
AddictedChem: A Data-Driven Integrated Platform for New Psychoactive Substance Identification

 , ,
, ,  , ,
, ,
Abstract
:1. Introduction
2. Results and Discussion
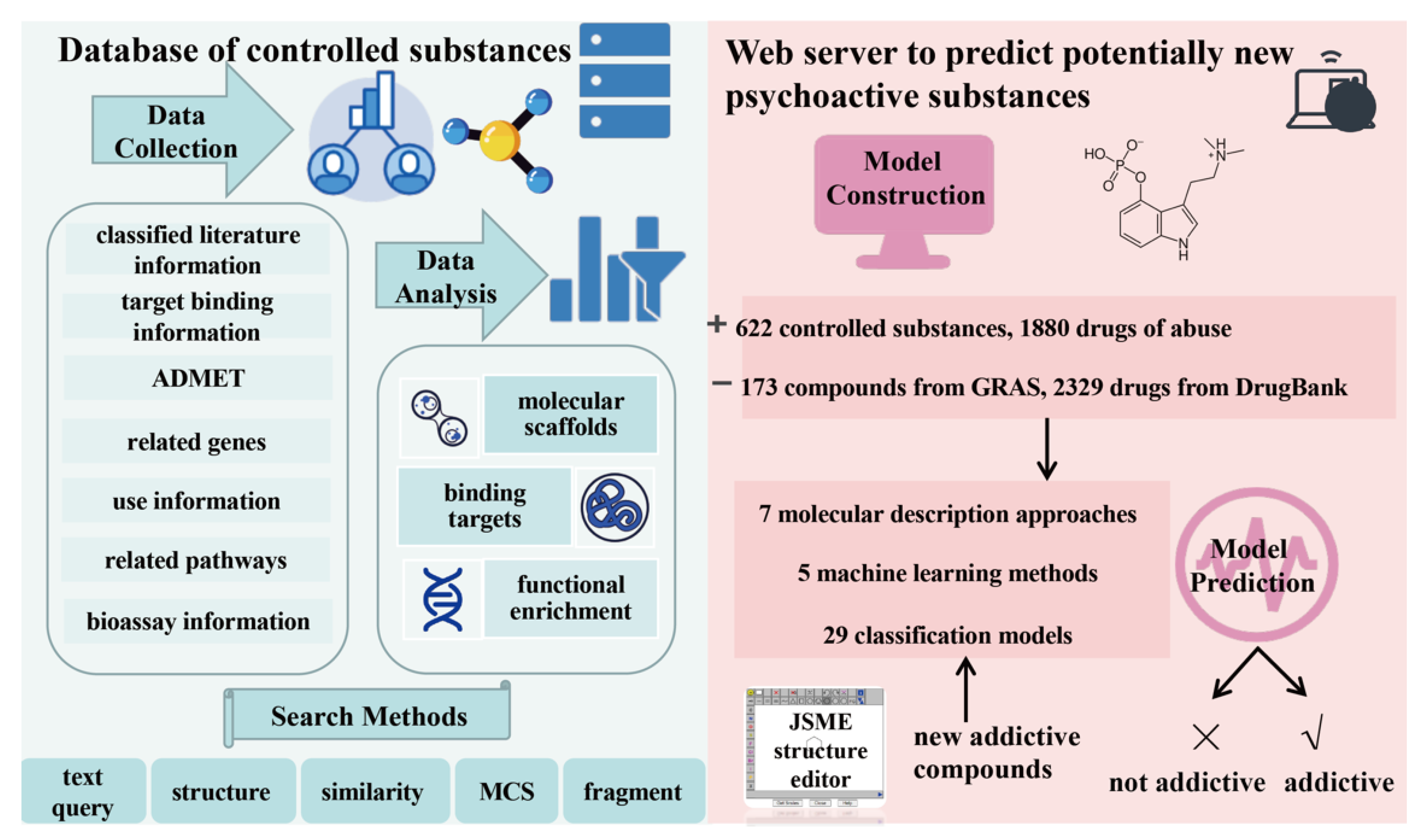
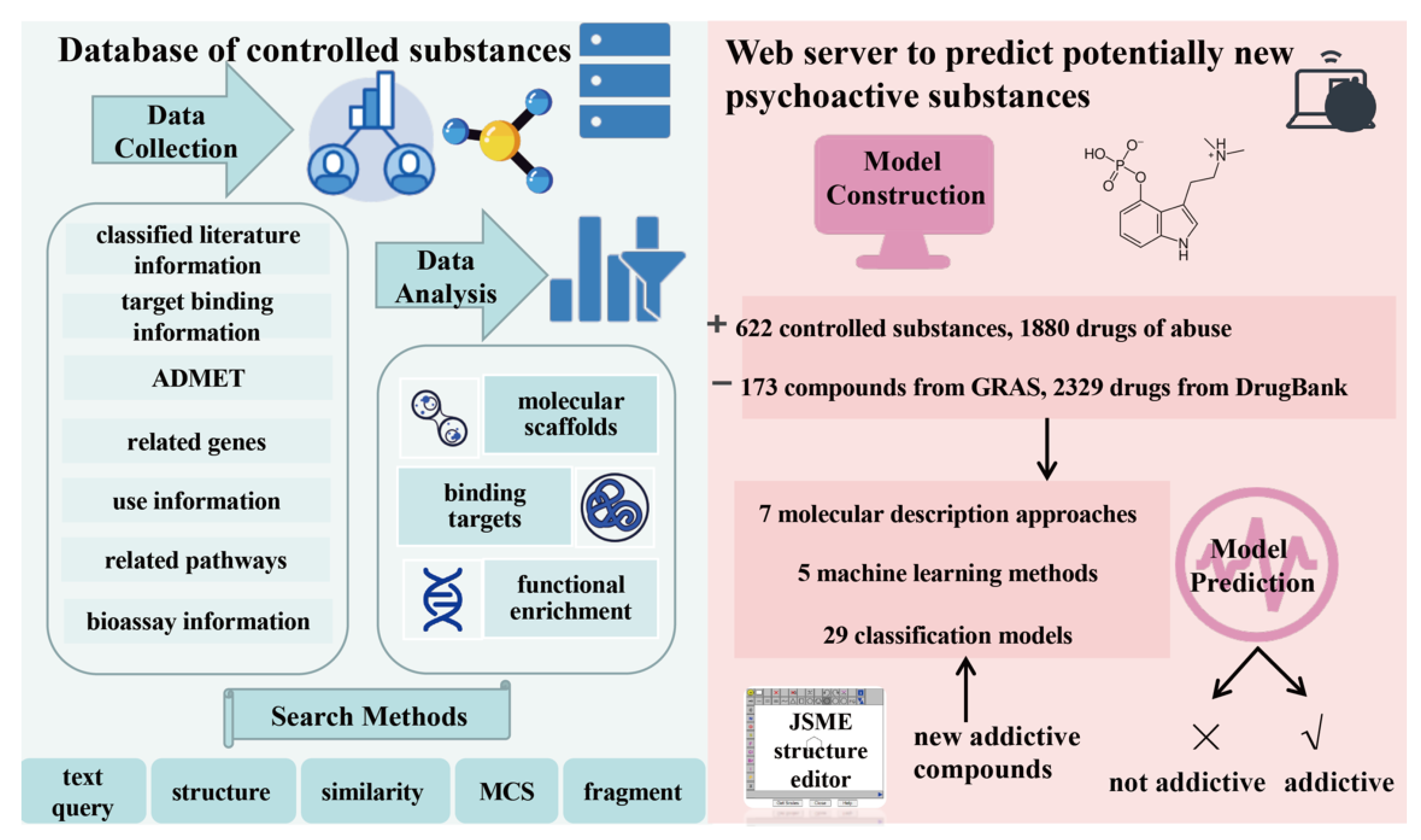
2.1. Overview of AddictedChem
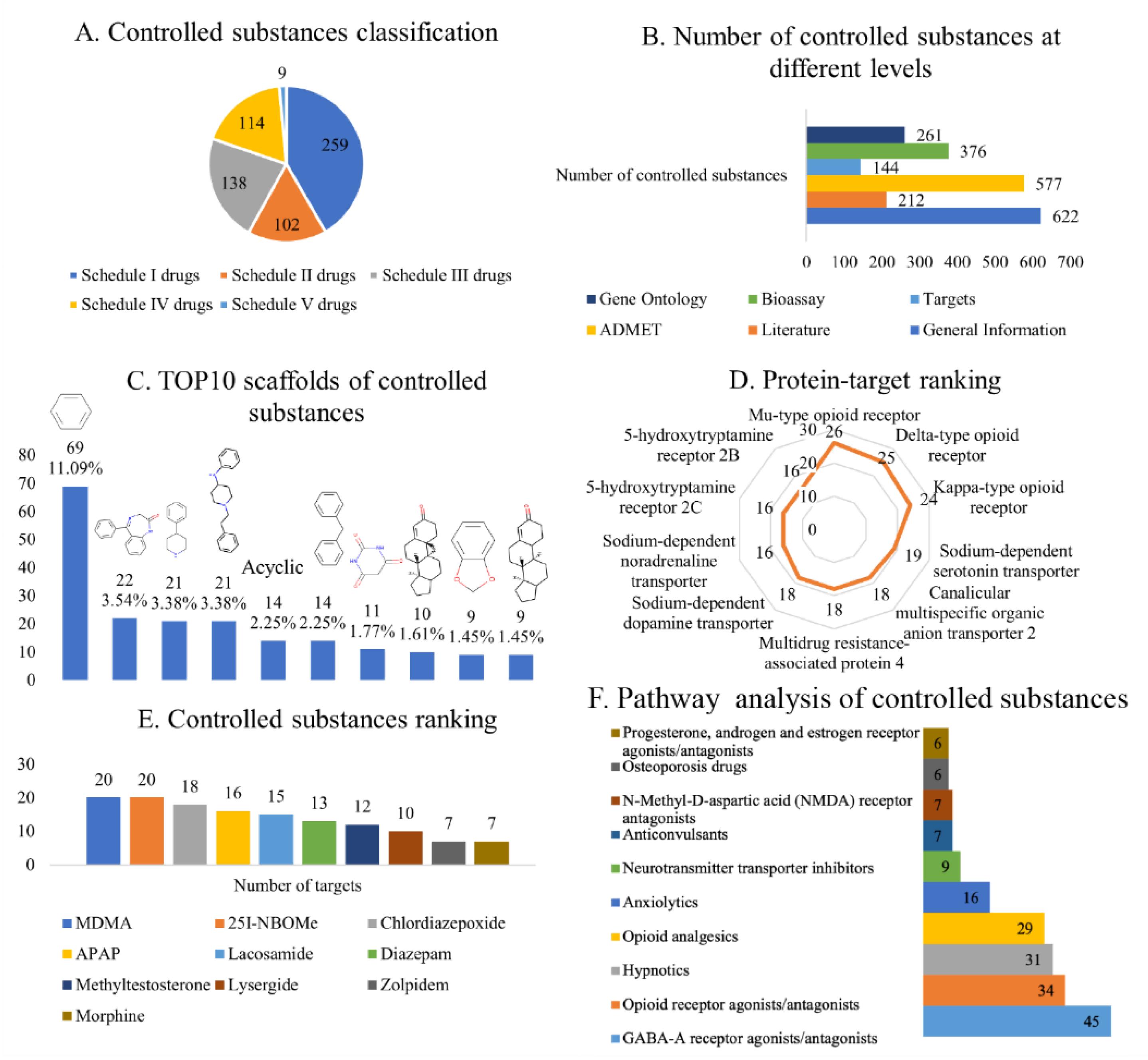
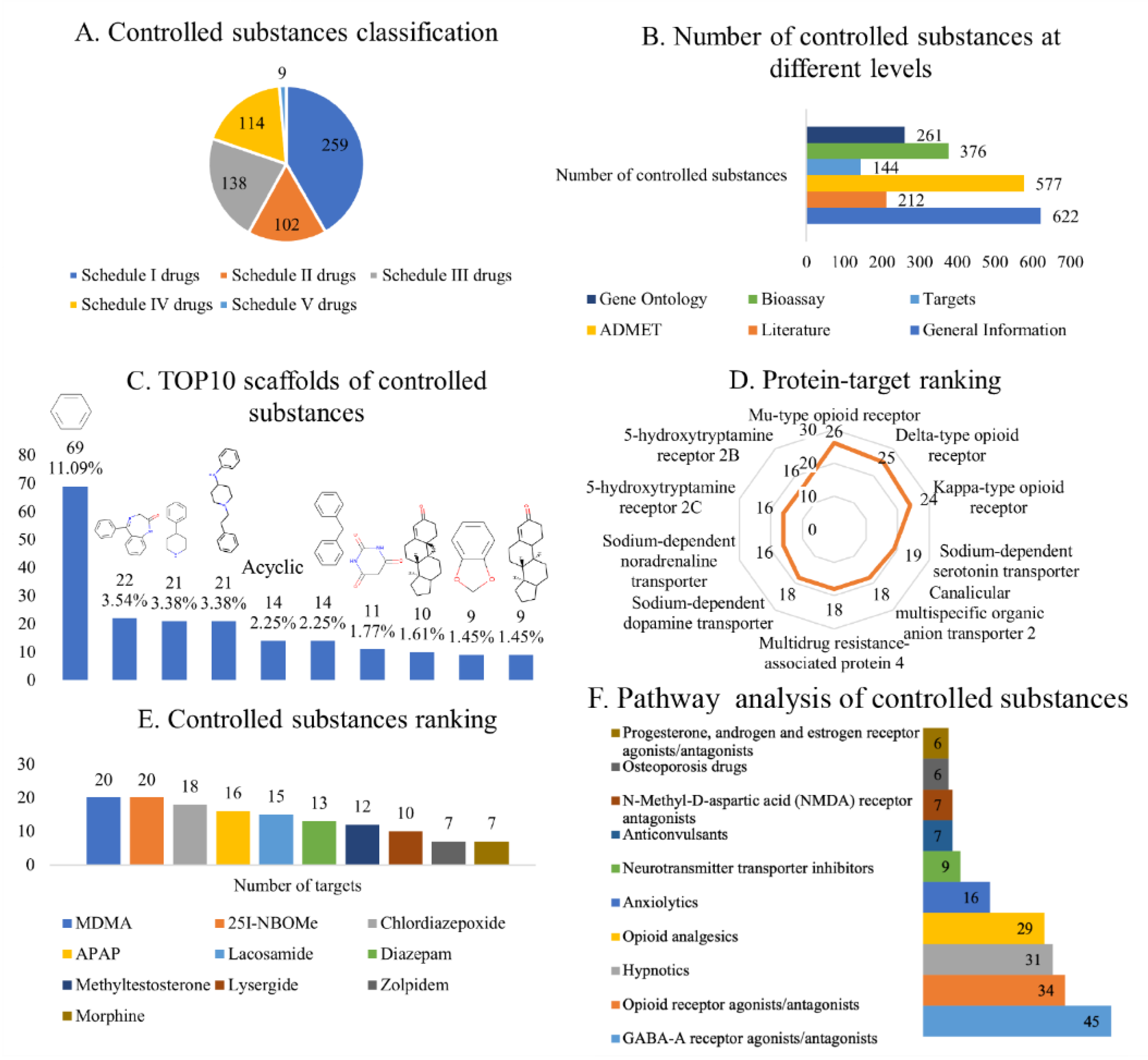
2.2. Scaffold Analysis of Controlled Substances
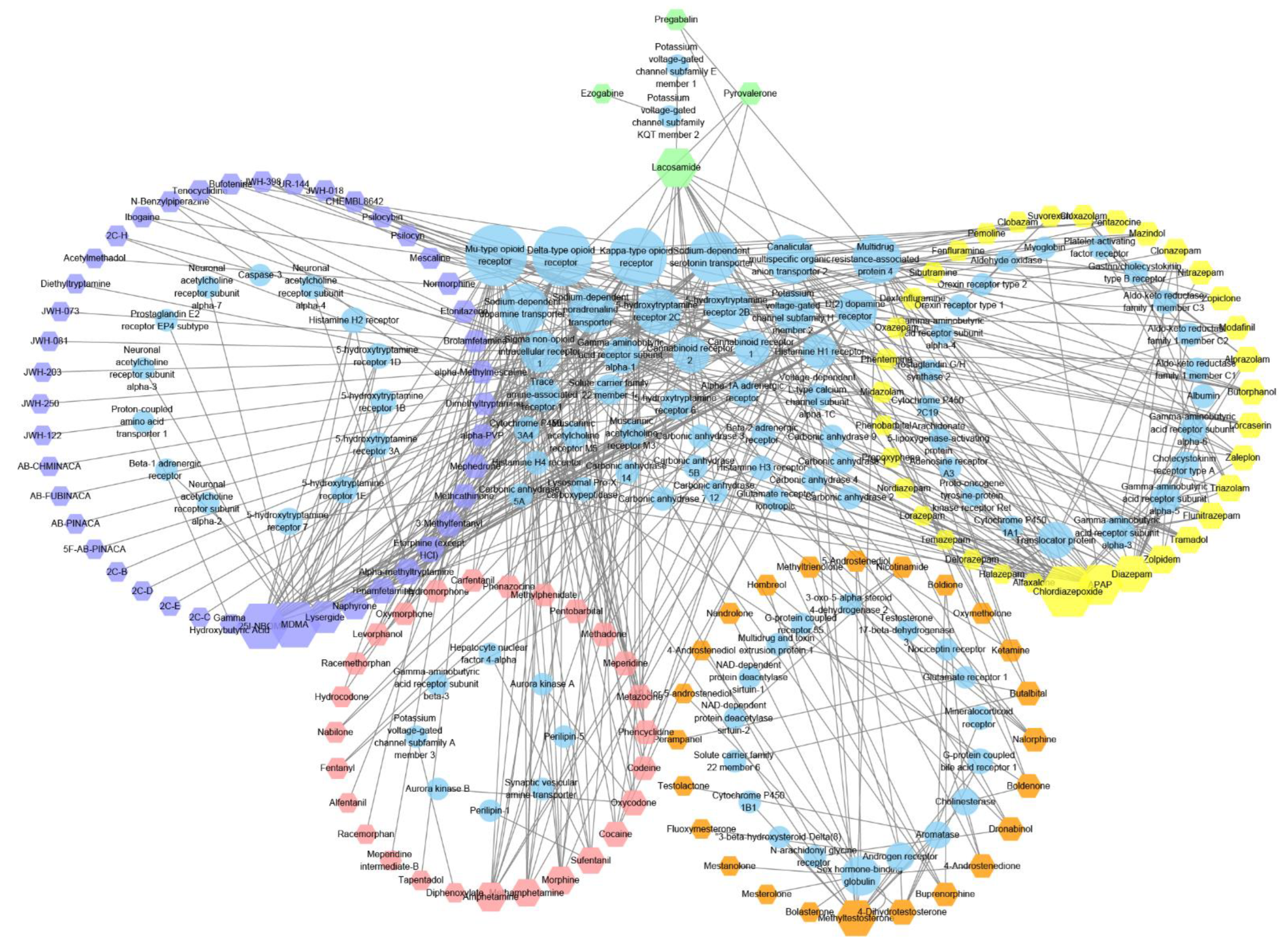
2.3. Analysis of Controlled Substance Targets
2.4. Functional Enrichment Analysis
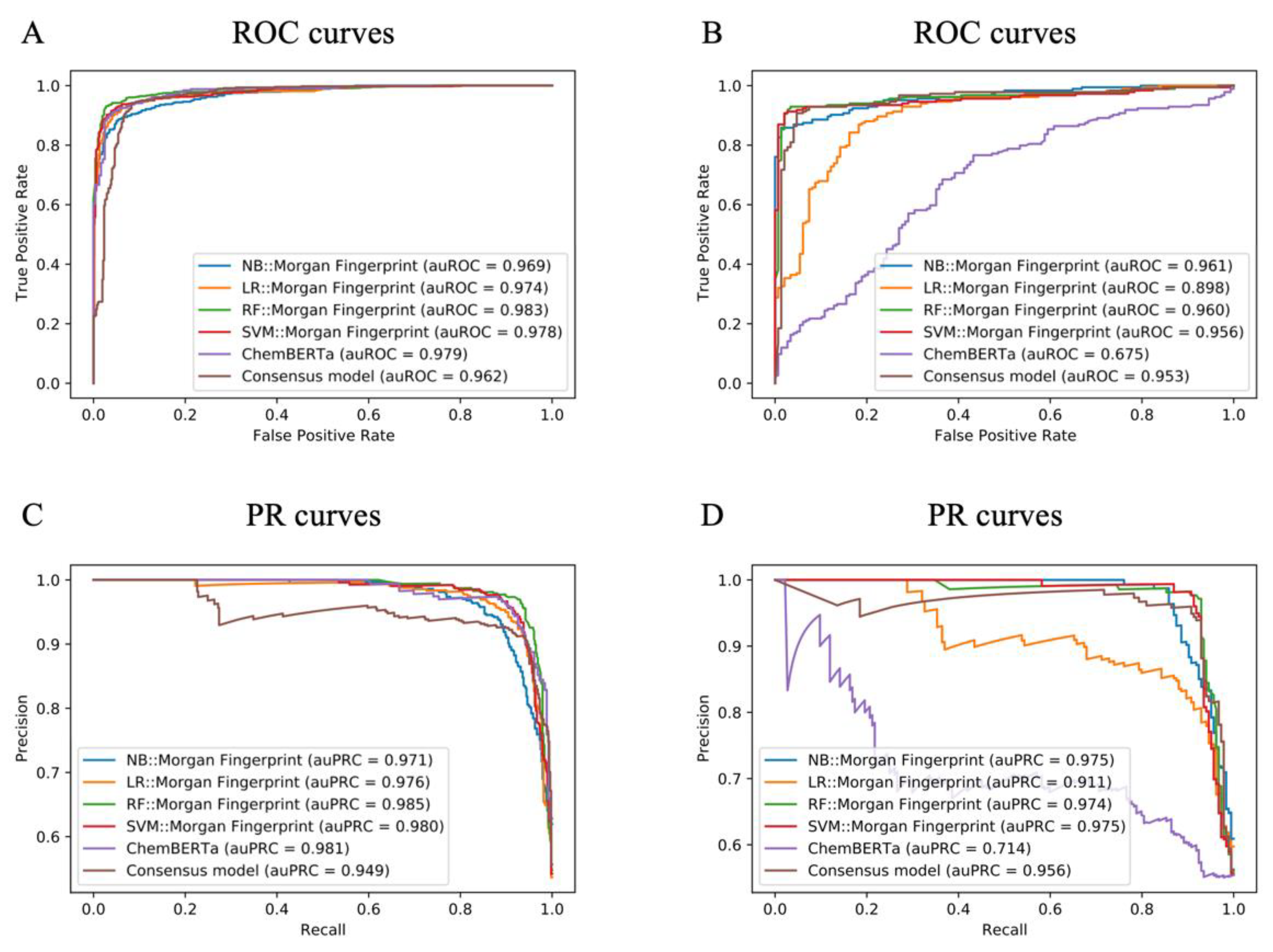
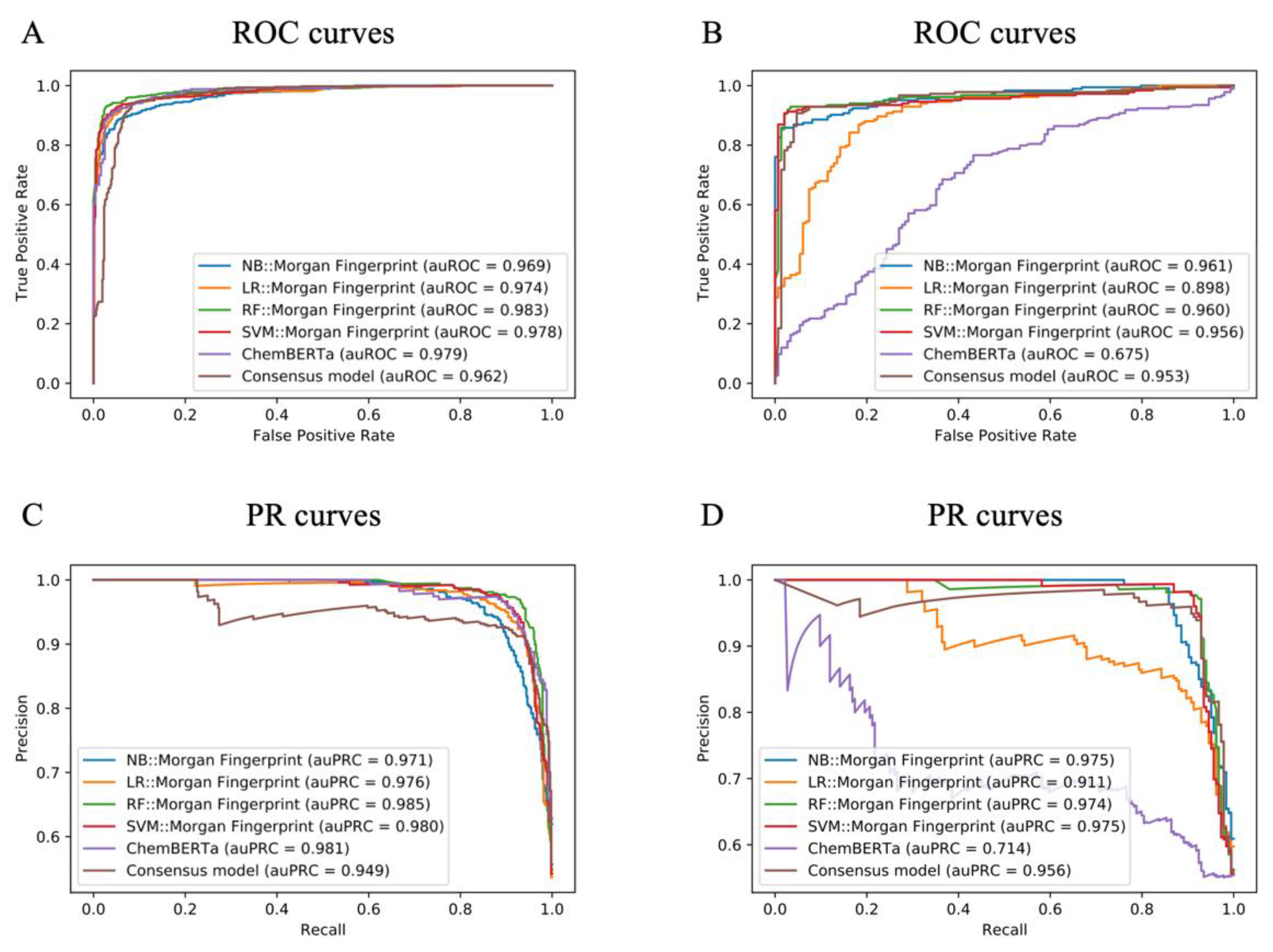
2.5. Model Performance for NPS Prediction
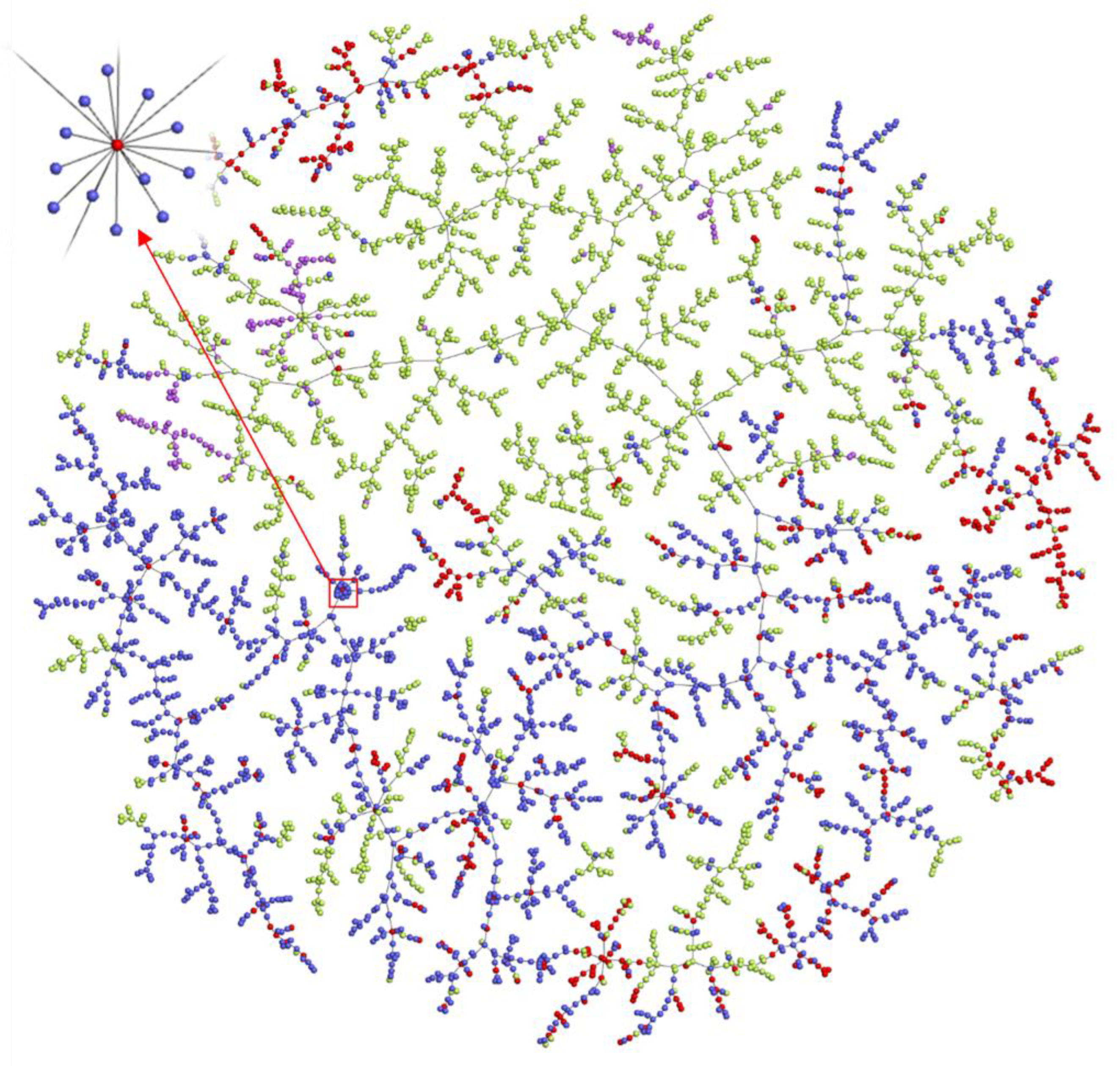
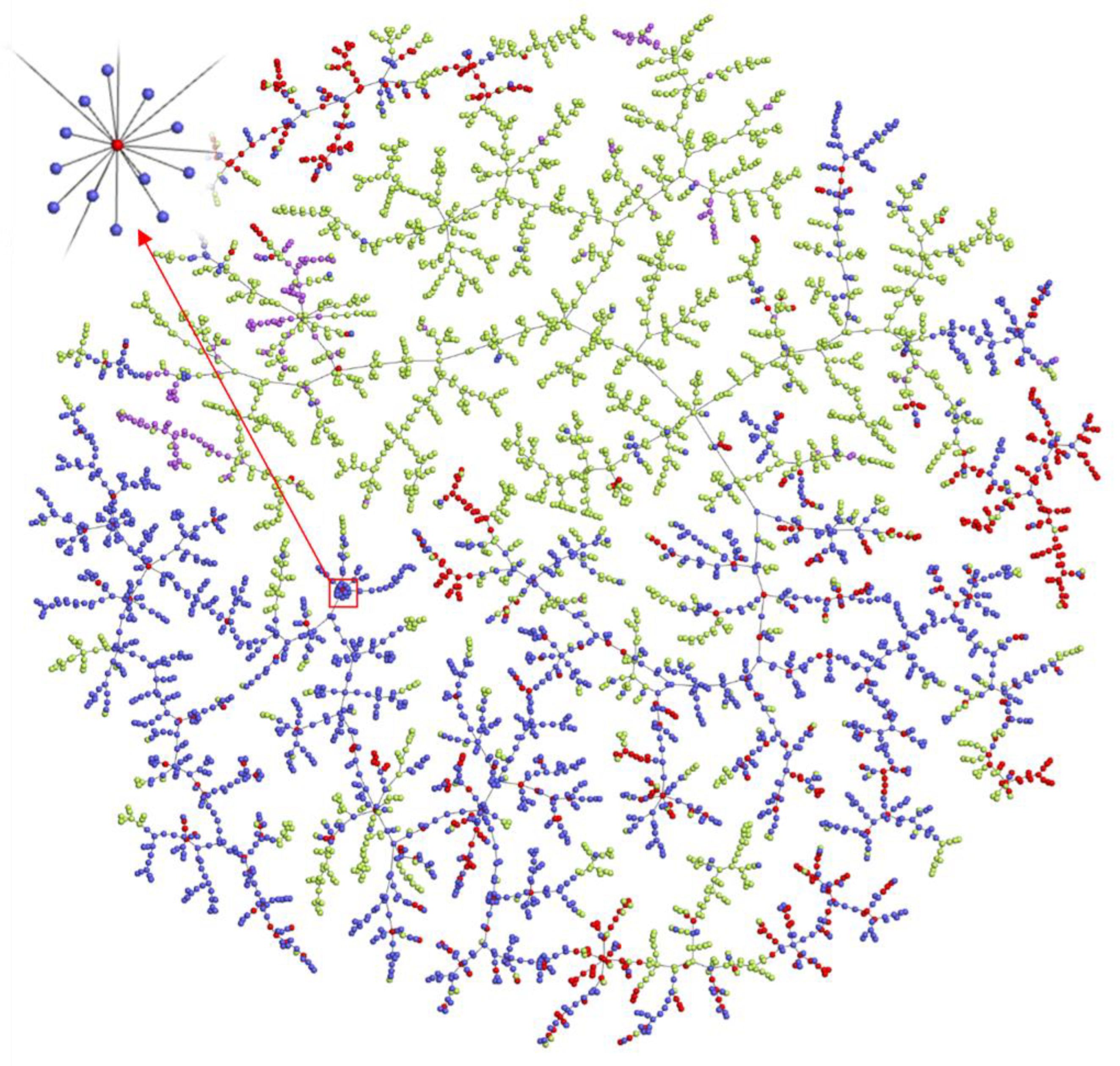
