8, 86–132. [Google Scholar] [CrossRef]
- Misrani, A.; Tabassum, S.; Yang, L. Mitochondrial Dysfunction and Oxidative Stress in Alzheimer’s Disease. Front. Aging Neurosci. 2021, 13, 617588. [Google Scholar] [CrossRef]
- Iqubal, A.; Syed, M.A.; Najmi, A.K.; Ali, J.; Haque, S.E. Ameliorative effect of nerolidol on cyclophosphamide-induced gonadal toxicity in Swiss Albino mice: Biochemical-, histological- and immunohistochemical-based evidences. Andrologia 2020, 52, e13535. [Google Scholar] [CrossRef] [PubMed]
- Regen, F.; Hellmann-Regen, J.; Costantini, E.; Reale, M. Neuroinflammation and Alzheimer’s Disease: Implications for Microglial Activation. Curr. Alzheimer Res. 2017, 14, 1140–1148. [Google Scholar] [CrossRef] [PubMed]
- Streit, W.J. Microglial activation and neuroinflammation in Alzheimer’s disease: A critical examination of recent history. Front. Aging Neurosci. 2010, 2, 22. [Google Scholar] [CrossRef] [PubMed]
- Schlachetzki, J.C.M.; Hüll, M. Microglial activation in Alzheimer’s disease. Curr. Alzheimer Res. 2009, 6, 554–563. [Google Scholar] [CrossRef]
- Cheignon, C.; Tomas, M.; Bonnefont-Rousselot, D.; Faller, P.; Hureau, C.; Collin, F. Oxidative stress and the amyloid beta peptide in Alzheimer’s disease. Redox Biol. 2018, 14, 450–464. [Google Scholar] [CrossRef]
- Murphy, M.P.; LeVine, H., 3rd. Alzheimer’s Disease and the Amyloid-β Peptide. J. Alzheimer’s Dis. 2010, 19, 311–323. [Google Scholar] [CrossRef]
- Zhang, Y.-W.; Thompson, R.; Zhang, H.; Xu, H. APP processing in Alzheimer’s disease. Mol. Brain 2011, 4, 3. [Google Scholar] [CrossRef]
- Simunkova, M.; Alwasel, S.H.; Alhazza, I.M.; Jomova, K.; Kollar, V.; Rusko, M.; Valko, M. Management of oxidative stress and other pathologies in Alzheimer’s disease. Arch. Toxicol. 2019, 93, 2491–2513. [Google Scholar] [CrossRef]
- Cassidy, L.; Fernandez, F.; Johnson, J.B.; Naiker, M.; Owoola, A.G.; Broszczak, D.A. Oxidative stress in alzheimer’s disease: A review on emergent natural polyphenolic therapeutics. Complement. Ther. Med. 2019, 49, 102294. [Google Scholar] [CrossRef]
- Wake, H.; Moorhouse, A.J.; Jinno, S.; Kohsaka, S.; Nabekura, J. Resting Microglia Directly Monitor the Functional State of Synapses In Vivo and Determine the Fate of Ischemic Terminals. J. Neurosci. 2009, 29, 3974–3980. [Google Scholar] [CrossRef]
- Aloisi, F. Immune function of microglia. Glia 2001, 36, 165–179. [Google Scholar] [CrossRef]
- Ekdahl, C.; Kokaia, Z.; Lindvall, O. Brain inflammation and adult neurogenesis: The dual role of microglia. Neuroscience 2009, 158, 1021–1029. [Google Scholar] [CrossRef]
- Gomes-Leal, W. Microglial physiopathology: How to explain the dual role of microglia after acute neural disorders? Brain Behav. 2012, 2, 345–356. [Google Scholar] [CrossRef]
- Parkhurst, C.N.; Yang, G.; Ninan, I.; Savas, J.N.; Yates, J.R., III; Lafaille, J.J.; Hempstead, B.L.; Littman, D.R.; Gan, W.-B. Microglia promote learning-dependent synapse formation through brain-derived neurotrophic factor. Cell 2013, 155, 1596–1609. [Google Scholar] [CrossRef]
- Ben Achour, S.; Pascual, O. Glia: The many ways to modulate synaptic plasticity. Neurochem. Int. 2010, 57, 440–445. [Google Scholar] [CrossRef]
- Kettenmann, H.; Hanisch, U.-K.; Noda, M.; Verkhratsky, A. Physiology of Microglia. Physiol. Rev. 2011, 91, 461–553. [Google Scholar] [CrossRef]
- Coull, J.A.M.; Beggs, S.; Boudreau, D.; Boivin, D.; Tsuda, M.; Inoue, K.; Gravel, C.; Salter, M.W.; De Koninck, Y. BDNF from microglia causes the shift in neuronal anion gradient underlying neuropathic pain. Nature 2005, 438, 1017–1021. [Google Scholar] [CrossRef]
- Pascual, O.; Ben Achour, S.; Rostaing, P.; Triller, A.; Bessis, A. Microglia activation triggers astrocyte-mediated modulation of excitatory neurotransmission. Proc. Natl. Acad. Sci. USA 2011, 109, E197–E205. [Google Scholar] [CrossRef]
- Tremblay, M.-E.; Zettel, M.L.; Ison, J.R.; Allen, P.D.; Majewska, A.K. Effects of aging and sensory loss on glial cells in mouse visual and auditory cortices. Glia 2012, 60, 541–558. [Google Scholar] [CrossRef]
- Yung, R.L.; Julius, A. Epigenetics, aging, and autoimmunity. Autoimmunity 2008, 41, 329–335. [Google Scholar] [CrossRef]
- Streit, W.J.; Sammons, N.W.; Kuhns, A.J.; Sparks, D.L. Dystrophic microglia in the aging human brain. Glia 2004, 45, 208–212. [Google Scholar] [CrossRef]
- Streit, W.J. Microglial senescence: Does the brain’s immune system have an expiration date? Trends Neurosci. 2006, 29, 506–510. [Google Scholar] [CrossRef]
- Godbout, J.P.; Johnson, R. Age and Neuroinflammation: A Lifetime of Psychoneuroimmune Consequences. Immunol. Allergy Clin. N. Am. 2009, 29, 321–337. [Google Scholar] [CrossRef]
