
Catalytic Properties of Caseinolytic Protease Subunit of Plasmodium knowlesi and Its Inhibition by a Member of δ-Lactone, Hyptolide

 , , and
, , and
Abstract
:1. Introduction
2. Results
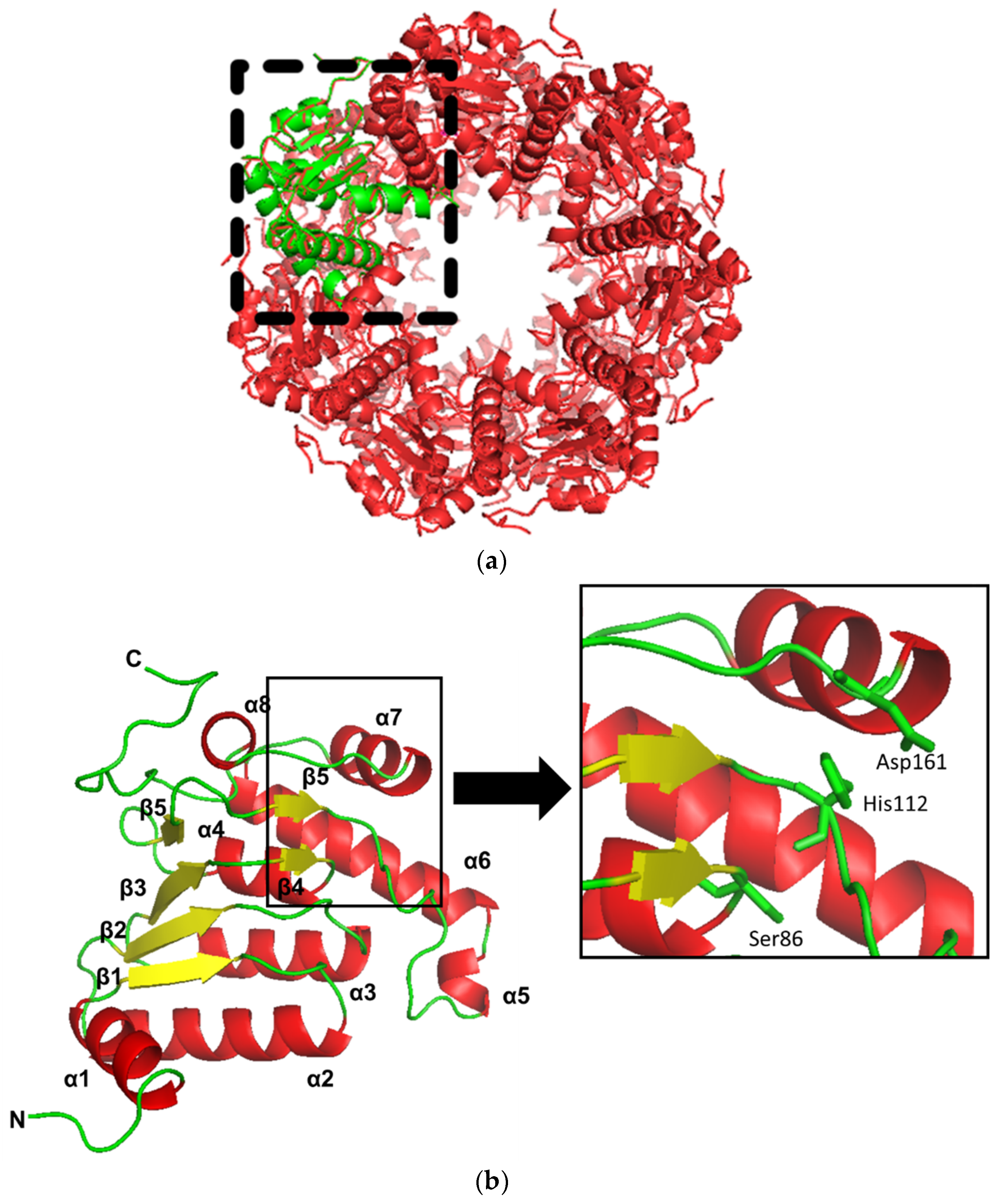
