
Advances on Delivery of Cytotoxic Enzymes as Anticancer Agents

 ,
,
Abstract
1. Introduction
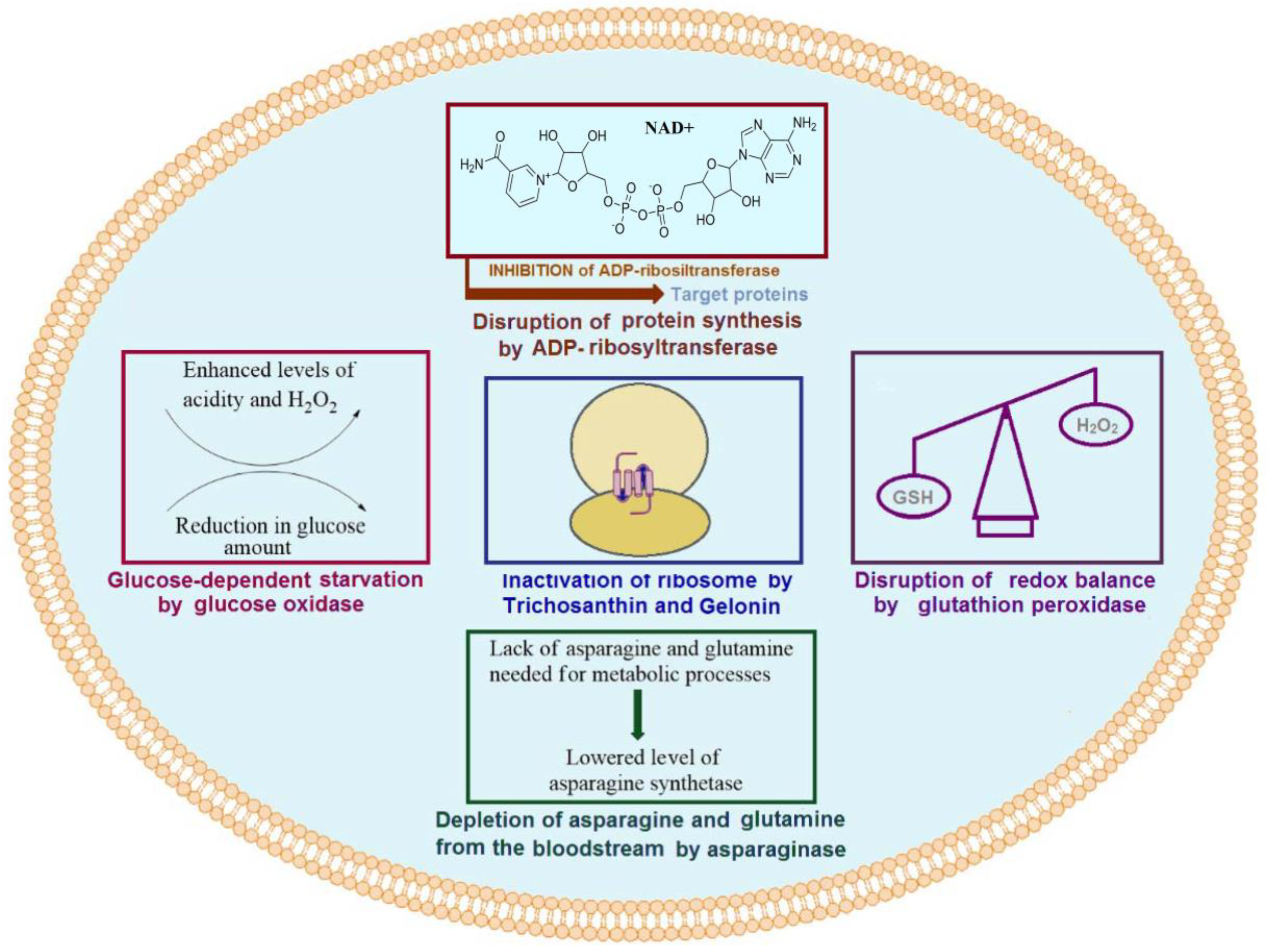
1.1. Ribosome-Inactivating Proteins
1.2. Oxidoreductases
1.3. Asparaginase
1.4. Bacterial Toxins
2. Trichosanthin

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Formulation | Results | IC50 Values | Refs. |
|---|---|---|---|
| Combination of TCS with Interleukin-2 | Combination with IL-2 resulted in synergistic effect on PC3 prostate cancer cells in vivo. | 50.6 µg/mL (PC3) | [27] |
| Protection of TCS-LMWP-MSP with PEG | The inclusion of MSP (MMP-2 substrate) between TCS-LMWP and PEG inhibited HT1080 tumor volume and weight twice more efficiently. | 0.13 µM (HT1080) | [41] |
| Protection of TCS-LMWP-MSP with PEG | No significant changes were observed in A549/T cells in vivo, but the combination with PTX in liposome totally inhibited the tumor volume. | 1.6 µM/mL (A549/T) | [42] |
| Fusion of TCS-CPP-MSP with Lactoferrin (LF) | The linkage of LF with TCS-CPP-MSP lowered the IC50 value and enhanced GL261 tumor inhibition significantly. | 0.37 µM (GL261) | [43] |
| Co-delivery of TCS and albendazole by self-assembly via BSA and Ag NPs | Number of metastatic nodules dramatically reduced. Tumor volume and weight in mice bearing multidrug-resistant A-549/T cell greatly inhibited, and thus concluded to prevent lung metastasis. | <0.1 µg/mL (A549/T and HCT8/ADR cells) | [44] |
| Development of recombinant ABD-PTN-TAT-TCS protein | Recombinant TCS capable of binding albumin greatly inhibited tumor in mice bearing 4T1 cells. | 1.7 µM (4T1) | [46] |
3. Gelonin
| Formulation | Results | IC50 Values | Refs. |
|---|---|---|---|
| ATTEMPTS rGel-TAT + T84.66-Hep + Protamine | Targeting by T84.66-Hep enhanced the drug tumor accumulation 43-fold compared to free recombinant form of Gel-TAT. | 29.2 nM in LS174T cells for Gel-TAT. | [51] |
| ATTEMPTS rGel-TAT + T84.66-Hep + Protamine | Developed ATTEMPTS system efficiently inhibited tumor volume in mice bearing LS174T cells. Targeting by T84.66 antibody and release by protamine and heparin resulted in two-fold efficiency in vivo. | 79 nM (CT26), 68 nM (LS174T), 61 nM (9L), 84 nM (PC3). | [52,53] |
| Combination of Gel-TAT with Hep + protamine | Similar IC50 values were observed with non cancer MDCK and 293 HEK cells. Gel-TAT and Gel-TAT/Hep + protamine treatment efficiently inhibited LS174T tumor volume with insignificant changes. | 72 nM (LS174T), 46 nM (U87 MG), 58 nM (9L), 66 nM (Hela). | [54] |
| Combined action of fusion of Gel-VEGF121 and PCI | Fusing with VEGF121 enhanced the Gel cytotoxicity in PAE/VEGFR-2 and PAE/VEGFR-1 cells more than 400- and 250-fold. | 44 pM (PAE/VEGFR-2), 26 pM, (PAE/VEGFR-1). | [57] |
| Construction of recombinant Gel fusion with EGFR targeting sequence | Despite great in vitro activity, no significant changes were observed in SCC-026 tumor volume in mice. The integration of PCI inhibited the tumor volume by ~45% compared to untreated control. | 60 nM (SCC-026), 3.7 nM (SCC-040), 19 nm (SCC-074). | [59] |
| Construction of recombinant Gel affibody with IGF-1R (Gel-IAFF) | Cell viability levels of U87 MG and U251 MG cells enhanced by more than 10-fold by the inclusion IGF-1R sequence. No significant changes were found 293T and LNCaP cells. | 0.18 nM (U87 MG), 0.14 nM (U251MG). | [64] |
| Fusing Gel with F3 peptides: F3-Gel, 2F3-Gel, and 3F3-Gel | 2F3-Gel and 3F3-Gel fusions inhibited LNCaP tumor volume twice more efficiently compared to free Gel. 2F3-Gel and 3F3-Gel revealed significantly lower IC50 values in HEK cells. | 63 nM (LNCaP), 99 nM (PC3), 73 nM (DU145) for 2F3-Gel. | [65] |
| Construction of recombinant Gel fusion F3 peptide | Fusing with F3 peptide lowered the IC50 value of Gel in HeLa LNCaP, 9L, and U87 MG cells at least 6-fold. The fusion inhibited U87 MG tumor volume 5-fold more efficiently than Gel. | 0.34 µM (HeLa), 0.41 µM (LNCaP), 0.39 (9L), 0.33 (U87 MG). | [66] |
| Construction of a recombinant Gel fusion with chlorotoxin (Gel-CLTX) | Obtained recombinant fusion increased Gel toxicity ~20-fold to U87 MG and 9L cells. No significant changes were observed in the toxicity level to non cancer 293 HEK and SVG p12 cells. | 180 nM in U87MG and 9L cells. | [67] |
| Fusion of Gel with BLyS that target BLyS receptors | Direct correlation was established between BAFF-R level and sensitivity to BLyS-Gel. No correlation was found with TACI protein. | 5–50 nm in ABC DLBCL cell lines | [69,71] |
| Fusion of Gel with B cell lymphocyte stimulator (BLyS) that target BLyS receptors | Generated BLyS-Gel fusion totally inhibited and reduced DLBCL xenograft tumor in mice. The fusion showed great targeting index in cells that overexpress BLyS receptors. | 7 pM (OCI-Ly10) 8 pM (OCI-Ly3) 0.1 nM (SUDHL-4) 5 nM (SUDHL-6) | [70,71] |
4. Peroxidase
5. Glucose Oxidase
| Formulation | Results | Study Method | Refs. |
|---|---|---|---|
| Fabrication of GOx-poly(FBMA-co-OEGMA) nanogels (NGs) | In a C8161 melanoma mouse model, NGs inhibited tumor growth 3.5-fold more effectively than the GOX (dose 100 mU) on 16 d post administration. NGs-treated mice exhibited 1.9-fold longer median survival times than GOX treated mice at the doses. | IC50 of GOx made 24.4 ng/mL in C8161 cells. | [96] |
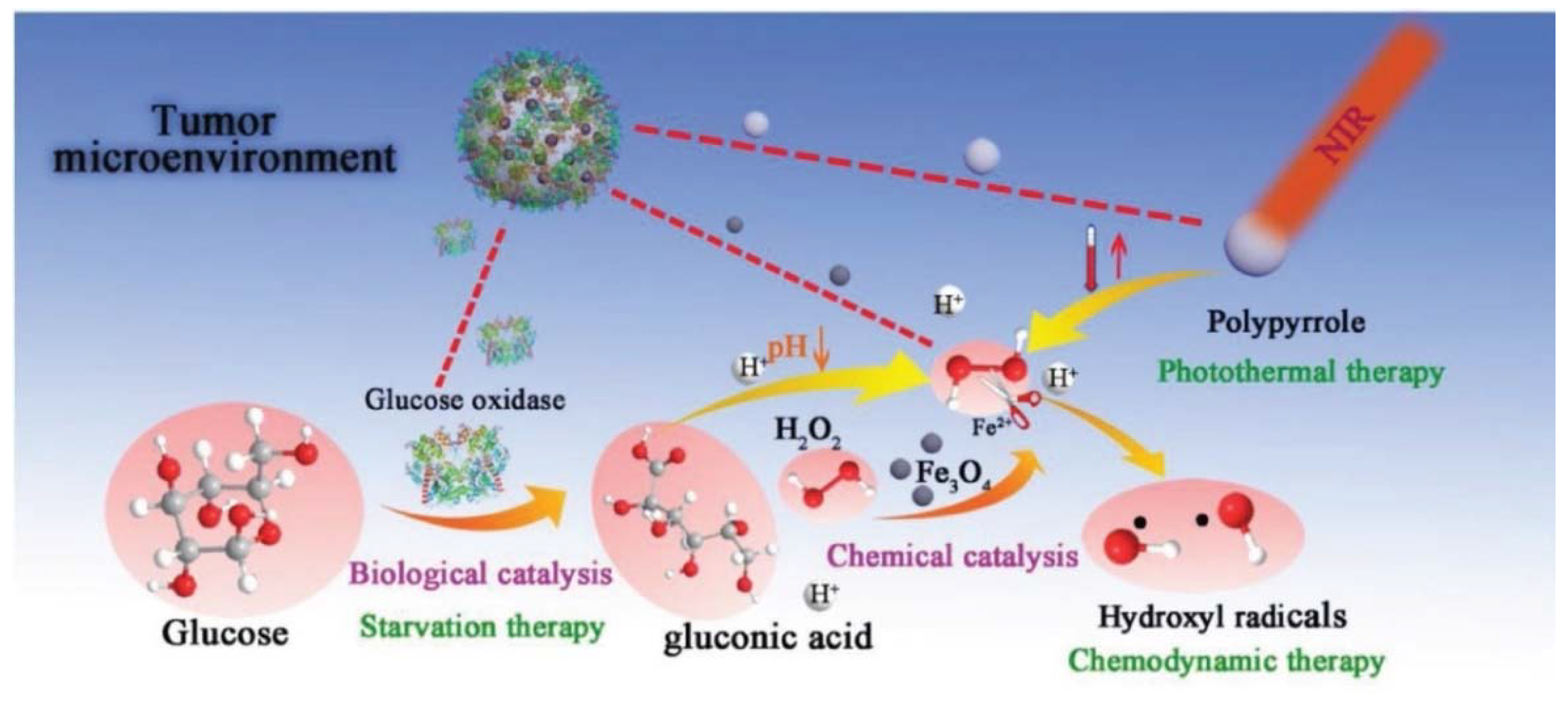
| Fabrication of Fe3O4@PPy@GOx nanocatalysts (NCs) | The NCs (163.5 nm) exhibited cytotoxic activity in 4T1, HeLa, HUVEC cells and 4T1 tumor-bearing mice. The GOX activity was improved by photothermal-enhanced sequential Fenton nanocatalytic effect. | 70 mg/kg dose of Fe3O4@PPy@GOx resulted in efficiency in vivo. | [100] |
| Fabrication of GOx@ZIF@MPN adenosine triphosphate (ATP)-responsive NPs | The NPs (180 nm) exhibited antitumor activity in 4T1 cells and 4T1 tumor-bearing mice. The GOX activity was improved by ATP-responsive autocatalysis and acceleration of the Fenton nanocatalysis. | 0.3 mg/kg GOx-contained NPs showed efficiency in 4T1 cells. | [101] |
| Fabrication of ZIF-8@GOx-AgNPs@MBN multifunctional nanoreactor (Nr) | The Nr (400 nm) showed high cytotoxic effect against HeLa cells and significant antitumor activity (96.8%, 200 μg/mL) in tumor-bearing mice. The Nr exhibited catalysis-enhanced synergistic starvation/metal ion poisoning cancer therapy. | 0.08 mg/mL of Nr showed 94% inhibitory effect in HeLa cells. | [102] |
| Fabrication of γ-PGA@GOx@Mn, Cu-CDs multifunctional NPs | The NPs (80 nm) exhibited in cytotoxic activity in 4T1 cells and tumor inhibition activity (~90–95%) in 4T1 tumor-bearing mice. The therapeutic action of GOX was improved by the PDT, PTT and checkpoint-blockade immunotherapy. | 100-150 μg/mL of NPs inhibited 4T1 cell by 82–91%. | [103] |
| Fabrication of PLL- and HA-modified GOx-loaded silica NPs | MSNs-GOx/PLL/HA NPs reduced the initial tumor volume in mice bearing HepG2 cells. Modification with PLL/HA caused significant reduction in tumor volume. | 40 µg/mL dose reduced cell viability to <20%. | [107] |
| Fabrication of complex the GOX, PLL-g-PEG, and anti-PSMA antibody | Modifying GOX with cationic copolymer and linking with anti-PSMA antibody efficiently inhibited PSMA-expressing prostate cancer cells. | 1 µg/mL dose reduced LNCaP cell viability to <40%. | [108] |
6. Asparaginase
| Formulation | Results | Size (nm) | EE and LC Level | IC 50 Value | Refs. |
|---|---|---|---|---|---|
| PTD-modified ATTEMPTS system for ASP | The inclusion of TAT peptide was not able to enhance the cytotoxic effect of ASP in vivo. | 0.0100 UI/mL (L5178Y) | [123] | ||
| Encapsulation in positive liposome | Enhanced level of physical stability and in vitro cytotoxicity. | 35.2 ± 4.5 | 2.39% (LC) | 50 µg/mL (EAC) | [125] |
| Formulation of chitosan modified lipid nanoparticles | Significantly (14%) lowered IC50 value on H446 lung carcinoma cells. | 426.60 ± 36.34 | 66.47 ± 2.81 (EE) | [126] | |
| Encapsulation in chitosan/TPP nanosystems | ASP with high LC became more resistant to high temperature and alkaline condition. | 340 ± 12 | 76.2% (EE) 47.6% (LC) | [127] | |
| β-cyclodextrin-chitosan-ASP nanobiocomposite | Obtained nanobiocomposite revealed a four-fold activity on PC3 over U937 cells. | 40–80 | 125 µg/mL (PC3) | [128] | |
| β-cyclodextrin-gelatin-ASP nanobiocomposite | Developed nanobiocomposite showed better activity on Hela than U87 cells. | 59–81.6 av. 74.1 | 62.5 µg/mL (Hela) | [129] | |
