
Molecular Analysis and Conformational Dynamics of Human MC4R Disease-Causing Mutations


 ,
,
Abstract
:
1. Introduction
2. Material and Methods
2.1. Sequence and Structural Data Information
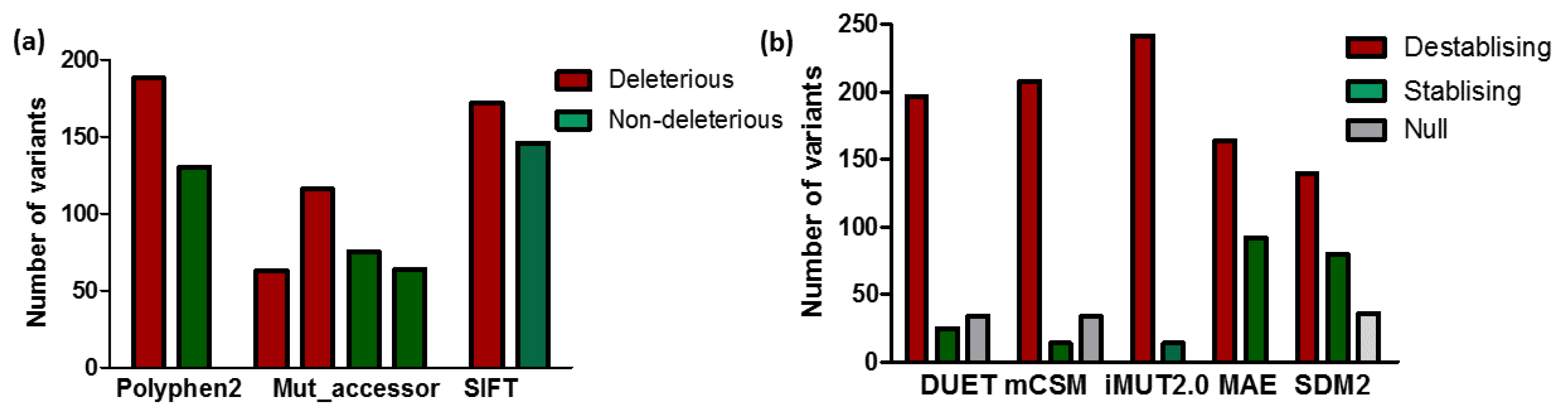
2.2. Sequence-Based Prediction of Deleterious Mutations
2.3. Structure-Based Predictors to Investigate Protein Stability/Destabilizing Mutations
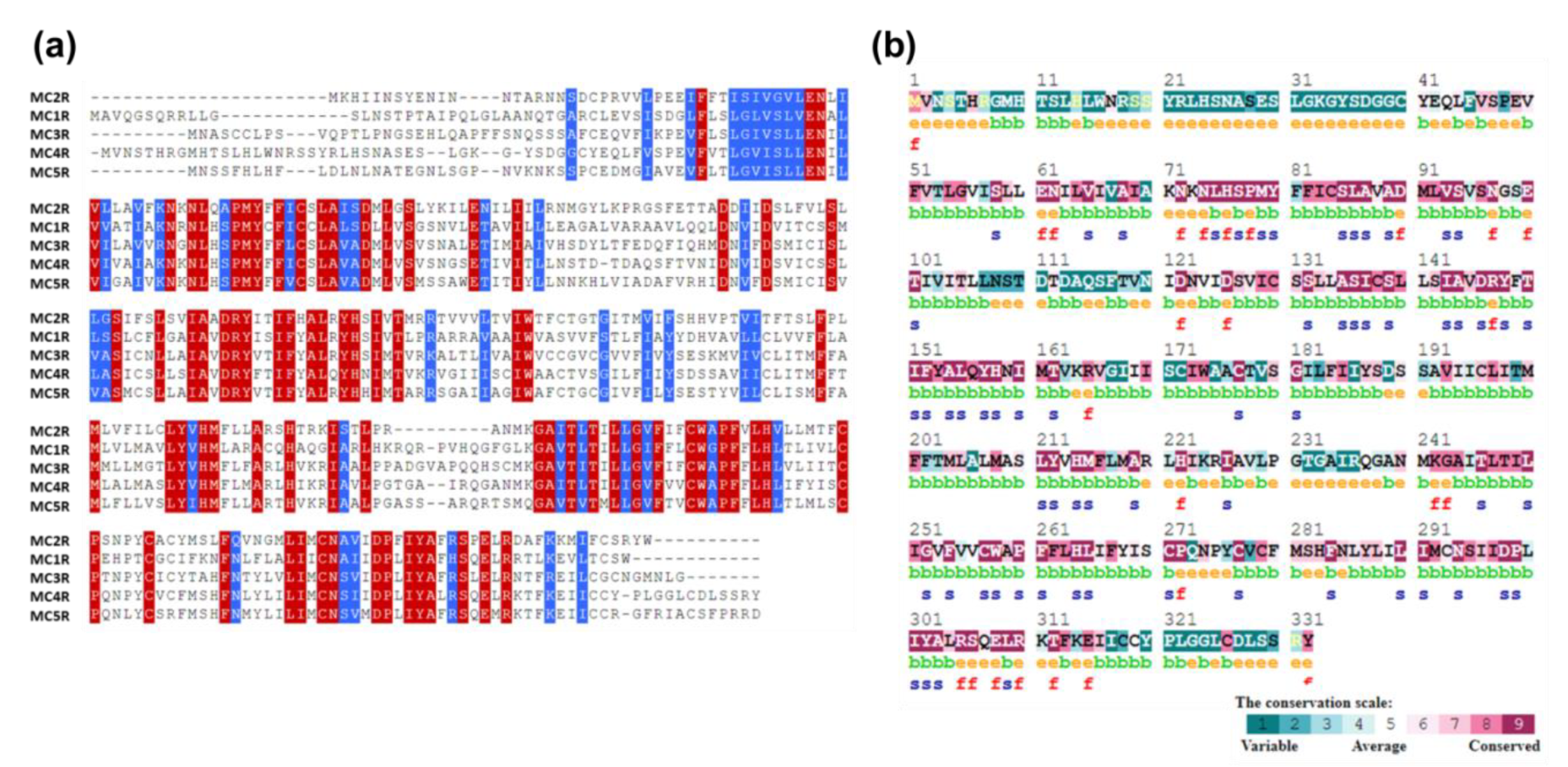
2.4. Conservation of Amino Acid Positions in the MC4R Sequence
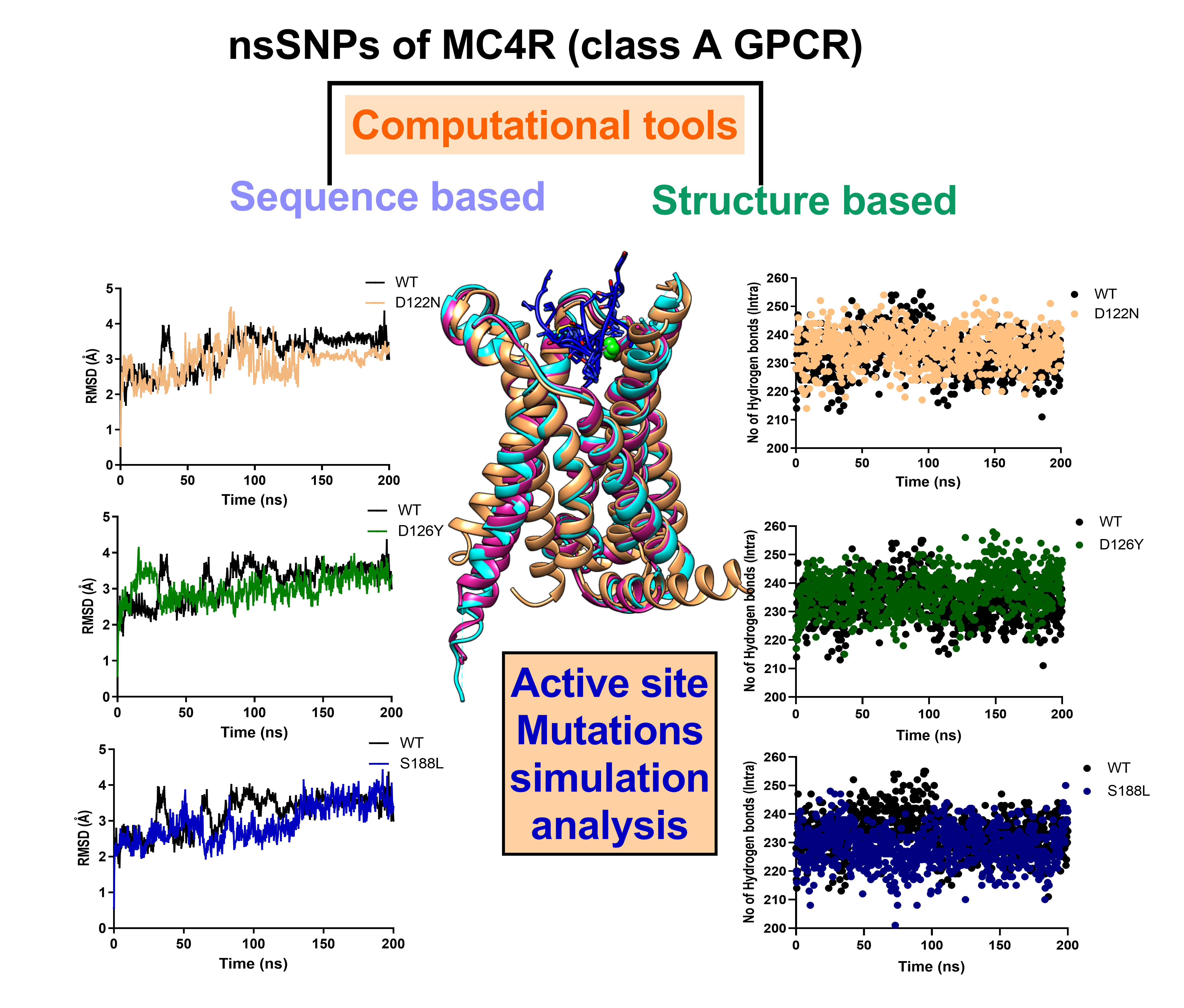
2.5. MD Simulation of MC4R Complex Structures
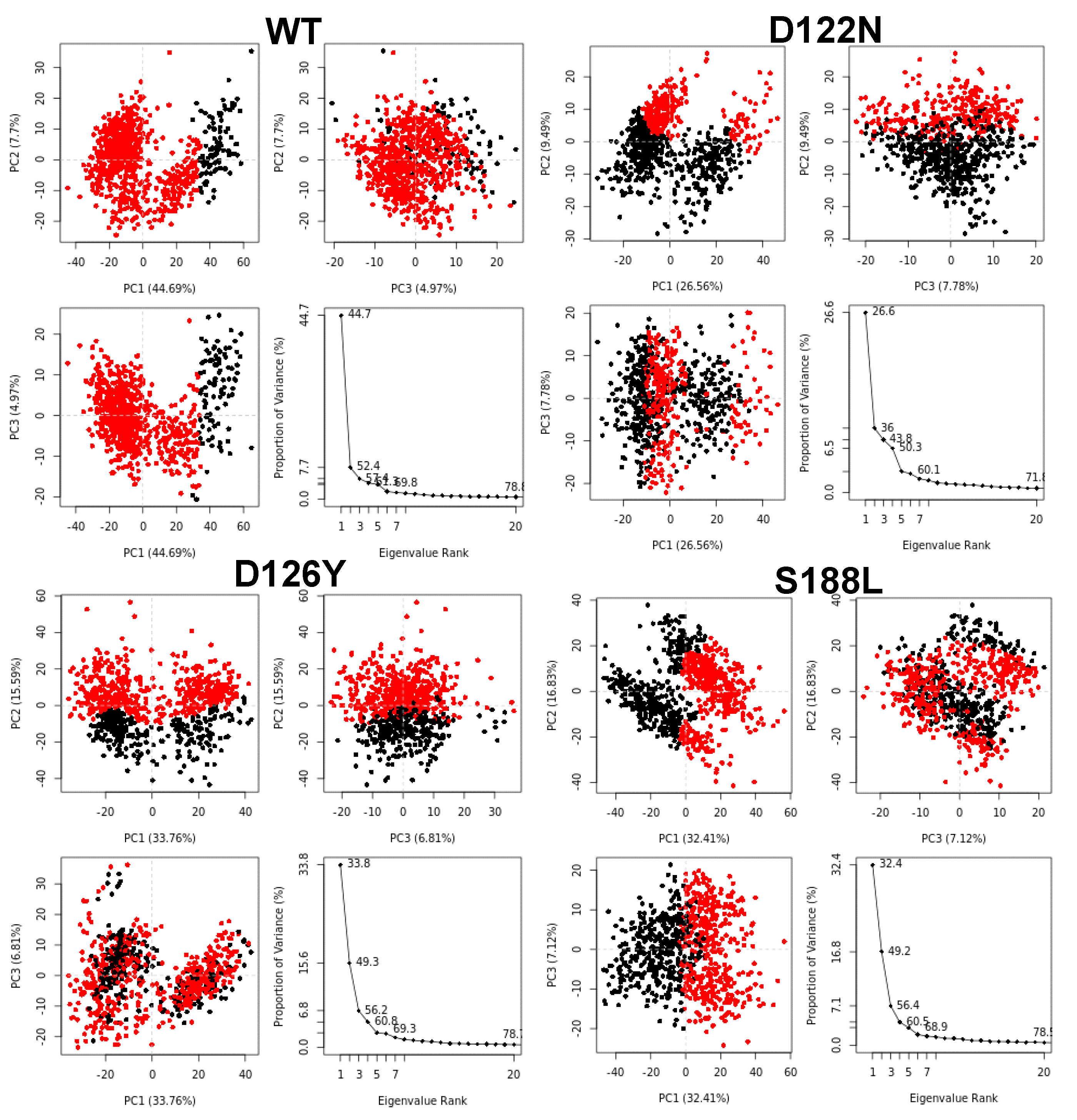
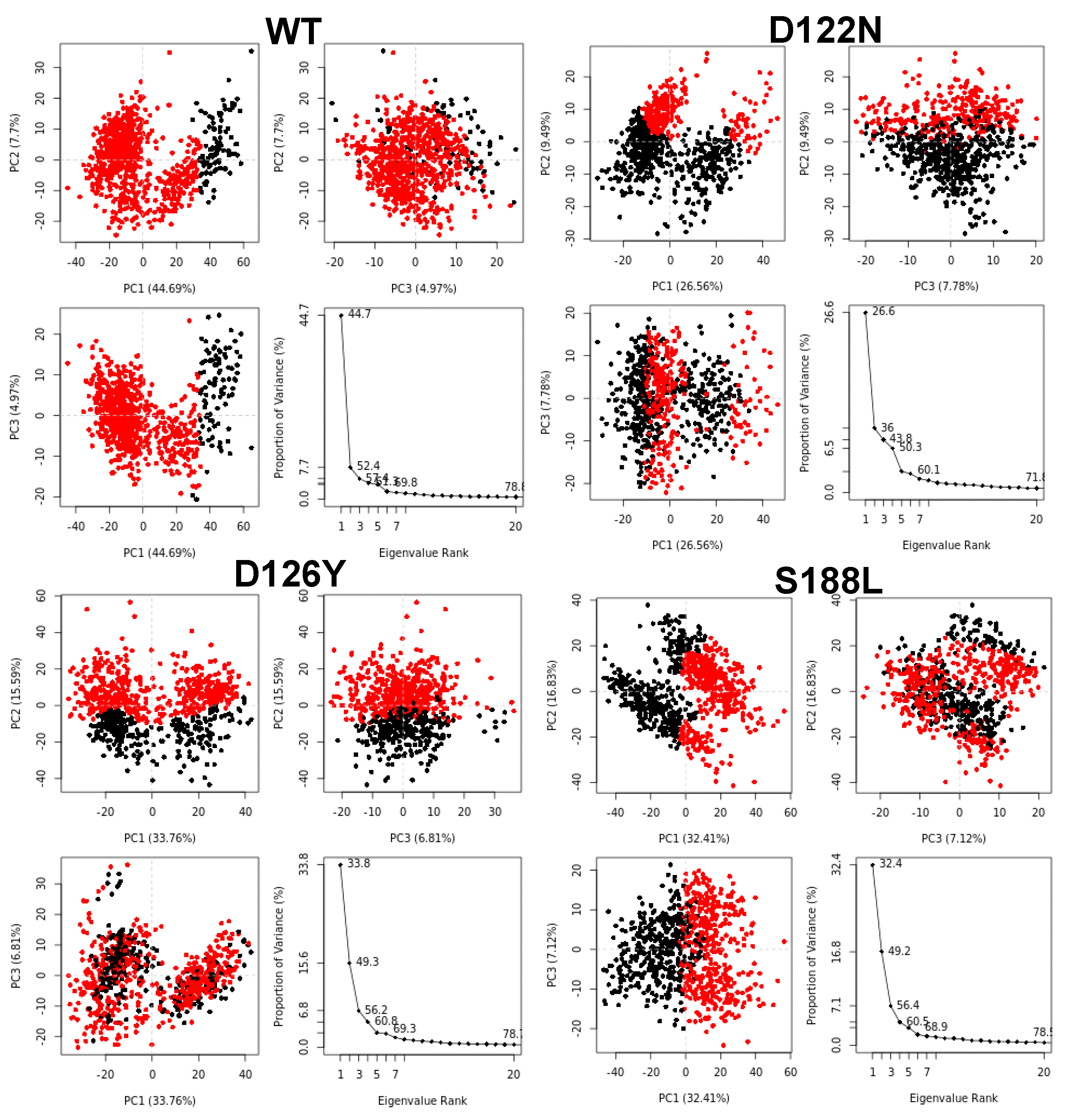
2.6. Principal Components Analysis (PCA)
3. Results and Discussion
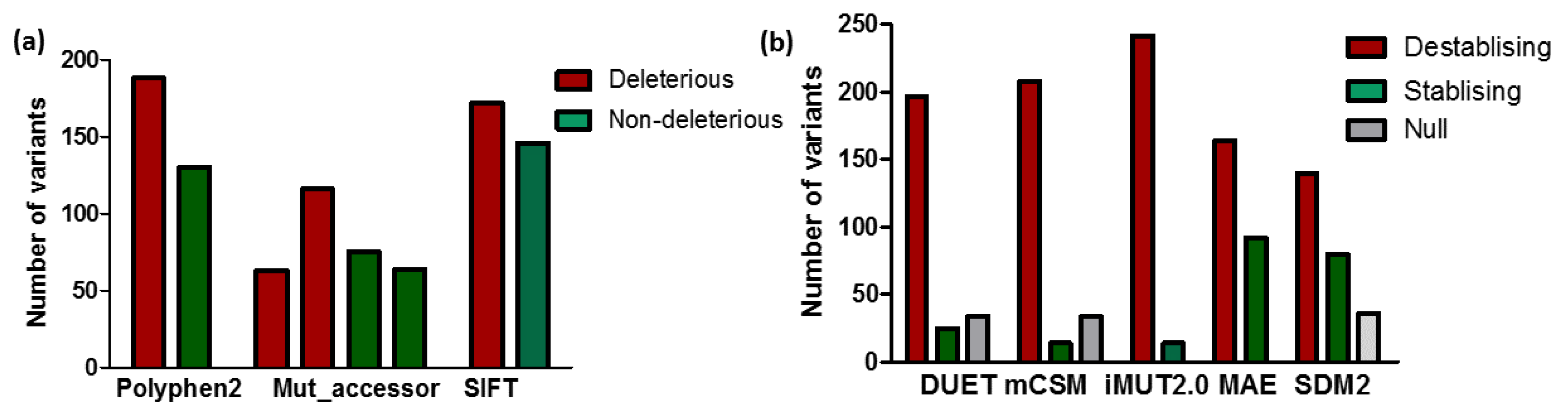
3.1. Predicting the Impact of nsSNPs of MC4R
3.2. Impact of Mutation on MC4R Stability
3.3. Sequence Conservation Analysis
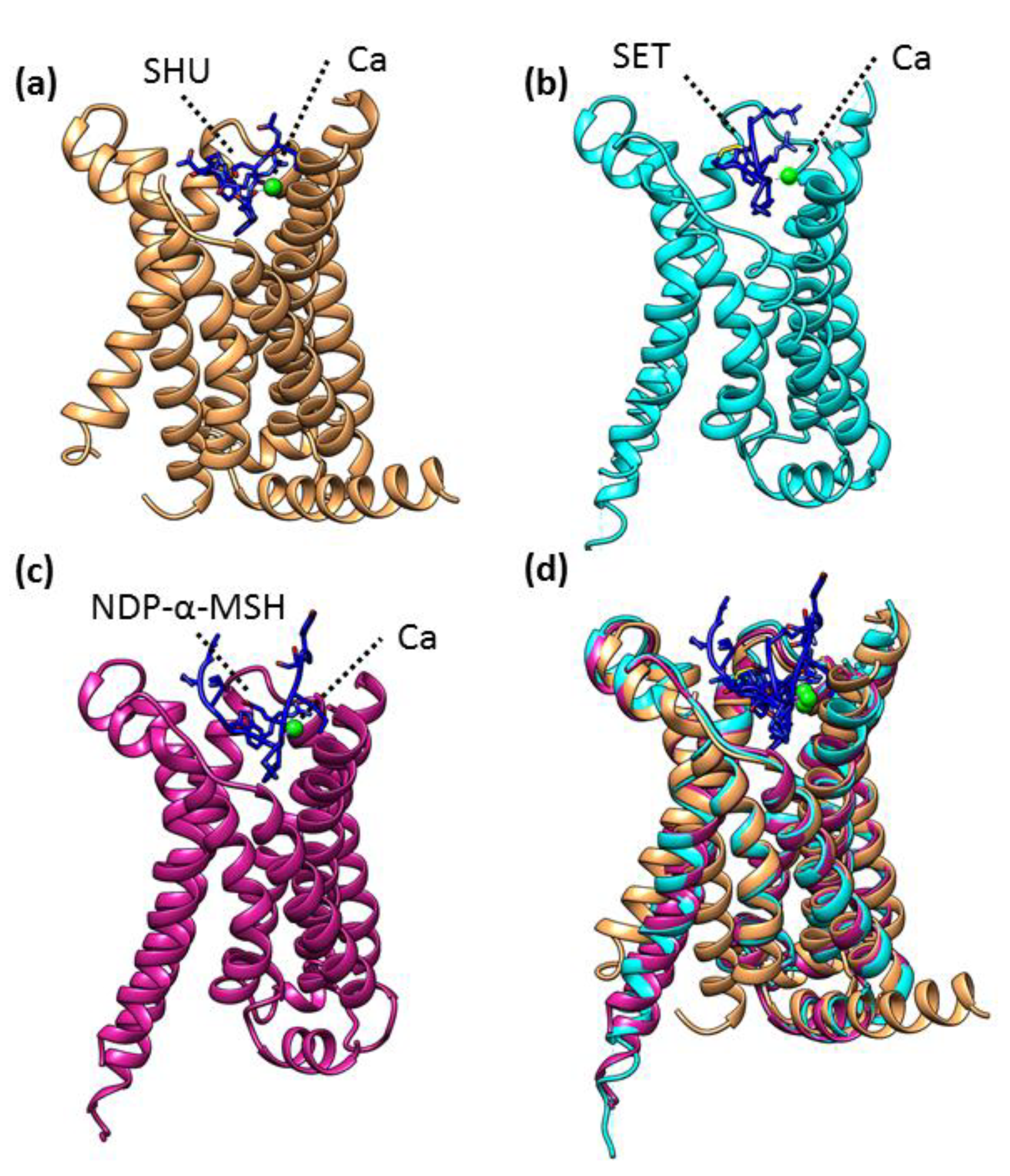
3.4. Structural Analysis of MC4R
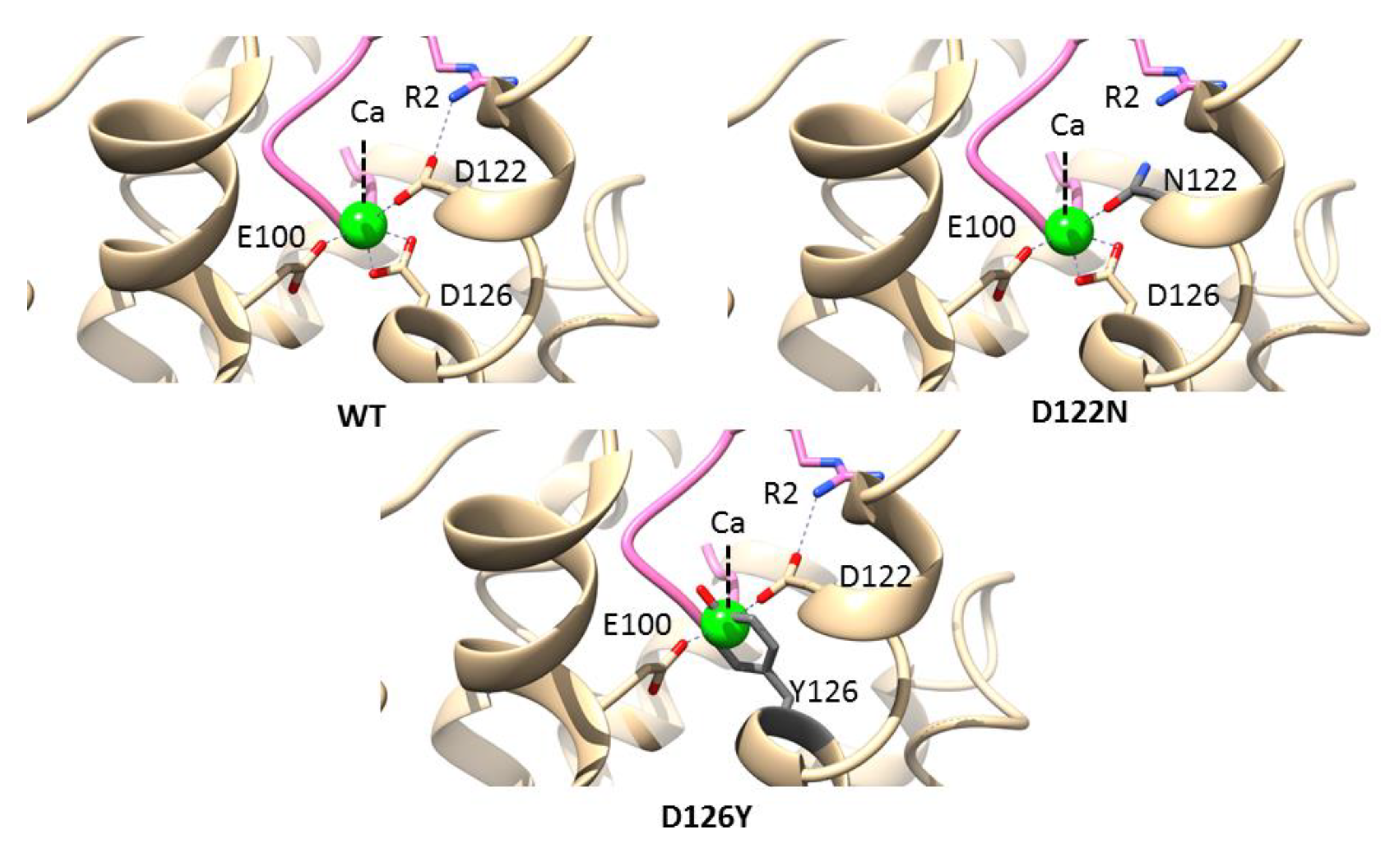
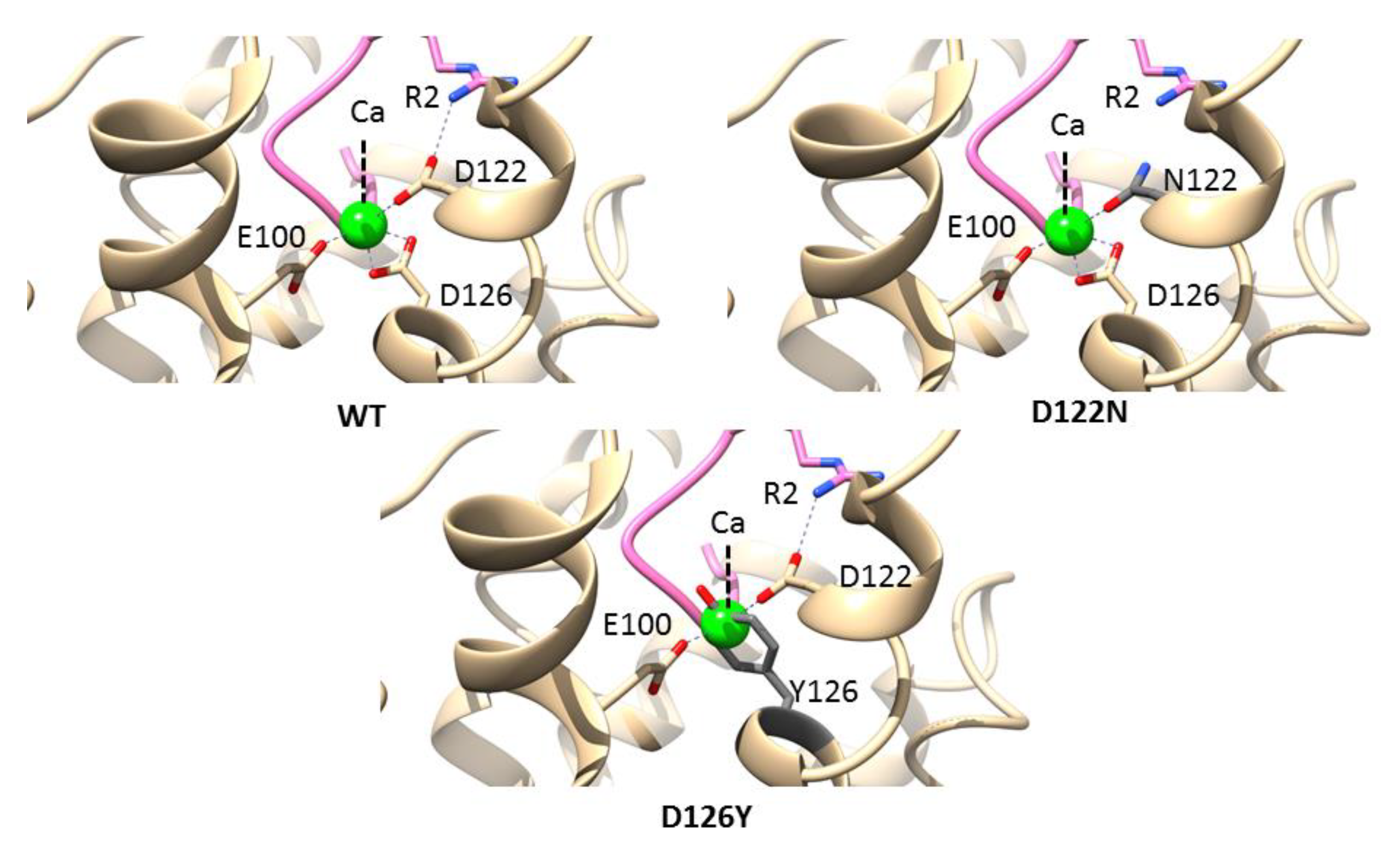
3.5. Mutations in the Calcium-Binding Site
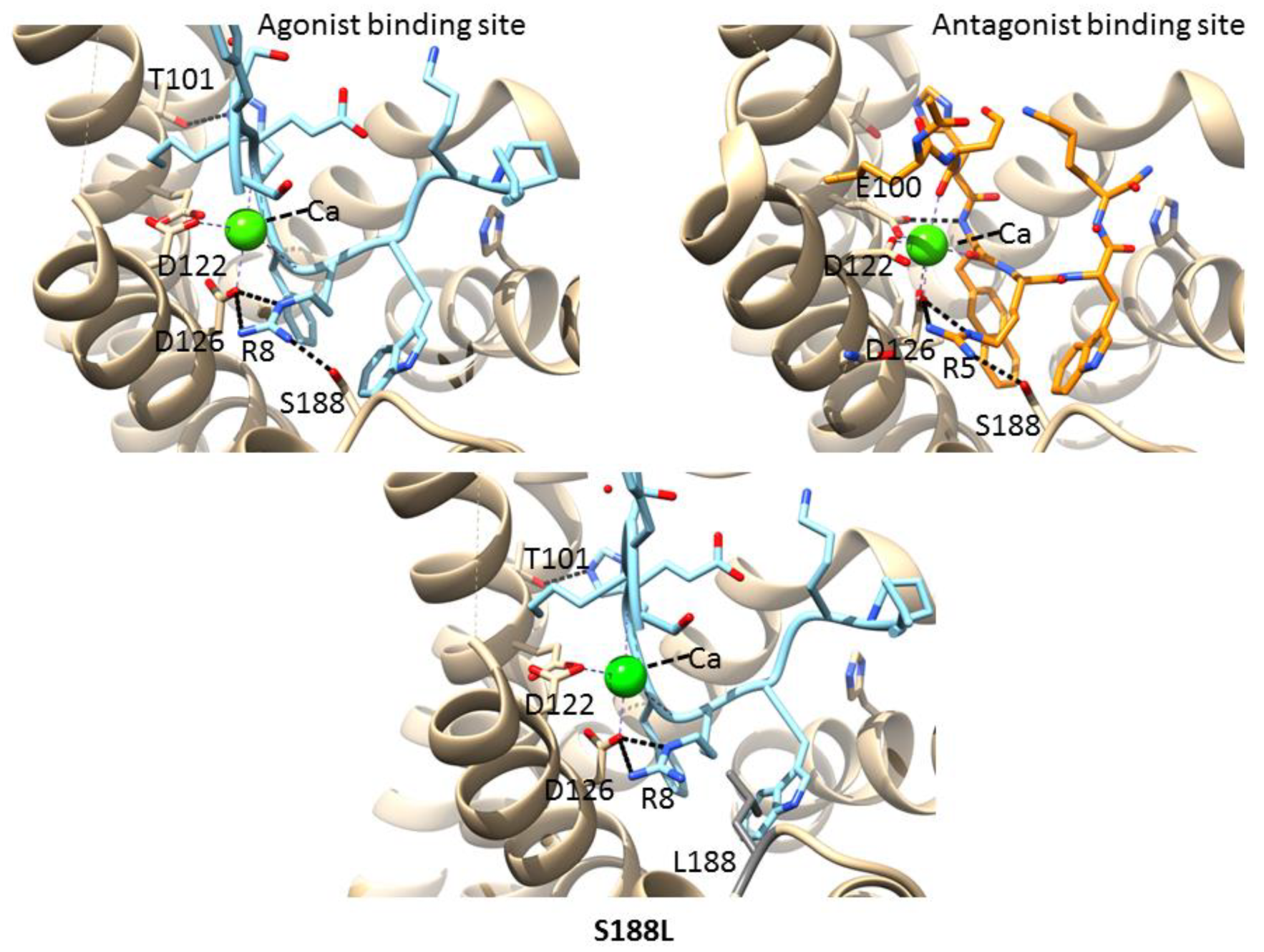
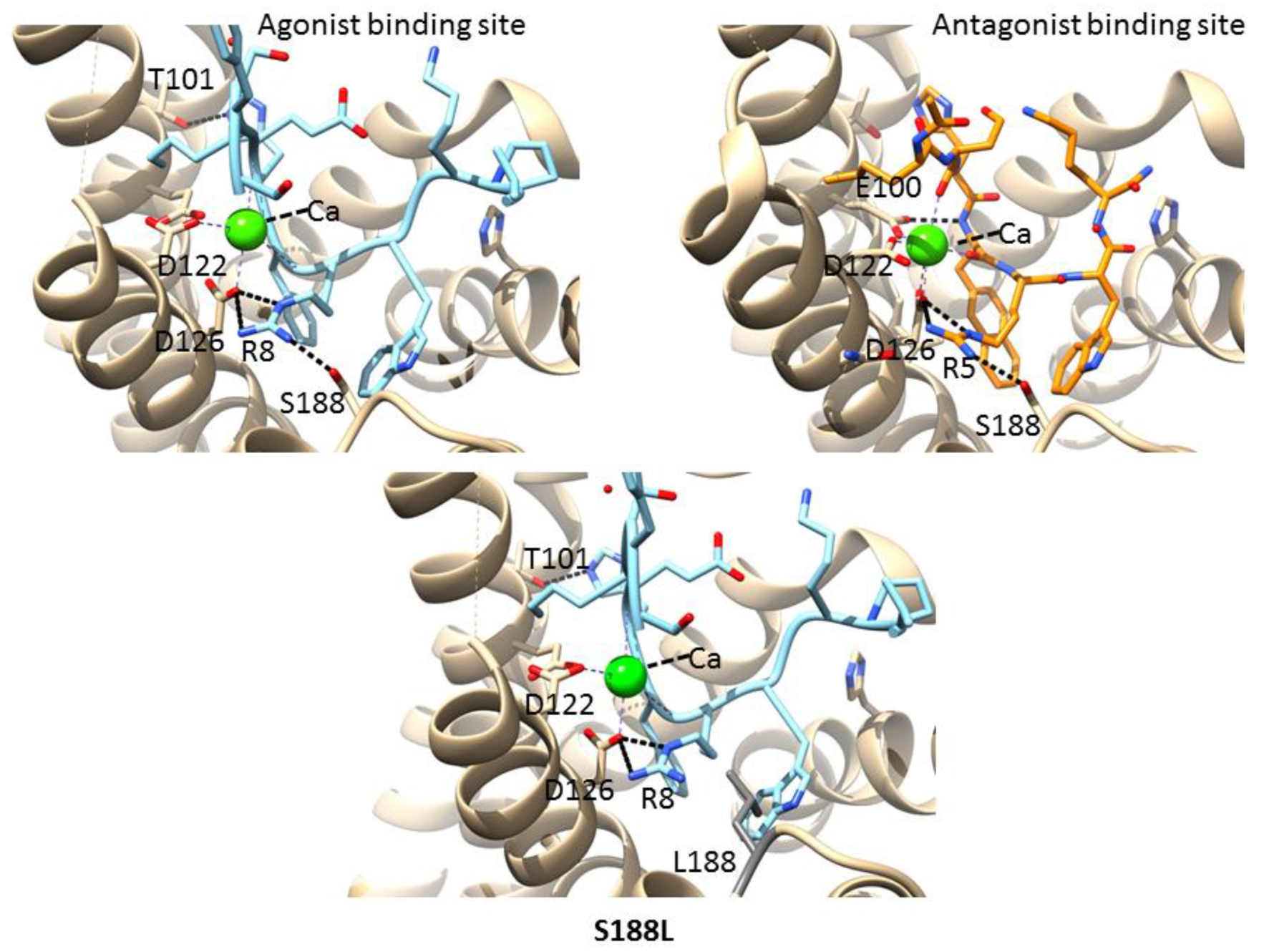
3.6. Mutations in the Agonist/Antagonist-Binding Pocket
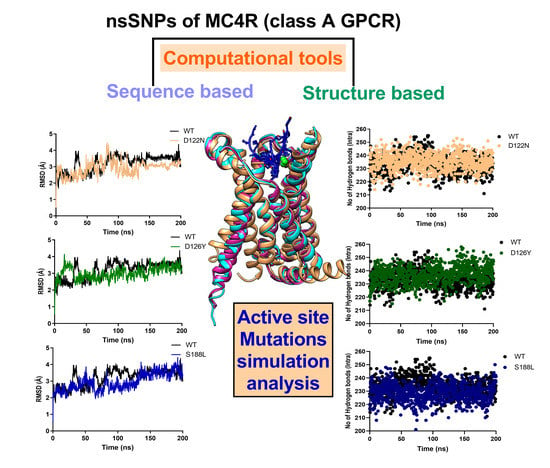
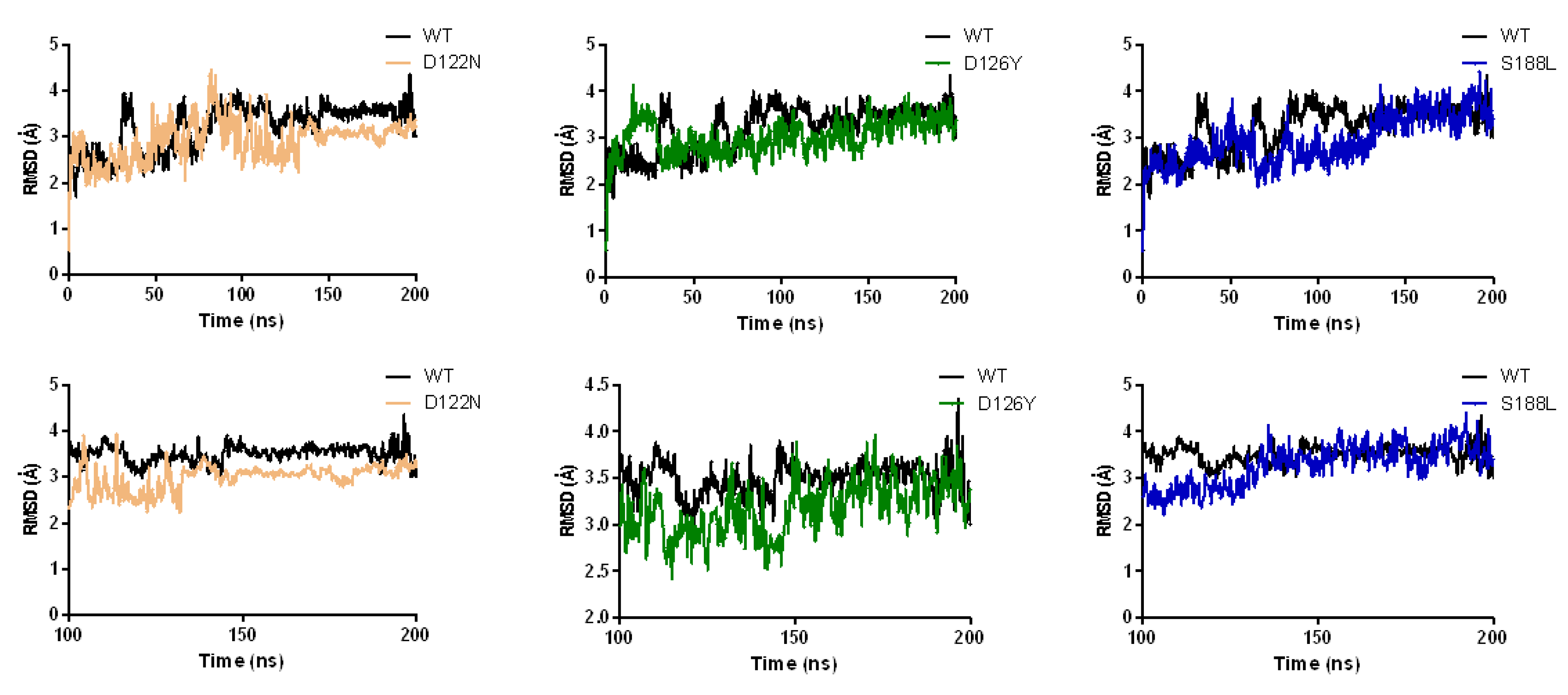
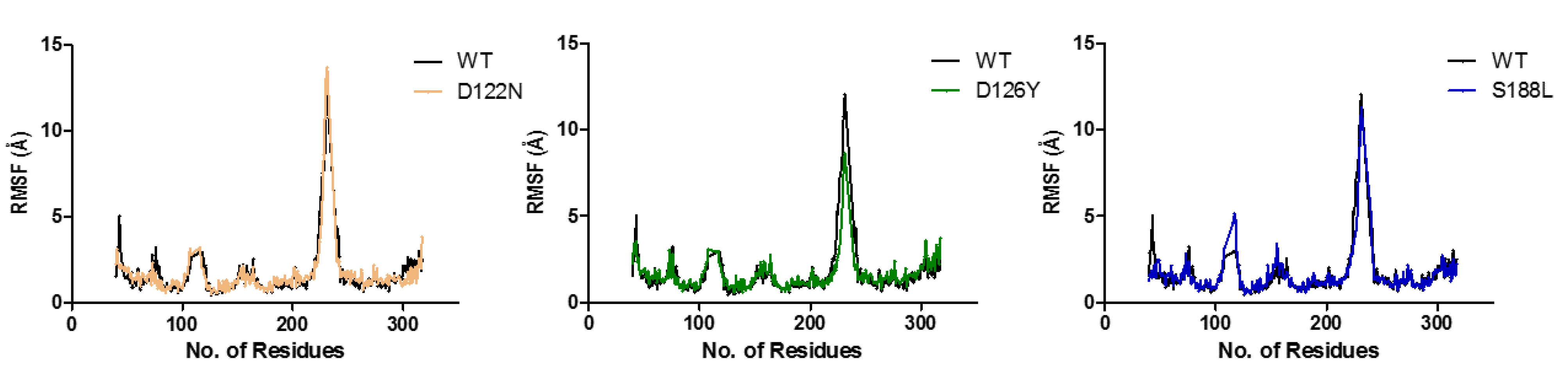
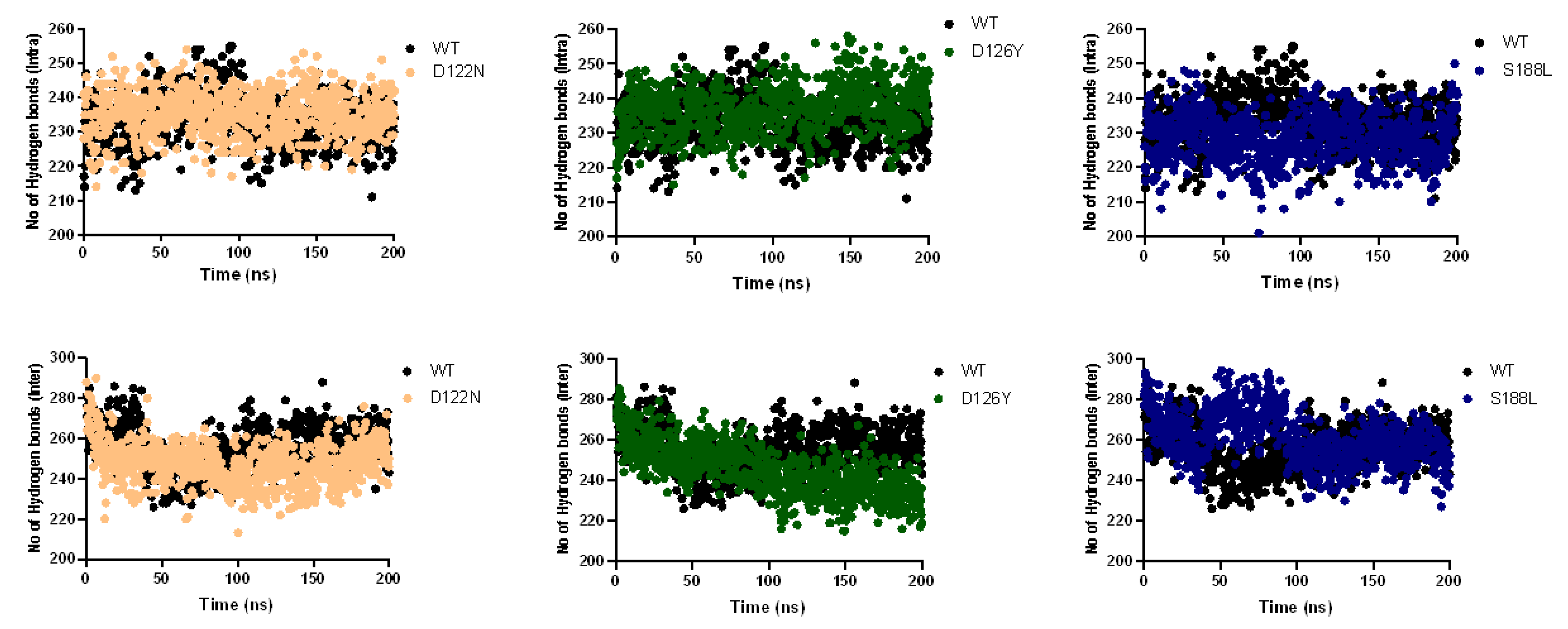
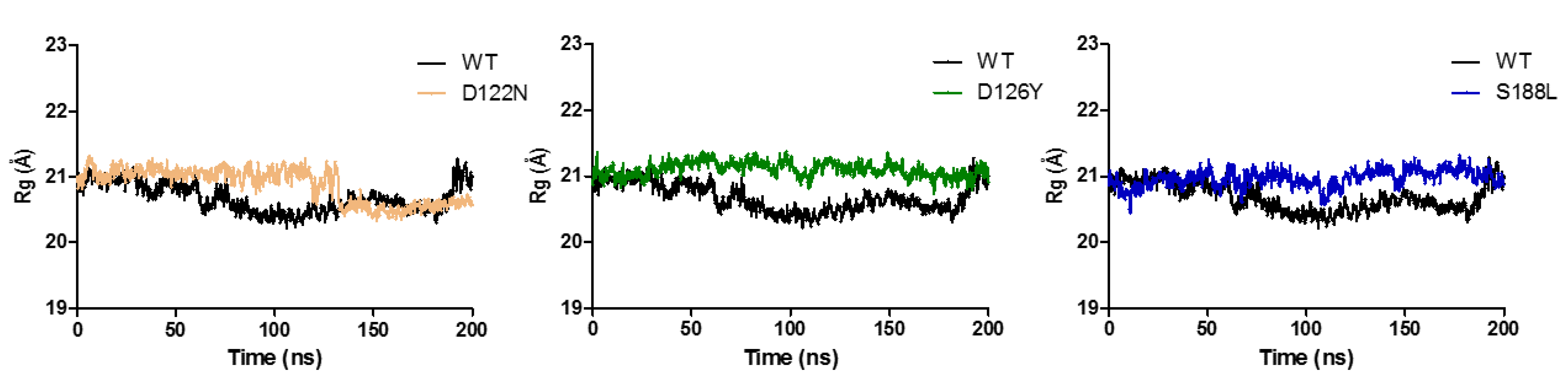
3.7. Dynamics of the Wild-Type and Selected Mutant Complexes
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Krashes, M.J.; Lowell, B.B.; Garfield, A.S. Melanocortin-4 Receptor-Regulated Energy Homeostasis. Nat. Neurosci. 2016, 19, 206–219. [Google Scholar] [CrossRef] [Green Version]