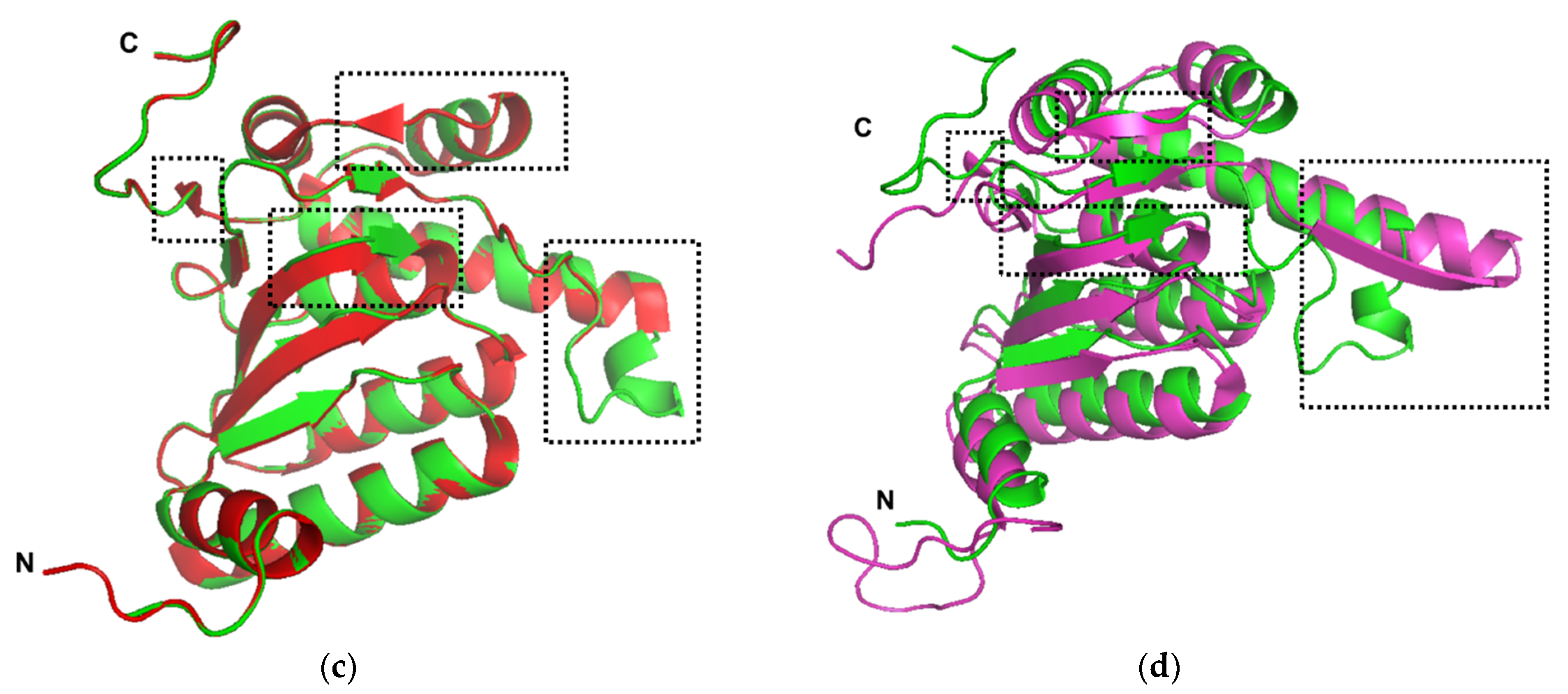
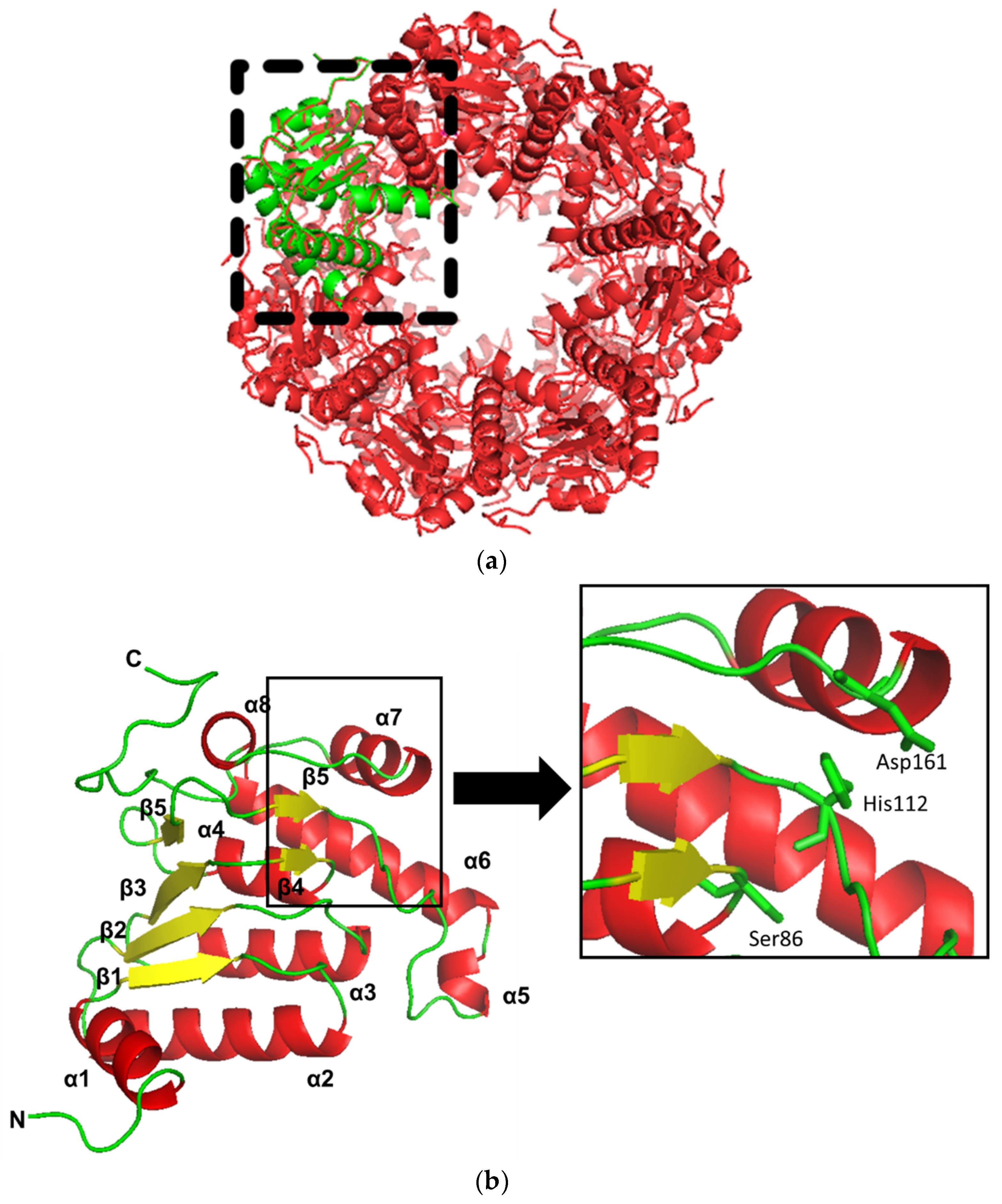
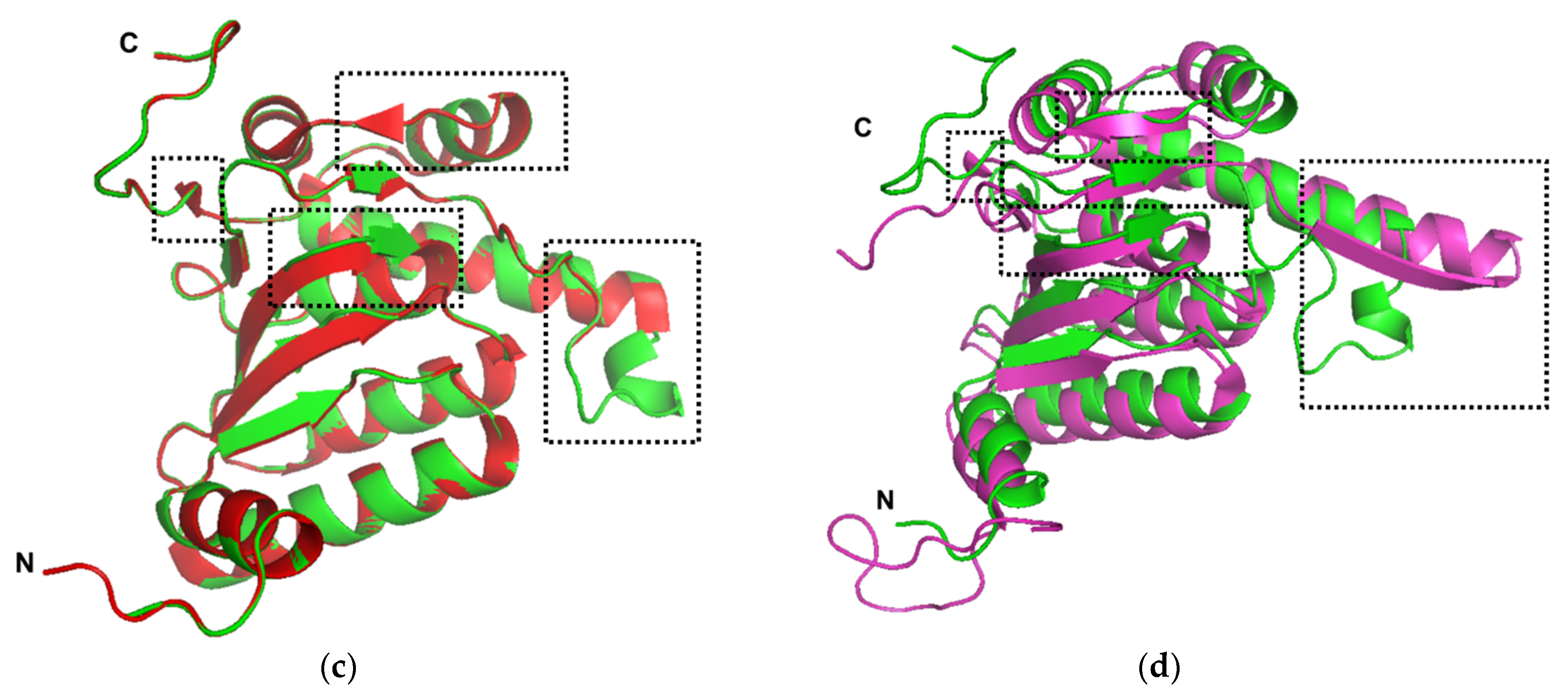