| Hollow NPs of Alg-g-PEG and cyclodextrin with ASP | Encapsulation significantly increased the enzyme stability in an acidic condition. | av. 467 | 37–80% (EE) | [130] | |
| Poly (lactic-co-glycolic) acid nanoformulation | Encapsulation in PLGA significantly inhibited EAT-tumor in mice. | 195 ± 0.2 | 80.23 (EE) 10% (LC) | [131] | |
| Encapsulation in liposome containing DSPE-PEG-200 | Encapsulation in liposomes significantly reduced LLC-tumor volume in mice. | 93.03 ± 0.49 | 53.99 (EE) | 0.23 UI/mL (LLC) | [132] |
| Immobilization of ASP-RGD on to Au NPs via PEG | RGD peptide-targeting enhanced anticancer efficacy on MCF-7 cancer cells. | 20–50 av. 29.24 | 89.8 µg/mL (MCF-7) | [137] | |
| Immobilization of ASP on to Au NPs | Obtained nanobiocomposite showed high toxicity to A549 and A2780 cancer cells. | 20–50 | 62.5 µg/mL (A549) | [138] | |
| Cerium-selenium nanobio-composite with ASP | Cerium-selenium nanobiocomposite caused synergistic effect with ASP. | 60–90 | 125 µg/mL (A549) | [139] | |
| Immobilization to magnetic NPs of SiO2, Fe3O4, PVDMA | Longer polymer chain was concluded to be more favourable for enzymatic reaction. | app. 20–30 | 32% (LC) | [140] |
7. Exotoxin
| Formulation | Results | Dose Actions | Refs. |
|---|---|---|---|
| Effect of PE exotoxin fused with ovalbumin on mice bearing EG7 cells | Tumor growth was greatly inhibited in ovalbumin expressing cells in vivo. No changes were determined in the tumor volume in non-ovalbumin expressing cells in vivo. | 100 µg dose led to ~20% lysis of EG7 cells. | [149] |
| Loading of PEIII in chitosan microparticles cross-linked TPP with ~1.09 µm size | Extension of cross-linking time reduced the drug release. The drug release was found to enhance with the increase in sonication power > 45 W. | 60–80% toxin was found released for the first few hours. | [150] |
| Phase I study of anti-mesothelin dsFv-PE38 (SS1P) in mesothelin-expressing cancers | Dose-limiting toxicity was linked normal pleural mesothelial cells that express mesothelin. No pericardial toxicity was observed despite mesothelin expression on normal pericardial cells. | Maximum tolerating dose made 45 µg/kg every other day | [158] |
| Fusion of PE24 with humanized SS1 Fab fragment (RG7787) | A three-cycle treatment with RG7787 led a reduction in initial 500 mm3 tumor volume by more than half for 110 days in NCI-H596 tumor model. | 2–3 mg/kg dose of RG7787 was more efficacy than SS1. | [159] |
| Phase II trial of recombinant RFB4(dsFv)-PE38 (BL22) in chemoresistant HCL | Single cycle of BL22 was found highly effective and without serious toxicity. Selective retreatment enhanced complete remission rate to 64% with no dose-limiting requirement. | 40 µg/kg dose (every other day) caused 25% complete remission rate | [160] |
| Phase I trial of PE 40 (BR96 sFv-PE40) with advanced solid tumor in patients | Rapid drug clearance made 11 days in many patients in association with Human Antitoxin Antibody. Partial tumor responses were observed for eight weeks. | 0.641 mg/m2 with gastrointestinal dose-limiting toxicity. | [167] |
| Phase I trial of Il-4-fused PE (NBI-3001) in tumors expressing IL-4 receptor | Hepatotoxicity was the main side-effect that prevented escalating dose of NBE 3001. Fatigue, headache, arthralgia were reported as main adverse events. The toxin was detected in less than 5% of patients. | 8–16 mg/m2 daily dose 5 times every 28 days caused no dose-limiting toxicity | [170] |
| Phase I trial of recombinant erb-38, containing Fv portion of e23 monoclonal antibody | Observed hepatotoxic effects were suggested linked with the expression of erbB2 on hepatocytes. Targeting with erbB2 was concluded to likely cause side effects in liver. | 1.0 and 2.0 µg/kg doses caused hepatotoxicity in patients. | [171] |
| Phase study of ScFv(FRP5)- ETA against erbB2-overexpressing tumor cells | Local therapy of ScFv(FRP5)- ETA was concluded to be effective against erbB2-expressing tumor. Retreatment was suggested to be effective as antibodies recover in patients. | 0.6–6mg dose caused shrink in 60% cases of the study. | [172] |
| Phase I study of scFv(FRP5)-ETA in erbB2-overexpressing metastatic cancers | Results indicated maximum tolerated dose 12.5 µg/kg can be administered to erbB2-overexpressing tumors. The main side-effect was hepatotoxicity due to erbB2 expression in hepatocytes. | 10 µg/kg dose was suggested no to induce side–effects. | [173] |
8. Diphtheria Toxin
| Formulation | Results | Dose Actions | Refs. |
|---|---|---|---|
| DT fusion with IL-2 (DAB389-IL2) that target IL-2-overexpressing hypoxic HCC cells | The combination DAB389-IL-2 and retinoic acid caused the suppression in hypoxic HCC compared to treatments with either DITOX-IL-2 or retinoic acid. | 5 µM (SNU-475) 20 µM (HepG2) caused significant changes | [186] |
| Construction of V6A derived from DAB-IL-2 with single amino acid substitution (sDAB-IL-2) | Combined action of sDAB-IL-2 with anti-PD-1 antibodies inhibited tumor growth ~5-fold more efficiently compared to sDAB-IL-2 treatment in B16F10 melanoma model. | 5 µg dose on day 7 and 10 significantly reduced B16F10 tumor volume | [189] |
| Plasmid of DT with H19 regulatory sequences that target ovarian cancer ascites fluid | DTA-H19 plasmid more significantly inhibited tumor volume in mice bearing ES-2 cells compared to luciferase-H19 plasmid complexed with PEI. | 25 µg dose was administered four times with two-day intervals | [195] |
| Construction of DITOX-H6 fusion with T22 that target CXCR4-overexpressing cells | T-22-DITOX-H6 reduced tumor size of CXCR4 expressing HeLa cells ~6-fold compared to control. T22 showed efficacy wih DITOX-H6 and PE-24 toxins. | By 10 µg (3 times a week) 8 doses in tumor model | [199] |
| Fusion of DITOX-H6 with T22 that target CXCR-4-overexpressing cells (T22-DITOX-H6) | Self-assembling T22-DITOX-H6 NPs efficiently targeted AML cell lines that overexpress CXCR-4 and thus revealed antineoplastic effect. | 10 µg dose (10 times) potently blocked AML in bone marrow | [200] |
| Construction of DT with VEGF That target vascular endothelial growth factor | The treatment with DT-VEGF significantly inhibited the tumor volume in mice bearing HPAF-2 (four-fold) and AsPC-1 (two-fold) in mice. | 100 µg/kg dose (every other day) after 3 days of tumor implantation | [201] |
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Correction Statement
References
- Gilabert-Oriol, R.; Weng, A.; Mallinckrodt, B.; Melzig, M.F.; Fuchs, H.; Thakur, M. Immunotoxins constructed with ribosome-inactivating proteins and their enhancers: A lethal cocktail with tumor specific efficacy. Curr. Pharm. Des. 2014, 20, 6584–6643. [Google Scholar] [CrossRef] [PubMed]
- Asrorov, A.M.; Gu, Z.; Min, K.A.; Shin, M.C.; Huang, Y. Advances on Tumor-Targeting Delivery of Cytotoxic Proteins. ACS Pharmacol. Transl. Sci. 2020, 3, 107–118. [Google Scholar] [CrossRef] [PubMed]
- Zeng, M.; Zheng, M.; Lu, D.; Wang, J.; Jiang, W.; Sha, O. Anti-tumor activities and apoptotic mechanism of ribosome-inactivating proteins. Chin. J. Cancer 2015, 34, 325–334. [Google Scholar] [CrossRef]
- Rust, A.; Partridge, L.J.; Davletov, B.; Hautbergue, G.M. The Use of Plant-Derived Ribosome Inactivating Proteins in Immunotoxin Development: Past, Present and Future Generations. Toxins 2017, 9, 344. [Google Scholar] [CrossRef] [PubMed]
- Tafazoli, S.; O’Brien, P.J. Peroxidases: A role in the metabolism and side effects of drugs. Drug Discov. Today 2005, 10, 617–625. [Google Scholar] [CrossRef]
- Wang, C.; Yang, J.; Dong, C.; Shi, S. Glucose Oxidase-Related Cancer Therapies. Adv. Ther. 2020, 3, 2000110. [Google Scholar] [CrossRef]
- Symons, M.C.; Rusakiewicz, S.; Rees, R.C.; Ahmad, S.I. Hydrogen peroxide: A potent cytotoxic agent effective in causing cellular damage and used in the possible treatment for certain tumours. Med. Hypotheses 2001, 57, 56–58. [Google Scholar] [CrossRef]
- Mycielska, M.E.; Mohr, M.T.J.; Schmidt, K.; Drexler, K.; Rümmele, P.; Haferkamp, S.; Schlitt, H.J.; Gaumann, A.; Adamski, J.; Geissler, E.K. Potential Use of Gluconate in Cancer Therapy. Front. Oncol. 2019, 9, 522. [Google Scholar] [CrossRef]
- Egler, R.A.; Ahuja, S.P.; Matloub, Y. L-asparaginase in the treatment of patients with acute lymphoblastic leukemia. J. Pharmacol. Pharmacother. 2016, 7, 62–71. [Google Scholar] [CrossRef]