- Fatima, M.T.; Ahmed, I.; Fakhro, K.A.; Akil, A.S.A.-S. Melanocortin-4 Receptor Complexity in Energy Homeostasis, Obesity and Drug Development Strategies. Diabetes Obes. Metab. 2022, 24, 583–598. [Google Scholar] [CrossRef]
- Baldini, G.; Phelan, K.D. The Melanocortin Pathway and Control of Appetite-Progress and Therapeutic Implications. J. Endocrinol. 2019, 241, R1–R33. [Google Scholar] [CrossRef] [PubMed]
- Huvenne, H.; Dubern, B.; Clément, K.; Poitou, C. Rare Genetic Forms of Obesity: Clinical Approach and Current Treatments in 2016. Obes. Facts 2016, 9, 158–173. [Google Scholar] [CrossRef]
- Gonçalves, J.P.L.; Palmer, D.; Meldal, M. MC4R Agonists: Structural Overview on Antiobesity Therapeutics. Trends Pharmacol. Sci. 2018, 39, 402–423. [Google Scholar] [CrossRef] [PubMed]
- Lotta, L.A.; Mokrosiński, J.; de Oliveira, E.M.; Li, C.; Sharp, S.J.; Luan, J.; Brouwers, B.; Ayinampudi, V.; Bowker, N.; Kerrison, N.; et al. Human Gain-of-Function MC4R Variants Show Signaling Bias and Protect against Obesity. Cell 2019, 177, 597–607.e9. [Google Scholar] [CrossRef] [Green Version]
- Tao, Y.-X. The Melanocortin-4 Receptor: Physiology, Pharmacology, and Pathophysiology. Endocr. Rev. 2010, 31, 506–543. [Google Scholar] [CrossRef] [Green Version]
- Kühnen, P.; Krude, H.; Biebermann, H. Melanocortin-4 Receptor Signalling: Importance for Weight Regulation and Obesity Treatment. Trends Mol. Med. 2019, 25, 136–148. [Google Scholar] [CrossRef]
- Namjou, B.; Stanaway, I.B.; Lingren, T.; Mentch, F.D.; Benoit, B.; Dikilitas, O.; Niu, X.; Shang, N.; Shoemaker, A.H.; Carey, D.J.; et al. Evaluation of the MC4R Gene across EMERGE Network Identifies Many Unreported Obesity-Associated Variants. Int. J. Obes. 2021, 45, 155–169. [Google Scholar] [CrossRef] [PubMed]
