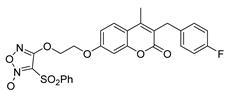
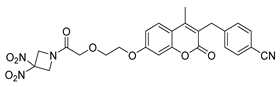
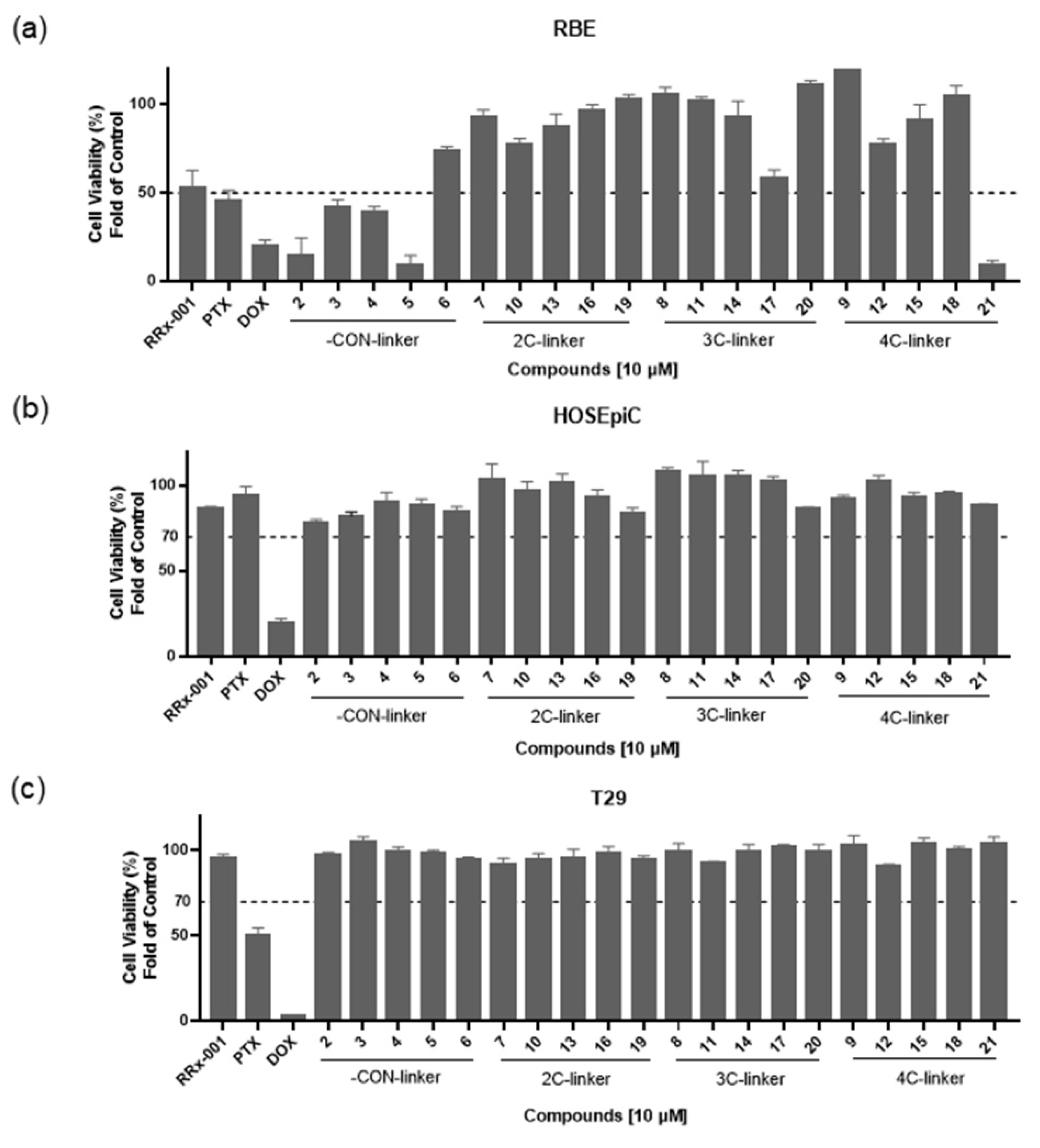
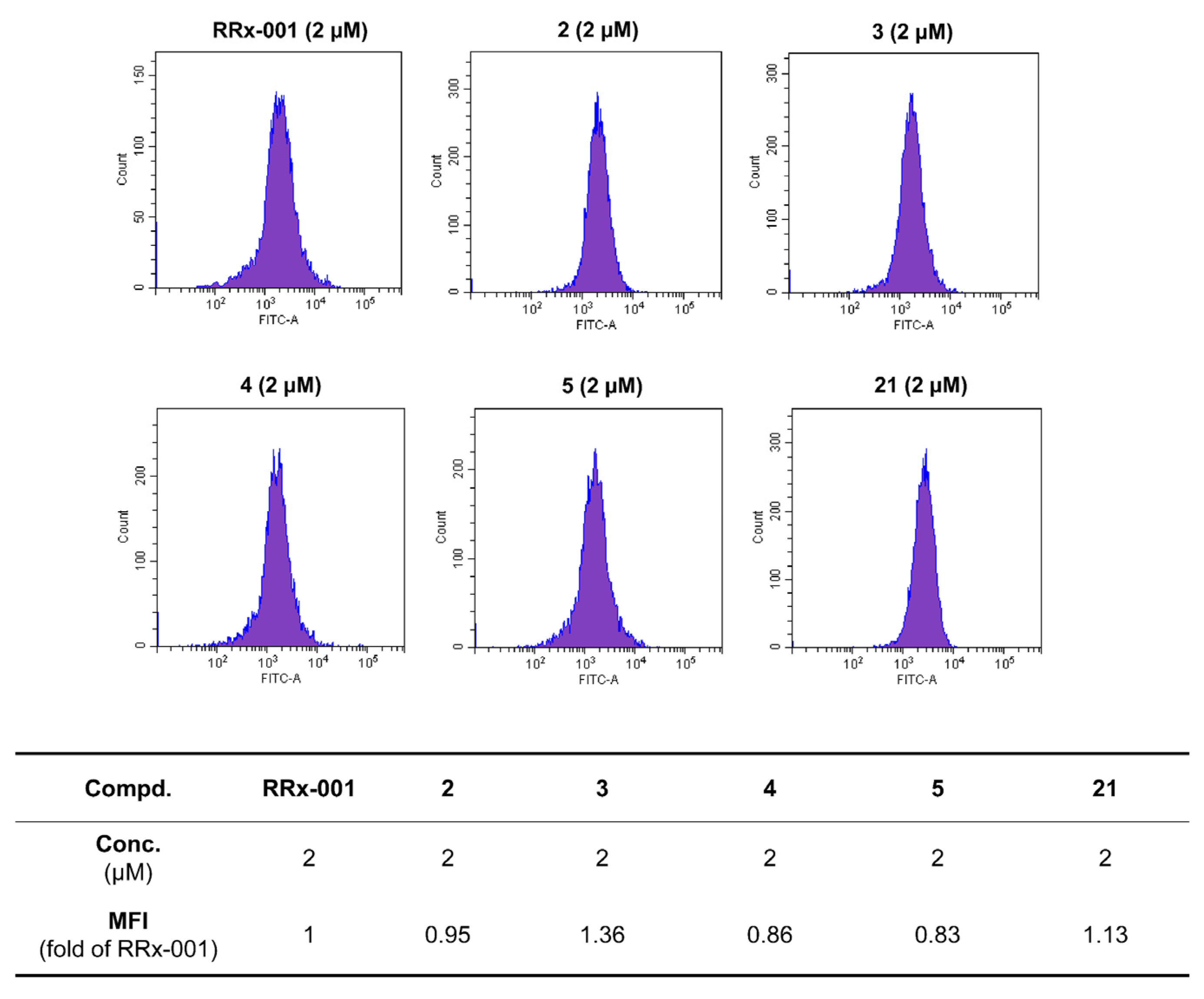
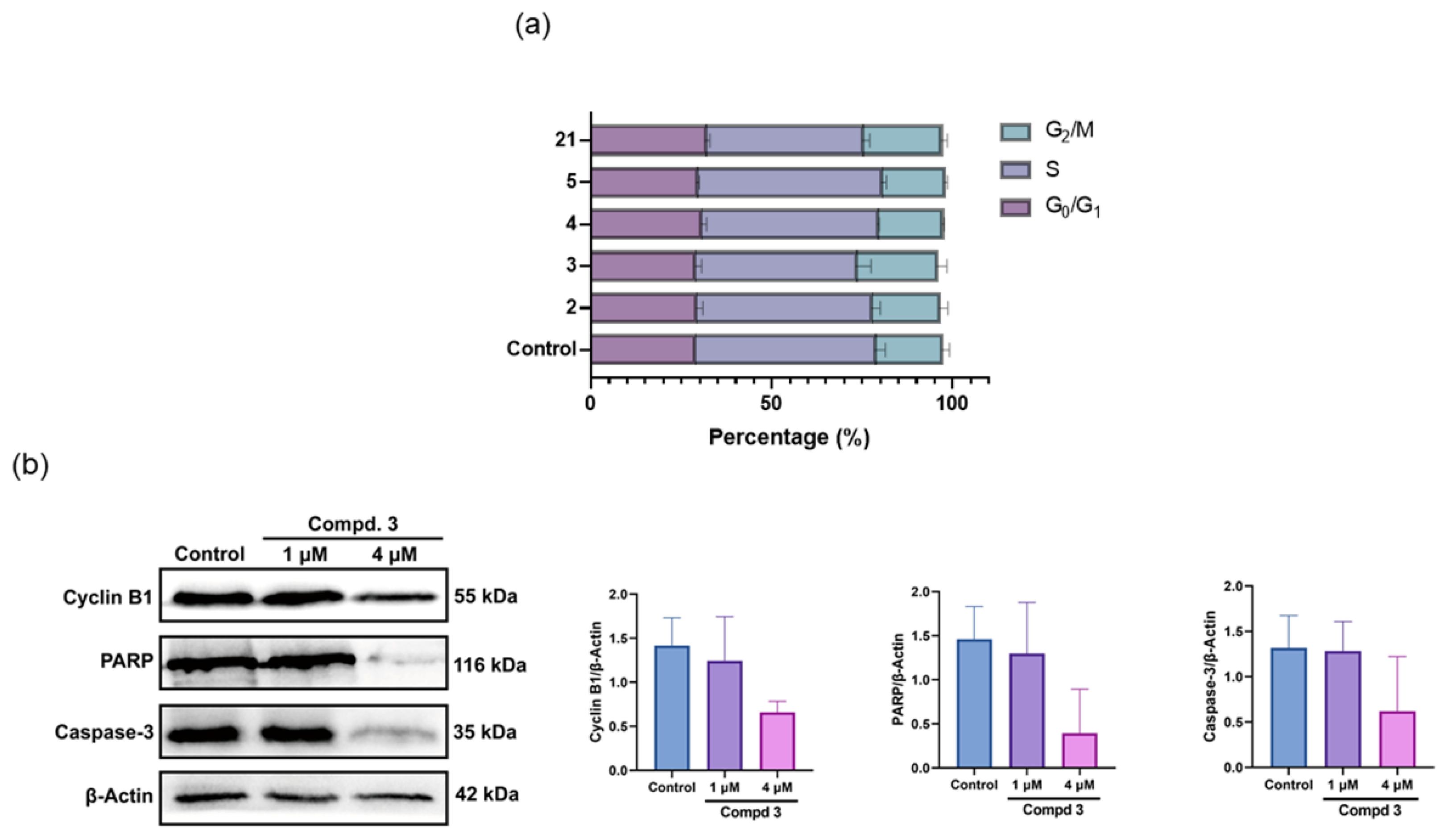
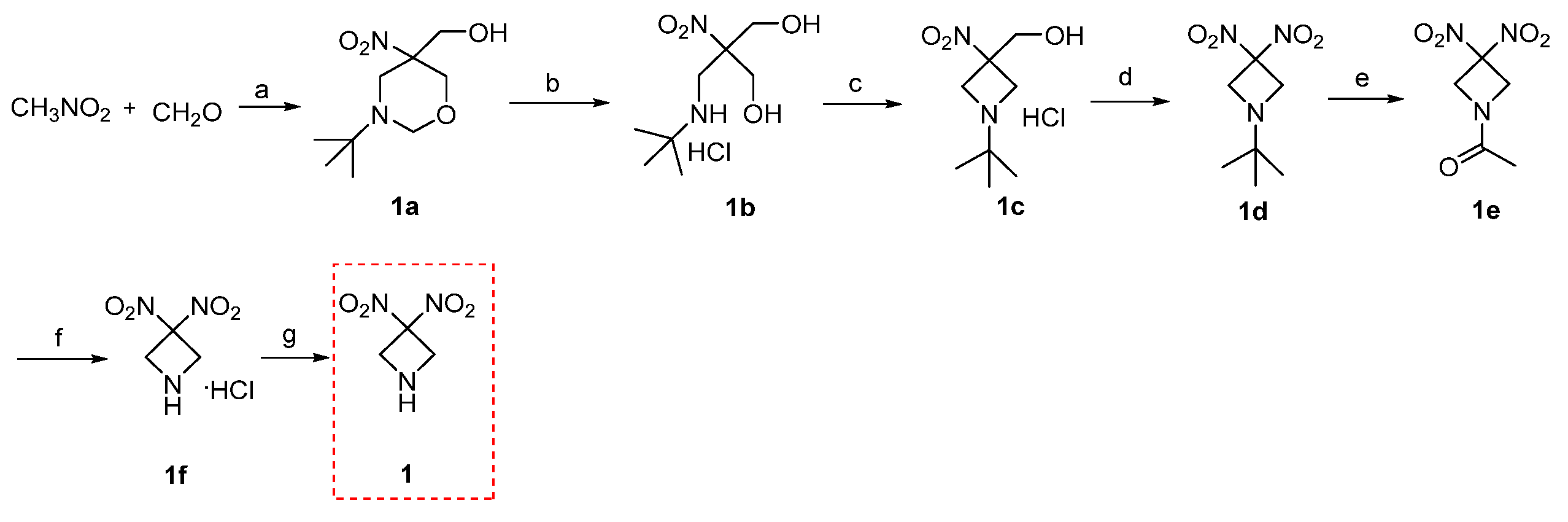
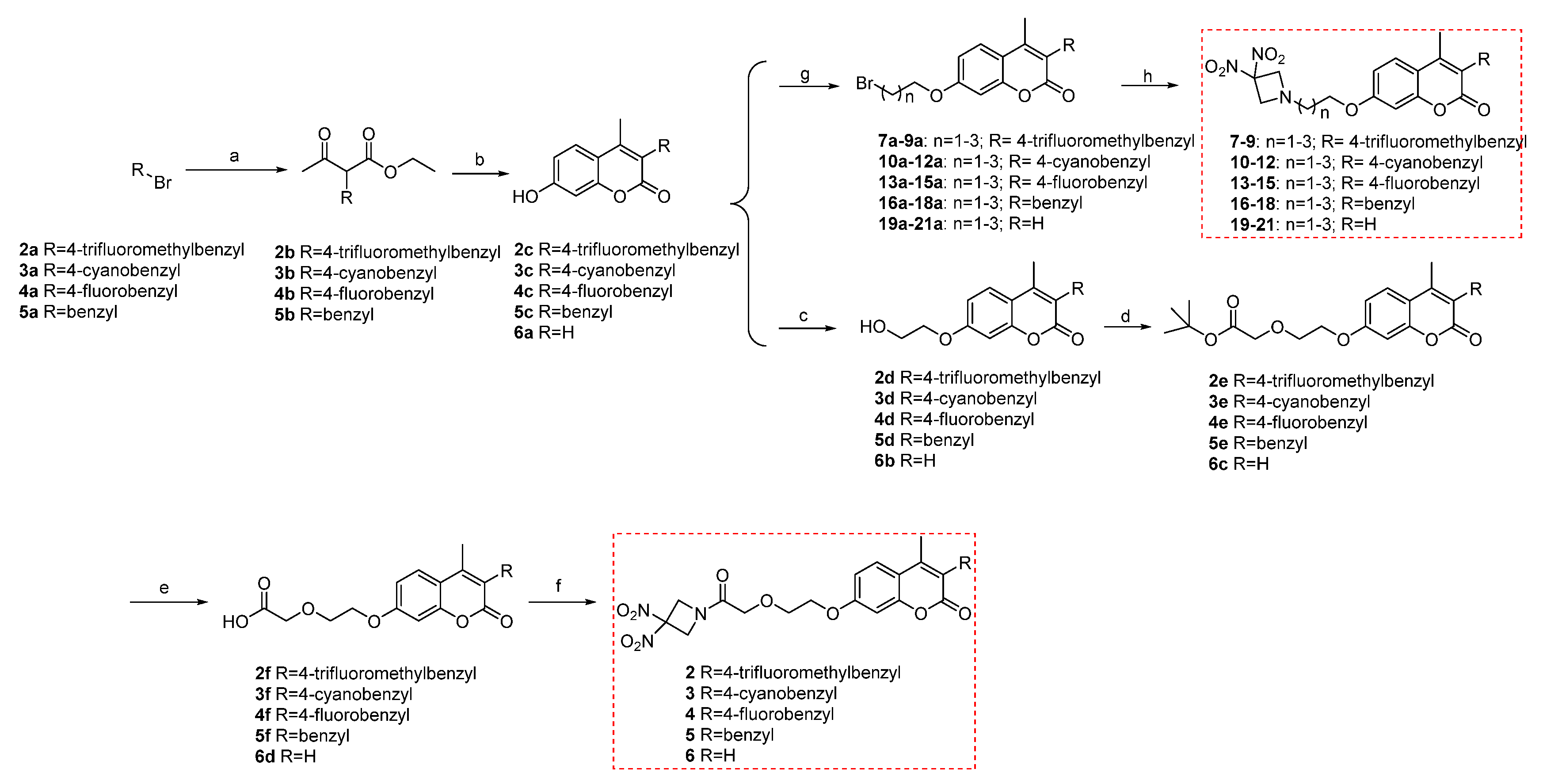
3.2. Synthesis
N-tert-butyl-5-hydroxymethyl-5-nitro-1,3-oxazine (1a): To aqueous solution of paraformaldehyde (24 g, 0.8 mol) and 40%NaOH (600 μL) in 120 mL of distilled water, nitromethane (10.5 mL, 0.195 mol) was added dropwise over 1 h at 40 °C. The reaction mixture was heated to 60 °C and stirred for 1 h. Then, the solution of tert-butylamine (20.3 mL, 0.262 mol) in distilled water (36 mL) was added dropwise slowly. The mixture was stirred for another 4 h, cooled to room temperature, and stirred for 1 h again. The precipitate was collected by vacuum filtration at room temperature, washed with distilled water, and vacuum freeze-dried to give 1a (yellow solid, 34 g, 78.8% yield). ESI-MS m/z 231.1 [M + H]+.