- Shrivastava, A.; Khan, A.A.; Khurshid, M.; Kalam, M.A.; Jain, S.K.; Singhal, P.K. Recent developments in L-asparaginase discovery and its potential as anticancer agent. Crit. Rev. Oncol./Hematol. 2016, 100, 1–10. [Google Scholar] [CrossRef]
- Fu, C.H.; Sakamoto, K.M. PEG-asparaginase. Expert Opin. Pharmacother. 2007, 8, 1977–1984. [Google Scholar] [CrossRef] [PubMed]
- Krueger, K.M.; Barbieri, J.T. The family of bacterial ADP-ribosylating exotoxins. Clin. Microbiol. Rev. 1995, 8, 34–47. [Google Scholar] [CrossRef] [PubMed]
- Wolf, P.; Elsässer-Beile, U. Pseudomonas exotoxin A: From virulence factor to anti-cancer agent. Int. J. Med. Microbiol. IJMM 2009, 299, 161–176. [Google Scholar] [CrossRef]
- Mateyak, M.K.; Kinzy, T.G. ADP-ribosylation of translation elongation factor 2 by diphtheria toxin in yeast inhibits translation and cell separation. J. Biol. Chem. 2013, 288, 24647–24655. [Google Scholar] [CrossRef]
- Wen, D.; Wang, J.; Yan, H.; Chen, J.; Xia, K.; Liu, J.; Zhang, A. Effect of Radix Trichosanthis and Trichosanthin on Hepatitis B Virus in HepG2.2.15 Cells. J. Nanosci. Nanotechnol. 2015, 15, 2094–2098. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Chan, H.; Wang, Y.Y.; Ouyang, D.Y.; Zheng, Y.T.; Tam, S.C. Trichosanthin suppresses the elevation of p38 MAPK, and Bcl-2 induced by HSV-1 infection in Vero cells. Life Sci. 2006, 79, 1287–1292. [Google Scholar] [CrossRef]
- Zhao, W.L.; Zhang, F.; Feng, D.; Wu, J.; Chen, S.; Sui, S.F. A novel sorting strategy of trichosanthin for hijacking human immunodeficiency virus type 1. Biochem. Biophys. Res. Commun. 2009, 384, 347–351. [Google Scholar] [CrossRef]
- Zhao, W.; Feng, D.; Sun, S.; Han, T.; Sui, S. The anti-viral protein of trichosanthin penetrates into human immunodeficiency virus type 1. Acta Biochim. Biophys. Sin. 2010, 42, 91–97. [Google Scholar] [CrossRef]
- Zhao, J.; Ben, L.H.; Wu, Y.L.; Hu, W.; Ling, K.; Xin, S.M.; Nie, H.L.; Ma, L.; Pei, G. Anti-HIV agent trichosanthin enhances the capabilities of chemokines to stimulate chemotaxis and G protein activation, and this is mediated through interaction of trichosanthin and chemokine receptors. J. Exp. Med. 1999, 190, 101–111. [Google Scholar] [CrossRef]
- Xu, J.; Gao, D.F.; Yan, G.L.; Fan, J.M. Induced apoptotic action of recombinant trichosanthin in human stomach adenocarcinoma MCG803 cells. Mol. Biol. Rep. 2009, 36, 1559–1564. [Google Scholar] [CrossRef]
- Fridman, J.S.; Lowe, S.W. Control of apoptosis by p53. Oncogene 2003, 22, 9030–9040. [Google Scholar] [CrossRef] [PubMed]
- Al Bitar, S.; Gali-Muhtasib, H. The Role of the Cyclin Dependent Kinase Inhibitor p21(cip1/waf1) in Targeting Cancer: Molecular Mechanisms and Novel Therapeutics. Cancers 2019, 11, 1475. [Google Scholar] [CrossRef] [PubMed]
- Kang, M.; Ou, H.; Wang, R.; Liu, W.; Mao, Y.; Tang, A. Effect of trichosanthin on apoptosis and telomerase activity of nasopharyngeal carcinomas in nude mice. J. BUON 2013, 18, 675–682. [Google Scholar] [PubMed]
- Li, J.; Xia, X.; Ke, Y.; Nie, H.; Smith, M.A.; Zhu, X. Trichosanthin induced apoptosis in HL-60 cells via mitochondrial and endoplasmic reticulum stress signaling pathways. Biochim. Biophys. Acta 2007, 1770, 1169–1180. [Google Scholar] [CrossRef]
- Cui, L.; Song, J.; Wu, L.; Huang, L.; Wang, Y.; Huang, Y.; Yu, H.; Huang, Y.; You, C.C.; Ye, J. Smac is another pathway in the anti-tumour activity of Trichosanthin and reverses Trichosanthin resistance in CaSki cervical cancer cells. Biomed. Pharmacother. 2015, 69, 119–124. [Google Scholar] [CrossRef]
- Huang, Y.; Song, H.; Hu, H.; Cui, L.; You, C.; Huang, L. Trichosanthin inhibits DNA methyltransferase and restores methylation-silenced gene expression in human cervical cancer cells. Mol. Med. Rep. 2012, 6, 872–878. [Google Scholar] [CrossRef]
- Li, J.; Li, H.; Zhang, Z.; Wang, N.; Zhang, Y. The anti-cancerous activity of recombinant trichosanthin on prostate cancer cell PC3. Biol. Res. 2016, 49, 21. [Google Scholar] [CrossRef]
- Kadam, C.Y.; Abhang, S.A. Apoptosis Markers in Breast Cancer Therapy. Adv. Clin. Chem. 2016, 74, 143–193. [Google Scholar]
- Shi, W.W.; Wong, K.B.; Shaw, P.C. Structural and Functional Investigation and Pharmacological Mechanism of Trichosanthin, a Type 1 Ribosome-Inactivating Protein. Toxins 2018, 10, 335. [Google Scholar] [CrossRef]
- Chan, W.Y.; Huang, H.; Tam, S.C. Receptor-mediated endocytosis of trichosanthin in choriocarcinoma cells. Toxicology 2003, 186, 191–203. [Google Scholar] [CrossRef]
- Jiao, Y.; Liu, W. Low-density lipoprotein receptor-related protein 1 is an essential receptor for trichosanthin in 2 choriocarcinoma cell lines. Biochem. Biophys. Res. Commun. 2010, 391, 1579–1584. [Google Scholar] [CrossRef] [PubMed]
- Miao, J.; Jiang, Y.; Wang, D.; Zhou, J.; Fan, C.; Jiao, F.; Liu, B.; Zhang, J.; Wang, Y.; Zhang, Q. Trichosanthin suppresses the proliferation of glioma cells by inhibiting LGR5 expression and the Wnt/beta-catenin signaling pathway. Oncol. Rep. 2015, 34, 2845–2852. [Google Scholar] [CrossRef] [PubMed]
- Wend, P.; Holland, J.D.; Ziebold, U.; Birchmeier, W. Wnt signaling in stem and cancer stem cells. Semin. Cell Dev. Biol. 2010, 21, 855–863. [Google Scholar] [CrossRef] [PubMed]
- Cai, Y.; Xiong, S.; Zheng, Y.; Luo, F.; Jiang, P.; Chu, Y. Trichosanthin enhances anti-tumor immune response in a murine Lewis lung cancer model by boosting the interaction between TSLC1 and CRTAM. Cell Mol. Immunol. 2011, 8, 359–367. [Google Scholar] [CrossRef]
- Wang, B.L.; Su, H.; Chen, Y.; Wang, J.; Xu, G.L. A role for trichosanthin in the expansion of CD4CD25 regulatory T cells. Scand. J. Immunol. 2010, 71, 258–266. [Google Scholar] [CrossRef]
- Jiang, Q.; Bai, T.; Shen, S.; Li, L.; Ding, H.; Wang, P. Increase of cytosolic calcium induced by trichosanthin suppresses cAMP/PKC levels through the inhibition of adenylyl cyclase activity in HeLa cells. Mol. Biol. Rep. 2011, 38, 2863–2868. [Google Scholar] [CrossRef]
- Wang, P.; Chen, L.L.; Yan, H.; Li, J.C. Trichosanthin suppresses HeLa cell proliferation through inhibition of the PKC/MAPK signaling pathway. Cell Biol. Toxicol. 2009, 25, 479–488. [Google Scholar] [CrossRef]
- Fang, E.F.; Zhang, C.Z.; Zhang, L.; Wong, J.H.; Chan, Y.S.; Pan, W.L.; Dan, X.L.; Yin, C.M.; Cho, C.H.; Ng, T.B. Trichosanthin inhibits breast cancer cell proliferation in both cell lines and nude mice by promotion of apoptosis. PLoS ONE 2012, 7, e41592. [Google Scholar] [CrossRef]
- Sha, O.; Niu, J.; Ng, T.B.; Cho, E.Y.; Fu, X.; Jiang, W. Anti-tumor action of trichosanthin, a type 1 ribosome-inactivating protein, employed in traditional Chinese medicine: A mini review. Cancer Chemother. Pharmacol. 2013, 71, 1387–1393. [Google Scholar] [CrossRef]
- Huang, Y.; Jiang, Y.; Wang, H.; Wang, J.; Shin, M.C.; Byun, Y.; He, H.; Liang, Y.; Yang, V.C. Curb challenges of the “Trojan Horse” approach: Smart strategies in achieving effective yet safe cell-penetrating peptide-based drug delivery. Adv. Drug Deliv. Rev. 2013, 65, 1299–1315. [Google Scholar] [CrossRef]
- Chen, Y.; Zhang, M.; Jin, H.; Tang, Y.; Wang, H.; Xu, Q.; Li, Y.; Li, F.; Huang, Y. Intein-mediated site-specific synthesis of tumor-targeting protein delivery system: Turning PEG dilemma into prodrug-like feature. Biomaterials 2017, 116, 57–68. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Zhang, M.; Jin, H.; Tang, Y.; Wu, A.; Xu, Q.; Huang, Y. Prodrug-Like, PEGylated Protein Toxin Trichosanthin for Reversal of Chemoresistance. Mol. Pharm. 2017, 14, 1429–1438. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Zhang, M.; Jin, H.; Li, D.; Xu, F.; Wu, A.; Wang, J.; Huang, Y. Glioma Dual-Targeting Nanohybrid Protein Toxin Constructed by Intein-Mediated Site-Specific Ligation for Multistage Booster Delivery. Theranostics 2017, 7, 3489–3503. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Liang, J.; Wu, A.; Chen, Y.; Zhao, P.; Lin, T.; Zhang, M.; Xu, Q.; Wang, J.; Huang, Y. Co-Delivery of Trichosanthin and Albendazole by Nano-Self-Assembly for Overcoming Tumor Multidrug-Resistance and Metastasis. ACS Appl. Mater. Interfaces 2017, 9, 26648–26664. [Google Scholar] [CrossRef] [PubMed]
- Lin, T.; Zhao, P.; Jiang, Y.; Tang, Y.; Jin, H.; Pan, Z.; He, H.; Yang, V.C.; Huang, Y. Blood-Brain-Barrier-Penetrating Albumin Nanoparticles for Biomimetic Drug Delivery via Albumin-Binding Protein Pathways for Antiglioma Therapy. ACS Nano 2016, 10, 9999–10012. [Google Scholar] [CrossRef]