- Aykut, A.; Özen, S.; Gökşen, D.; Ata, A.; Onay, H.; Atik, T.; Darcan, Ş.; Özkinay, F. Melanocortin 4 Receptor (MC4R) Gene Variants in Children and Adolescents Having Familial Early-Onset Obesity: Genetic and Clinical Characteristics. Eur. J. Pediatr. 2020, 179, 1445–1452. [Google Scholar] [CrossRef]
- Lubrano-Berthelier, C.; Cavazos, M.; Dubern, B.; Shapiro, A.; Stunff, C.L.E.; Zhang, S.; Picart, F.; Govaerts, C.; Froguel, P.; Bougneres, P.; et al. Molecular Genetics of Human Obesity-Associated MC4R Mutations. Ann. N. Y. Acad. Sci. 2003, 994, 49–57. [Google Scholar] [CrossRef]
- MacKenzie, R.G. Obesity-Associated Mutations in the Human Melanocortin-4 Receptor Gene. Peptides 2006, 27, 395–403. [Google Scholar] [CrossRef]
- Huszar, D.; Lynch, C.A.; Fairchild-Huntress, V.; Dunmore, J.H.; Fang, Q.; Berkemeier, L.R.; Gu, W.; Kesterson, R.A.; Boston, B.A.; Cone, R.D.; et al. Targeted Disruption of the Melanocortin-4 Receptor Results in Obesity in Mice. Cell 1997, 88, 131–141. [Google Scholar] [CrossRef] [Green Version]
- Büch, T.R.H.; Heling, D.; Damm, E.; Gudermann, T.; Breit, A. Pertussis Toxin-Sensitive Signaling of Melanocortin-4 Receptors in Hypothalamic GT1-7 Cells Defines Agouti-Related Protein as a Biased Agonist. J. Biol. Chem. 2009, 284, 26411–26420. [Google Scholar] [CrossRef] [Green Version]
- Kühnen, P.; Clément, K.; Wiegand, S.; Blankenstein, O.; Gottesdiener, K.; Martini, L.L.; Mai, K.; Blume-Peytavi, U.; Grüters, A.; Krude, H. Proopiomelanocortin Deficiency Treated with a Melanocortin-4 Receptor Agonist. N. Engl. J. Med. 2016, 375, 240–246. [Google Scholar] [CrossRef]
- Clément, K.; Biebermann, H.; Farooqi, I.S.; Van der Ploeg, L.; Wolters, B.; Poitou, C.; Puder, L.; Fiedorek, F.; Gottesdiener, K.; Kleinau, G.; et al. MC4R Agonism Promotes Durable Weight Loss in Patients with Leptin Receptor Deficiency. Nat. Med. 2018, 24, 551–555. [Google Scholar] [CrossRef]