N-(tert-butylamino)methyl-2-nitro-1,3-propandiolhydrochloride (1b): To a solution of concentrated hydrochloric acid (6.71 mL, 81 mmol) in methanol (62.5 mL), 1a (17.4 g, 80 mmol) was added. The reaction solution was refluxed for 20 h. The solvent was removed through vacuum evaporation and the residue was dissolved in isopropyl alcohol (25 mL). The solution was recrystallized below 0 °C and the precipitate was filtered, washed with isopropanol, and vacuum freeze-dried to give 1b (white solid, 10.4 g, 54% yield). ESI-MS m/z 207.0 [M + H]+.
N-tert-butyl-3-hydroxymethyl-3-nitroazetidine hydrochloride (1c): To a solution of DIAD (5.15 mL, 25.98 mmol) and 1b (5.0 g, 20.60 mmol) in butanone (40 mL), Ph3P (29.89 g, 0.132 mol) in butanone was added dropwise over 1 h at 50 °C. The reaction mixture was stirred at 50 °C for 4 h, filtered, washed with cold butanone (30 mL), and vacuum freeze-dried to give 1c (white solid,3.0 g, 65% yield, m.p. 161.8–163.5 °C). ESI-MS m/z 189.0 [M + H]+.
N-tert-butyl-3,3-dinitroazetidine (1d): To a solution of 1c (1.35 g, 6 mmol) in distilled water (6 mL), NaOH aqueous solution (3 mL, 717 mg, 17.9 mmol) was added and was stirred for 3 h at room temperature. After cooling to 8 °C, cold NaNO2 solution (4.5 mL, 1.65 g, 23.9 mmol) and K3Fe(CN)6 (197 mg, 6 mmol) in distilled water were added slowly. Then, Na2S2O8 (1.78 g, 7.5 mmol) was added. The yellow solution was stirred for another 1 h at room temperature and extracted with dichloromethane (150 mL). The organic layer was dried with MgSO4 and the solvent was removed via vacuum evaporation to give 1d (yellow liquid, 858 mg, 70.5% yield). ESI-MS m/z 203.9 [M + H]+.
N-acetyl-3,3-dintroazetidine (1e): Compound 1d (1.0 g, 4.92 mmol) and acetic anhydride (1.8 mL, 19.98 mmol) were slowly added into the reaction system, followed by injecting boron trifluoride etherate (1.31 mL, 0.492 mmol) using syringe. The mixture was reacted at 115–125 °C for 3 h under nitrogen atmosphere. Excess acetic anhydride was removed by vacuum distillation. The residue was recrystallized in chloroform to give 1e (white crystal, 312 mg, 33.6% yield, m.p. 112.3–114.5 °C). ESI-MS m/z 189.9 [M + H]+. 1H NMR (600 MHz, DMSO) δ 5.07 (s, 2H), 4.74 (s, 2H), 1.87 (s, 3H).
3,3-Dintroazetidine hydrochloride (1f): To a solution of 1e (150 mg, 0.817 mmol) in distilled water (20 mL), 10% hydrochloric acid (1.5 mL) was added dropwise. The solution was stirred and refluxed for 4 h. The solvent was removed by vacuum evaporation to give 1f (white solid, 100 mg, 66.7% yield). ESI-MS m/z 148.1 [M + H]+.
3,3-Dintroazetidine (1): The solution of 1f (1.0 g, 5.45 mmol) in distilled water (32 mL) was heated to 50 °C, and 10% NaHCO3 was slowly added dropwise until pH-9. The mixture was extracted with chloroform (3 × 20 mL), and the organic layer was dried with MgSO4. The solvent was removed under lowered pressure to give intermediate 1 (light yellow oil, 400 mg, 50% yield). ESI-MS m/z 148.1 [M + H]+. 1H NMR (600 MHz, DMSO) δ 4.34 (s, 4H).
General Procedure for the Preparation of 2–5b. In an ice bath, a reaction solution of 60% NaH (680 mg, 17 mmol) in dry THF (40 mL) was stirred for 10 min. Ethyl acetoacetate (2.2 mL, 17 mmol) was added dropwise. After stirring the reaction for 30 min, starting materials 2–5a (15.3 mmol) were added and continued reaction at room temperature in the TLC monitor. After the reaction finished, the solids were filtered out, most of the solvents were removed, and water (100 mL) was added. The mixture was extracted with ethyl acetate, washed with saturated salt, dried over anhydrous sodium sulfate, and evaporated in vacuo to get compounds 2–5b. These products were directly used as the next reactants without any further purification.
Ethyl 3-oxo-2-(4-(trifluoromethyl)benzyl)butanoate (2b). The title compound was obtained starting from 2a and ethyl acetoacetate. Analytical data for 2b (a yellow oily substance, 4.2 g, 95% yield): ESI-MS m/z 289.2 [M + H]+.
Ethyl 2-(4-cyanobenzyl)-3-oxobutanoate (3b). The title compound was obtained starting from 3a and ethyl acetoacetate. Analytical data for 3b (a yellow oily substance, 3.7 g, 98% yield): ESI-MS m/z 245.9 [M + H]+.
Ethyl 2-(4-fluorobenzyl)-3-oxobutanoate (4b). The title compound was obtained starting from 4a and ethyl acetoacetate. Analytical data for 4b (a yellow oily substance, 3.6 g, 97% yield): ESI-MS m/z 260.9 [M + Na]+.
Ethyl 2-benzyl-3-oxobutanoate (5b). The title compound was obtained starting from 5a and ethyl acetoacetate. Analytical data for 5b (a yellow oily substance, 3.3 g, 98% yield): ESI-MS m/z 221.0 [M + H]+.
General Procedure for the Preparation of 2–5c. Resorcinol (1.35 g, 12.250 mmol) was added to a reaction flask containing 2–5b and 70% H2SO4 (30 mL) in the ice bath. After 30 min, the ice bath was removed and the reaction continued at room temperature. After the reaction finished, the solution was slowly added into ice water (300 mL) and stirred for 30 min to precipitate a large number of solids. After extraction, filtration, and drying, compounds 2–5c were obtained.
7-Hydroxy-4-methyl-3-(4-(trifluoromethyl)benzyl)-2H-chromen-2-one (2c). The title compound was obtained starting from 2b and resorcinol. Analytical data for 2c (white solid, 3.4 g 82% yield): ESI-MS m/z 335.2 [M + H]+.
4-((7-Hydroxy-4-methyl-2-oxo-2H-chromen-3-yl)methyl)benzonitrile (3c). The title compound was obtained starting from 3b and resorcinol. Analytical data for 3c (white solid, 3.0 g, 83% yield): ESI-MS m/z 292.3 [M + H]+.