- Sierra, A.; Gottfried-Blackmore, A.C.; McEwen, B.S.; Bulloch, K. Microglia derived from aging mice exhibit an altered inflammatory profile. Glia 2007, 55, 412–424. [Google Scholar] [CrossRef]
- Wynne, A.M.; Henry, C.J.; Godbout, J.P. Immune and behavioral consequences of microglial reactivity in the aged brain. Integr. Comp. Biol. 2009, 49, 254–266. [Google Scholar] [CrossRef]
- Dilger, R.; Johnson, R.W. Aging, microglial cell priming, and the discordant central inflammatory response to signals from the peripheral immune system. J. Leukoc. Biol. 2008, 84, 932–939. [Google Scholar] [CrossRef]
- Chen, J.; Buchanan, J.B.; Sparkman, N.L.; Godbout, J.P.; Freund, G.G.; Johnson, R.W. Neuroinflammation and disruption in working memory in aged mice after acute stimulation of the peripheral innate immune system. Brain Behav. Immun. 2008, 22, 301–311. [Google Scholar] [CrossRef]
- Zhang, G.; Wang, Z.; Hu, H.; Zhao, M.; Sun, L. Microglia in Alzheimer’s Disease: A Target for Therapeutic Intervention. Front. Cell. Neurosci. 2021, 15, 749587. [Google Scholar] [CrossRef]
- Zhang, S.; Hu, L.; Jiang, J.; Li, H.; Wu, Q.; Ooi, K.; Wang, J.; Feng, Y.; Zhu, D.; Xia, C. HMGB1/RAGE axis mediates stress-induced RVLM neuroinflammation in mice via impairing mitophagy flux in microglia. J. Neuroinflamm. 2020, 17, 15. [Google Scholar] [CrossRef]
- Nguyen, A.T.; Wang, K.; Hu, G.; Wang, X.; Miao, Z.; Azevedo, J.A.; Suh, E.; Van Deerlin, V.M.; Choi, D.; Roeder, K.; et al. APOE and TREM2 regulate amyloid-responsive microglia in Alzheimer’s disease. Acta Neuropathol. 2020, 140, 477–493. [Google Scholar] [CrossRef]
- Deane, R.; Singh, I.; Sagare, A.P.; Bell, R.D.; Ross, N.T.; LaRue, B.; Love, R.; Perry, S.; Paquette, N.; Deane, R.J.; et al. A multimodal RAGE-specific inhibitor reduces amyloid β–mediated brain disorder in a mouse model of Alzheimer disease. J. Clin. Investig. 2012, 122, 1377–1392. [Google Scholar] [CrossRef] [PubMed]
- Frenkel, D.; Wilkinson, K.; Zhao, L.; Hickman, S.E.; Means, T.K.; Puckett, L.; Farfara, D.; Kingery, N.D.; Weiner, H.L.; El Khoury, J. Scara1 deficiency impairs clearance of soluble amyloid-β by mononuclear phagocytes and accelerates Alzheimer’s-like disease progression. Nat. Commun. 2013, 4, 2030. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.-M.; Mun, B.-R.; Lee, S.-J.; Joh, Y.; Lee, H.-Y.; Ji, K.-Y.; Choi, H.-R.; Lee, E.-H.; Kim, E.-M.; Jang, J.-H.; et al. TREM2 promotes Aβ phagocytosis by upregulating C/EBPα-dependent CD36 expression in microglia. Sci. Rep. 2017, 7, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Ulland, T.K.; Song, W.M.; Huang, S.C.-C.; Ulrich, J.D.; Sergushichev, A.; Beatty, W.L.; Loboda, A.A.; Zhou, Y.; Cairns, N.J.; Kambal, A.; et al. TREM2 Maintains Microglial Metabolic Fitness in Alzheimer’s Disease. Cell 2017, 170, 649–663.e13. [Google Scholar] [CrossRef] [PubMed]
- Crehan, H.; Hardy, J.; Pocock, J. Blockage of CR1 prevents activation of rodent microglia. Neurobiol. Dis. 2013, 54, 139–149. [Google Scholar] [CrossRef]
- Aikawa, T.; Ren, Y.; Yamazaki, Y.; Tachibana, M.; Johnson, M.R.; Anderson, C.T.; Martens, Y.A.; Holm, M.-L.; Asmann, Y.W.; Saito, T.; et al. ABCA7 haplodeficiency disturbs microglial immune responses in the mouse brain. Proc. Natl. Acad. Sci. USA 2019, 116, 23790–23796. [Google Scholar] [CrossRef]
- Cho, S.-H.; Sun, B.; Zhou, Y.; Kauppinen, T.M.; Halabisky, B.; Wes, P.; Ransohoff, R.M.; Gan, L. CX3CR1 Protein Signaling Modulates Microglial Activation and Protects against Plaque-independent Cognitive Deficits in a Mouse Model of Alzheimer Disease. J. Biol. Chem. 2011, 286, 32713–32722. [Google Scholar] [CrossRef]
- Heneka, M.T.; Kummer, M.P.; Stutz, A.; Delekate, A.; Schwartz, S.; Vieira-Saecker, A.; Griep, A.; Axt, D.; Remus, A.; Tzeng, T.-C.; et al. NLRP3 is activated in Alzheimer’s disease and contributes to pathology in APP/PS1 mice. Nature 2013, 493, 674–678. [Google Scholar] [CrossRef]
- Ruganzu, J.B.; Zheng, Q.; Wu, X.; He, Y.; Peng, X.; Jin, H.; Zhou, J.; Ma, R.; Ji, S.; Ma, Y.; et al. TREM2 overexpression rescues cognitive deficits in APP/PS1 transgenic mice by reducing neuroinflammation via the JAK/STAT/SOCS signaling pathway. Exp. Neurol. 2020, 336, 113506. [Google Scholar] [CrossRef]
- Leyns, C.E.G.; Gratuze, M.; Narasimhan, S.; Jain, N.; Koscal, L.J.; Jiang, H.; Manis, M.; Colonna, M.; Lee, V.M.Y.; Ulrich, J.D.; et al. TREM2 function impedes tau seeding in neuritic plaques. Nat. Neurosci. 2019, 22, 1217–1222. [Google Scholar] [CrossRef]
- Shi, Y.; Initiative, A.D.N.; Yamada, K.; Liddelow, S.A.; Smith, S.T.; Zhao, L.; Luo, W.; Tsai, R.M.; Spina, S.; Grinberg, L.T.; et al. ApoE4 markedly exacerbates tau-mediated neurodegeneration in a mouse model of tauopathy. Nature 2017, 549, 523–527. [Google Scholar] [CrossRef]