2.1. Sequence Analysis and Structural Homology Modeling
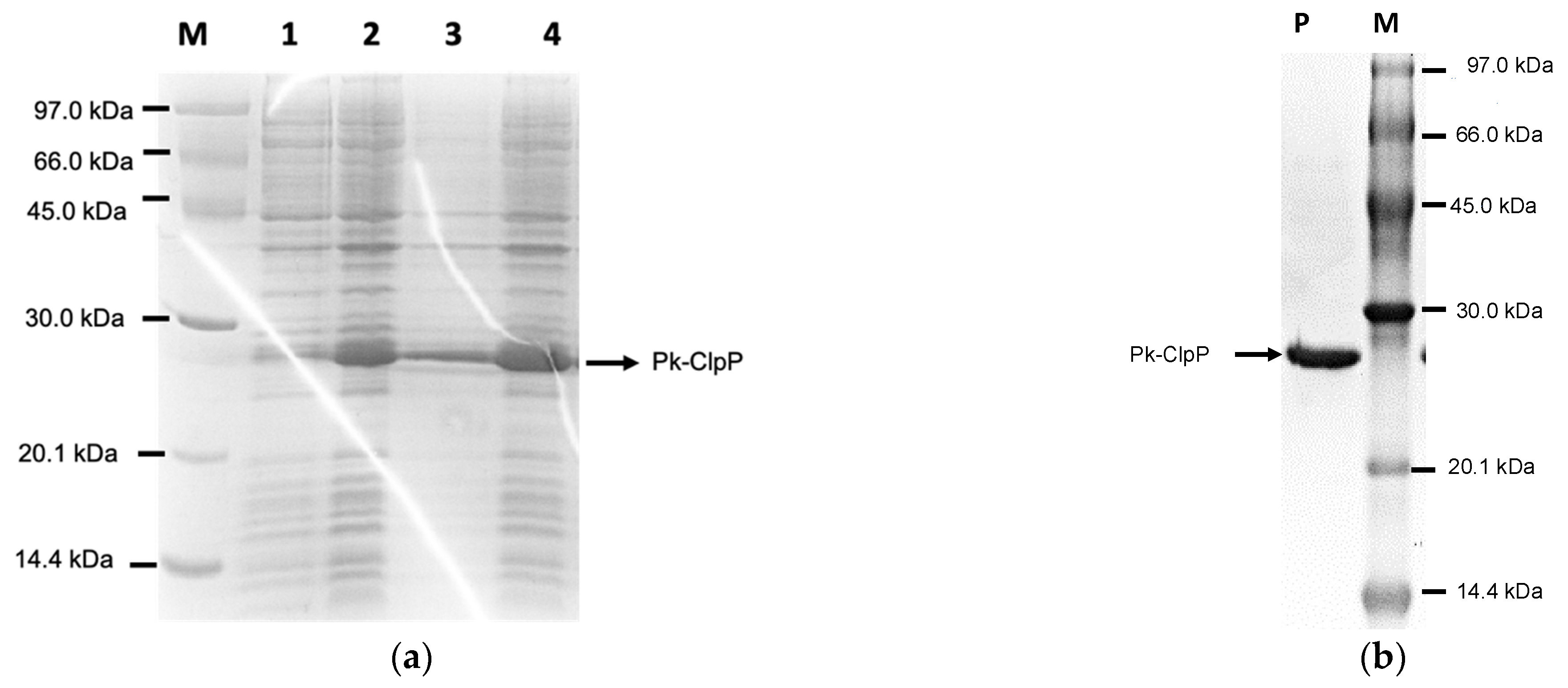
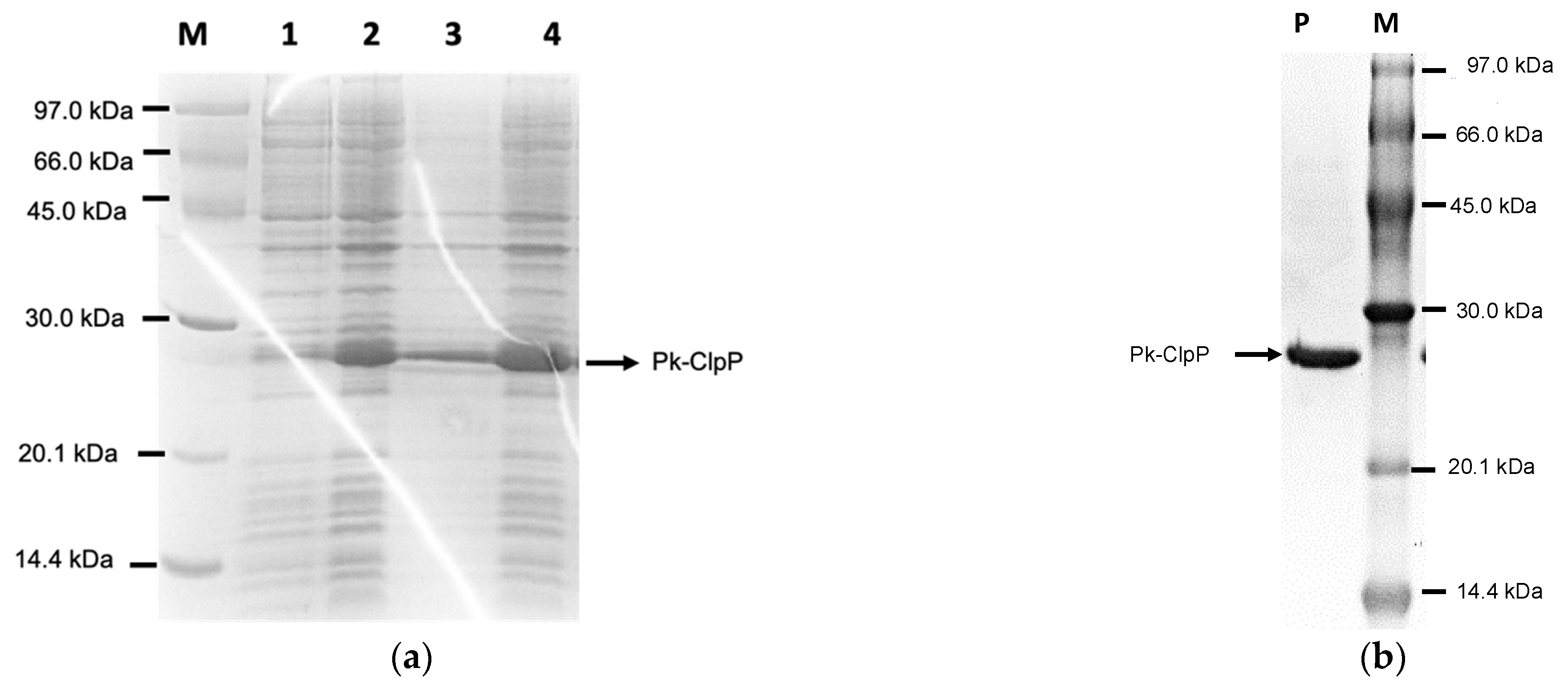