3-(4-Fluorobenzyl)-7-hydroxy-4-methyl-2H-chromen-2-one (4c). The title compound was obtained starting from 4b and resorcinol. Analytical data for 4c (yellow solid, 3.2 g, 91% yield): ESI-MS m/z 284.9 [M + H]+.
3-Benzyl-7-hydroxy-4-methyl-2H-chromen-2-one (5c). The title compound was obtained starting from 5b and resorcinol. Analytical data for 5c (yellow solid, 2.7 g, 83% yield): ESI-MS m/z 266.9 [M + H]+.
General Procedure for the Preparation of 2–5d and 6b. To a stirred solution of 2–5c and 6a (3.517 mmol) in DMF (15 mL) at room temperature, corresponding halo alcohol (10.553 mmol), NaI (1.05 mmol), and K2CO3 (10.553 mmol) were added. The mixture was refluxed for 2–5 h and then poured into ice water (50 mL). After filtration, the residue was washed with water (3 × 10 mL) and dried to obtain 2–5d and 6b.
7-(2-hydroxyethoxy)-4-methyl-3-(4-(trifluoromethyl)benzyl)-2H-chromen-2-one (2d). The title compound was obtained starting from 2c and ethylene chlorohydrin. Analytical data for 2d (white solid, 1.1 g, 81% yield, m.p. 104–106 °C): ESI-MS m/z 379.1 [M + H]+.
4-((7-(2-hydroxyethoxy)-4-methyl-2-oxo-2H-chromen-3-yl)methyl)benzonitrile (3d). The title compound was obtained starting from 3c and ethylene chlorohydrin. Analytical data for 3d (white solid, 978 mg, 83% yield, m.p. 80–82 °C): ESI-MS m/z 336.3 [M + H]+.
3-(4-fluorobenzyl)-7-(2-hydroxyethoxy)-4-methyl-2H-chromen-2-one (4d). The title compound was obtained starting from 4c and ethylene chlorohydrin. Analytical data for 4d (white solid, 992 mg, 81% yield): ESI-MS m/z 329.1 [M + H]+.
3-benzyl-7-(2-hydroxyethoxy)-4-methyl-2H-chromen-2-one (5d). The title compound was obtained starting from 5c and ethylene chlorohydrin. Analytical data for 5d (white solid, 861 mg, 81% yield): ESI-MS m/z 311.1 [M + H]+.
7-(2-Hydroxyethoxy)-4-methyl-2H-chromen-2-one (6b). The title compound was obtained starting from 6a and ethylene chlorohydrin. Analytical data for 6b (white solid, 619 mg, 81% yield): ESI-MS m/z 221.1 [M + H]+.
General Procedure for the Preparation of 2–5e and 6c. To a stirred solution of 2–5d and 6b (4.54 mmol) in anhydrous DMF (15 mL) at 0 °C, NaH (60%, 363 mg, 9.08 mmol) was added. The mixture was stirred for 20 min and t-butylbromoacetate (1.34 mL, 9.08 mmol) was added. The reaction mixture was warmed to room temperature and stirred for 1.5 h, and then poured into a saturated aqueous solution of NH4Cl (10 mL) and extracted with ethyl acetate (3 × 30 mL). The combined extracts were washed with brine, dried over Na2SO4, filtered, and evaporated in vacuo. The residue was purified by column chromatography on silica gel to give compound 2–5e and 6c.
T-butyl 2-(2-((4-methyl-2-oxo-3-(4-(trifluoromethyl)benzyl)-2H-chromen-7-yl)oxy) ethoxy)acetate (2e): The title compound was obtained starting from 2d. Analytical data for 2e (colorless liquid, 446 mg, 25% yield): ESI-MS m/z 436.8 [M + H]+; 1H NMR (600 MHz, DMSO) δ 7.67 (d, J = 7.7 Hz, 2H), 7.57 (d, J = 7.7 Hz, 2H), 6.92 (d, J = 8.1 Hz, 1H), 6.47 (d, J = 13.2 Hz, 2H), 4.23 (s, 2H), 3.96 (s, 2H), 3.93 (s, 2H), 3.70 (d, J = 4.5 Hz, 2H), 2.05 (s, 3H), 1.43 (s, 9H).
Tert-butyl 2-(2-((3-(4-cyanobenzyl)-4-methyl-2-oxo-2H-chromen-7-yl)oxy)ethoxy) acetate (3e): The title compound was obtained starting from 3d. Analytical data for 3e (white acicular crystal, 239 mg, 13.4% yield, m.p. 90.5–92.4 °C): ESI-MS m/z 393.9 [M + H]+; 1H NMR (600 MHz, DMSO) δ 7.76 (t, J = 13.1 Hz, 3H), 7.44 (d, J = 7.5 Hz, 2H), 7.01 (dd, J = 20.5, 11.6 Hz, 2H), 4.24 (s, 2H), 4.07 (s, 2H), 4.04 (s, 2H), 3.84 (s, 2H), 2.44 (s, 3H), 1.43 (s, 9H).
Tert-butyl 2-(2-((3-(4-fluorobenzyl)-4-methyl-2-oxo-2H-chromen-7-yl)oxy)ethoxy) acetate (4e): The title compound was obtained starting from 4d. Analytical data for 4e (yellow liquid, 301 mg, 14.8% yield): ESI-MS m/z 442.9 [M + H]+, 1H NMR (600 MHz, DMSO) δ 7.35 (dd, J = 8.2, 5.8 Hz, 2H), 7.12 (t, J = 8.8 Hz, 2H), 6.89 (d, J = 8.2 Hz, 1H), 6.49–6.42 (m, 2H), 4.23 (s, 2H), 3.95 (t, J = 4.9 Hz, 2H), 3.81 (s, 2H), 3.70 (dd, J = 10.3, 5.2 Hz, 2H), 2.05 (s, 3H), 1.43 (s, 9H).
Tert-butyl 2-(2-((3-benzyl-4-methyl-2-oxo-2H-chromen-7-yl)oxy)ethoxy)acetate (5e): The title compound was obtained starting from 5d. Analytical data for 5e (yellow liquid, 442 mg, 23% yield): ESI-MS m/z 424.9 [M + H]+; 1H NMR (600 MHz, DMSO) δ 7.32 (t, J = 5.4 Hz, 2H), 7.29 (d, J = 7.8 Hz, 1H), 7.19 (t, J = 6.9 Hz, 1H), 6.89 (d, J = 8.2 Hz, 1H), 6.46 (dd, J = 12.7, 4.3 Hz, 2H), 4.23 (s, 2H), 3.95 (t, J = 4.9 Hz, 2H), 3.83 (s, 2H), 3.70 (dd, J = 10.2, 5.1 Hz, 2H), 2.05 (s, 3H), 1.32 (s, 9H).
Tert-butyl 2-(2-((4-methyl-2-oxo-2H-chromen-7-yl)oxy)ethoxy)acetate (6c): The title compound was obtained starting from 6b. Analytical data for 6c (white solid, 240 mg, 19% yield, m.p. 79.0–80.9 °C): ESI-MS m/z 279.1 [M + H]+; 1H NMR (600 MHz, DMSO) δ 7.69 (d, J = 8.7 Hz, 1H), 7.04–6.95 (m, 2H), 6.22 (s, 1H), 4.24 (s, 2H), 4.07 (s, 2H), 3.84 (s, 2H), 2.40 (s, 3H), 1.43 (s, 9H).
General Procedure for the Preparation of 2–5f and 6d. To a stirred solution of 2–5e and 6c (50 mg) in DCM (5 mL) at 0 °C was added TFA (100 μL). The reaction mixture was warmed to room temperature and stirred for 2 h. After the reaction finished, the solvent and unreacted TFA were evaporated in vacuo to get compounds 2–5f and 6d.
2-(2-((4-methyl-2-oxo-3-(4-(trifluoromethyl)benzyl)-2H-chromen-7-yl)oxy)ethoxy) acetic acid (2f): The title compound was obtained starting from 2e. Analytical data for 2f (brown solid, 48 mg, 99% yield): ESI-MS m/z 436.8 [M + H]+.
2-(2-((3-(4-cyanobenzyl)-4-methyl-2-oxo-2H-chromen-7-yl)oxy)ethoxy)acetic acid (3f): The title compound was obtained starting from 3e. Analytical data for 3f (brown solid, 43 mg, 100% yield): ESI-MS m/z 393.8 [M + H]+.
2-(2-((3-(4-fluorobenzyl)-4-methyl-2-oxo-2H-chromen-7-yl)oxy)ethoxy)acetic acid (4f): The title compound was obtained starting from 4e. Analytical data for 4f (brown solid, 39 mg, 88.8% yield): ESI-MS m/z 386.9 [M + H]+.