- Mancuso, R.; Daele, J.V.D.; Fattorelli, N.; Wolfs, L.; Balusu, S.; Burton, O.; Liston, A.; Sierksma, A.; Fourne, Y.; Poovathingal, S.; et al. Stem-cell-derived human microglia transplanted in mouse brain to study human disease. Nat. Neurosci. 2019, 22, 2111–2116. [Google Scholar] [CrossRef]
- Mancuso, R.; Fryatt, G.; Cleal, M.; Obst, J.; Pipi, E.; Monzón-Sandoval, J.; Ribe, E.; Winchester, L.; Webber, C.; Nevado, A.; et al. CSF1R inhibitor JNJ-40346527 attenuates microglial proliferation and neurodegeneration in P301S mice. Brain 2019, 142, 3243–3264. [Google Scholar] [CrossRef]
- Lautrup, S.; Lou, G.; Aman, Y.; Nilsen, H.; Tao, J.; Fang, E.F. Microglial mitophagy mitigates neuroinflammation in Alzheimer’s disease. Neurochem. Int. 2019, 129, 104469. [Google Scholar] [CrossRef]
- Berg, J.V.; Prokop, S.; Miller, K.R.; Obst, J.; E Kälin, R.; Lopategui-Cabezas, I.; Wegner, A.; Mair, F.; Schipke, C.G.; Peters, O.; et al. Inhibition of IL-12/IL-23 signaling reduces Alzheimer’s disease–like pathology and cognitive decline. Nat. Med. 2012, 18, 1812–1819. [Google Scholar] [CrossRef]
- Martín-Maestro, P.; Gargini, R.; Sproul, A.A.; García, E.; Antón, L.C.; Noggle, S.; Arancio, O.; Avila, J.; García-Escudero, V. Mitophagy failure in fibroblasts and iPSC-derived neurons of alzheimer’s disease-associated presenilin 1 mutation. Front. Mol. Neurosci. 2017, 10, 291. [Google Scholar] [CrossRef]
- Scheibye-Knudsen, M.; Fang, E.F.; Croteau, D.L.; Wilson, D.M.; Bohr, V.A. Protecting the mitochondrial powerhouse. Trends Cell Biol. 2014, 25, 158–170. [Google Scholar] [CrossRef]
- Fang, E.F.; Scheibye-Knudsen, M.; Brace, L.; Kassahun, H.; SenGupta, T.; Nilsen, H.; Mitchell, J.R.; Croteau, D.L.; Bohr, V.A. Defective Mitophagy in XPA via PARP-1 Hyperactivation and NAD+/SIRT1 Reduction. Cell 2014, 157, 882–896. [Google Scholar] [CrossRef]
- Fang, E.F.; Kassahun, H.; Croteau, D.L.; Scheibye-Knudsen, M.; Marosi, K.; Lu, H.; Shamanna, R.A.; Kalyanasundaram, S.; Bollineni, R.C.; Wilson, M.A.; et al. NAD + Replenishment Improves Lifespan and Healthspan in Ataxia Telangiectasia Models via Mitophagy and DNA Repair. Cell Metab. 2016, 24, 566–581. [Google Scholar] [CrossRef]
- Li, M.; Pisalyaput, K.; Galvan, M.; Tenner, A.J. Macrophage colony stimulatory factor and interferon-gamma trigger distinct mechanisms for augmentation of beta-amyloid-induced microglia-mediated neurotoxicity. J. Neurochem. 2004, 91, 623–633. [Google Scholar] [CrossRef]
- Wildburger, N.C.; Gyngard, F.; Guillermier, C.; Patterson, B.W.; Elbert, D.; Mawuenyega, K.G.; Schneider, T.; Green, K.; Roth, R.; Schmidt, R.E.; et al. Amyloid-β Plaques in Clinical Alzheimer’s Disease Brain Incorporate Stable Isotope Tracer In Vivo and Exhibit Nanoscale Heterogeneity. Front. Neurol. 2018, 9, 169. [Google Scholar] [CrossRef]
- Takeuchi, H.; Jin, S.; Wang, J.; Zhang, G.; Kawanokuchi, J.; Kuno, R.; Sonobe, Y.; Mizuno, T.; Suzumura, A. Tumor Necrosis Factor-α Induces Neurotoxicity via Glutamate Release from Hemichannels of Activated Microglia in an Autocrine Manner. J. Biol. Chem. 2006, 281, 21362–21368. [Google Scholar] [CrossRef]
- Bordji, K.; Becerril-Ortega, J.; Nicole, O.; Buisson, A. Activation of Extrasynaptic, But Not Synaptic, NMDA Receptors Modifies Amyloid Precursor Protein Expression Pattern and Increases Amyloid- Production. J. Neurosci. 2010, 30, 15927–15942. [Google Scholar] [CrossRef]
- Wilkinson, K.; El Khoury, J. Microglial Scavenger Receptors and Their Roles in the Pathogenesis of Alzheimer’s Disease. Int. J. Alzheimer’s Dis. 2012, 2012, 489456. [Google Scholar] [CrossRef]
- El Khoury, J.; Toft, M.; Hickman, S.E.; Means, T.K.; Terada, K.; Geula, C.; Luster, A.D. Ccr2 deficiency impairs microglial accumulation and accelerates progression of Alzheimer-like disease. Nat. Med. 2007, 13, 432–438. [Google Scholar] [CrossRef]
- Weiner, H.L.; Frenkel, D. Immunology and immunotherapy of Alzheimer’s disease. Nat. Rev. Immunol. 2006, 6, 404–416. [Google Scholar] [CrossRef]
- Jaworski, T.; Lechat, B.; Demedts, D.; Gielis, L.; Devijver, H.; Borghgraef, P.; Duimel, H.; Verheyen, F.; Kügler, S.; Van Leuven, F. Dendritic Degeneration, Neurovascular Defects, and Inflammation Precede Neuronal Loss in a Mouse Model for Tau-Mediated Neurodegeneration. Am. J. Pathol. 2011, 179, 2001–2015. [Google Scholar] [CrossRef]
- Azevedo, E.P.; Ledo, J.H.; Barbosa, G.; Sobrinho, M.; Diniz, L.; Fonseca, A.C.C.; Gomes, F.; Romão, L.; Lima, F.R.S.; Palhano, F.L.; et al. Activated microglia mediate synapse loss and short-term memory deficits in a mouse model of transthyretin-related oculoleptomeningeal amyloidosis. Cell Death Dis. 2013, 4, e789. [Google Scholar] [CrossRef][Green Version]