- UniProt Consortium UniProt: The Universal Protein Knowledgebase in 2021. Nucleic Acids Res. 2021, 49, D480–D489. [CrossRef]
- Hubbard, T.; Barker, D.; Birney, E.; Cameron, G.; Chen, Y.; Clark, L.; Cox, T.; Cuff, J.; Curwen, V.; Down, T.; et al. The Ensembl Genome Database Project. Nucleic Acids Res. 2002, 30, 38–41. [Google Scholar] [CrossRef] [Green Version]
- Kumar, P.; Henikoff, S.; Ng, P.C. Predicting the Effects of Coding Non-Synonymous Variants on Protein Function Using the SIFT Algorithm. Nat. Protoc. 2009, 4, 1073–1081. [Google Scholar] [CrossRef]
- Adzhubei, I.A.; Schmidt, S.; Peshkin, L.; Ramensky, V.E.; Gerasimova, A.; Bork, P.; Kondrashov, A.S.; Sunyaev, S.R. A Method and Server for Predicting Damaging Missense Mutations. Nat. Methods 2010, 7, 248–249. [Google Scholar] [CrossRef] [Green Version]
- Reva, B.; Antipin, Y.; Sander, C. Predicting the Functional Impact of Protein Mutations: Application to Cancer Genomics. Nucleic Acids Res. 2011, 39, e118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pires, D.E.V.; Ascher, D.B.; Blundell, T.L. MCSM: Predicting the Effects of Mutations in Proteins Using Graph-Based Signatures. Bioinformatics 2014, 30, 335–342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, M.D.S.; Bava, K.A.; Gromiha, M.M.; Prabakaran, P.; Kitajima, K.; Uedaira, H.; Sarai, A. ProTherm and ProNIT: Thermodynamic Databases for Proteins and Protein-Nucleic Acid Interactions. Nucleic Acids Res. 2006, 34, D204–D206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moal, I.H.; Fernández-Recio, J. SKEMPI: A Structural Kinetic and Energetic Database of Mutant Protein Interactions and Its Use in Empirical Models. Bioinformatics 2012, 28, 2600–2607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pandurangan, A.P.; Ochoa-Montaño, B.; Ascher, D.B.; Blundell, T.L. SDM: A Server for Predicting Effects of Mutations on Protein Stability. Nucleic Acids Res. 2017, 45, W229–W235. [Google Scholar] [CrossRef] [Green Version]