2.2. Over-Expression and Purification
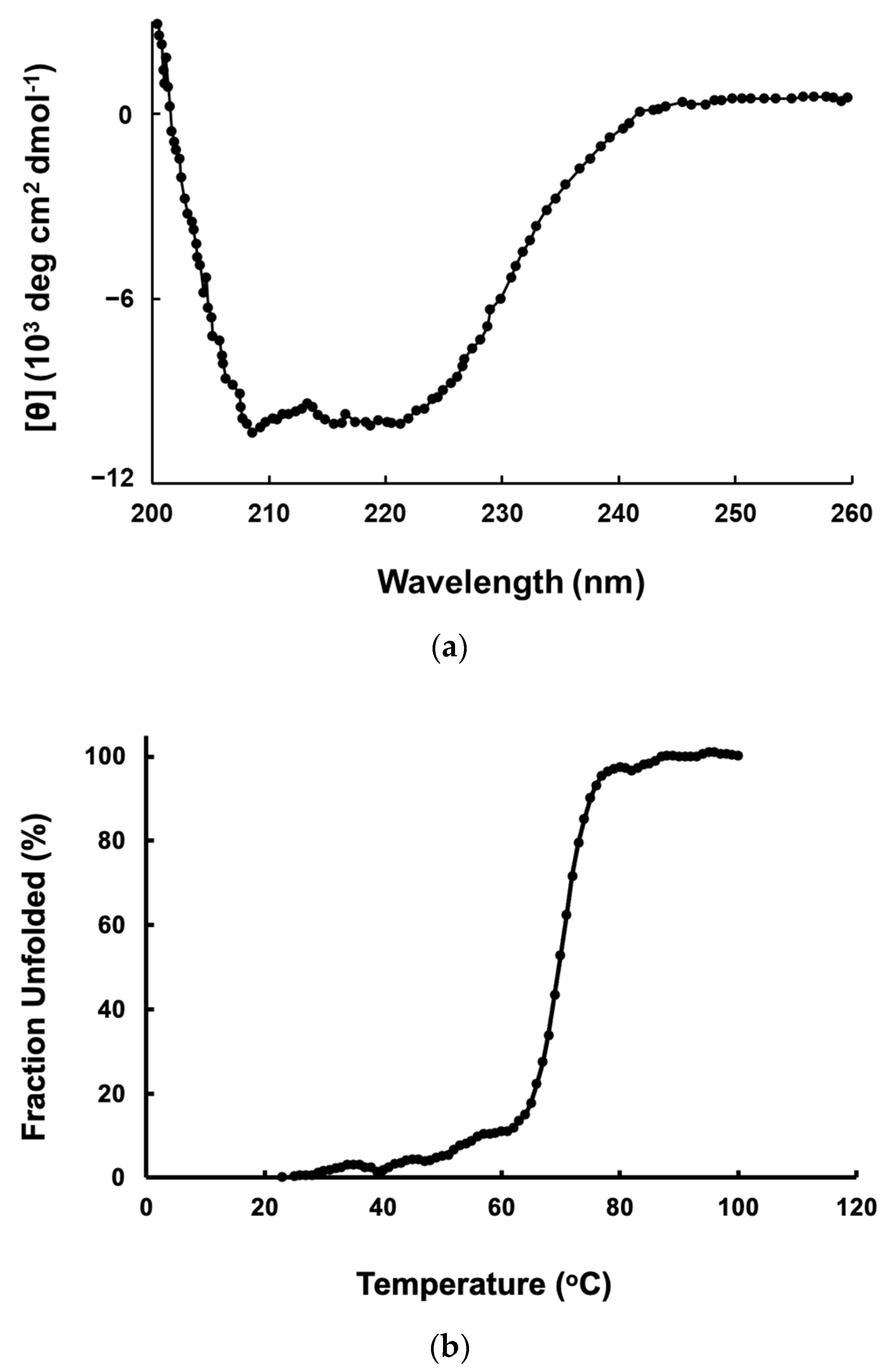