- Chang, Y.; Yao, S.; Chen, Y.; Huang, J.; Wu, A.; Zhang, M.; Xu, F.; Li, F.; Huang, Y. Genetically-engineered protein prodrug-like nanoconjugates for tumor-targeting biomimetic delivery via a SHEATH strategy. Nanoscale 2019, 11, 611–621. [Google Scholar] [CrossRef]
- Rosenblum, M.G.; Kohr, W.A.; Beattie, K.L.; Beattie, W.G.; Marks, W.; Toman, P.D.; Cheung, L. Amino acid sequence analysis, gene construction, cloning, and expression of gelonin, a toxin derived from Gelonium multiflorum. J. Interferon Cytokine Res. 1995, 15, 547–555. [Google Scholar] [CrossRef]
- Lyu, M.A.; Cao, Y.J.; Mohamedali, K.A.; Rosenblum, M.G. Cell-targeting fusion constructs containing recombinant gelonin. Methods Enzymol. 2012, 502, 167–214. [Google Scholar]
- Stirpe, F.; Olsnes, S.; Pihl, A. Gelonin, a new inhibitor of protein synthesis, nontoxic to intact cells. Isolation, characterization, and preparation of cytotoxic complexes with concanavalin A. J. Biol. Chem. 1980, 255, 6947–6953. [Google Scholar] [CrossRef]
- Nolan, P.A.; Garrison, D.A.; Better, M. Cloning and expression of a gene encoding gelonin, a ribosome-inactivating protein from Gelonium multiflorum. Gene 1993, 134, 223–227. [Google Scholar] [CrossRef]
- Shin, M.C.; Zhang, J.; Min, K.A.; He, H.; David, A.E.; Huang, Y.; Yang, V.C. PTD-Modified ATTEMPTS for Enhanced Toxin-based Cancer Therapy: An In Vivo Proof-of-Concept Study. Pharm. Res. 2015, 32, 2690–2703. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Zhang, M.; Min, K.A.; Wang, H.; Shin, M.C.; Li, F.; Yang, V.C.; Huang, Y. Improved Protein Toxin Delivery Based on ATTEMPTS Systems. Curr. Drug Targets 2018, 19, 380–392. [Google Scholar] [CrossRef]
- Wang, H.; Moon, C.; Shin, M.C.; Wang, Y.; He, H.; Yang, V.C.; Huang, Y. Heparin-Regulated Prodrug-Type Macromolecular Theranostic Systems for Cancer Therapy. Nanotheranostics 2017, 1, 114–130. [Google Scholar] [CrossRef] [PubMed]
- Shin, M.C.; Zhao, J.; Zhang, J.; Huang, Y.; He, H.; Wang, M.; Min, K.A.; Yang, V.C. Recombinant TAT-gelonin fusion toxin: Synthesis and characterization of heparin/protamine-regulated cell transduction. J. Biomed. Mater. Res. A 2015, 103, 409–419. [Google Scholar] [CrossRef] [PubMed]
- Shin, M.C.; Zhang, J.; Min, K.A.; Lee, K.; Byun, Y.; David, A.E.; He, H.; Yang, V.C. Cell-penetrating peptides: Achievements and challenges in application for cancer treatment. J. Biomed. Mater. Res. A 2014, 102, 575–587. [Google Scholar] [CrossRef]
- Veenendaal, L.M.; Jin, H.; Ran, S.; Cheung, L.; Navone, N.; Marks, J.W.; Waltenberger, J.; Thorpe, P.; Rosenblum, M.G. In vitro and in vivo studies of a VEGF121/rGelonin chimeric fusion toxin targeting the neovasculature of solid tumors. Proc. Natl. Acad. Sci. USA 2002, 99, 7866–7871. [Google Scholar] [CrossRef]
- Weyergang, A.; Fremstedal, A.S.; Skarpen, E.; Peng, Q.; Mohamedali, K.A.; Eng, M.S.; Cheung, L.H.; Rosenblum, M.G.; Waltenberger, J.; Berg, K. Light-enhanced VEGF121/rGel: A tumor targeted modality with vascular and immune-mediated efficacy. J. Control. Release 2018, 288, 161–172. [Google Scholar] [CrossRef]
- Shin, M.C.; Min, K.A.; Cheong, H.; Moon, C.; Huang, Y.; He, H.; Yang, V.C. Preparation and Characterization of Gelonin-Melittin Fusion Biotoxin for Synergistically Enhanced Anti-Tumor Activity. Pharm. Res. 2016, 33, 2218–2228. [Google Scholar] [CrossRef]
- Berstad, M.B.; Cheung, L.H.; Berg, K.; Peng, Q.; Fremstedal, A.S.; Patzke, S.; Rosenblum, M.G.; Weyergang, A. Design of an EGFR-targeting toxin for photochemical delivery: In vitro and in vivo selectivity and efficacy. Oncogene 2015, 34, 5582–5592. [Google Scholar] [CrossRef]
- Amin, D.N.; Hida, K.; Bielenberg, D.R.; Klagsbrun, M. Tumor endothelial cells express epidermal growth factor receptor (EGFR) but not ErbB3 and are responsive to EGF and to EGFR kinase inhibitors. Cancer Res. 2006, 66, 2173–2180. [Google Scholar] [CrossRef]
- Bull-Hansen, B.; Berstad, M.B.; Berg, K.; Cao, Y.; Skarpen, E.; Fremstedal, A.S.; Rosenblum, M.G.; Peng, Q.; Weyergang, A. Photochemical activation of MH3-B1/rGel: A HER2-targeted treatment approach for ovarian cancer. Oncotarget 2015, 6, 12436–12451. [Google Scholar] [CrossRef] [PubMed]
- Berg, K.; Selbo, P.K.; Prasmickaite, L.; Tjelle, T.E.; Sandvig, K.; Moan, J.; Gaudernack, G.; Fodstad, O.; Kjolsrud, S.; Anholt, H.; et al. Photochemical internalization: A novel technology for delivery of macromolecules into cytosol. Cancer Res. 1999, 59, 1180–1183. [Google Scholar] [PubMed]
- Martinez de Pinillos Bayona, A.; Moore, C.M.; Loizidou, M.; MacRobert, A.J.; Woodhams, J.H. Enhancing the efficacy of cytotoxic agents for cancer therapy using photochemical internalisation. Int. J. Cancer 2016, 138, 1049–1057. [Google Scholar] [CrossRef] [PubMed]
- Ham, S.; Min, K.A.; Yang, J.W.; Shin, M.C. Fusion of gelonin and anti-insulin-like growth factor-1 receptor (IGF-1R) affibody for enhanced brain cancer therapy. Arch. Pharm. Res. 2017, 40, 1094–1104. [Google Scholar] [CrossRef]
- Shin, M.C.; Min, K.A.; Cheong, H.; Moon, C.; Huang, Y.; He, H.; Yang, V.C. Tandem-multimeric F3-gelonin fusion toxins for enhanced anti-cancer activity for prostate cancer treatment. Int. J. Pharm. 2017, 524, 101–110. [Google Scholar] [CrossRef]
- Ham, S.-H.; Min, K.A.; Shin, M.C. Molecular tumor targeting of gelonin by fusion with F3 peptide. Acta Pharmacol. Sin. 2017, 38, 897–906. [Google Scholar] [CrossRef][Green Version]
- Park, T.; Min, K.A.; Cheong, H.; Moon, C.; Shin, M.C. Genetic engineering and characterisation of chlorotoxin-fused gelonin for enhanced glioblastoma therapy. J. Drug Target. 2019, 27, 950–958. [Google Scholar] [CrossRef]
- Nimmanapalli, R.; Lyu, M.A.; Du, M.; Keating, M.J.; Rosenblum, M.G.; Gandhi, V. The growth factor fusion construct containing B-lymphocyte stimulator (BLyS) and the toxin rGel induces apoptosis specifically in BAFF-R-positive CLL cells. Blood 2007, 109, 2557–2564. [Google Scholar] [CrossRef]
- Lyu, M.A.; Sung, B.; Cheung, L.H.; Marks, J.W.; Aggarwal, B.B.; Aguiar, R.C.; Rosenblum, M.G. The rGel/BLyS fusion toxin inhibits STAT3 signaling via down-regulation of interleukin-6 receptor in diffuse large B-cell lymphoma. Biochem. Pharmacol. 2010, 80, 1335–1342. [Google Scholar] [CrossRef]
- Lyu, M.A.; Rai, D.; Ahn, K.S.; Sung, B.; Cheung, L.H.; Marks, J.W.; Aggarwal, B.B.; Aguiar, R.C.; Gandhi, V.; Rosenblum, M.G. The rGel/BLyS fusion toxin inhibits diffuse large B-cell lymphoma growth in vitro and in vivo. Neoplasia 2010, 12, 366–375. [Google Scholar] [CrossRef]
- Lyu, M.A.; Cheung, L.H.; Hittelman, W.N.; Marks, J.W.; Aguiar, R.C.; Rosenblum, M.G. The rGel/BLyS fusion toxin specifically targets malignant B cells expressing the BLyS receptors BAFF-R, TACI, and BCMA. Mol. Cancer Ther. 2007, 6, 460–470. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Marks, J.D.; Marks, J.W.; Cheung, L.H.; Kim, S.; Rosenblum, M.G. Construction and characterization of novel, recombinant immunotoxins targeting the Her2/neu oncogene product: In vitro and in vivo studies. Cancer Res. 2009, 69, 8987–8995. [Google Scholar] [CrossRef] [PubMed]
- Provoda, C.J.; Stier, E.M.; Lee, K.D. Tumor cell killing enabled by listeriolysin O-liposome-mediated delivery of the protein toxin gelonin. J. Biol. Chem. 2003, 278, 35102–35108. [Google Scholar] [CrossRef]
- Shin, M.C.; Zhang, J.; David, A.E.; Trommer, W.E.; Kwon, Y.M.; Min, K.A.; Kim, J.H.; Yang, V.C. Chemically and biologically synthesized CPP-modified gelonin for enhanced anti-tumor activity. J. Control. Release 2013, 172, 169–178. [Google Scholar] [CrossRef]
- Spadiut, O.; Herwig, C. Production and purification of the multifunctional enzyme horseradish peroxidase. Pharm. Bioprocess. 2013, 1, 283–295. [Google Scholar] [CrossRef] [PubMed]
- Folkes, L.K.; Wardman, P. Oxidative activation of indole-3-acetic acids to cytotoxic species- a potential new role for plant auxins in cancer therapy. Biochem. Pharmacol. 2001, 61, 129–136. [Google Scholar] [CrossRef]
- Kim, D.S.; Jeon, S.E.; Jeong, Y.M.; Kim, S.Y.; Kwon, S.B.; Park, K.C. Hydrogen peroxide is a mediator of indole-3-acetic acid/horseradish peroxidase-induced apoptosis. FEBS Lett. 2006, 580, 1439–1446. [Google Scholar] [CrossRef]
- Doskey, C.M.; Buranasudja, V.; Wagner, B.A.; Wilkes, J.G.; Du, J.; Cullen, J.J.; Buettner, G.R. Tumor cells have decreased ability to metabolize H2O2: Implications for pharmacological ascorbate in cancer therapy. Redox Biol. 2016, 10, 274–284. [Google Scholar] [CrossRef]