- Pires, D.E.V.; Ascher, D.B.; Blundell, T.L. DUET: A Server for Predicting Effects of Mutations on Protein Stability Using an Integrated Computational Approach. Nucleic Acids Res. 2014, 42, W314–W319. [Google Scholar] [CrossRef] [PubMed]
- Capriotti, E.; Fariselli, P.; Casadio, R. I-Mutant2.0: Predicting Stability Changes upon Mutation from the Protein Sequence or Structure. Nucleic Acids Res. 2005, 33, W306–W310. [Google Scholar] [CrossRef] [Green Version]
- Laimer, J.; Hiebl-Flach, J.; Lengauer, D.; Lackner, P. MAESTROweb: A Web Server for Structure-Based Protein Stability Prediction. Bioinformatics 2016, 32, 1414–1416. [Google Scholar] [CrossRef] [PubMed]
- Madeira, F.; Park, Y.M.; Lee, J.; Buso, N.; Gur, T.; Madhusoodanan, N.; Basutkar, P.; Tivey, A.R.N.; Potter, S.C.; Finn, R.D.; et al. The EMBL-EBI Search and Sequence Analysis Tools APIs in 2019. Nucleic Acids Res. 2019, 47, W636–W641. [Google Scholar] [CrossRef] [Green Version]
- Ashkenazy, H.; Erez, E.; Martz, E.; Pupko, T.; Ben-Tal, N. ConSurf 2010: Calculating Evolutionary Conservation in Sequence and Structure of Proteins and Nucleic Acids. Nucleic Acids Res. 2010, 38, W529–W533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krieger, E.; Vriend, G. YASARA View—Molecular Graphics for All Devices—From Smartphones to Workstations. Bioinformatics 2014, 30, 2981–2982. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. Ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from Ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef] [Green Version]