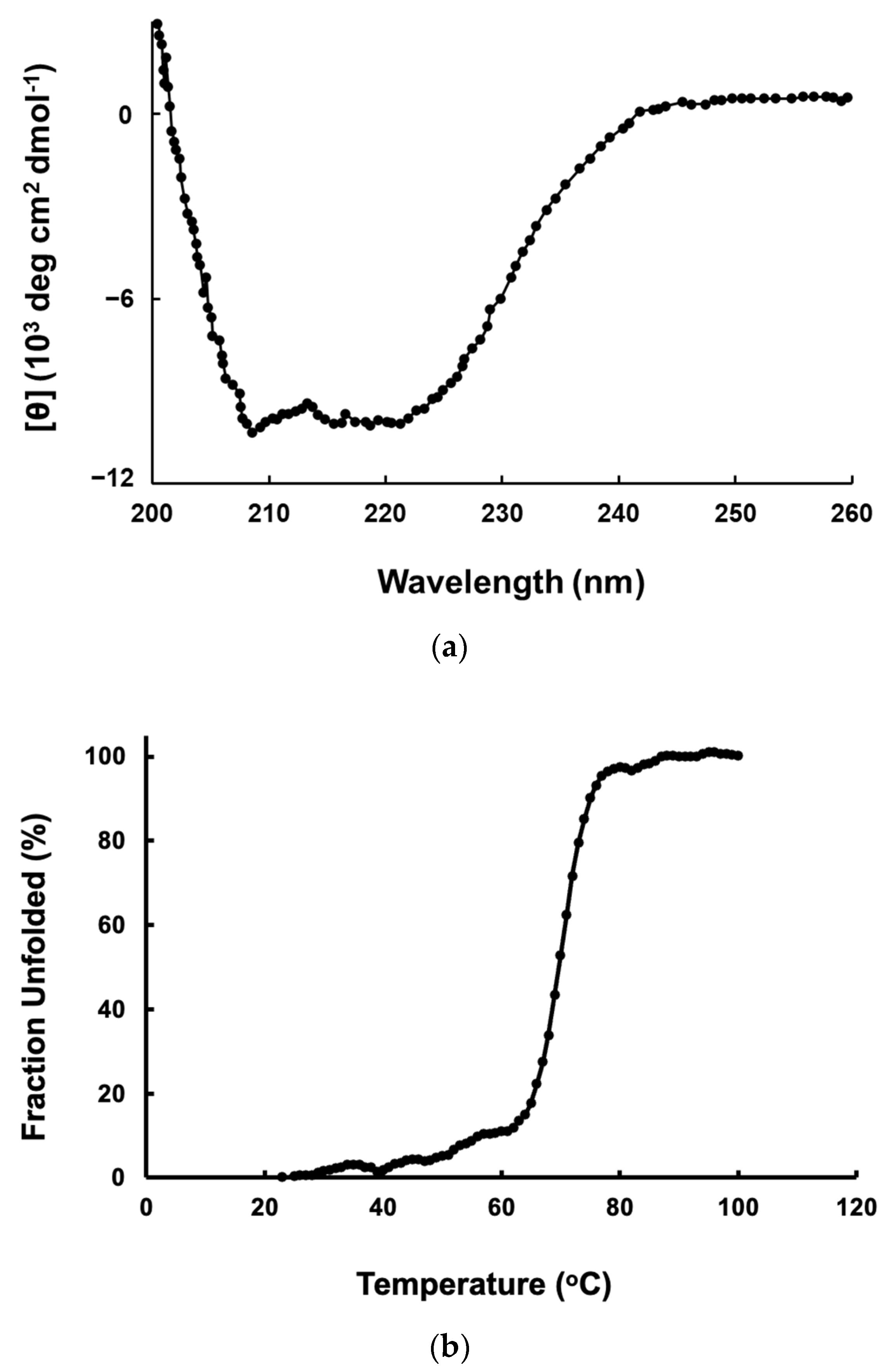
2.3. CD Spectra
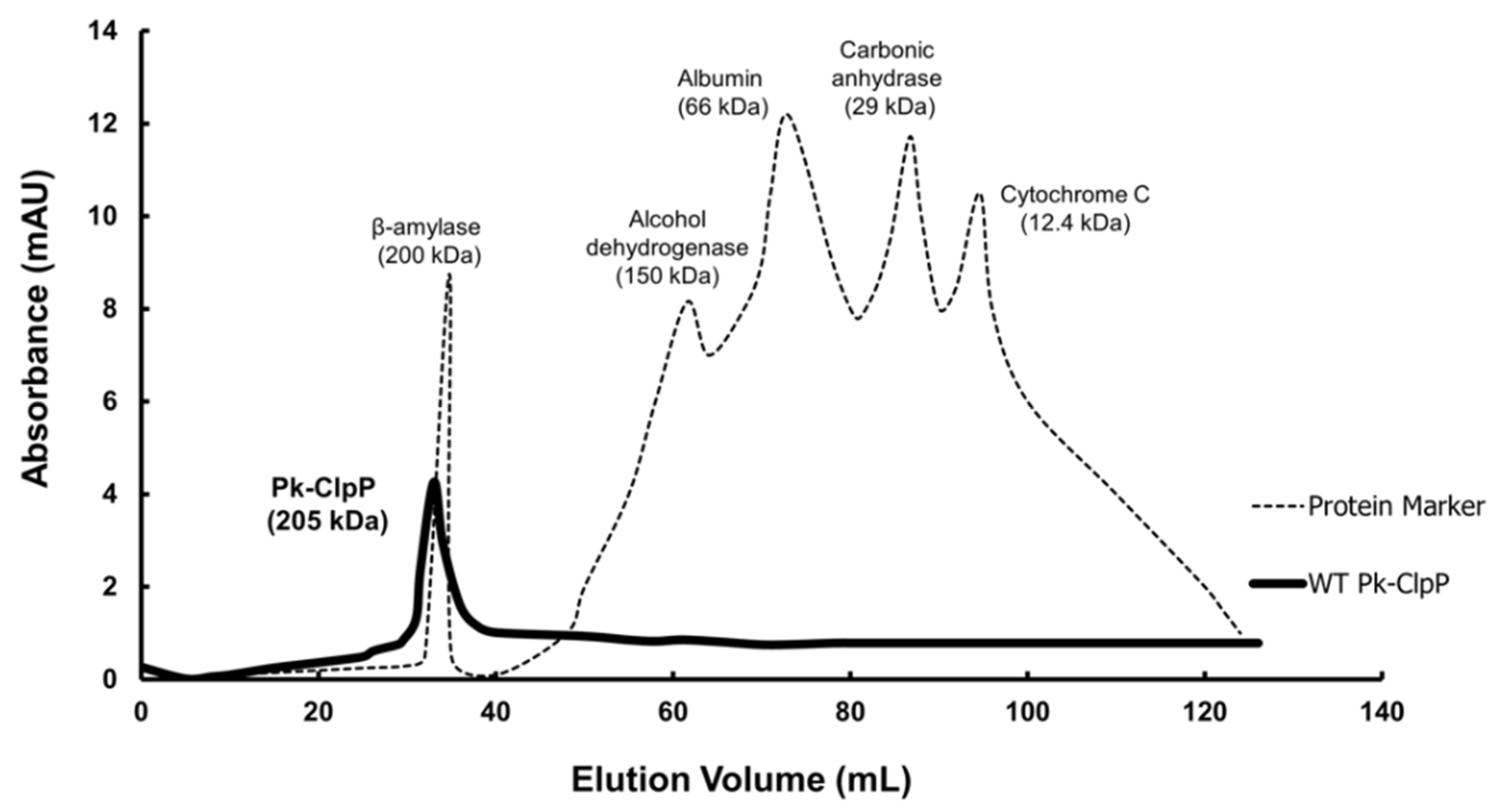
2.4. Oligomerization
2.5. Catalytic Activity