- Tasmine, A.; Hininger-Favier, I.; Carriere, I.; Arnaud, J.; Gourlet, V.; Roussel, A.-M.; Berr, C. Plasma Selenium Over Time and Cognitive Decline in the Elderly. Epidemiology 2007, 18, 52–58. [Google Scholar] [CrossRef]
- Sy, M.; Kitazawa, M.; Medeiros, R.; Whitman, L.; Cheng, D.; Lane, T.E.; LaFerla, F.M. Inflammation Induced by Infection Potentiates Tau Pathological Features in Transgenic Mice. Am. J. Pathol. 2011, 178, 2811–2822. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhao, Y.; Zhang, J.; Yang, G. Mechanisms of NLRP3 Inflammasome Activation: Its Role in the Treatment of Alzheimer’s Disease. Neurochem. Res. 2020, 45, 2560–2572. [Google Scholar] [CrossRef] [PubMed]
- Severini, C.; Barbato, C.; Di Certo, M.G.; Gabanella, F.; Petrella, C.; Di Stadio, A.; De Vincentiis, M.; Polimeni, A.; Ralli, M.; Greco, A. Alzheimer’s Disease: New Concepts on the Role of Autoimmunity and NLRP3 Inflammasome in the Pathogenesis of the Disease. Curr. Neuropharmacol. 2021, 19, 498–512. [Google Scholar] [CrossRef] [PubMed]
- Tarkowski, E.; Andreasen, N.; Blennow, K. Intrathecal inflammation precedes development of Alzheimer’s disease. J. Neurol. Neurosurg. Psychiatry 2003, 74, 1200–1205. [Google Scholar] [CrossRef] [PubMed]
- Wright, A.; Zinn, R.; Hohensinn, B.; Konen, L.M.; Beynon, S.B.; Tan, R.P.; Clark, I.A.; Abdipranoto, A.; Vissel, B. Neuroinflammation and Neuronal Loss Precede Aβ Plaque Deposition in the hAPP-J20 Mouse Model of Alzheimer’s Disease. PLoS ONE 2013, 8, e59586. [Google Scholar] [CrossRef]
- Ng, A.; Tam, W.W.; Zhang, M.W.; Ho, C.S.; Husain, S.F.; McIntyre, R.S.; Ho, R. IL-1β, IL-6, TNF- α and CRP in Elderly Patients with Depression or Alzheimer’s disease: Systematic Review and Meta-Analysis. Sci. Rep. 2018, 8, 12050. [Google Scholar] [CrossRef]
- Schroder, K.; Tschopp, J. The Inflammasomes. Cell 2010, 140, 821–832. [Google Scholar] [CrossRef]
- Gustin, A.; Kirchmeyer, M.; Koncina, E.; Felten, P.; Losciuto, S.; Heurtaux, T.; Tardivel, A.; Heuschling, P.; Dostert, C. NLRP3 Inflammasome Is Expressed and Functional in Mouse Brain Microglia but Not in Astrocytes. PLoS ONE 2015, 10, e0130624. [Google Scholar] [CrossRef]
- Saresella, M.; La Rosa, F.; Piancone, F.; Zoppis, M.; Marventano, I.; Calabrese, E.; Rainone, V.; Nemni, R.; Mancuso, R.; Clerici, M. The NLRP3 and NLRP1 inflammasomes are activated in Alzheimer’s disease. Mol. Neurodegener. 2016, 11, 23. [Google Scholar] [CrossRef]
- Halle, A.; Hornung, V.; Petzold, G.C.; Stewart, C.R.; Monks, B.G.; Reinheckel, T.; Fitzgerald, K.A.; Latz, E.; Moore, K.J.; Golenbock, D.T. The NALP3 inflammasome is involved in the innate immune response to amyloid-β. Nat. Immunol. 2008, 9, 857–865. [Google Scholar] [CrossRef]
- Streit, W.J.; Mrak, R.E.; Griffin, W.S.T. Microglia and neuroinflammation: A pathological perspective. J. Neuroinflamm. 2004, 1, 14. [Google Scholar] [CrossRef][Green Version]
- Graeber, M.B.; Li, W.; Rodriguez, M.L. Role of microglia in CNS inflammation. FEBS Lett. 2011, 585, 3798–3805. [Google Scholar] [CrossRef]
- Chen, H.; Cúrdia, V.; Ferrero, A. The Macroeconomic Effects of Large-scale Asset Purchase Programmes. Econ. J. 2012, 122, F289–F315. [Google Scholar] [CrossRef]
- Hughes, E.; Bergles, D.E. Hidden Progenitors Replace Microglia in the Adult Brain. Neuron 2014, 82, 253–255. [Google Scholar] [CrossRef]
- Rosczyk, H.; Sparkman, N.; Johnson, R. Neuroinflammation and cognitive function in aged mice following minor surgery. Exp. Gerontol. 2008, 43, 840–846. [Google Scholar] [CrossRef]
- Ponomarev, E.D.; Shriver, L.P.; Maresz, K.; Dittel, B.N. Microglial cell activation and proliferation precedes the onset of CNS autoimmunity. J. Neurosci. Res. 2005, 81, 374–389. [Google Scholar] [CrossRef]
- Rock, R.B.; Gekker, G.; Hu, S.; Sheng, W.S.; Cheeran, M.; Lokensgard, J.R.; Peterson, P.K. Role of Microglia in Central Nervous System Infections. Clin. Microbiol. Rev. 2004, 17, 942–964. [Google Scholar] [CrossRef]
- Kamphuis, W.; Mamber, C.; Moeton, M.; Kooijman, L.; Sluijs, J.A.; Jansen, A.H.P.; Verveer, M.; De Groot, L.R.; Smith, V.D.; Rangarajan, S.; et al. GFAP Isoforms in Adult Mouse Brain with a Focus on Neurogenic Astrocytes and Reactive Astrogliosis in Mouse Models of Alzheimer Disease. PLoS ONE 2012, 7, e42823. [Google Scholar] [CrossRef]
- Fraser, H.B.; Moses, A.M.; Schadt, E.E. Evidence for widespread adaptive evolution of gene expression in budding yeast. Proc. Natl. Acad. Sci. USA 2010, 107, 2977–2982. [Google Scholar] [CrossRef]
- Inoue, K.; Tsuda, M. Purinergic systems, neuropathic pain and the role of microglia. Exp. Neurol. 2011, 234, 293–301. [Google Scholar] [CrossRef]