- Gould, I.R.; Skjevik, A.A.; Dickson, C.J.; Madej, B.D.; Walker, R.C. Lipid17: A Comprehensive AMBER Force Field for the Simulation of Zwitterionic and Anionic Lipids. Manuscr. Prep. 2018, in press. [Google Scholar]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and Testing of a General Amber Force Field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef]
- Jakalian, A.; Jack, D.B.; Bayly, C.I. Fast, Efficient Generation of High-Quality Atomic Charges. AM1-BCC Model: II. Parameterization and Validation. J. Comput. Chem. 2002, 23, 1623–1641. [Google Scholar] [CrossRef] [PubMed]
- Krieger, E.; Nielsen, J.E.; Spronk, C.A.E.M.; Vriend, G. Fast Empirical PKa Prediction by Ewald Summation. J. Mol. Graph. Model. 2006, 25, 481–486. [Google Scholar] [CrossRef]
- Krieger, E.; Darden, T.; Nabuurs, S.B.; Finkelstein, A.; Vriend, G. Making Optimal Use of Empirical Energy Functions: Force-Field Parameterization in Crystal Space. Proteins 2004, 57, 678–683. [Google Scholar] [CrossRef] [PubMed]
- Krieger, E.; Vriend, G. New Ways to Boost Molecular Dynamics Simulations. J. Comput. Chem. 2015, 36, 996–1007. [Google Scholar] [CrossRef] [PubMed]
- Berendsen, H.J.C.; Postma, J.P.M.; van Gunsteren, W.F.; DiNola, A.; Haak, J.R. Molecular Dynamics with Coupling to an External Bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef] [Green Version]
- Ichiye, T.; Karplus, M. Collective Motions in Proteins: A Covariance Analysis of Atomic Fluctuations in Molecular Dynamics and Normal Mode Simulations. Proteins 1991, 11, 205–217. [Google Scholar] [CrossRef] [PubMed]
- Shlens, J. A Tutorial on Principal Component Analysis. arXiv 2014, arXiv:1404.1100. Available online: https://arxiv.org/abs/1404.1100 (accessed on 1 April 2022).
- Chai, B.-X.; Pogozheva, I.D.; Lai, Y.-M.; Li, J.-Y.; Neubig, R.R.; Mosberg, H.I.; Gantz, I. Receptor-Antagonist Interactions in the Complexes of Agouti and Agouti-Related Protein with Human Melanocortin 1 and 4 Receptors. Biochemistry 2005, 44, 3418–3431. [Google Scholar] [CrossRef]