- Jiao, Y.; Wang, Y.; Guo, S.; Wang, G. Glutathione peroxidases as oncotargets. Oncotarget 2017, 8, 80093–80102. [Google Scholar] [CrossRef]
- Diehn, M.; Cho, R.W.; Lobo, N.A.; Kalisky, T.; Dorie, M.J.; Kulp, A.N.; Qian, D.; Lam, J.S.; Ailles, L.E.; Wong, M.; et al. Association of reactive oxygen species levels and radioresistance in cancer stem cells. Nature 2009, 458, 780–783. [Google Scholar] [CrossRef]
- Ye, X.Q.; Li, Q.; Wang, G.H.; Sun, F.F.; Huang, G.J.; Bian, X.W.; Yu, S.C.; Qian, G.S. Mitochondrial and energy metabolism-related properties as novel indicators of lung cancer stem cells. Int. J. Cancer 2011, 129, 820–831. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, L.; Sandhu, J.K.; Harper, M.E.; Cuperlovic-Culf, M. Role of Glutathione in Cancer: From Mechanisms to Therapies. Biomolecules 2020, 10, 1429. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.; Choi, A.; Jun, Y.; Kim, N.; Yook, J.I.; Kim, S.Y.; Lee, S.; Kang, S.W. Glutathione peroxidase-1 regulates adhesion and metastasis of triple-negative breast cancer cells via FAK signaling. Redox Biol. 2020, 29, 101391. [Google Scholar] [CrossRef] [PubMed]
- Greco, O.; Folkes, L.K.; Wardman, P.; Tozer, G.M.; Dachs, G.U. Development of a novel enzyme/prodrug combination for gene therapy of cancer: Horseradish peroxidase/indole-3-acetic acid. Cancer Gene Ther. 2000, 7, 1414–1420. [Google Scholar] [CrossRef]
- Bonifert, G.; Folkes, L.; Gmeiner, C.; Dachs, G.; Spadiut, O. Recombinant horseradish peroxidase variants for targeted cancer treatment. Cancer Med. 2016, 5, 1194–1203. [Google Scholar] [CrossRef]
- Melo, M.N.; Pereira, F.M.; Rocha, M.A.; Ribeiro, J.G.; Diz, F.M.; Monteiro, W.F.; Ligabue, R.A.; Severino, P.; Fricks, A.T. Immobilization and characterization of horseradish peroxidase into chitosan and chitosan/PEG nanoparticles: A comparative study. Process Biochem. 2020, 98, 160–171. [Google Scholar] [CrossRef]
- Sotokawa, S.; Kitamura, T.; Takahashi, D.; Toshima, K. An anthraquinone-enzyme-peptide hybrid as a photo-switchable enzyme. Chem. Commun. 2018, 54, 10614–10617. [Google Scholar] [CrossRef] [PubMed]
- Bankar, S.B.; Bule, M.V.; Singhal, R.S.; Ananthanarayan, L. Glucose oxidase—An overview. Biotechnol Adv. 2009, 27, 489–501. [Google Scholar] [CrossRef]
- Libertino, S.; Aiello, V.; Scandurra, A.; Renis, M.; Sinatra, F. Immobilization of the Enzyme Glucose Oxidase on Both Bulk and Porous SiO2 Surfaces. Sensors 2008, 8, 5637–5648. [Google Scholar] [CrossRef]
- Mano, N. Engineering glucose oxidase for bioelectrochemical applications. Bioelectrochemistry 2019, 128, 218–240. [Google Scholar] [CrossRef]
- Wong, C.M.; Wong, K.H.; Chen, X.D. Glucose oxidase: Natural occurrence, function, properties and industrial applications. Appl. Microbiol. Biotechnol. 2008, 78, 927–938. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Wang, L.; Zhang, Y.; Xiao, S.; Bi, F.; Zhao, J.; Gai, G.; Ding, J. Glucose Oxidase-Based Glucose-Sensitive Drug Delivery for Diabetes Treatment. Polymers 2017, 9, 255. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wang, Z.; Yu, J.; Kahkoska, A.R.; Buse, J.B.; Gu, Z. Glucose-Responsive Insulin and Delivery Systems: Innovation and Translation. Adv. Mater. 2020, 32, e1902004. [Google Scholar] [CrossRef] [PubMed]
- Volpatti, L.R.; Matranga, M.A.; Cortinas, A.B.; Delcassian, D.; Daniel, K.B.; Langer, R.; Anderson, D.G. Glucose-Responsive Nanoparticles for Rapid and Extended Self-Regulated Insulin Delivery. ACS Nano 2020, 14, 488–497. [Google Scholar] [CrossRef]
- Fu, L.H.; Qi, C.; Hu, Y.R.; Lin, J.; Huang, P. Glucose Oxidase-Instructed Multimodal Synergistic Cancer Therapy. Adv. Mater. 2020, 32, e2003130. [Google Scholar] [CrossRef]
- Vaupel, P.; Schmidberger, H.; Mayer, A. The Warburg effect: Essential part of metabolic reprogramming and central contributor to cancer progression. Int. J. Radiat. Biol. 2019, 95, 912–919. [Google Scholar] [CrossRef]
- Wang, M.; Wang, D.; Chen, Q.; Li, C.; Li, Z.; Lin, J. Recent Advances in Glucose-Oxidase-Based Nanocomposites for Tumor Therapy. Small 2019, 15, e1903895. [Google Scholar] [CrossRef]
- Fu, L.H.; Qi, C.; Lin, J.; Huang, P. Catalytic chemistry of glucose oxidase in cancer diagnosis and treatment. Chem. Soc. Rev. 2018, 47, 6454–6472. [Google Scholar] [CrossRef]
- Zhao, W.; Hu, J.; Gao, W. Glucose Oxidase-Polymer Nanogels for Synergistic Cancer-Starving and Oxidation Therapy. ACS Appl Mater. Interfaces 2017, 9, 23528–23535. [Google Scholar] [CrossRef]
- Feng, W.; Han, X.; Wang, R.; Gao, X.; Hu, P.; Yue, W.; Chen, Y.; Shi, J. Nanocatalysts-Augmented and Photothermal-Enhanced Tumor-Specific Sequential Nanocatalytic Therapy in Both NIR-I and NIR-II Biowindows. Adv. Mater. 2019, 31, e1805919. [Google Scholar] [CrossRef]
- Zhang, L.; Wan, S.S.; Li, C.X.; Xu, L.; Cheng, H.; Zhang, X.Z. An Adenosine Triphosphate-Responsive Autocatalytic Fenton Nanoparticle for Tumor Ablation with Self-Supplied H2O2 and Acceleration of Fe(III)/Fe(II) Conversion. Nano Lett. 2018, 18, 7609–7618. [Google Scholar] [CrossRef] [PubMed]
- Sun, D.; Qi, G.; Ma, K.; Qu, X.; Xu, W.; Xu, S.; Jin, Y. Tumor Microenvironment-Activated Degradable Multifunctional Nanoreactor for Synergistic Cancer Therapy and Glucose SERS Feedback. iScience 2020, 23, 101274. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Wang, W.; Wu, F.; Zheng, T.; Ashley, J.; Mohammadniaei, M.; Zhang, Q.; Wang, M.; Li, L.; Shen, J.; et al. Biodegradable Poly(gamma-glutamic acid)@glucose oxidase@carbon dot nanoparticles for simultaneous multimodal imaging and synergetic cancer therapy. Biomaterials 2020, 252, 120106. [Google Scholar] [CrossRef] [PubMed]
- He, T.; Xu, H.; Zhang, Y.; Yi, S.; Cui, R.; Xing, S.; Wei, C.; Lin, J.; Huang, P. Glucose Oxidase-Instructed Traceable Self-Oxygenation/Hyperthermia Dually Enhanced Cancer Starvation Therapy. Theranostics 2020, 10, 1544–1554. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Rauth, A.M.; Wu, X.Y. Immobilization and bioactivity of glucose oxidase in hydrogel microspheres formulated by an emulsification-internal gelation-adsorption-polyelectrolyte coating method. Int. J. Pharm. 2007, 339, 148–156. [Google Scholar] [CrossRef]
- Cheng, J.; Liu, Q.; Shuhendler, A.J.; Rauth, A.M.; Wu, X.Y. Optimizing the design and in vitro evaluation of bioreactive glucose oxidase-microspheres for enhanced cytotoxicity against multidrug resistant breast cancer cells. Colloids Surf. B Biointerfaces 2015, 130, 164–172. [Google Scholar] [CrossRef]
- Du, X.; Zhang, T.; Ma, G.; Gu, X.; Wang, G.; Li, J. Glucose-responsive mesoporous silica nanoparticles to generation of hydrogen peroxide for synergistic cancer starvation and chemistry therapy. Int. J. Nanomed. 2019, 14, 2233–2251. [Google Scholar] [CrossRef] [PubMed]
- Flynn, N.; Ranjan, A.; Ramsey, J.D. Intracellular Delivery of Glucose Oxidase for Enhanced Cytotoxicity toward PSMA-Expressing Prostate Cancer Cells. Macromol. Biosci. 2019, 19, e1900183. [Google Scholar] [CrossRef]
- Zhu, H.; Cao, G.; Fu, Y.; Fang, C.; Chu, Q.; Li, X.; Wu, Y.; Han, G. ATP-responsive hollow nanocapsules for DOX/GOx delivery to enable tumor inhibition with suppressed P-glycoprotein. Nano Res. 2021, 14, 222–231. [Google Scholar] [CrossRef]
- Chan, W.K.; Lorenzi, P.L.; Anishkin, A.; Purwaha, P.; Rogers, D.M.; Sukharev, S.; Rempe, S.B.; Weinstein, J.N. The glutaminase activity of L-asparaginase is not required for anticancer activity against ASNS-negative cells. Blood 2014, 123, 3596–3606. [Google Scholar] [CrossRef]
- Covini, D.; Tardito, S.; Bussolati, O.; Chiarelli, L.R.; Pasquetto, M.V.; Digilio, R.; Valentini, G.; Scotti, C. Expanding targets for a metabolic therapy of cancer: L-asparaginase. Recent Pat. Anticancer Drug Discov. 2012, 7, 4–13. [Google Scholar] [CrossRef] [PubMed]
- Brumano, L.P.; da Silva, F.V.S.; Costa-Silva, T.A.; Apolinario, A.C.; Santos, J.; Kleingesinds, E.K.; Monteiro, G.; Rangel-Yagui, C.O.; Benyahia, B.; Junior, A.P. Development of L-Asparaginase Biobetters: Current Research Status and Review of the Desirable Quality Profiles. Front. Bioeng. Biotechnol. 2018, 6, 212. [Google Scholar] [CrossRef] [PubMed]
- Pavlova, N.N.; Hui, S.; Ghergurovich, J.M.; Fan, J.; Intlekofer, A.M.; White, R.M.; Rabinowitz, J.D.; Thompson, C.B.; Zhang, J. As Extracellular Glutamine Levels Decline, Asparagine Becomes an Essential Amino Acid. Cell Metab. 2018, 27, 428–438.e5. [Google Scholar] [CrossRef] [PubMed]