2.6. Exploration of the Chemical Space of Addictive Compounds
3. Materials and Methods
3.1. Data Sources
3.2. Data Curation
3.3. Data Analysis
3.3.1. Scaffold Analysis of Controlled Substances
3.3.2. Target Analysis of Controlled Substances
3.3.3. Functional Enrichment Analysis
3.4. Model Construction
3.5. Performance Evaluation
3.6. Chemical Space Exploration
3.7. Database and Webserver
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- United Nations Office on Drugs and Crime. World Drug Report 2021; United Nations Office on Drugs and Crime: Vienna, Austria, 2021. [Google Scholar]
- United Nations. Single Convention on Narcotic Drugs of 1961 as Amended by the 1972 Protocol Amending the Single Convention on Narcotic Drugs 1961; United Nations: New York, NY, USA, 1961. [Google Scholar]
- United Nations. Convention on Psychotropic Substances of 1971; United Nations: New York, NY, USA, 1971. [Google Scholar]
- United Nations. United Nations Convention against Illicit Traffic in Narcotic Drugs and Psychotropic Substances; United Nations: New York, NY, USA, 1988. [Google Scholar]
- Congressional Research Service. The Controlled Substances Act (CSA):A Legal Overview for the 117th Congress; Congressional Research Service: Washington, DC, USA, 2021.
- Giannini, A.J. An approach to drug abuse, intoxication and withdrawal. Am. Fam. Physician 2000, 61, 2763–2774. [Google Scholar] [PubMed]