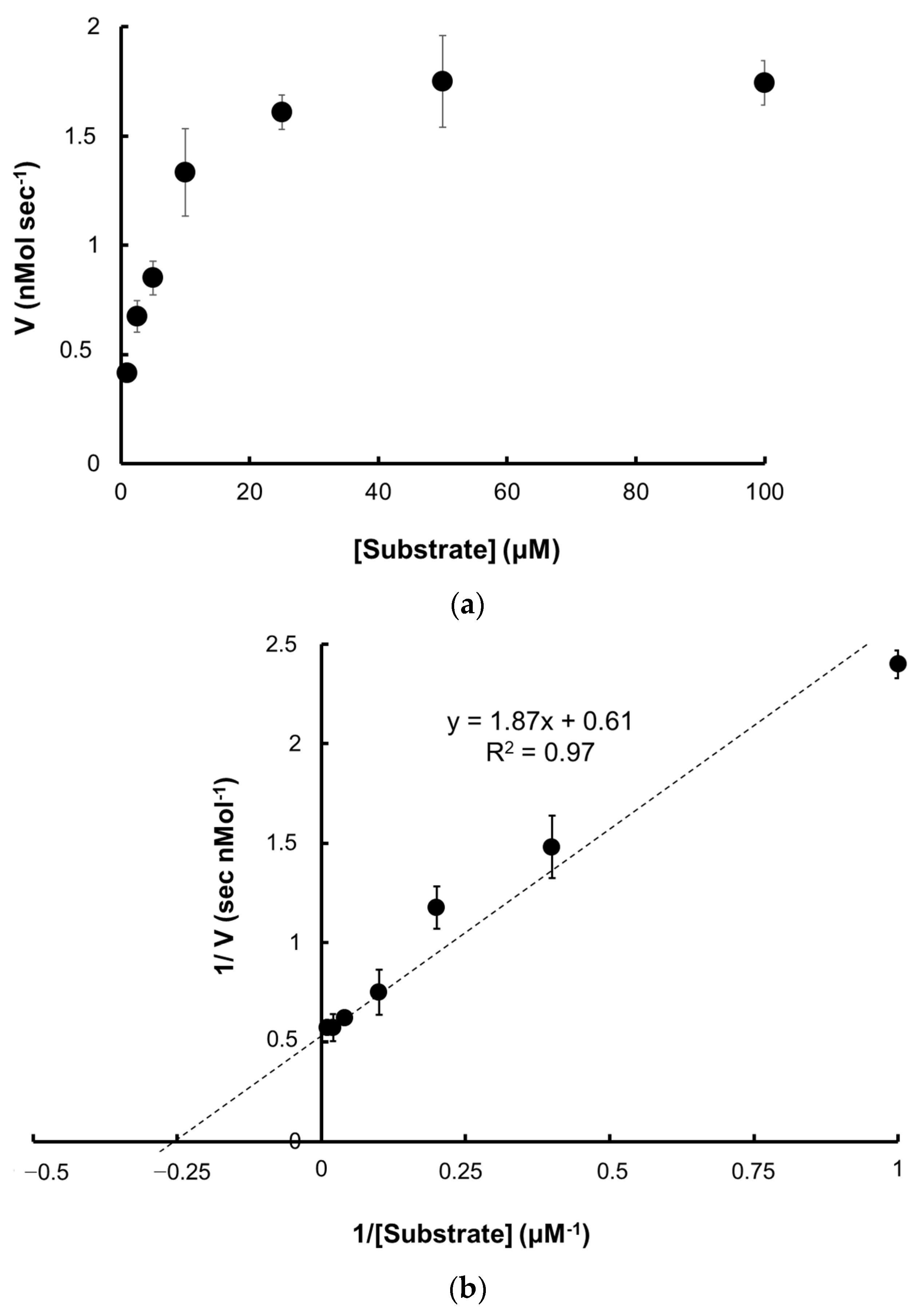
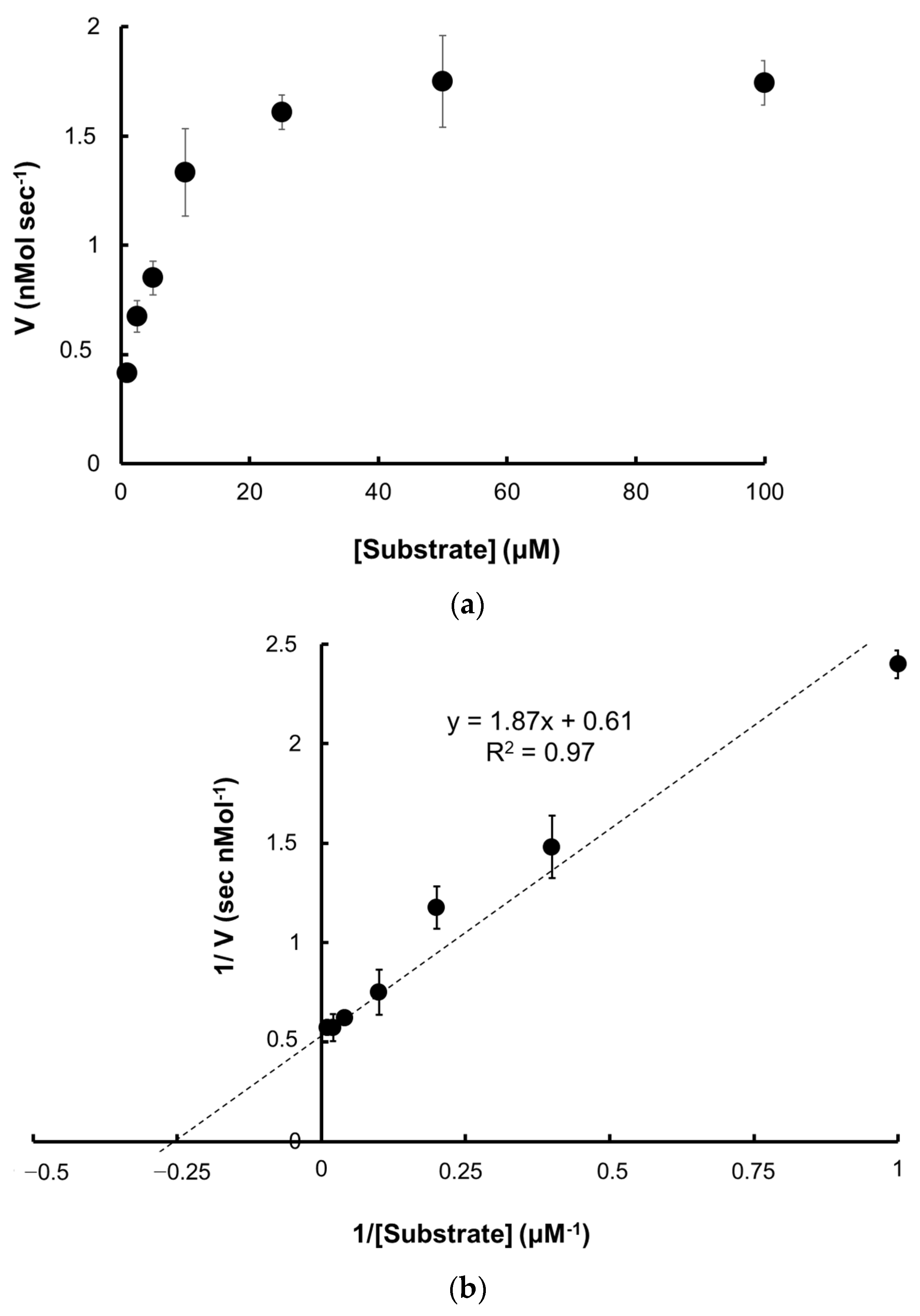
2.5.1. Kinetic Parameters