- Vervliet, T.; Parys, J.B. L-asparaginase-induced apoptosis in ALL cells involves IP3 receptor signaling. Cell Calcium 2019, 83, 102076. [Google Scholar] [CrossRef]
- Tallal, L.; Tan, C.; Oettgen, H.; Wollner, N.; McCarthy, M.; Helson, L.; Burchenal, J.; Karnofsky, D.; Murphy, M.L.E. coli L-asparaginase in the treatment of leukemia and solid tumors in 131 children. Cancer 1970, 25, 306–320. [Google Scholar] [CrossRef]
- Sobat, M.; Asad, S.; Kabiri, M.; Mehrshad, M. Metagenomic discovery and functional validation of L-asparaginases with anti-leukemic effect from the Caspian Sea. iScience 2021, 24, 101973. [Google Scholar] [CrossRef]
- Vidya, J.; Sajitha, S.; Ushasree, M.V.; Sindhu, R.; Binod, P.; Madhavan, A.; Pandey, A. Genetic and metabolic engineering approaches for the production and delivery of L-asparaginases: An overview. Bioresour. Technol. 2017, 245 Pt B, 1775–1781. [Google Scholar] [CrossRef]
- Nunes, J.C.F.; Cristovao, R.O.; Freire, M.G.; Santos-Ebinuma, V.C.; Faria, J.L.; Silva, C.G.; Tavares, A.P.M. Recent Strategies and Applications for l-Asparaginase Confinement. Molecules 2020, 25, 5827. [Google Scholar] [CrossRef]
- Kotzia, G.A.; Labrou, N.E. Engineering thermal stability of L-asparaginase by in vitro directed evolution. FEBS J. 2009, 276, 1750–1761. [Google Scholar] [CrossRef]
- Mehta, R.K.; Verma, S.; Pati, R.; Sengupta, M.; Khatua, B.; Jena, R.K.; Sethy, S.; Kar, S.K.; Mandal, C.; Roehm, K.H.; et al. Mutations in subunit interface and B-cell epitopes improve antileukemic activities of Escherichia coli asparaginase-II: Evaluation of immunogenicity in mice. J. Biol. Chem. 2014, 289, 3555–3570. [Google Scholar] [CrossRef]
- Kotzia, G.A.; Lappa, K.; Labrou, N.E. Tailoring structure-function properties of L-asparaginase: Engineering resistance to trypsin cleavage. Biochem. J. 2007, 404, 337–343. [Google Scholar] [CrossRef] [PubMed]
- Burke, P.W.; Hoelzer, D.; Park, J.H.; Schmiegelow, K.; Douer, D. Managing toxicities with asparaginase-based therapies in adult ALL: Summary of an ESMO Open-Cancer Horizons roundtable discussion. ESMO Open 2020, 5, e000858. [Google Scholar] [CrossRef] [PubMed]
- Kwon, Y.M.; Li, Y.T.; Liang, J.F.; Park, Y.J.; Chang, L.C.; Yang, V.C. PTD-modified ATTEMPTS system for enhanced asparaginase therapy: A proof-of-concept investigation. J. Control. Release 2008, 130, 252–258. [Google Scholar] [CrossRef] [PubMed]
- Cruz, M.E.M.; Gaspar, M.M.; Lopes, F.; Jorge, J.S.; Perez-Soler, R. Liposomal l-asparaginase: In vitro evaluation. Int. J. Pharm. 1993, 96, 67–77. [Google Scholar] [CrossRef]
- De, A.; Venkatesh, D.N. Design and evaluation of liposomal delivery system for L-Asparaginese. J. Appl. Pharm. Sci. 2012, 2, 112–117. [Google Scholar]
- Wan, S.; He, D.; Yuan, Y.; Yan, Z.; Zhang, X.; Zhang, J. Chitosan-modified lipid nanovesicles for efficient systemic delivery of l-asparaginase. Colloids Surf. B Biointerfaces 2016, 143, 278–284. [Google Scholar] [CrossRef]
- Bahreini, E.; Aghaiypour, K.; Abbasalipourkabir, R.; Mokarram, A.R.; Goodarzi, M.T.; Saidijam, M. Preparation and nanoencapsulation of l-asparaginase II in chitosan-tripolyphosphate nanoparticles and in vitro release study. Nanoscale Res. Lett. 2014, 9, 340. [Google Scholar] [CrossRef]
- Baskar, G.; Supria Sree, N. Synthesis, characterization and anticancer activity of β-cyclodextrin-Asparaginase nanobiocomposite on prostate and lymphoma cancer cells. J. Drug Deliv. Sci. Technol. 2020, 55, 101417. [Google Scholar] [CrossRef]
- Baskar, G.; Supria Sree, N. Anticancer activity of gelatin-asparaginase nanobiocomposite against cervical and brain cancer cell lines. J. Drug Deliv. Sci. Technol. 2020, 57, 101689. [Google Scholar] [CrossRef]
- Ha, W.; Meng, X.-W.; Li, Q.; Fan, M.-M.; Peng, S.-L.; Ding, L.-S.; Tian, X.; Zhang, S.; Li, B.-J. Self-assembly hollow nanosphere for enzyme encapsulation. Soft Matter. 2010, 6, 1405–1408. [Google Scholar] [CrossRef]
- Singh, M.; Hassan, N.; Verma, D.; Thakur, P.; Panda, B.P.; Panda, A.K.; Sharma, R.K.; Mirza, A.; Mansoor, S.; Alrokayan, S.H.; et al. Design of expert guided investigation of native L-asparaginase encapsulated long-acting cross-linker-free poly (lactic-co-glycolic) acid nanoformulation in an Ehrlich ascites tumor model. Saudi Pharm. J. 2020, 28, 719–728. [Google Scholar] [CrossRef] [PubMed]
- Do, T.T.; Do, T.P.; Nguyen, T.N.; Nguyen, T.C.; Vu, T.T.P.; Nguyen, T.G.A. Nanoliposomal L-Asparaginase and Its Antitumor Activities in Lewis Lung Carcinoma Tumor-Induced BALB/c Mice. Adv. Mater. Sci. Eng. 2019, 2019, 3534807. [Google Scholar] [CrossRef]
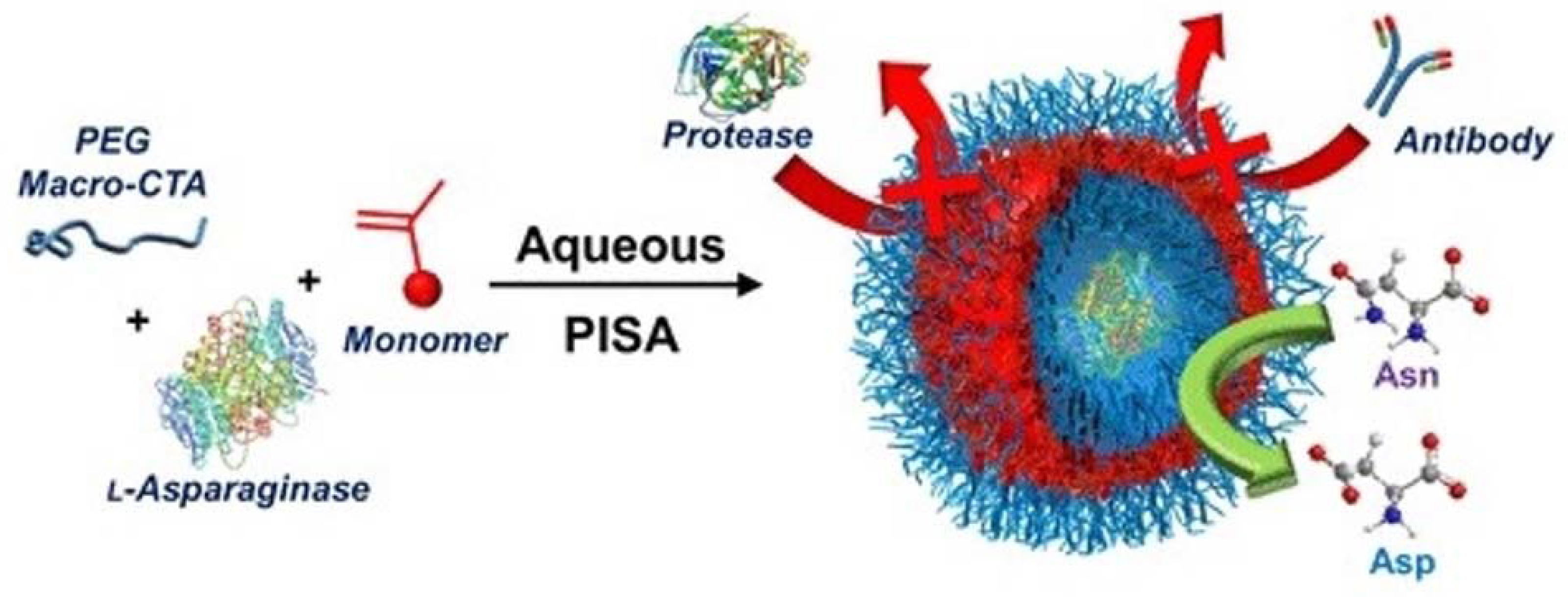
- Blackman, L.D.; Varlas, S.; Arno, M.C.; Houston, Z.H.; Fletcher, N.L.; Thurecht, K.J.; Hasan, M.; Gibson, M.I.; O’Reilly, R.K. Confinement of Therapeutic Enzymes in Selectively Permeable Polymer Vesicles by Polymerization-Induced Self-Assembly (PISA) Reduces Antibody Binding and Proteolytic Susceptibility. ACS Cent. Sci. 2018, 4, 718–723. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.; Jeong, J.H.; Lim, D.; Hong, Y.; Lim, H.J.; Kim, G.J.; Shin, S.R.; Lee, J.J.; Yun, M.; Harris, R.A.; et al. L-Asparaginase delivered by Salmonella typhimurium suppresses solid tumors. Mol. Ther. Oncolytics 2015, 2, 15007. [Google Scholar] [CrossRef] [PubMed]
- Asrorov, A.M.; Gu, Z.; Li, F.; Liu, L.; Huang, Y. Biomimetic camouflage delivery strategies for cancer therapy. Nanoscale 2021, 13, 8693–8706. [Google Scholar] [CrossRef] [PubMed]
- Bailly, J.; Sezanne, V.; Godfrin, Y. L-Asparaginase Loaded Red Blood Cells: From Prescription to Injection. Blood 2011, 118, 4252. [Google Scholar] [CrossRef]
- Al-Dulimi, A.G.; Al-Saffar, A.Z.; Sulaiman, G.M.; Khalil, K.A.A.; Khashan, K.S.; Al-Shmgani, H.S.A.; Ahmed, E.M. Immobilization of l-asparaginase on gold nanoparticles for novel drug delivery approach as anti-cancer agent against human breast carcinoma cells. J. Mater. Res. Technol. 2020, 9, 15394–15411. [Google Scholar] [CrossRef]
- Baskar, G.; Garrick, B.G.; Lalitha, K.; Chamundeeswari, M. Gold nanoparticle mediated delivery of fungal asparaginase against cancer cells. J. Drug Deliv. Sci. Technol. 2018, 44, 498–504. [Google Scholar] [CrossRef]
- Baskar, G.; Lalitha, K.; Aiswarya, R.; Naveenkumar, R. Synthesis, characterization and synergistic activity of cerium-selenium nanobiocomposite of fungal l-asparaginase against lung cancer. Mater. Sci. Eng. C Mater. Biol. Appl. 2018, 93, 809–815. [Google Scholar] [CrossRef]
- Mu, X.; Qiao, J.; Qi, L.; Dong, P.; Ma, H. Poly(2-vinyl-4,4-dimethylazlactone)-functionalized magnetic nanoparticles as carriers for enzyme immobilization and its application. ACS Appl. Mater. Interfaces 2014, 6, 21346–21354. [Google Scholar] [CrossRef]
- Mohamadzadeh, M. Microbial Toxins: Current Research and Future Trends. Expert Rev. Anti-Infect. Ther. 2009, 7, 695–696. [Google Scholar] [CrossRef][Green Version]
- Sharma, P.C.; Sharma, D.; Sharma, A.; Bhagat, M.; Ola, M.; Thakur, V.K.; Bhardwaj, J.K.; Goyal, R.K. Recent advances in microbial toxin-related strategies to combat cancer. Semin. Cancer Biol. 2021, 7, 7. [Google Scholar] [CrossRef] [PubMed]
- Callahan, L.T., 3rd. Purification and characterization of Pseudomonas aeruginosa exotoxin. Infect. Immun. 1974, 9, 113–118. [Google Scholar] [CrossRef] [PubMed]
- Kreitman, R.J.; Siegall, C.B.; Chaudhary, V.K.; FitzGerald, D.J.; Pastan, I. Properties of chimeric toxins with two recognition domains: Interleukin 6 and transforming growth factor alpha at different locations in Pseudomonas exotoxin. Bioconj. Chem. 1992, 3, 63–68. [Google Scholar] [CrossRef]
- Robbins, D.H.; Margulies, I.; Stetler-Stevenson, M.; Kreitman, R.J. Hairy cell leukemia, a B-cell neoplasm that is particularly sensitive to the cytotoxic effect of anti-Tac(Fv)-PE38 (LMB-2). Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2000, 6, 693–700. [Google Scholar]
- Kuan, C.T.; Wang, Q.C.; Pastan, I. Pseudomonas exotoxin A mutants. Replacement of surface exposed residues in domain II with cysteine residues that can be modified with polyethylene glycol in a site-specific manner. J. Biol. Chem. 1994, 269, 7610–7616. [Google Scholar] [CrossRef]
- Boland, E.L.; Van Dyken, C.M.; Duckett, R.M.; McCluskey, A.J.; Poon, G.M. Structural complementation of the catalytic domain of pseudomonas exotoxin A. J. Mol. Biol. 2014, 426, 645–655. [Google Scholar] [CrossRef]