- Perry, V.H.; Teeling, J. Microglia and macrophages of the central nervous system: The contribution of microglia priming and systemic inflammation to chronic neurodegeneration. Semin. Immunopathol. 2013, 35, 601–612. [Google Scholar] [CrossRef]
- Selkoe, D.J. Alzheimer’s Disease Is a Synaptic Failure. Science 2002, 298, 789–791. [Google Scholar] [CrossRef]
- Di Filippo, M.; Tozzi, A.; Costa, C.; Belcastro, V.; Tantucci, M.; Picconi, B.; Calabresi, P. Plasticity and repair in the post-ischemic brain. Neuropharmacology 2008, 55, 353–362. [Google Scholar] [CrossRef]
- Von Bernhardi, R.; Eugenín-von Bernhardi, L.; Eugenín, J. Microglial cell dysregulation in brain aging and neurodegeneration. Front. Aging Neurosci. 2015, 7, 124. [Google Scholar] [CrossRef]
- Cagnin, A.; Brooks, D.J.; Kennedy, A.M.; Gunn, R.N.; Myers, R.; Turkheimer, F.E.; Jones, T.; Banati, R.B. In-vivo measurement of activated microglia in dementia. Lancet 2001, 358, 461–467. [Google Scholar] [CrossRef]
- Kim, S.U.; De Vellis, J. Microglia in health and disease. J. Neurosci. Res. 2005, 81, 302–313. [Google Scholar] [CrossRef]
- Neumann, M.; Sampathu, D.M.; Kwong, L.K.; Truax, A.C.; Micsenyi, M.C.; Chou, T.T.; Bruce, J.; Schuck, T.; Grossman, M.; Clark, C.M.; et al. Ubiquitinated TDP-43 in Frontotemporal Lobar Degeneration and Amyotrophic Lateral Sclerosis. Science 2006, 314, 130–133. [Google Scholar] [CrossRef]
- Kitamura, R.; Tsukamoto, K.; Harada, K.; Shimizu, A.; Shimada, S.; Kobayashi, T.; Imokawa, G. Mechanisms underlying the dysfunction of melanocytes in vitiligo epidermis: Role of SCF/KIT protein interactions and the downstream effector, MITF-M. J. Pathol. 2004, 202, 463–475. [Google Scholar] [CrossRef]
- Jack, C.R., Jr.; Knopman, D.S.; Jagust, W.J.; Shaw, L.M.; Aisen, P.S.; Weiner, M.W.; Petersen, R.C.; Trojanowski, J.Q. Hypothetical model of dynamic biomarkers of the Alzheimer’s pathological cascade. Lancet Neurol. 2010, 9, 119–128. [Google Scholar] [CrossRef]
- Liddelow, S.A.; Guttenplan, K.A.; Clarke, L.E.; Bennett, F.C.; Bohlen, C.J.; Schirmer, L.; Bennett, M.L.; Münch, A.E.; Chung, W.-S.; Peterson, T.C.; et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 2017, 541, 481–487. [Google Scholar] [CrossRef]
- Asai, H.; Ikezu, S.; Tsunoda, S.; Medalla, M.; Luebke, J.; Haydar, T.; Wolozin, B.; Butovsky, O.; Kügler, S.; Ikezu, T. Depletion of microglia and inhibition of exosome synthesis halt tau propagation. Nat. Neurosci. 2015, 18, 1584–1593. [Google Scholar] [CrossRef]
- Sosna, J.; Philipp, S.; Albay, R.I.; Reyes-Ruiz, J.M.; Baglietto-Vargas, D.; LaFerla, F.M.; Glabe, C.G. Early long-term administration of the CSF1R inhibitor PLX3397 ablates microglia and reduces accumulation of intraneuronal amyloid, neuritic plaque deposition and pre-fibrillar oligomers in 5XFAD mouse model of Alzheimer’s disease. Mol. Neurodegener. 2018, 13, 11. [Google Scholar] [CrossRef] [PubMed]
- Spangenberg, E.; Severson, P.L.; Hohsfield, L.A.; Crapser, J.; Zhang, J.; Burton, E.A.; Zhang, Y.; Spevak, W.; Lin, J.; Phan, N.Y.; et al. Sustained microglial depletion with CSF1R inhibitor impairs parenchymal plaque development in an Alzheimer’s disease model. Nat. Commun. 2019, 10, 3758. [Google Scholar] [CrossRef] [PubMed]
- Casali, B.T.; MacPherson, K.P.; Reed-Geaghan, E.G.; Landreth, G.E. Microglia depletion rapidly and reversibly alters amyloid pathology by modification of plaque compaction and morphologies. Neurobiol. Dis. 2020, 142, 104956. [Google Scholar] [CrossRef] [PubMed]
- Chiozzi, P.; Sarti, A.C.; Sanz, J.M.; Giuliani, A.L.; Adinolfi, E.; Vultaggio-Poma, V.; Falzoni, S.; Di Virgilio, F. Amyloid β-dependent mitochondrial toxicity in mouse microglia requires P2X7 receptor expression and is prevented by nimodipine. Sci. Rep. 2019, 9, 6475. [Google Scholar] [CrossRef]
- Jiao, S.-S.; Yao, X.-Q.; Liu, Y.-H.; Wang, Q.-H.; Zeng, F.; Lu, J.-J.; Liu, J.; Zhu, C.; Shen, L.-L.; Liu, C.-H.; et al. Edaravone alleviates Alzheimer’s disease-type pathologies and cognitive deficits. Proc. Natl. Acad. Sci. USA 2015, 112, 5225–5230. [Google Scholar] [CrossRef]
- Yang, R.; Wang, Q.; Li, F.; Li, J.; Liu, X. Edaravone injection ameliorates cognitive deficits in rat model of Alzheimer’s disease. Neurol. Sci. 2015, 36, 2067–2072. [Google Scholar] [CrossRef]
- Yin, J.; Zhao, F.; Chojnacki, J.; Fulp, J.; Klein, W.L.; Zhang, S.; Zhu, X. NLRP3 Inflammasome Inhibitor Ameliorates Amyloid Pathology in a Mouse Model of Alzheimer’s Disease. Mol. Neurobiol. 2017, 55, 1977–1987. [Google Scholar] [CrossRef]
- Lučiūnaitė, A.; McManus, R.M.; Jankunec, M.; Rácz, I.; Dansokho, C.; Dalgėdienė, I.; Schwartz, S.; Brosseron, F.; Heneka, M.T. Soluble Aβ oligomers and protofibrils induce NLRP3 inflammasome activation in microglia. J. Neurochem. 2019, 155, 650–661. [Google Scholar] [CrossRef]