- Arias-Carrión, O.; Stamelou, M.; Murillo-Rodríguez, E.; Menéndez-González, M.; Pöppel, E. Dopaminergic reward system: A short integrative review. Int. Arch. Med. 2010, 3, 24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wise, R.A.; Rompre, P.P. Brain dopamine and reward. Annu. Rev. Psychol. 1989, 40, 191–225. [Google Scholar] [CrossRef] [PubMed]
- Johnson, S.W.; North, R.A. Opioids excite dopamine neurons by hyperpolarization of local interneurons. J. Neurosci. 1992, 12, 483–488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lüscher, C.; Ungless, M.A. The mechanistic classification of addictive drugs. PLoS Med. 2006, 3, e437. [Google Scholar] [CrossRef]
- Wishart, D.S.; Feunang, Y.D.; Guo, A.C.; Lo, E.J.; Marcu, A.; Grant, J.R.; Sajed, T.; Johnson, D.; Li, C.; Sayeeda, Z.; et al. DrugBank 5.0: A major update to the DrugBank database for 2018. Nucleic Acids Res. 2018, 46, D1074–D1082. [Google Scholar] [CrossRef]
- Yang, H.; Lou, C.; Sun, L.; Li, J.; Cai, Y.; Wang, Z.; Li, W.; Liu, G.; Tang, Y. admetSAR 2.0: Web-service for prediction and optimization of chemical ADMET properties. Bioinformatics 2019, 35, 1067–1069. [Google Scholar] [CrossRef]
- Gilson, M.K.; Liu, T.; Baitaluk, M.; Nicola, G.; Hwang, L.; Chong, J. BindingDB in 2015: A public database for medicinal chemistry, computational chemistry and systems pharmacology. Nucleic Acids Res. 2016, 44, D1045–D1053. [Google Scholar] [CrossRef]
- Davis, A.P.; Grondin, C.J.; Johnson, R.J.; Sciaky, D.; Wiegers, J.; Wiegers, T.C.; Mattingly, C.J. Comparative Toxicogenomics Database (CTD): Update 2021. Nucleic Acids Res. 2021, 49, D1138–D1143. [Google Scholar] [CrossRef]
- European Monitoring Centre for Drugs and Drug Addiction. European Drug Report 2021: Trends and Developments; European Monitoring Centre for Drugs and Drug Addiction: Lisbon, Portugal, 2021; p. 60.
- Schifano, F.; Chiappini, S.; Corkery, J.M.; Guirguis, A. Abuse of Prescription Drugs in the Context of Novel Psychoactive Substances (NPS): A Systematic Review. Brain Sci. 2018, 8, 73. [Google Scholar] [CrossRef] [Green Version]
- UNODC. Early Warning Advisory on New Psychoactive Substances. 2022. Available online: https://www.unodc.org/LSS/Page/NPS (accessed on 19 April 2022).