- El-Behaedi, S.; Landsman, R.; Rudloff, M.; Kolyvas, E.; Albalawy, R.; Zhang, X.; Bera, T.; Collins, K.; Kozlov, S.; Alewine, C. Protein Synthesis Inhibition Activity of Mesothelin Targeting Immunotoxin LMB-100 Decreases Concentrations of Oncogenic Signaling Molecules and Secreted Growth Factors. Toxins 2018, 10, 447. [Google Scholar] [CrossRef]
- Becerra, J.C.; Arthur, J.F.; Landucci, G.R.; Forthal, D.N.; Theuer, C.P. CD8+ T-cell mediated tumor protection by Pseudomonas exotoxin fused to ovalbumin in C57BL/6 mice. Surgery 2003, 133, 404–410. [Google Scholar] [CrossRef]
- Taranejoo, S.; Janmaleki, M.; Rafienia, M.; Kamali, M.; Mansouri, M. Chitosan microparticles loaded with exotoxin A subunit antigen for intranasal vaccination against Pseudomonas aeruginosa: An in vitro study. Carbohydr. Polym. 2011, 83, 1854–1861. [Google Scholar] [CrossRef]
- Sharma, A.K.; FitzGerald, D. Pseudomonas exotoxin kills Drosophila S2 cells via apoptosis. Toxicon 2010, 56, 1025–1034. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Leamon, C.P.; Pastan, I.; Low, P.S. Cytotoxicity of folate-Pseudomonas exotoxin conjugates toward tumor cells. Contribution of translocation domain. J. Biol. Chem. 1993, 268, 24847–24854. [Google Scholar] [CrossRef]
- Kreitman, R.J.; Pastan, I. Recombinant toxins containing human granulocyte-macrophage colony-stimulating factor and either pseudomonas exotoxin or diphtheria toxin kill gastrointestinal cancer and leukemia cells. Blood 1997, 90, 252–259. [Google Scholar] [CrossRef] [PubMed]
- Amoozadeh, S.; Hemmati, M.; Farajollahi, M.M.; Akbari, N.; Tarighi, P. Preparation of Diphtheria and Pseudomonas Exotoxin A Immunotoxins and Evaluation of Their Cytotoxicity Effect on SK-BR-3, BT-474, and MDA-MB-231 Breast Cancer Cell Lines. Cancer Investig. 2019, 37, 546–557. [Google Scholar] [CrossRef]
- Klausz, K.; Kellner, C.; Derer, S.; Valerius, T.; Staudinger, M.; Burger, R.; Gramatzki, M.; Peipp, M. The novel multispecies Fc-specific Pseudomonas exotoxin A fusion protein α-Fc-ETA’ enables screening of antibodies for immunotoxin development. J. Immunol. Methods 2015, 418, 75–83. [Google Scholar] [CrossRef]
- Kellner, C.; Bleeker, W.K.; Lammerts van Bueren, J.J.; Staudinger, M.; Klausz, K.; Derer, S.; Glorius, P.; Muskulus, A.; de Goeij, B.E.; van de Winkel, J.G.; et al. Human kappa light chain targeted Pseudomonas exotoxin A—Identifying human antibodies and Fab fragments with favorable characteristics for antibody-drug conjugate development. J. Immunol. Methods 2011, 371, 122–133. [Google Scholar] [CrossRef]
- Alderson, R.F.; Kreitman, R.J.; Chen, T.; Yeung, P.; Herbst, R.; Fox, J.A.; Pastan, I. CAT-8015: A second-generation pseudomonas exotoxin A-based immunotherapy targeting CD22-expressing hematologic malignancies. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2009, 15, 832–839. [Google Scholar] [CrossRef]
- Hassan, R.; Bullock, S.; Premkumar, A.; Kreitman, R.J.; Kindler, H.; Willingham, M.C.; Pastan, I. Phase I study of SS1P, a recombinant anti-mesothelin immunotoxin given as a bolus I.V. infusion to patients with mesothelin-expressing mesothelioma, ovarian, and pancreatic cancers. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2007, 13, 5144–5149. [Google Scholar] [CrossRef]
- Bauss, F.; Lechmann, M.; Krippendorff, B.F.; Staack, R.; Herting, F.; Festag, M.; Imhof-Jung, S.; Hesse, F.; Pompiati, M.; Kollmorgen, G.; et al. Characterization of a re-engineered, mesothelin-targeted Pseudomonas exotoxin fusion protein for lung cancer therapy. Mol. Oncol. 2016, 10, 1317–1329. [Google Scholar] [CrossRef]
- Kreitman, R.J.; Stetler-Stevenson, M.; Margulies, I.; Noel, P.; Fitzgerald, D.J.P.; Wilson, W.H.; Pastan, I. Phase II trial of recombinant immunotoxin RFB4(dsFv)-PE38 (BL22) in patients with hairy cell leukemia. J. Clin. Oncol. 2009, 27, 2983–2990. [Google Scholar] [CrossRef]
- Fleming, B.D.; Ho, M. Development of Glypican-3 targeting immunotoxins for the treatment of liver cancer: An update. Biomolecules. 2020, 10, 934. [Google Scholar] [CrossRef] [PubMed]
- Kreitman, R.J.; Wilson, W.H.; Bergeron, K.; Raggio, M.; Stetler-Stevenson, M.; FitzGerald, D.J.; Pastan, I. Efficacy of the anti-CD22 recombinant immunotoxin BL22 in chemotherapy-resistant hairy-cell leukemia. N. Engl. J. Med. 2001, 345, 241–247. [Google Scholar] [CrossRef] [PubMed]
- Kreitman, R.J.; Squires, D.R.; Stetler-Stevenson, M.; Noel, P.; FitzGerald, D.J.; Wilson, W.H.; Pastan, I. Phase I trial of recombinant immunotoxin RFB4(dsFv)-PE38 (BL22) in patients with B-cell malignancies. J. Clin. Oncol. 2005, 23, 6719–6729. [Google Scholar] [CrossRef]
- Kreitman, R.J.; Wilson, W.H.; White, J.D.; Stetler-Stevenson, M.; Jaffe, E.S.; Giardina, S.; Waldmann, T.A.; Pastan, I. Phase I trial of recombinant immunotoxin anti-Tac(Fv)-PE38 (LMB-2) in patients with hematologic malignancies. J. Clin. Oncol. 2000, 18, 1622–1636. [Google Scholar] [CrossRef] [PubMed]
- Pai, L.H.; Gallo, M.G.; FitzGerald, D.J.; Pastan, I. Antitumor activity of a transforming growth factor alpha-Pseudomonas exotoxin fusion protein (TGF-alpha-PE40). Cancer Res. 1991, 51, 2808–2812. [Google Scholar] [PubMed]
- Pai, L.H.; Wittes, R.; Setser, A.; Willingham, M.C.; Pastan, I. Treatment of advanced solid tumors with immunotoxin LMB-1: An antibody linked to Pseudomonas exotoxin. Nat. Med. 1996, 2, 350–353. [Google Scholar] [CrossRef] [PubMed]
- Posey, J.A.; Khazaeli, M.B.; Bookman, M.A.; Nowrouzi, A.; Grizzle, W.E.; Thornton, J.; Carey, D.E.; Lorenz, J.M.; Sing, A.P.; Siegall, C.B.; et al. A phase I trial of the single-chain immunotoxin SGN-10 (BR96 sFv-PE40) in patients with advanced solid tumors. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2002, 8, 3092–3099. [Google Scholar]
- Rand, R.W.; Kreitman, R.J.; Patronas, N.; Varricchio, F.; Pastan, I.; Puri, R.K. Intratumoral administration of recombinant circularly permuted interleukin-4-Pseudomonas exotoxin in patients with high-grade glioma. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2000, 6, 2157–2165. [Google Scholar]
- Weber, F.; Asher, A.; Bucholz, R.; Berger, M.; Prados, M.; Chang, S.; Bruce, J.; Hall, W.; Rainov, N.G.; Westphal, M.; et al. Safety, tolerability, and tumor response of IL4-Pseudomonas exotoxin (NBI-3001) in patients with recurrent malignant glioma. J. Neuro-Oncol. 2003, 64, 125–137. [Google Scholar] [CrossRef]
- Garland, L.; Gitlitz, B.; Ebbinghaus, S.; Pan, H.; de Haan, H.; Puri, R.K.; Von Hoff, D.; Figlin, R. Phase I trial of intravenous IL-4 pseudomonas exotoxin protein (NBI-3001) in patients with advanced solid tumors that express the IL-4 receptor. J. Immunother. 2005, 28, 376–381. [Google Scholar] [CrossRef]
- Pai-Scherf, L.H.; Villa, J.; Pearson, D.; Watson, T.; Liu, E.; Willingham, M.C.; Pastan, I. Hepatotoxicity in cancer patients receiving erb-38, a recombinant immunotoxin that targets the erbB2 receptor. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 1999, 5, 2311–2315. [Google Scholar]
- Azemar, M.; Djahansouzi, S.; Jäger, E.; Solbach, C.; Schmidt, M.; Maurer, A.B.; Mross, K.; Unger, C.; von Minckwitz, G.; Dall, P.; et al. Regression of cutaneous tumor lesions in patients intratumorally injected with a recombinant single-chain antibody-toxin targeted to ErbB2/HER2. Breast Cancer Res. Treat. 2003, 82, 155–164. [Google Scholar] [CrossRef] [PubMed]
- von Minckwitz, G.; Harder, S.; Hövelmann, S.; Jäger, E.; Al-Batran, S.E.; Loibl, S.; Atmaca, A.; Cimpoiasu, C.; Neumann, A.; Abera, A.; et al. Phase I clinical study of the recombinant antibody toxin scFv(FRP5)-ETA specific for the ErbB2/HER2 receptor in patients with advanced solid malignomas. Breast Cancer Res. BCR 2005, 7, R617–R626. [Google Scholar] [CrossRef] [PubMed]
- Sampson, J.H.; Akabani, G.; Archer, G.E.; Bigner, D.D.; Berger, M.S.; Friedman, A.H.; Friedman, H.S.; Herndon, J.E., 2nd; Kunwar, S.; Marcus, S.; et al. Progress report of a Phase I study of the intracerebral microinfusion of a recombinant chimeric protein composed of transforming growth factor (TGF)-alpha and a mutated form of the Pseudomonas exotoxin termed PE-38 (TP-38) for the treatment of malignant brain tumors. J. Neuro-Oncol. 2003, 65, 27–35. [Google Scholar]