- Garcez, M.L.; Mina, F.; Bellettini-Santos, T.; Da Luz, A.P.; Schiavo, G.L.; Macieski, J.M.C.; Medeiros, E.B.; Marques, A.O.; Magnus, N.Q.; Budni, J. The Involvement of NLRP3 on the Effects of Minocycline in an AD-Like Pathology Induced by β-Amyloid Oligomers Administered to Mice. Mol. Neurobiol. 2018, 56, 2606–2617. [Google Scholar] [CrossRef]
- Parikh, A.; Kathawala, K.; Li, J.; Chen, C.; Shan, Z.; Cao, X.; Wang, Y.-J.; Garg, S.; Zhou, X.-F. Self-nanomicellizing solid dispersion of edaravone: Part II: In vivo assessment of efficacy against behavior deficits and safety in Alzheimer’s disease model. Drug Des. Dev. Ther. 2018, 12, 2111–2128. [Google Scholar] [CrossRef]
- McLarnon, J.G.; Ryu, M.J.K.; Walker, D.G.; Choi, B.H.B. Upregulated Expression of Purinergic P2X7Receptor in Alzheimer Disease and Amyloid-β Peptide-Treated Microglia and in Peptide-Injected Rat Hippocampus. J. Neuropathol. Exp. Neurol. 2006, 65, 1090–1097. [Google Scholar] [CrossRef]
- Ryu, J.K.; McLarnon, J.G. Block of purinergic P2X7 receptor is neuroprotective in an animal model of Alzheimer’s disease. NeuroReport 2008, 19, 1715–1719. [Google Scholar] [CrossRef]
- Geldmacher, D.S.; Fritsch, T.; McClendon, M.J.; Landreth, G. A Randomized Pilot Clinical Trial of the Safety of Pioglitazone in Treatment of Patients with Alzheimer Disease. Arch. Neurol. 2011, 68, 45–50. [Google Scholar] [CrossRef]
- Yan, Q.; Zhang, J.; Liu, H.; Babu-Khan, S.; Vassar, R.; Biere, A.L.; Citron, M.; Landreth, G. Anti-Inflammatory Drug Therapy Alters β-Amyloid Processing and Deposition in an Animal Model of Alzheimer’s Disease. J. Neurosci. 2003, 23, 7504–7509. [Google Scholar] [CrossRef]
- Sato, T.; Hanyu, H.; Hirao, K.; Kanetaka, H.; Sakurai, H.; Iwamoto, T. Efficacy of PPAR-γ agonist pioglitazone in mild Alzheimer disease. Neurobiol. Aging 2011, 32, 1626–1633. [Google Scholar] [CrossRef]
- Cheng, Q.; Danao, J.; Talreja, S.; Wen, P.; Yin, J.; Sun, N.; Li, C.-M.; Chui, D.; Tran, D.; Koirala, S.; et al. TREM2-activating antibodies abrogate the negative pleiotropic effects of the Alzheimer’s disease variant Trem2R47H on murine myeloid cell function. J. Biol. Chem. 2018, 293, 12620–12633. [Google Scholar] [CrossRef]
- Price, B.R.; Sudduth, T.L.; Weekman, E.M.; Johnson, S.; Hawthorne, D.; Woolums, A.; Wilcock, D.M. Therapeutic Trem2 activation ameliorates amyloid-beta deposition and improves cognition in the 5XFAD model of amyloid deposition. J. Neuroinflammation 2020, 17, 238. [Google Scholar] [CrossRef]
- Wang, S.; Mustafa, M.; Yuede, C.M.; Salazar, S.V.; Kong, P.; Long, H.; Ward, M.; Siddiqui, O.; Paul, R.; Gilfillan, S.; et al. Anti-human TREM2 induces microglia proliferation and reduces pathology in an Alzheimer’s disease model. J. Exp. Med. 2020, 217, e20200785. [Google Scholar] [CrossRef]
- Schlepckow, K.; Monroe, K.M.; Kleinberger, G.; Cantuti-Castelvetri, L.; Parhizkar, S.; Xia, D.; Willem, M.; Werner, G.; Pettkus, N.; Brunner, B.; et al. Enhancing protective microglial activities with a dual function TREM 2 antibody to the stalk region. EMBO Mol. Med. 2020, 12, e11227. [Google Scholar] [CrossRef]
- Zhang, M.; Schmitt-Ulms, G.; Sato, C.; Xi, Z.; Zhang, Y.; Zhou, Y.; George-Hyslop, P.S.; Rogaeva, E. Drug Repositioning for Alzheimer’s Disease Based on Systematic ‘omics’ Data Mining. PLoS ONE 2016, 11, e0168812. [Google Scholar] [CrossRef]
- Miles, L.A.; Hermans, S.J.; Crespi, G.A.; Gooi, J.; Doughty, L.; Nero, T.L.; Markulić, J.; Ebneth, A.; Wroblowski, B.; Oehlrich, D.; et al. Small Molecule Binding to Alzheimer Risk Factor CD33 Promotes Aβ Phagocytosis. iScience 2019, 19, 110–118. [Google Scholar] [CrossRef]
- Muffat, J.; Li, Y.; Yuan, B.; Mitalipova, M.; Omer, A.; Corcoran, S.; Bakiasi, G.; Tsai, L.-H.; Aubourg, P.; Ransohoff, R.M.; et al. Efficient derivation of microglia-like cells from human pluripotent stem cells. Nat. Med. 2016, 22, 1358–1367. [Google Scholar] [CrossRef]
- Douvaras, P.; Sun, B.; Wang, M.; Kruglikov, I.; Lallos, G.; Zimmer, M.; Terrenoire, C.; Zhang, B.; Gandy, S.; Schadt, E.; et al. Directed Differentiation of Human Pluripotent Stem Cells to Microglia. Stem Cell Rep. 2017, 8, 1516–1524. [Google Scholar] [CrossRef]
- Haenseler, W.; Sansom, S.N.; Buchrieser, J.; Newey, S.E.; Moore, C.S.; Nicholls, F.J.; Chintawar, S.; Schnell, C.; Antel, J.P.; Allen, N.D.; et al. A Highly Efficient Human Pluripotent Stem Cell Microglia Model Displays a Neuronal-Co-culture-Specific Expression Profile and Inflammatory Response. Stem Cell Rep. 2017, 8, 1727–1742. [Google Scholar] [CrossRef]