- Heyder, N.A.; Kleinau, G.; Speck, D.; Schmidt, A.; Paisdzior, S.; Szczepek, M.; Bauer, B.; Koch, A.; Gallandi, M.; Kwiatkowski, D.; et al. Structures of Active Melanocortin-4 Receptor-Gs-Protein Complexes with NDP-α-MSH and Setmelanotide. Cell Res. 2021, 31, 1176–1189. [Google Scholar] [CrossRef]
- Collet, T.-H.; Dubern, B.; Mokrosinski, J.; Connors, H.; Keogh, J.M.; de Oliveira, E.M.; Henning, E.; Poitou-Bernert, C.; Oppert, J.-M.; Tounian, P.; et al. Evaluation of a Melanocortin-4 Receptor (MC4R) Agonist (Setmelanotide) in MC4R Deficiency. Mol. Metab. 2017, 6, 1321–1329. [Google Scholar] [CrossRef] [PubMed]
- Stutzmann, F.; Tan, K.; Vatin, V.; Dina, C.; Jouret, B.; Tichet, J.; Balkau, B.; Potoczna, N.; Horber, F.; O’Rahilly, S.; et al. Prevalence of Melanocortin-4 Receptor Deficiency in Europeans and Their Age-Dependent Penetrance in Multigenerational Pedigrees. Diabetes 2008, 57, 2511–2518. [Google Scholar] [CrossRef] [Green Version]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A Visualization System for Exploratory Research and Analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [Green Version]
- Israeli, H.; Degtjarik, O.; Fierro, F.; Chunilal, V.; Gill, A.K.; Roth, N.J.; Botta, J.; Prabahar, V.; Peleg, Y.; Chan, L.F.; et al. Structure Reveals the Activation Mechanism of the MC4 Receptor to Initiate Satiation Signaling. Science 2021, 372, 808–814. [Google Scholar] [CrossRef]
- Yu, J.; Gimenez, L.E.; Hernandez, C.C.; Wu, Y.; Wein, A.H.; Han, G.W.; McClary, K.; Mittal, S.R.; Burdsall, K.; Stauch, B.; et al. Determination of the Melanocortin-4 Receptor Structure Identifies Ca2+ as a Cofactor for Ligand Binding. Science 2020, 368, 428–433. [Google Scholar] [CrossRef] [PubMed]