- Goldberg, M.R.; Heimbrook, D.C.; Russo, P.; Sarosdy, M.F.; Greenberg, R.E.; Giantonio, B.J.; Linehan, W.M.; Walther, M.; Fisher, H.A.; Messing, E.; et al. Phase I clinical study of the recombinant oncotoxin TP40 in superficial bladder cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 1995, 1, 57–61. [Google Scholar]
- Mesri, E.A.; Ono, M.; Kreitman, R.J.; Klagsbrun, M.; Pastan, I. The heparin-binding domain of heparin-binding EGF-like growth factor can target Pseudomonas exotoxin to kill cells exclusively through heparan sulfate proteoglycans. J. Cell Sci. 1994, 107 Pt 9, 2599–2608. [Google Scholar] [CrossRef]
- Wu, T.; Zhu, J. Recent development and optimization of Pseudomonas aeruginosa exotoxin immunotoxins in cancer therapeutic applications. Int. Immunopharmacol. 2021, 96, 107759. [Google Scholar] [CrossRef]
- Alewine, C.; Ahmad, M.; Peer, C.J.; Hu, Z.I.; Lee, M.J.; Yuno, A.; Kindrick, J.D.; Thomas, A.; Steinberg, S.M.; Trepel, J.B.; et al. Phase I/II Study of the Mesothelin-targeted Immunotoxin LMB-100 with Nab-Paclitaxel for Patients with Advanced Pancreatic Adenocarcinoma. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2020, 26, 828–836. [Google Scholar] [CrossRef]
- Mossoba, M.E.; Onda, M.; Taylor, J.; Massey, P.R.; Treadwell, S.; Sharon, E.; Hassan, R.; Pastan, I.; Fowler, D.H. Pentostatin plus cyclophosphamide safely and effectively prevents immunotoxin immunogenicity in murine hosts. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2011, 17, 3697–3705. [Google Scholar] [CrossRef]
- Biggers, K.; Scheinfeld, N. VB4-845, a conjugated recombinant antibody and immunotoxin for head and neck cancer and bladder cancer. Curr. Opin. Mol. Ther. 2008, 10, 176–186. [Google Scholar]
- Bell, C.E.; Eisenberg, D. Crystal Structure of Diphtheria Toxin Bound to Nicotinamide Adenine Dinucleotide. Biochemistry 1996, 35, 1137–1149. [Google Scholar] [CrossRef] [PubMed]
- Choe, S.; Bennett, M.J.; Fujii, G.; Curmi, P.M.; Kantardjieff, K.A.; Collier, R.J.; Eisenberg, D. The crystal structure of diphtheria toxin. Nature 1992, 357, 216–222. [Google Scholar] [CrossRef] [PubMed]
- Bell, C.E.; Eisenberg, D. Crystal structure of nucleotide-free diphtheria toxin. Biochemistry 1997, 36, 481–488. [Google Scholar] [CrossRef] [PubMed]
- Bennett, M.J.; Eisenberg, D. Refined structure of monomelic diphtheria toxin at 2.3 Å resolution. Protein Sci. 1994, 3, 1464–1475. [Google Scholar] [CrossRef] [PubMed]
- Potala, S.; Sahoo, S.K.; Verma, R.S. Targeted therapy of cancer using diphtheria toxin-derived immunotoxins. Drug Discov. Today 2008, 13, 807–815. [Google Scholar] [CrossRef]
- Kim, B.H.; Yoon, J.H.; Myung, S.J.; Lee, J.H.; Lee, S.H.; Lee, S.M.; Lee, H.S. Enhanced interleukin-2 diphtheria toxin conjugate-induced growth suppression in retinoic acid-treated hypoxic hepatocellular carcinoma cells. Cancer Lett. 2009, 274, 259–265. [Google Scholar] [CrossRef]
- Lewis, D.J.; Dao, H., Jr.; Nagarajan, P.; Duvic, M. Primary cutaneous anaplastic large-cell lymphoma: Complete remission for 13 years after denileukin diftitox. JAAD Case Rep. 2017, 3, 501–504. [Google Scholar] [CrossRef]
- Attia, P.; Maker, A.V.; Haworth, L.R.; Rogers-Freezer, L.; Rosenberg, S.A. Inability of a Fusion Protein of IL-2 and Diphtheria Toxin (Denileukin Diftitox, DAB389IL-2, ONTAK) to Eliminate Regulatory T Lymphocytes in Patients with Melanoma. J. Immunother. 2005, 28, 582–592. [Google Scholar] [CrossRef]
- Cheung, L.S.; Fu, J.; Kumar, P.; Kumar, A.; Urbanowski, M.E.; Ihms, E.A.; Parveen, S.; Bullen, C.K.; Patrick, G.J.; Harrison, R.; et al. Second-generation IL-2 receptor-targeted diphtheria fusion toxin exhibits antitumor activity and synergy with anti-PD-1 in melanoma. Proc. Natl. Acad. Sci. USA 2019, 116, 3100–3105. [Google Scholar] [CrossRef]
- Chen, M.; Ho, C.; Teng, M.; Chen, H. 53 Adeno-associated virus (AAV) carrying diphtheria toxin a gene for pancreatic cancer therapy. Eur. J. Cancer 2014, 50, 22. [Google Scholar] [CrossRef]
- Peng, W.; Verbitsky, A.; Bao, Y.; Sawicki, J. Regulated expression of diphtheria toxin in prostate cancer cells. Mol. Ther. J. Am. Soc. Gene Ther. 2002, 6, 537–545. [Google Scholar] [CrossRef] [PubMed]
- Lidor, Y.J.; Lee, W.E.; Nilson, J.H.; Maxwell, I.H.; Su, L.J.; Brand, E.; Glode, L.M. In vitro expression of the diphtheria toxin A-chain gene under the control of human chorionic gonadotropin gene promoters as a means of directing toxicity to ovarian cancer cell lines. Am. J. Obstet. Gynecol. 1997, 177, 579–585. [Google Scholar] [CrossRef]
- Qiao, J.; Caruso, M. PG13 packaging cells produce recombinant retroviruses carrying a diphtheria toxin mutant which kills cancer cells. J. Virol. 2002, 76, 7343–7348. [Google Scholar] [CrossRef] [PubMed][Green Version]
- He, M.; Wang, Y.; Chen, X.; Zhao, Y.; Lou, K.; Wang, Y.; Huang, L.; Hou, X.; Xu, J.; Cai, X.; et al. Spatiotemporally controllable diphtheria toxin expression using a light-switchable transgene system combining multifunctional nanoparticle delivery system for targeted melanoma therapy. J. Control. Release 2020, 319, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Mizrahi, A.; Czerniak, A.; Levy, T.; Amiur, S.; Gallula, J.; Matouk, I.; Abu-lail, R.; Sorin, V.; Birman, T.; de Groot, N.; et al. Development of targeted therapy for ovarian cancer mediated by a plasmid expressing diphtheria toxin under the control of H19 regulatory sequences. J. Transl. Med. 2009, 7, 69. [Google Scholar] [CrossRef]
- Scaiewicz, V.; Sorin, V.; Fellig, Y.; Birman, T.; Mizrahi, A.; Galula, J.; Abu-Lail, R.; Shneider, T.; Ohana, P.; Buscail, L.; et al. Use of H19 Gene Regulatory Sequences in DNA-Based Therapy for Pancreatic Cancer. J. Oncol. 2010, 2010, 178174. [Google Scholar] [CrossRef]
- Hasenpusch, G.; Pfeifer, C.; Aneja, M.K.; Wagner, K.; Reinhardt, D.; Gilon, M.; Ohana, P.; Hochberg, A.; Rudolph, C. Aerosolized BC-819 inhibits primary but not secondary lung cancer growth. PLoS ONE 2011, 6, e20760. [Google Scholar] [CrossRef]
- Gofrit, O.N.; Benjamin, S.; Halachmi, S.; Leibovitch, I.; Dotan, Z.; Lamm, D.L.; Ehrlich, N.; Yutkin, V.; Ben-Am, M.; Hochberg, A. DNA based therapy with diphtheria toxin-A BC-819: A phase 2b marker lesion trial in patients with intermediate risk nonmuscle invasive bladder cancer. J. Urol. 2014, 191, 1697–1702. [Google Scholar] [CrossRef]
- Sánchez-García, L.; Serna, N.; Álamo, P.; Sala, R.; Céspedes, M.V.; Roldan, M.; Sánchez-Chardi, A.; Unzueta, U.; Casanova, I.; Mangues, R.; et al. Self-assembling toxin-based nanoparticles as self-delivered antitumoral drugs. J. Control. Release 2018, 274, 81–92. [Google Scholar] [CrossRef]
- Pallarès, V.; Núñez, Y.; Sánchez-García, L.; Falgàs, A.; Serna, N.; Unzueta, U.; Gallardo, A.; Alba-Castellón, L.; Álamo, P.; Sierra, J.; et al. Antineoplastic effect of a diphtheria toxin-based nanoparticle targeting acute myeloid leukemia cells overexpressing CXCR4. J. Control. Release 2021, 335, 117–129. [Google Scholar] [CrossRef]
- Hotz, H.G.; Gill, P.S.; Masood, R.; Hotz, B.; Buhr, H.J.; Foitzik, T.; Hines, O.J.; Reber, H.A. Specific targeting of tumor vasculature by diphtheria toxin-vascular endothelial growth factor fusion protein reduces angiogenesis and growth of pancreatic cancer. J. Gastrointest. Surg. Off. J. Soc. Surg. Aliment. Tract. 2002, 6, 159–166; discussion 166. [Google Scholar] [CrossRef]
- Agarwal, M.; Mondal, T.; Bose, B. Peptides derived from a short stretch of diphtheria toxin bind to heparin-binding epidermal growth factor-like growth factor. Toxicon 2019, 169, 109–116. [Google Scholar] [CrossRef] [PubMed]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Asrorov, A.M.; Muhitdinov, B.; Tu, B.; Mirzaakhmedov, S.; Wang, H.; Huang, Y. Advances on Delivery of Cytotoxic Enzymes as Anticancer Agents. Molecules 2022, 27, 3836. https://doi.org/10.3390/molecules27123836
Asrorov AM, Muhitdinov B, Tu B, Mirzaakhmedov S, Wang H, Huang Y. Advances on Delivery of Cytotoxic Enzymes as Anticancer Agents. Molecules. 2022; 27(12):3836. https://doi.org/10.3390/molecules27123836
Chicago/Turabian StyleAsrorov, Akmal M., Bahtiyor Muhitdinov, Bin Tu, Sharafitdin Mirzaakhmedov, Huiyuan Wang, and Yongzhuo Huang. 2022. "Advances on Delivery of Cytotoxic Enzymes as Anticancer Agents" Molecules 27, no. 12: 3836. https://doi.org/10.3390/molecules27123836
APA StyleAsrorov, A. M., Muhitdinov, B., Tu, B., Mirzaakhmedov, S., Wang, H., & Huang, Y. (2022). Advances on Delivery of Cytotoxic Enzymes as Anticancer Agents. Molecules, 27(12), 3836. https://doi.org/10.3390/molecules27123836