- Kim, S.; Chang, K.-A.; Kim, J.A.; Park, H.-G.; Ra, J.C.; Kim, H.-S.; Suh, Y.-H. The Preventive and Therapeutic Effects of Intravenous Human Adipose-Derived Stem Cells in Alzheimer’s Disease Mice. PLoS ONE 2012, 7, e45757. [Google Scholar] [CrossRef]
- Kim, K.-S.; Kim, H.S.; Park, J.-M.; Kim, H.W.; Park, M.-K.; Lee, H.-S.; Lim, D.S.; Lee, T.H.; Chopp, M.; Moon, J. Long-term immunomodulatory effect of amniotic stem cells in an Alzheimer’s disease model. Neurobiol. Aging 2013, 34, 2408–2420. [Google Scholar] [CrossRef]
- A Kim, J.; Ha, S.; Shin, K.Y.; Kim, S.; Lee, K.J.; Chong, Y.H.; Chang, K.-A.; Suh, Y.-H. Neural stem cell transplantation at critical period improves learning and memory through restoring synaptic impairment in Alzheimer’s disease mouse model. Cell Death Dis. 2015, 6, e1789. [Google Scholar] [CrossRef]
- Lee, I.-S.; Jung, K.; Kim, I.-S.; Lee, H.; Kim, M.; Yun, S.; Hwang, K.; Shin, J.E.; Park, K.I. Human neural stem cells alleviate Alzheimer-like pathology in a mouse model. Mol. Neurodegener. 2015, 10, 38. [Google Scholar] [CrossRef]
- Rojo, R.; Raper, A.; Ozdemir, D.D.; Lefevre, L.; Grabert, K.; Wollscheid-Lengeling, E.; Bradford, B.; Caruso, M.; Gazova, I.; Sánchez, A.; et al. Deletion of a Csf1r enhancer selectively impacts CSF1R expression and development of tissue macrophage populations. Nat. Commun. 2019, 10, 3215. [Google Scholar] [CrossRef]
- Bruttger, J.; Karram, K.; Wörtge, S.; Regen, T.; Marini, F.; Hoppmann, N.; Klein, M.; Blank, T.; Yona, S.; Wolf, Y.; et al. Genetic Cell Ablation Reveals Clusters of Local Self-Renewing Microglia in the Mammalian Central Nervous System. Immunity 2015, 43, 92–106. [Google Scholar] [CrossRef]
- Wang, Y.; Cella, M.; Mallinson, K.; Ulrich, J.D.; Young, K.L.; Robinette, M.L.; Gilfillan, S.; Krishnan, G.M.; Sudhakar, S.; Zinselmeyer, B.; et al. TREM2 Lipid Sensing Sustains the Microglial Response in an Alzheimer’s Disease Model. Cell 2015, 160, 1061–1071. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L. CD33 in Alzheimer’s Disease—Biology, Pathogenesis, and Therapeutics: A Mini-Review. Gerontology 2018, 65, 323–331. [Google Scholar] [CrossRef] [PubMed]
- Lim, G.P.; Yang, F.; Chu, T.; Chen, P.; Beech, W.; Teter, B.; Tran, T.; Ubeda, O.; Ashe, K.H.; Frautschy, S.A.; et al. Ibuprofen Suppresses Plaque Pathology and Inflammation in a Mouse Model for Alzheimer’s Disease. J. Neurosci. 2000, 20, 5709–5714. [Google Scholar] [CrossRef] [PubMed]
- Heneka, M.T.; Sastre, M.; Dumitrescu-Ozimek, L.; Hanke, A.; Dewachter, I.; Kuiperi, C.; O’Banion, M.K.; Klockgether, T.; Van Leuven, F.; Landreth, G.E. Acute treatment with the PPARγ agonist pioglitazone and ibuprofen reduces glial inflammation and Aβ1–42 levels in APPV717I transgenic mice. Brain 2005, 128, 1442–1453. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, B.L.; Cramer, P.E.; Varvel, N.H.; Reed-Geaghan, E.; Jiang, Q.; Szabo, A.; Herrup, K.; Lamb, B.T.; Landreth, G.E. Ibuprofen attenuates oxidative damage through NOX2 inhibition in Alzheimer’s disease. Neurobiol. Aging 2012, 33, 197.e21–197.e32. [Google Scholar] [CrossRef]
- Galimberti, D.; Scarpini, E. Pioglitazone for the treatment of Alzheimer’s disease. Expert Opin. Investig. Drugs 2016, 26, 97–101. [Google Scholar] [CrossRef]
- Dong, Y.; Li, X.; Cheng, J.; Hou, L. Drug Development for Alzheimer’s Disease: Microglia Induced Neuroinflammation as a Target? Int. J. Mol. Sci. 2019, 20, 558. [Google Scholar] [CrossRef]
- Thawkar, B.S.; Kaur, G. Inhibitors of NF-κB and P2X7/NLRP3/Caspase 1 pathway in microglia: Novel therapeutic opportunities in neuroinflammation induced early-stage Alzheimer’s disease. J. Neuroimmunol. 2019, 326, 62–74. [Google Scholar] [CrossRef]
- Dempsey, C.; Rubio-Araiz, A.; Bryson, K.J.; Finucane, O.; Larkin, C.; Mills, E.L.; Robertson, A.A.B.; Cooper, M.A.; O’Neill, L.A.J.; Lynch, M.A. Inhibiting the NLRP3 inflammasome with MCC950 promotes non-phlogistic clearance of amyloid-β and cognitive function in APP/PS1 mice. Brain, Behav. Immun. 2017, 61, 306–316. [Google Scholar] [CrossRef]
- Howard, R.; Zubko, O.; Bradley, R.; Harper, E.; Pank, L.; O’Brien, J.; Fox, C.; Tabet, N.; Livingston, G.; Bentham, P.; et al. Minocycline at 2 Different Dosages vs Placebo for Patients with Mild Alzheimer Disease: A Randomized Clinical Trial. JAMA Neurol. 2020, 77, 164–174. [Google Scholar] [CrossRef]
- Wang, H.-M.; Zhang, T.; Huang, J.-K.; Xiang, J.-Y.; Chen, J.-J.; Fu, J.-L.; Zhao, Y.-W. Edaravone Attenuates the Proinflammatory Response in Amyloid-β-Treated Microglia by Inhibiting NLRP3 Inflammasome-Mediated IL-1β Secretion. Cell. Physiol. Biochem. 2017, 43, 1113–1125. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.G.; Won, S.M.; Gwag, B.J.; Lee, Y.B. Microglial P2X7receptor expression is accompanied by neuronal damage in the cerebral cortex of the APPswe/PS1dE9 mouse model of Alzheimer’s disease. Exp. Mol. Med. 2011, 43, 7–14. [Google Scholar] [CrossRef] [PubMed]