- Amadei, A.; Linssen, A.B.; Berendsen, H.J. Essential Dynamics of Proteins. Proteins 1993, 17, 412–425. [Google Scholar] [CrossRef]
- Kitao, A.; Go, N. Investigating Protein Dynamics in Collective Coordinate Space. Curr. Opin. Struct. Biol. 1999, 9, 164–169. [Google Scholar] [CrossRef]
- García Large-Amplitude Nonlinear Motions in Proteins. Phys. Rev. Lett. 1992, 68, 2696–2699. [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| S. No. | Variants | Predicted ΔΔG (I-Mutant2.0) | Predicted ΔΔG (mCSM-Membrane) | Outcome |
|---|---|---|---|---|
| 1 | WT | 00 | 00 | - |
| 2 | D122N | −2.64 | −0.575 | Highly destabilizing |
| 3 | D126Y | −2.41 | −0.913 | Highly destabilizing |
| 4 | S188L | −2.20 | −0.648 | Highly destabilizing |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fatima, M.T.; Islam, Z.; Kolatkar, P.R.; Al-Shabeeb Akil, A.S. Molecular Analysis and Conformational Dynamics of Human MC4R Disease-Causing Mutations. Molecules 2022, 27, 4037. https://doi.org/10.3390/molecules27134037
Fatima MT, Islam Z, Kolatkar PR, Al-Shabeeb Akil AS. Molecular Analysis and Conformational Dynamics of Human MC4R Disease-Causing Mutations. Molecules. 2022; 27(13):4037. https://doi.org/10.3390/molecules27134037
Chicago/Turabian StyleFatima, Munazza Tamkeen, Zeyaul Islam, Prasanna R. Kolatkar, and Ammira Sarah Al-Shabeeb Akil. 2022. "Molecular Analysis and Conformational Dynamics of Human MC4R Disease-Causing Mutations" Molecules 27, no. 13: 4037. https://doi.org/10.3390/molecules27134037
APA StyleFatima, M. T., Islam, Z., Kolatkar, P. R., & Al-Shabeeb Akil, A. S. (2022). Molecular Analysis and Conformational Dynamics of Human MC4R Disease-Causing Mutations. Molecules, 27(13), 4037. https://doi.org/10.3390/molecules27134037