2-(2-((3-(4-fluorobenzyl)-4-methyl-2-oxo-2H-chromen-7-yl)oxy)ethoxy)acetic acid (5f): The title compound was obtained starting from 5e. Analytical data for 5f (yellow solid, 43 mg, 99% yield): ESI-MS m/z 368.9 [M + H]+.
2-(2-((4-methyl-2-oxo-2H-chromen-7-yl)oxy)ethoxy)acetic acid (6d): The title compound was obtained starting from 6c. Analytical data for 6d (brown solid, 41 mg, 98% yield): ESI-MS m/z 278.9 [M + H]+.
General Procedure for the Preparation of 2–6. Substituted carboxylic acid 2–5f and 6d (0.2031 mmol) were dissolved in DCM (10 mL) and stirred for 30 min at room temperature. DIPEA (71 μL, 0.4062 mmol) and HATU (154 mg, 0.4062 mmol) were added to the solution and stirred for 40 min at room temperature. After being added intermediate 1 (60 mg, 0.4062 mmol), the mixture was further stirred at room temperature for another 4 h, then water was added. The mixture was extracted with ethyl acetate, washed with saturated salt, and dried over anhydrous Na2SO4 to obtain crude product, which was purified by column chromatography on silica gel to give compounds 2–6.
7-(2-(2-(3,3-Dinitroazetidin-1-yl)-2-oxoethoxy)ethoxy)-4-methyl-3-(4-(trifluoromethyl)benzyl)-2H-chromen-2-one (2): The title compound was obtained starting from 2f and 3,3-dinitroazetidine. Analytical data for 2 (yellow solid, 34 mg, 30.7% yield, m.p. 65.3–67.0 °C): ESI-MS m/z 565.7 [M + H]+; 1H NMR (600 MHz, DMSO) δ 7.67 (d, J = 7.9 Hz, 2H), 7.55 (d, J = 7.9 Hz, 2H), 6.97 (d, J = 8.8 Hz, 1H), 6.52 (s, 2H), 5.13 (s, 2H), 4.91 (s, 2H), 4.37 (s, 2H), 3.97 (t, J = 4.7 Hz, 2H), 3.94 (s, 2H), 3.72 (t, J = 9.8, 4.8 Hz, 2H), 2.07 (s, 3H). 13C NMR (151 MHz, DMSO) δ 168.98, 167.58, 158.96, 154.28, 146.09, 143.92, 128.57, 127.09, 125.08, 125.06, 124.41, 107.41, 107.09, 105.59, 99.92, 69.41, 66.15, 60.43, 59.37, 34.52, 21.91.
4-((7-(2-(2-(3,3-Dinitroazetidin-1-yl)-2-oxoethoxy)ethoxy)-4-methyl-2-oxo-2H-chromen-3-yl)methyl)benzonitrile (3): The title compound was obtained starting from 3f and 3,3-dinitroazetidine. Analytical data for 3 (yellow solid, 30 mg, 28% yield, m.p. 74.9–76.3 °C): ESI-MS m/z 522.7 [M + H]+; 1H NMR (600 MHz, DMSO) δ 7.82–7.70 (m, 3H), 7.44 (d, J = 6.0 Hz, 2H), 7.07–6.97 (m, 2H), 5.15 (s, 2H), 4.80 (s, 2H), 4.28 (s, 2H), 4.18 (s, 2H), 4.05 (s, 2H), 3.83 (s, 2H), 2.44 (s, 3H). 13C NMR (151 MHz, DMSO) δ 170.16, 160.98, 160.79, 153.28, 149.02, 145.27, 132.18, 128.98, 126.66, 119.82, 118.74, 113.42, 112.35, 108.80, 107.68, 100.90, 69.54, 68.93, 67.36, 59.56, 56.77, 32.22, 15.10.
7-(2-(2-(3,3-Dinitroazetidin-1-yl)-2-oxoethoxy)ethoxy)-3-(4-fluorobenzyl)-4-methyl-2H-chromen-2-one (4): The title compound was obtained starting from 4f and 3,3-dinitroazetidine. Analytical data for 4 (yellow solid, 37 mg, 35% yield, m.p. 70.0–71.9 °C): ESI-MS m/z 515.8 [M + H]+; 1H NMR (600 MHz, DMSO) δ 7.39–7.31 (m, 2H), 7.11 (t, J = 8.6 Hz, 2H), 6.94 (d, J = 8.0 Hz, 1H), 6.51 (d, J = 8.5 Hz, 2H), 5.12 (s, 2H), 4.89 (s, 2H), 4.36 (s, 2H), 3.97 (t, J = 4.6 Hz, 2H), 3.82 (s, 2H), 3.71 (t, J = 4.7 Hz, 2H), 2.07 (s, 3H). 13C NMR (151 MHz, DMSO) δ 168.92, 167.60, 166.89, 158.92, 154.32, 144.95, 134.90, 129.57, 128.66, 128.07, 124.49, 114.96, 114.82, 107.43, 107.10, 105.59, 99.94, 69.40, 66.14, 60.42, 59.37, 58.95, 56.91, 33.87, 21.68.
3-Benzyl-7-(2-(2-(3,3-dinitroazetidin-1-yl)-2-oxoethoxy)ethoxy)-4-methyl-2H-chromen-2-one (5): The title compound was obtained starting from 5f and 3,3-dinitroazetidine. Analytical data for 5 (white solid, 30 mg, 30% yield, m.p. 82.5–84.5 °C): ESI-MS m/z 497.8 [M + H]+; 1H NMR (600 MHz, DMSO) δ 7.35–7.25 (m, 5H), 6.94 (d, J = 8.0 Hz, 1H), 6.51 (d, J = 7.8 Hz, 2H), 5.13 (s, 2H), 4.86 (s, 2H), 4.72 (s, 2H), 4.35 (s, 2H), 3.97 (t, J = 4.9 Hz, 2H), 3.84 (s, 2H), 3.71 (dd, J = 10.0, 5.0 Hz, 2H), 2.08 (s, 3H). 13C NMR (151 MHz, DMSO) δ 168.92, 167.63, 158.91, 154.36, 144.78, 138.83, 128.73, 128.23, 127.83, 125.81, 124.57, 107.45, 107.11, 105.61, 99.96, 69.40, 66.21, 60.47, 59.37, 34.70, 21.66.
7-(2-(2-(3,3-dinitroazetidin-1-yl)-2-oxoethoxy)ethoxy)-4-methyl-2H-chromen-2-one (6): The title compound was obtained starting from 5f and 3,3-dinitroazetidine. Analytical data for 5 (white solid, 37 mg, 30% yield, m.p. 82.5–84.5 °C): ESI-MS m/z 408.1 [M + H]+; 1H NMR (600 MHz, DMSO) δ 7.70 (d, J = 8.8 Hz, 1H), 7.06–6.95 (m, 2H), 6.22 (s, 1H), 5.16 (s, 2H), 4.80 (s, 2H), 4.33–4.24 (m, 2H), 4.18 (s, 2H), 3.87–3.78 (m, 2H), 2.41 (s, 3H). 13C NMR (151 MHz, DMSO) δ 170.16, 161.26, 159.97, 154.57, 153.25, 126.35, 113.08, 112.22, 111.05, 107.68, 101.11, 69.54, 68.91, 67.37, 59.56, 56.77, 17.97.
General Procedure for the Preparation of 7–21a. Compounds 2–5c and 6a (500 mg, 1.76 mmol) were dissolved in CH3CN (15 mL) in a three-necked flask. Then K2CO3 (1.2 g, 8.8 mmol) and various dibromo alkane (8.8 mmol) were added to the reaction mixture, which was heated to 80 °C and stirred for 7 h. After finishing, the mixture was cooled to room temperature and filtered. The filtrate was concentrated in vacuo; then, DCM (20 mL) and H2O (10 mL) were added. The mixture was extracted with DCM, washed with saturated salt and dried over anhydrous Na2SO4 to obtain compounds 7–21a.
7-(2-Bromoethoxy)-4-methyl-3-(4-(trifluoromethyl)benzyl)-2H-chromen-2-one (7a): The title compound was obtained starting from 2c and 1,2-dibromoethane. Analytical data for 7a (white solid, 658 mg, 85% yield): ESI-MS m/z 440.7 [M + H]+.
7-(3-Bromopropoxy)-4-methyl-3-(4-(trifluoromethyl)benzyl)-2H-chromen-2-one (8a): The title compound was obtained starting from 2c and 1,3-dibromopropane. Analytical data for 8a (white solid, 663 mg, 83% yield): ESI-MS m/z 454.7 [M + H]+.