- Glennon, R.A. Bath salts, mephedrone, and methylenedioxypyrovalerone as emerging illicit drugs that will need targeted therapeutic intervention. Adv. Pharmacol. 2014, 69, 581–620. [Google Scholar] [PubMed] [Green Version]
- Peacock, A.; Bruno, R.; Gisev, N.; Degenhardt, L.; Hall, W.; Sedefov, R.; White, J.; Thomas, K.V.; Farrell, M.; Griffiths, P. New psychoactive substances: Challenges for drug surveillance, control, and public health responses. Lancet 2019, 394, 1668–1684. [Google Scholar] [CrossRef]
- Melo, E.B.D.; Martins, J.; Rodrigues, C.H.; Bruni, A.T. In silico risk assessment studies of new psychoactive substances derived from amphetamines and cathinones. J. Braz. Chem. Soc. 2020, 31, 927–940. [Google Scholar]
- Bienfait, B.; Ertl, P. JSME: A free molecule editor in JavaScript. J. Cheminformatics 2013, 5, 24. [Google Scholar] [CrossRef] [Green Version]
- Bajusz, D.; Rácz, A.; Héberger, K. Why is Tanimoto index an appropriate choice for fingerprint-based similarity calculations? J. Cheminformatics 2015, 7, 20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, Y.; Jiang, T.; Girke, T. A maximum common substructure-based algorithm for searching and predicting drug-like compounds. Bioinformatics 2008, 24, i366–i374. [Google Scholar] [CrossRef] [Green Version]
- Barnard, J.M. Substructure searching methods: Old and new. J. Chem. Inf. Comput. Sci. 1993, 33, 532–538. [Google Scholar] [CrossRef]
- Velkoborsky, J.; Hoksza, D. Scaffold analysis of PubChem database as background for hierarchical scaffold-based visualization. J. Cheminformatics 2016, 8, 74. [Google Scholar] [CrossRef] [Green Version]
- Oeri, H.E. Beyond ecstasy: Alternative entactogens to 3,4-methylenedioxymethamphetamine with potential applications in psychotherapy. J. Psychopharmacol. 2021, 35, 512–536. [Google Scholar] [CrossRef]
- Skerritt, J.H.; Johnston, G.A. Enhancement of GABA binding by benzodiazepines and related anxiolytics. Eur. J. Pharmacol. 1983, 89, 193–198. [Google Scholar] [CrossRef]
- Alert from NDEWS Web Monitoring Team: Increases in Reddit Discussions of DMXE October 2020–March 2021. 2021. Available online: https://ndews.org/?wysija-page=1&controller=email&action=view&email_id=125&wysijap=subscriptions (accessed on 26 March 2021).
- Probst, D.; Reymond, J.L. Visualization of very large high-dimensional data sets as minimum spanning trees. J. Cheminformatics 2020, 12, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.; Thiessen, P.A.; Bolton, E.E.; Chen, J.; Fu, G.; Gindulyte, A.; Han, L.; He, J.; He, S.; Shoemaker, B.A.; et al. PubChem Substance and Compound Databases. Nucleic Acids Res. 2016, 44, D1202–D1213. [Google Scholar] [CrossRef] [PubMed]
- Dalby, A.; Nourse, J.G.; Hounshell, W.D.; Gushurst, A.K.; Grier, D.L.; Leland, B.A.; Laufer, J. Description of several chemical structure file formats used by computer programs developed at Molecular Design Limited. J. Chem. Inf. Comput. Sci. 1992, 32, 244–255. [Google Scholar] [CrossRef]
- U.S. Drug Enforcement Administration. 2017–2020 NFLIS-Drug Substance List; D.C.D.; U.S. Drug Enforcement Administration: Springfield, VA, USA, 2021.
- Kanehisa, M.; Furumichi, M.; Tanabe, M.; Sato, Y.; Morishima, K. KEGG: New perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 2017, 45, D353–D361. [Google Scholar] [CrossRef] [Green Version]
- Jewison, T.; Su, Y.; Disfany, F.M.; Liang, Y.; Knox, C.; Maciejewski, A.; Poelzer, J.; Huynh, J.; Zhou, Y.; Arndt, D.; et al. SMPDB 2.0: Big Improvements to the Small Molecule Pathway Database. Nucleic Acids Res. 2014, 42, D478–D484. [Google Scholar] [CrossRef] [Green Version]
- Landrum, G. RDKit: Open-Source Cheminformatics; Gregory Landrum: Zurich, Switzerland, 2006. [Google Scholar]
- Bemis, G.W.; Murcko, M.A. The properties of known drugs. 1. Molecular frameworks. J. Med. Chem. 1996, 39, 2887–2893. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Rogers, F.B. Medical subject headings. Bull. Med. Libr. Assoc. 1963, 51, 114–116. [Google Scholar]
- Yu, G.; Wang, L.G.; Han, Y.; He, Q.Y. clusterProfiler: An R package for comparing biological themes among gene clusters. Omics J. Integr. Biol. 2012, 16, 284–287. [Google Scholar] [CrossRef]
- Kier, L.B.; Hall, L.H. An electrotopological-state index for atoms in molecules. Pharm. Res. 1990, 7, 801–807. [Google Scholar] [CrossRef]
- Durant, J.L.; Leland, B.A.; Henry, D.R.; Nourse, J.G. Reoptimization of MDL keys for use in drug discovery. J. Chem. Inf. Comput. Sci. 2002, 42, 1273–1280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rogers, D.; Hahn, M. Extended-connectivity fingerprints. J. Chem. Inf. Model. 2010, 50, 742–754. [Google Scholar] [CrossRef] [PubMed]
- Probst, D.; Reymond, J.L. A probabilistic molecular fingerprint for big data settings. J. Cheminformatics 2018, 10, 66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaeger, S.; Fulle, S.; Turk, S. Mol2vec: Unsupervised machine learning approach with chemical intuition. J. Chem. Inf. Model. 2018, 58, 27–35. [Google Scholar] [CrossRef]