- Verheijen, J.; Sleegers, K. Understanding Alzheimer Disease at the Interface between Genetics and Transcriptomics. Trends Genet. 2018, 34, 434–447. [Google Scholar] [CrossRef] [PubMed]
- Liddelow, S.A.; Barres, B.A. Reactive Astrocytes: Production, Function, and Therapeutic Potential. Immunity 2017, 46, 957–967. [Google Scholar] [CrossRef]
- Zhao, N.; Francis, N.L.; Calvelli, H.R.; Moghe, P.V. Microglia-targeting nanotherapeutics for neurodegenerative diseases. APL Bioeng. 2020, 4, 030902. [Google Scholar] [CrossRef]
- Hemonnot, A.-L.; Hua, J.; Ulmann, L.; Hirbec, H. Microglia in Alzheimer Disease: Well-Known Targets and New Opportunities. Front. Aging Neurosci. 2019, 11, 233. [Google Scholar] [CrossRef]



| Event | Mediator | Effect on Microglial Function | References |
|---|---|---|---|
| Microglial mitophagy | High mobility group box 1/receptor for advanced glycation endproducts signaling mechanisms | Significant blockage of late-stage mitophagy in microglia | [79] |
| Role of microglia in amyloid beta (Aβ) | Apolipoprotein E Gene | Apolipoprotein E ε4 genotype is related to diminished Aβ plaques | [80] |
| Receptor for advanced glycation end products | Exerts dual effects in Aβ phagocytosis | [81] | |
| Scavenger receptor class A | Mediates microglial adhesion to Aβ and elevates the level of Aβ uptake by microglia | [82] | |
| Class B scavenger receptor | Exerts dual effects in Aβ phagocytosis | [83] | |
| Triggering receptor expressed on myeloid cells 2 Gene | Exerts dual effects in Aβ phagocytosis | [84] | |
| Complement C3b/C4b Receptor 1 Gene | Mediates microglia-mediated Aβ phagocytosis | [85] | |
| CD33 Gene | Decreases microglia-mediated Aβ phagocytosis | [31] | |
| ATP Binding Cassette Subfamily A Member 7 Gene | Mediates microglia-mediated Aβ phagocytosis | [86] | |
| Role of microglia in neuroinflammation | C-X3-C Motif Chemokine Receptor 1 | Deficiency of this inflammatory adipose chemokine system deteriorates tau phosphorylation | [87] |
| NOD-like receptor family pyrin domain-containing 3 | Exacerbates inflammatory response mediated by microglia | [88] | |
| Suppressors of cytokine signaling | Shows protective properties by balancing the level of inflammatory response | [89] | |
| Role of microglia in tau pathology | Triggering receptor expressed on myeloid cells 2 Gene | Mediates intraneuronal tau aggregation | [90] |
| Apolipoprotein E Gene | Apolipoprotein ε4 genotype significantly worsens neurodegeneration mediated by tau | [91] | |
| Colony-stimulating factor 1 receptor | Suppression of colony-stimulating factor 1 receptor results in the reduction of tau-mediated neurodegeneration | [92,93] |
| Therapeutic Approaches | Therapeutics | Mechanisms | References |
|---|---|---|---|
| Therapies targeting inflammatory response in microglia | Nimodipine, edaravone, minocycline, JC-124, MCC950, pioglitazone, ibuprofen | Amelioration of over-activated microglia and suppression of microglia-linked inflammatory responses | [104,144,145,146,147,148,149,150,151,152,153,154,155] |
| Therapies targeting microglial immunoreceptors | AL002c, AL002a, AL002, monoclonal antibody 4D9 | Improvement of TREM2 function to elevate microglial reactions towards Aβ | [156,157,158,159] |
| Lintuzumab, P22 | Suppression of CD33 function to elevate the level of Aβ phagocytosis | [160,161] | |
| Microglia modifying therapies | Inhibitors of colony-stimulating factor 1 receptor: PLX5622, PLX3397 | Reducing dysfunctional microglia | [141,142] |
| Stem cell therapy | Resupplying healthy microglia | [92,162,163,164,165,166,167,168] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Althafar, Z.M. Targeting Microglia in Alzheimer’s Disease: From Molecular Mechanisms to Potential Therapeutic Targets for Small Molecules. Molecules 2022, 27, 4124. https://doi.org/10.3390/molecules27134124
Althafar ZM. Targeting Microglia in Alzheimer’s Disease: From Molecular Mechanisms to Potential Therapeutic Targets for Small Molecules. Molecules. 2022; 27(13):4124. https://doi.org/10.3390/molecules27134124
Chicago/Turabian StyleAlthafar, Ziyad M. 2022. "Targeting Microglia in Alzheimer’s Disease: From Molecular Mechanisms to Potential Therapeutic Targets for Small Molecules" Molecules 27, no. 13: 4124. https://doi.org/10.3390/molecules27134124
APA StyleAlthafar, Z. M. (2022). Targeting Microglia in Alzheimer’s Disease: From Molecular Mechanisms to Potential Therapeutic Targets for Small Molecules. Molecules, 27(13), 4124. https://doi.org/10.3390/molecules27134124