7-(4-Bromobutoxy)-4-methyl-3-(4-(trifluoromethyl)benzyl)-2H-chromen-2-one (9a): The title compound was obtained starting from 2c and 1,4-dibromobutane. Analytical data for 9a (white solid, 750 mg, 91% yield): ESI-MS m/z 468.7 [M + H]+.
4-((7-(2-Bromoethoxy)-4-methyl-2-oxo-2H-chromen-3-yl)methyl)benzonitrile (10a): The title compound was obtained starting from 3c and 1,2-dibromoethane. Analytical data for 10a (white solid, 613 mg, 88% yield): ESI-MS m/z 397.0 [M + H]+.
4-((7-(3-Bromopropoxy)-4-methyl-2-oxo-2H-chromen-3-yl)methyl)benzonitrile (11a): The title compound was obtained starting from 3c and 1,3-dibromopropane. Analytical data for 11a (white solid, 434 mg, 83% yield): ESI-MS m/z 411.8 [M + H]+.
4-((7-(4-Bromobutoxy)-4-methyl-2-oxo-2H-chromen-3-yl)methyl)benzonitrile (12a): The title compound was obtained starting from 3c and 1,4-dibromobutane. Analytical data for 12a (yellow solid, 598 mg, 80% yield): ESI-MS m/z 425.9 [M + H]+.
7-(2-Bromoethoxy)-3-(4-fluorobenzyl)-4-methyl-2H-chromen-2-one (13a): The title compound was obtained starting from 4c and 1,2-dibromoethane. Analytical data for 13a (yellow solid, 665 mg, 97.3% yield): ESI-MS m/z 390.8 [M + H]+.
7-(3-Bromopropoxy)-3-(4-fluorobenzyl)-4-methyl-2H-chromen-2-one (14a): The title compound was obtained starting from 4c and 1,3-dibromopropane. Analytical data for 14a (white solid, 633 mg, 89% yield): ESI-MS m/z 404.8 [M + H]+.
7-(4-Bromobutoxy)-3-(4-fluorobenzyl)-4-methyl-2H-chromen-2-one (15a): The title compound was obtained starting from 4c and 1,4-dibromobutane. Analytical data for 15a (white solid, 625 mg, 77% yield): ESI-MS m/z 418.8 [M + H]+.
3-Benzyl-7-(2-bromoethoxy)-4-methyl-2H-chromen-2-one (16a): The title compound was obtained starting from 5c and 1,2-dibromoethane. Analytical data for 16a (white solid, 478 mg, 81% yield): ESI-MS m/z 336.3 [M + H]+.
3-Benzyl-7-(3-bromopropoxy)-4-methyl-2H-chromen-2-one (17a): The title compound was obtained starting from 5c and 1,3-dibromopropane. Analytical data for 17a (white solid, 530 mg, 78% yield): ESI-MS m/z 387.0 [M + H]+.
3-Benzyl-7-(4-bromobutoxy)-4-methyl-2H-chromen-2-one (18a): The title compound was obtained starting from 5c and 1,4-dibromobutane. Analytical data for 18a (white solid, 543 mg, 77% yield): ESI-MS m/z 402.4 [M + H]+.
7-(2-Bromoethoxy)-4-methyl-2H-chromen-2-one (19a): The title compound was obtained starting from 6a and 1,2-dibromoethane. Analytical data for 19a (white solid, 402 mg, 81% yield): ESI-MS m/z 283.0 [M + H]+.
7-(3-Bromopropoxy)-4-methyl-2H-chromen-2-one (20a): The title compound was obtained starting from 6a and 1,3-dibromopropane. Analytical data for 20a (white solid, 425 mg, 81% yield): ESI-MS m/z 298.9 [M + H]+.
7-(4-Bromobutoxy)-4-methyl-2H-chromen-2-one (21a): The title compound was obtained starting from 6a and 1,4-dibromobutane. Analytical data for 21a (white solid, 387 mg, 71% yield): ESI-MS m/z 310.8 [M + H]+.
General Procedure for the Preparation of 7–21. Compounds 7–21a (0.34 mmol), DMAP (166 mg, 1.36 mmol) and 3,3-dinitroazetidine (150 mg, 1.02 mmol) were dissolved in DMF (10 mL) in a three-necked flask. This mixture was heated to 40–80 °C and stirred for 6–8 h. After finishing, the mixture was cooled to room temperature and added water. Then, the reaction mixture was extracted with ethyl acetate, washed with saturated salt and dried over anhydrous Na2SO4 to obtain crude product, which was purified by column chromatography on silica gel to give the target compounds 7–21.
7-(2-(3,3-Dinitroazetidin-1-yl)ethoxy)-4-methyl-3-(4-(trifluoromethyl)benzyl)-2H-chromen-2-one (7): The title compound was obtained starting from 7a and 3,3-dinitroazetidine. Analytical data for 7 (yellow solid, 43 mg, 25% yield, m.p. 98.0–99.8 °C): ESI-MS m/z 507.8 [M + H]+; 1H NMR (600 MHz, DMSO) δ 7.76 (d, J = 8.8 Hz, 1H), 7.64 (d, J = 8.0 Hz, 2H), 7.46 (d, J = 7.9 Hz, 2H), 7.04–6.97 (m, 2H), 4.28 (s, 4H), 4.14 (t, J = 4.8 Hz, 2H), 4.05 (s, 2H), 3.04 (t, J = 4.8 Hz, 2H), 2.45 (s, 3H). 13C NMR (151 MHz, DMSO) δ 160.99, 160.62, 153.25, 148.86, 144.17, 128.67, 126.61, 125.09, 120.10, 113.44, 112.43, 109.34, 100.89, 67.24, 61.10, 55.14, 31.92, 15.09.
7-(3-(3,3-Dinitroazetidin-1-yl)propoxy)-4-methyl-3-(4-(trifluoromethyl)benzyl)-2H-chromen-2-one (8): The title compound was obtained starting from 8a and 3,3-dinitroazetidine. Analytical data for 8 (yellow liquid, 50 mg, 28% yield): ESI-MS m/z 521.7 [M + H]+; 1H NMR (600 MHz, DMSO) δ 7.75 (d, J = 8.3 Hz, 1H), 7.64 (d, J = 7.5 Hz, 2H), 7.46 (d, J = 7.5 Hz, 2H), 7.03–6.93 (m, 2H), 4.19 (s, 4H), 4.10 (t, 2H), 4.05 (s, 2H), 2.76 (t, J = 5.9 Hz, 2H), 2.45 (s, 3H), 1.85–1.77 (m, 2H). 13C NMR (151 MHz, DMSO) δ 161.02, 160.91, 153.29, 148.86, 144.18, 128.68, 126.59, 125.09, 120.01, 113.30, 112.35, 108.94, 100.83, 65.88, 60.40, 53.73, 31.91, 26.33, 15.08.
7-(4-(3,3-Dinitroazetidin-1-yl)butoxy)-4-methyl-3-(4-(trifluoromethyl)benzyl)-2H-chromen-2-one (9): The title compound was obtained starting from 9a and 3,3-dinitroazetidine. Analytical data for 9 (yellow liquid, 51 mg, 28% yield): ESI-MS m/z 536.2 [M + H]+; 1H NMR (600 MHz, DMSO) δ 7.74 (d, J = 8.0 Hz, 1H), 7.64 (d, J = 6.5 Hz, 2H), 7.46 (d, J = 6.5 Hz, 2H), 6.98 (d, J = 13.9 Hz, 2H), 5.76 (s, 1H), 4.15 (s, 4H), 4.08 (t, 2H), 4.05 (s, 2H), 2.65 (t, 2H), 2.45 (s, 3H), 1.79–1.71 (m, 2H), 1.51–1.43 (m, 2H). 13C NMR (151 MHz, DMSO) δ 161.03, 153.31, 148.88, 144.19, 128.68, 126.56, 125.11, 119.94, 113.22, 112.41, 109.01, 100.78, 67.81, 60.35, 56.72, 31.91, 25.86, 23.00, 15.08.
4-((7-(2-(3,3-Dinitroazetidin-1-yl)ethoxy)-4-methyl-2-oxo-2H-chromen-3-yl)methyl) benzonitrile (10): The title compound was obtained starting from 10a and 3,3-dinitroazetidine. Analytical data for 10 (yellow solid, 19 mg, 12% yield, m.p. 122.5–124.1 °C): ESI-MS m/z 464.8 [M + H]+; 1H NMR (600 MHz, DMSO) δ 7.83–7.72 (m, 3H), 7.44 (d, J = 7.7 Hz, 2H), 7.08–6.96 (m, 2H), 4.83 (s, 4H), 4.40 (t, J = 2.2 Hz, 2H), 4.33 (t, 2H), 4.04 (s, 2H), 2.44 (s, 3H). 13C NMR (151 MHz, DMSO) δ 160.96, 160.59, 153.24, 148.98, 145.25, 132.18, 128.98, 126.69, 119.93, 118.74, 113.56, 112.43, 108.81, 106.79, 101.06, 66.50, 63.67, 61.10, 32.22, 15.11.