- Wright, R.E. Logistic regression. In Reading and Understanding Multivariate Statistics; American Psychological Association: Washington, DC, USA, 1995; pp. 217–244. [Google Scholar]
- Zhang, H.J.A. The optimality of naive Bayes. In Proceedings of the 17th International Florida Artificial Intelligence Research Society Conference, Miami Beach, FL, USA, 12–14 May 2004; pp. 562–567. [Google Scholar]
- Breiman, L. Machine Learning. Mach. Learn. 2001, 45, 5–32. [Google Scholar] [CrossRef] [Green Version]
- Cortes, C.; Vapnik, V. Support-vector networks. Mach. Learn. 1995, 20, 273–297. [Google Scholar] [CrossRef]
- Pedregosa, F.; Varoquaux, G.; Gramfort, A.; Michel, V.; Thirion, B.; Grisel, O.; Blondel, M.; Prettenhofer, P.; Weiss, R.; Dubourg, V.; et al. Scikit-Learn: Machine Learning in Python. J. Mach. Learn. Res. 2011, 12, 2825–2830. [Google Scholar]
- Chithrananda, S. ChemBERTa: Large-scale self-supervised pretraining for molecular property prediction. arXiv 2020, arXiv:2010.09885. [Google Scholar]
- Bawa, M.; Condie, T.; Ganesan, P. LSH Forest: Self-Tuning indexes for similarity search. In Proceedings of the 14th International Conference on World Wide Web, WWW 2005, Chiba, Japan, 10–14 May 2005; pp. 651–660. [Google Scholar]
- Fix, E.; Hodges, J.L. Discriminatory analysis. Nonparametric discrimination: Consistency properties. Int. Stat. Rev. Rev. Int. Stat. 1989, 57, 238–247. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Test Set | External Validation Set | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| BA | MCC | F1 | AUC | BA | MCC | F1 | AUC | ||
| NB | RDKit Descriptors+E-state | 0.810 | 0.627 | 0.817 | 0.897 | 0.780 | 0.572 | 0.820 | 0.858 |
| MACCS | 0.794 | 0.595 | 0.803 | 0.887 | 0.806 | 0.629 | 0.844 | 0.895 | |
| RDKit fingerprint | 0.794 | 0.588 | 0.786 | 0.865 | 0.844 | 0.687 | 0.835 | 0.920 | |
| Morgan fingerprint | 0.909 | 0.818 | 0.905 | 0.969 | 0.911 | 0.817 | 0.912 | 0.961 | |
| SECFP | 0.886 | 0.772 | 0.882 | 0.960 | 0.919 | 0.834 | 0.921 | 0.968 | |
| MHFP6 | 0.787 | 0.586 | 0.800 | 0.893 | 0.844 | 0.685 | 0.856 | 0.894 | |
| Mol2vec | 0.714 | 0.472 | 0.754 | 0.779 | 0.5 | 0.0 | 0.0 | 0.606 | |
| LR | RDKit Descriptors+E-state | 0.899 | 0.797 | 0.897 | 0.966 | 0.656 | 0.310 | 0.670 | 0.727 |
| MACCS | 0.894 | 0.787 | 0.891 | 0.962 | 0.741 | 0.480 | 0.760 | 0.837 | |
| RDKit fingerprint | 0.919 | 0.838 | 0.917 | 0.967 | 0.903 | 0.805 | 0.913 | 0.964 | |
| Morgan fingerprint | 0.931 | 0.862 | 0.929 | 0.974 | 0.837 | 0.683 | 0.864 | 0.898 | |
| SECFP | 0.921 | 0.842 | 0.919 | 0.969 | 0.888 | 0.775 | 0.899 | 0.931 | |
| MHFP6 | 0.896 | 0.793 | 0.895 | 0.948 | 0.919 | 0.834 | 0.921 | 0.964 | |
| Mol2vec | 0.894 | 0.787 | 0.892 | 0.959 | 0.774 | 0.544 | 0.784 | 0.848 | |
| RF | RDKit Descriptors+E-state | 0.944 | 0.888 | 0.942 | 0.985 | 0.815 | 0.640 | 0.847 | 0.887 |
| MACCS | 0.940 | 0.880 | 0.938 | 0.986 | 0.892 | 0.786 | 0.906 | 0.950 | |
| RDKit fingerprint | 0.942 | 0.887 | 0.940 | 0.973 | 0.951 | 0.898 | 0.953 | 0.965 | |
| Morgan fingerprint | 0.950 | 0.900 | 0.948 | 0.983 | 0.921 | 0.842 | 0.929 | 0.960 | |
| SECFP | 0.942 | 0.884 | 0.940 | 0.979 | 0.943 | 0.885 | 0.948 | 0.964 | |
| MHFP6 | 0.927 | 0.854 | 0.924 | 0.974 | 0.491 | −0.019 | 0.574 | 0.500 | |
| Mol2vec | 0.921 | 0.842 | 0.918 | 0.973 | 0.702 | 0.478 | 0.795 | 0.871 | |
| SVM | RDKit Descriptors+E-state | 0.919 | 0.838 | 0.916 | 0.975 | 0.806 | 0.629 | 0.844 | 0.858 |
| MACCS | 0.921 | 0.842 | 0.918 | 0.980 | 0.860 | 0.720 | 0.875 | 0.927 | |
| RDKit fingerprint | 0.942 | 0.884 | 0.939 | 0.975 | 0.930 | 0.856 | 0.933 | 0.959 | |
| Morgan fingerprint | 0.933 | 0.870 | 0.929 | 0.978 | 0.928 | 0.854 | 0.934 | 0.956 | |
| SECFP | 0.934 | 0.871 | 0.931 | 0.977 | 0.939 | 0.874 | 0.942 | 0.960 | |
| MHFP6 | 0.890 | 0.780 | 0.889 | 0.955 | 0.747 | 0.546 | 0.664 | 0.949 | |
| Mol2vec | 0.907 | 0.814 | 0.906 | 0.971 | 0.799 | 0.629 | 0.847 | 0.910 | |
| ChemBERTa | - | 0.934 | 0.868 | 0.935 | 0.979 | 0.629 | 0.256 | 0.644 | 0.675 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Han, M.; Liu, S.; Zhang, D.; Zhang, R.; Liu, D.; Xing, H.; Sun, D.; Gong, L.; Cai, P.; Tu, W.; et al. AddictedChem: A Data-Driven Integrated Platform for New Psychoactive Substance Identification. Molecules 2022, 27, 3931. https://doi.org/10.3390/molecules27123931
Han M, Liu S, Zhang D, Zhang R, Liu D, Xing H, Sun D, Gong L, Cai P, Tu W, et al. AddictedChem: A Data-Driven Integrated Platform for New Psychoactive Substance Identification. Molecules. 2022; 27(12):3931. https://doi.org/10.3390/molecules27123931
Chicago/Turabian StyleHan, Mengying, Sheng Liu, Dachuan Zhang, Rui Zhang, Dongliang Liu, Huadong Xing, Dandan Sun, Linlin Gong, Pengli Cai, Weizhong Tu, and et al. 2022. "AddictedChem: A Data-Driven Integrated Platform for New Psychoactive Substance Identification" Molecules 27, no. 12: 3931. https://doi.org/10.3390/molecules27123931
APA StyleHan, M., Liu, S., Zhang, D., Zhang, R., Liu, D., Xing, H., Sun, D., Gong, L., Cai, P., Tu, W., Chen, J., & Hu, Q.-N. (2022). AddictedChem: A Data-Driven Integrated Platform for New Psychoactive Substance Identification. Molecules, 27(12), 3931. https://doi.org/10.3390/molecules27123931