4-((7-(3-(3,3-Dinitroazetidin-1-yl)propoxy)-4-methyl-2-oxo-2H-chromen-3-yl) methyl)benzonitrile (11): The title compound was obtained starting from 11a and 3,3-dinitroazetidine. Analytical data for 11 (yellow liquid, 16 mg, 10% yield): ESI-MS m/z 478.8 [M + H]+; 1H NMR (600 MHz, DMSO) δ 7.74 (d, J = 8.1 Hz, 3H), 7.43 (d, J = 8.1 Hz, 2H), 6.97 (dd, J = 11.4, 2.2 Hz, 2H), 4.19 (s, 4H), 4.10 (t, J = 6.2 Hz, 2H), 4.04 (s, 2H), 2.76 (t, J = 6.9 Hz, 2H), 2.43 (s, 3H), 1.81 (p, J = 6.5 Hz, 2H). 13C NMR (151 MHz, DMSO) δ 160.99, 160.93, 153.30, 149.02, 145.29, 132.18, 128.98, 126.62, 119.72, 118.74, 113.28, 112.36, 108.94, 108.80, 100.83, 65.88, 60.40, 53.73, 32.21, 26.32, 15.09.
4-((7-(4-(3,3-Dinitroazetidin-1-yl)butoxy)-4-methyl-2-oxo-2H-chromen-3-yl)methyl) benzonitrile (12): The title compound was obtained starting from 12a and 3,3-dinitroazetidine. Analytical data for 12 (yellow solid, 48 mg, 29% yield, m.p. 135.1–136.3 °C): ESI-MS m/z 492.8 [M + H]+; 1H NMR (600 MHz, DMSO) δ 7.74 (d, J = 8.1 Hz, 3H), 7.43 (d, J = 7.8 Hz, 2H), 6.97 (d, J = 11.1 Hz, 2H), 4.14 (s, 4H), 4.08 (t, J = 6.3 Hz, 2H), 4.04 (s, 2H), 2.65 (t, J = 6.9 Hz, 2H), 2.43 (s, 3H), 1.74 (dd, J = 13.5, 6.6 Hz, 2H), 1.52–1.43 (m, 2H). 13C NMR (151 MHz, DMSO) δ 161.06, 161.00, 153.32, 149.03, 145.30, 132.17, 128.97, 126.57, 119.66, 118.74, 113.20, 112.41, 109.01, 108.80, 100.77, 67.81, 60.35, 56.73, 32.21, 25.86, 23.00, 15.09.
7-(2-(3,3-Dinitroazetidin-1-yl)ethoxy)-3-(4-fluorobenzyl)-4-methyl-2H-chromen-2-one (13): The title compound was obtained starting from 13a and 3,3-dinitroazetidine. Analytical data for 13 (white solid, 47 mg, 30% yield, m.p. 137.9–139.2 °C): ESI-MS m/z 457.8 [M + H]+; 1H NMR (600 MHz, DMSO) δ 7.74 (d, J = 8.8 Hz, 1H), 7.30–7.22 (m, 2H), 7.09 (t, J = 8.6 Hz, 2H), 7.04–6.94 (m, 2H), 4.28 (s, 4H), 4.14 (t, J = 4.7 Hz, 2H), 3.93 (s, 2H), 3.04 (t, J = 4.7 Hz, 2H), 2.44 (s, 3H). 13C NMR (151 MHz, DMSO) δ 161.02, 160.51, 159.75, 153.17, 148.28, 135.25, 129.68, 129.63, 126.53, 120.89, 114.99, 114.85, 113.50, 112.37, 109.34, 100.86, 67.21, 61.09, 55.15, 31.18, 15.01.
7-(3-(3,3-Dinitroazetidin-1-yl)propoxy)-3-(4-fluorobenzyl)-4-methyl-2H-chromen-2-one (14): The title compound was obtained starting from 14a and 3,3-dinitroazetidine. Analytical data for 14 (yellow liquid, 16 mg, 10% yield): ESI-MS m/z 471.8 [M + H]+; 1H NMR (600 MHz, DMSO) δ 7.73 (d, J = 8.7 Hz, 1H), 7.28–7.24 (m, 2H), 7.08 (t, J = 8.7 Hz, 2H), 6.96 (d, J = 11.2 Hz, 2H), 4.18 (s, 4H), 4.09 (t, J = 6.2 Hz, 2H), 3.92 (s, 2H), 2.75 (t, J = 6.9 Hz, 2H), 2.43 (s, 3H), 1.80 (dt, J = 13.1, 6.6 Hz, 2H). 13C NMR (151 MHz, DMSO) δ 161.98, 161.68, 161.44, 153.85, 148.92, 135.90, 130.32, 130.27, 127.15, 121.44, 115.63, 115.49, 113.99, 112.93, 109.58, 101.44, 66.49, 61.03, 54.37, 31.81, 26.96, 15.64.
7-(4-(3,3-Dinitroazetidin-1-yl)butoxy)-3-(4-fluorobenzyl)-4-methyl-2H-chromen-2-one (15): The title compound was obtained starting from 15a and 3,3-dinitroazetidine. Analytical data for 15 (white solid, 45 mg, 27% yield, m.p. 113.3–115.1 °C): ESI-MS m/z 486.2 [M + H]+; 1H NMR (600 MHz, DMSO) δ 7.73 (s, 1H), 7.26 (d, 2H), 7.09 (d, 2H), 6.97 (d, 2H), 4.14 (s, 4H), 4.08 (t, 2H), 3.93 (s, 2H), 2.65 (t, 2H), 2.43 (s, 3H), 1.81–1.67 (m, 2H), 1.55–1.40 (m, 2H). 13C NMR (151 MHz, DMSO) δ 161.35, 161.05, 160.93, 153.23, 148.30, 135.28, 129.68, 129.63, 126.47, 120.74, 114.99, 114.85, 113.28, 112.35, 109.01, 100.75, 67.79, 60.35, 56.72, 31.18, 25.86, 23.00, 15.00.
3-Benzyl-7-(2-(3,3-dinitroazetidin-1-yl)ethoxy)-4-methyl-2H-chromen-2-one (16): The title compound was obtained starting from 16a and 3,3-dinitroazetidine. Analytical data for 16 (white solid, 31 mg, 21% yield, m.p. 173–175 °C): ESI-MS m/z 439.8 [M + H]+; 1H NMR (600 MHz, DMSO) δ 7.74 (d, J = 8.9 Hz, 1H), 7.27 (t, J = 7.5 Hz, 2H), 7.22 (d, J = 7.5 Hz, 2H), 7.18 (t, J = 7.2 Hz, 1H), 7.02–6.96 (m, 2H), 4.28 (s, 4H), 4.14 (t, J = 5.0 Hz, 2H), 3.95 (s, 2H), 3.04 (t, J = 5.0 Hz, 2H), 2.43 (s, 3H). 13C NMR (151 MHz, DMSO) δ 161.05, 160.48, 153.16, 148.20, 139.14, 128.26, 127.85, 126.49, 125.91, 120.96, 113.52, 112.35, 109.35, 100.86, 67.20, 61.09, 55.15, 31.97, 15.04.
3-Benzyl-7-(3-(3,3-dinitroazetidin-1-yl)propoxy)-4-methyl-2H-chromen-2-one (17): The title compound was obtained starting from 17a and 3,3-dinitroazetidine. Analytical data for 17 (white solid, 34 mg, 22% yield, m.p. 115.1–116.8 °C): ESI-MS m/z 453.8 [M + H]+; 1H NMR (600 MHz, DMSO) δ 7.73 (d, J = 8.7 Hz, 1H), 7.27 (t, J = 7.5 Hz, 2H), 7.22 (d, J = 7.4 Hz, 2H), 7.18 (t, J = 7.2 Hz, 1H), 7.00–6.92 (m, 2H), 4.19 (s, 4H), 4.09 (t, J = 6.3 Hz, 2H), 3.95 (s, 2H), 2.76 (t, J = 7.0 Hz, 2H), 2.43 (s, 3H), 1.84–1.77 (m, 2H). 13C NMR (151 MHz, DMSO) δ 161.08, 160.76, 153.20, 148.20, 139.15, 128.26, 127.85, 126.47, 125.90, 120.87, 113.38, 112.27, 108.94, 100.80, 65.84, 60.39, 54.74, 31.96, 26.32, 13.92.
3-Benzyl-7-(4-(3,3-dinitroazetidin-1-yl)butoxy)-4-methyl-2H-chromen-2-one (18): The title compound was obtained starting from 18a and 3,3-dinitroazetidine. Analytical data for 18 (yellow liquid, 22 mg, 14% yield): ESI-MS m/z 467.9 [M + H]+; 1H NMR (600 MHz, DMSO) δ 7.71 (d, J = 8.6 Hz, 1H), 7.26 (t, J = 7.3 Hz, 2H), 7.22 (d, J = 7.5 Hz, 2H), 7.17 (t, J = 7.1 Hz, 1H), 6.95 (d, J = 13.7 Hz, 2H), 4.14 (s, 4H), 4.07 (t, J = 6.3 Hz, 2H), 3.94 (s, 2H), 2.64 (t, J = 6.9 Hz, 2H), 2.42 (s, 3H), 1.78–1.69 (m, 2H), 1.52–1.42 (m, 2H). 13C NMR (151 MHz, DMSO) δ 161.09, 160.88, 153.21, 148.22, 139.16, 128.26, 127.85, 126.42, 125.90, 120.81, 113.30, 112.33, 108.94, 100.74, 67.77, 60.34, 56.69, 31.96, 25.85, 22.95, 15.02.
7-(2-(3,3-Dinitroazetidin-1-yl)ethoxy)-4-methyl-2H-chromen-2-one (19): The title compound was obtained starting from 19a and 3,3-dinitroazetidine. Analytical data for 19 (white solid, 18 mg, 15% yield, m.p. 91–93 °C): ESI-MS m/z 350.1 [M + H]+; 1H NMR (600 MHz, CDCl3) δ 7.51 (d, J = 8.8 Hz, 1H), 6.83 (dd, J = 8.8, 2.4 Hz, 1H), 6.78 (d, J = 2.3 Hz, 1H), 6.15 (s, 1H), 4.29 (s, 4H), 4.13 (t, 2H), 3.09 (t, 2H), 2.40 (s, 3H). 13C NMR (151 MHz, CDCl3) δ 160.47, 154.61, 151.80, 125.14, 113.48, 111.86, 111.75, 107.95, 100.61, 67.13, 61.77, 55.60, 18.06.
7-(3-(3,3-Dinitroazetidin-1-yl)propoxy)-4-methyl-2H-chromen-2-one (20): The title compound was obtained starting from 20a and 3,3-dinitroazetidine. Analytical data for 20 (yellow solid, 33 mg, 27% yield, m.p. 78.5–79.6 °C): ESI-MS m/z 364.1 [M + H]+; 1H NMR (600 MHz, DMSO) δ 7.69 (t, J = 8.3 Hz, 1H), 6.97 (t, J = 9.1 Hz, 2H), 6.22 (d, J = 7.0 Hz, 1H), 4.37 (s, 4H), 4.10 (t, J = 6.2 Hz, 2H), 2.76 (t, J = 6.9 Hz, 2H), 2.40 (s, 3H), 1.85–1.76 (m, 2H). 13C NMR (151 MHz, DMSO) δ 161.42, 159.99, 154.59, 153.26, 126.32, 112.22, 110.97, 108.94, 101.06, 65.89, 60.39, 53.74, 26.32, 17.97.
7-(4-(3,3-Dinitroazetidin-1-yl)butoxy)-4-methyl-2H-chromen-2-one (21): The title compound was obtained starting from 21a and 3,3-dinitroazetidine. Analytical data for 21 (yellow solid, 28 mg, 22% yield, m.p. 85.8–87.2 °C): ESI-MS m/z 378.1 [M + H]+; 1H NMR (600 MHz, DMSO) δ 7.68 (d, J = 8.7 Hz, 1H), 7.02–6.94 (m, 2H), 6.21 (s, 1H), 4.77 (s, 4H), 4.14 (t, 2H), 4.11 (t, 2H), 2.40 (s, 3H), 1.85–1.79 (m, 2H), 1.79–1.72 (m, 2H). 13C NMR (151 MHz, DMSO) δ 161.49, 160.00, 156.24, 154.60, 153.26, 126.29, 112.92, 112.27, 110.94, 106.85, 101.01, 67.66, 64.88, 24.92, 24.74, 17.96.


 ,
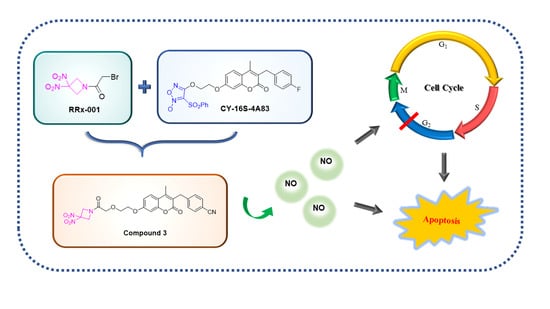
, 
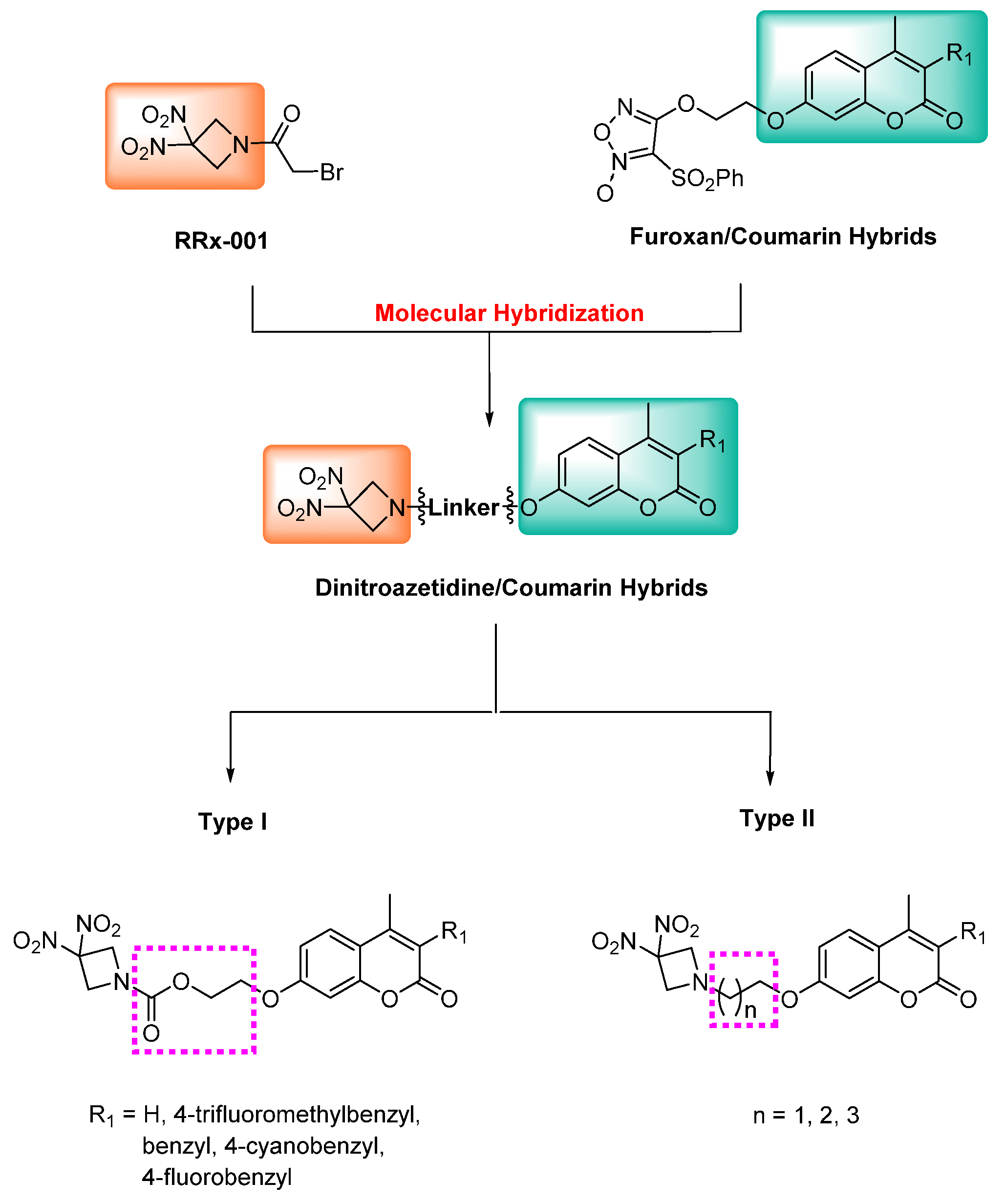




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}