2.5.2. Temperature and pH-Dependence Activities
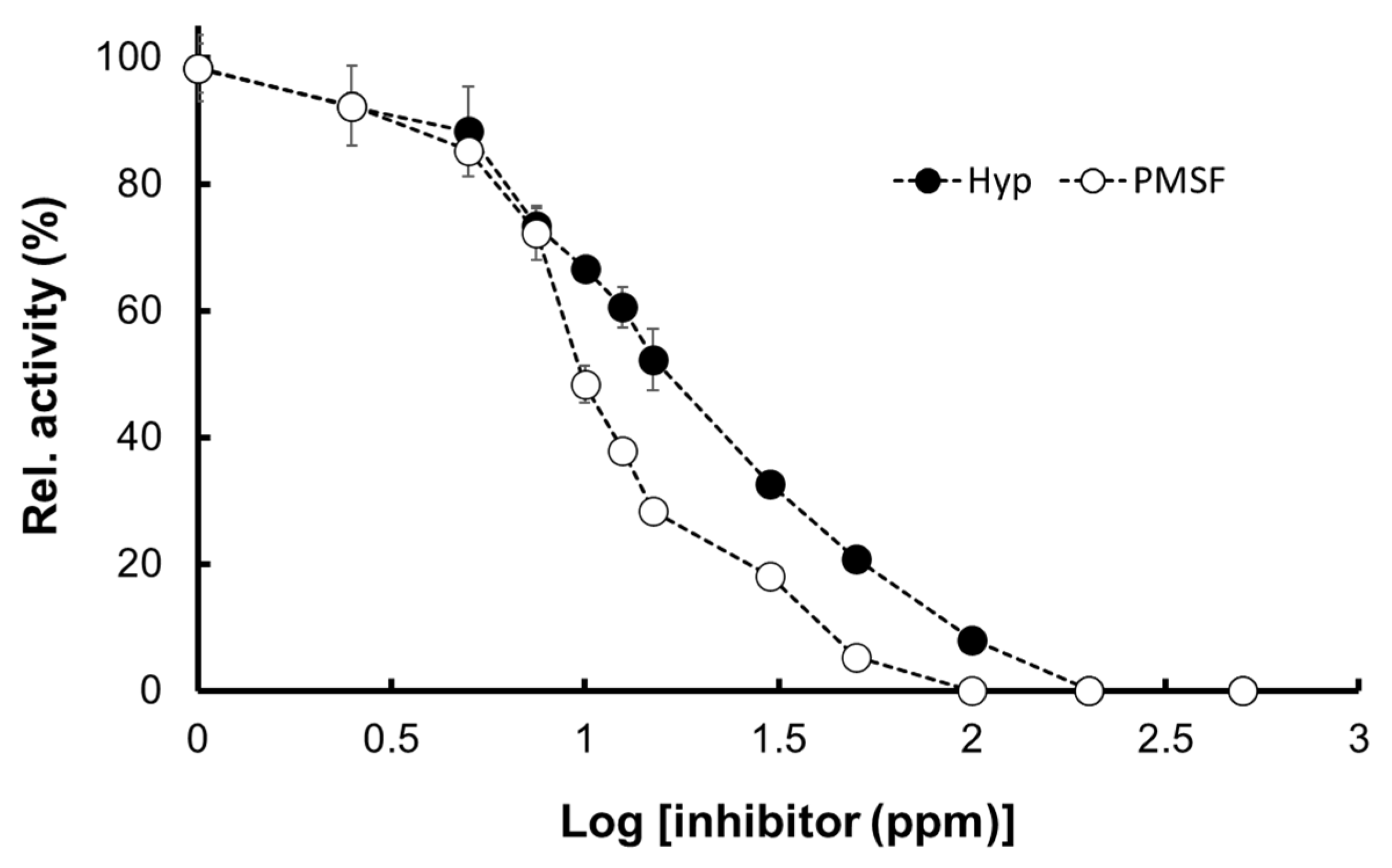
2.5.3. Inhibition by Hyptolide
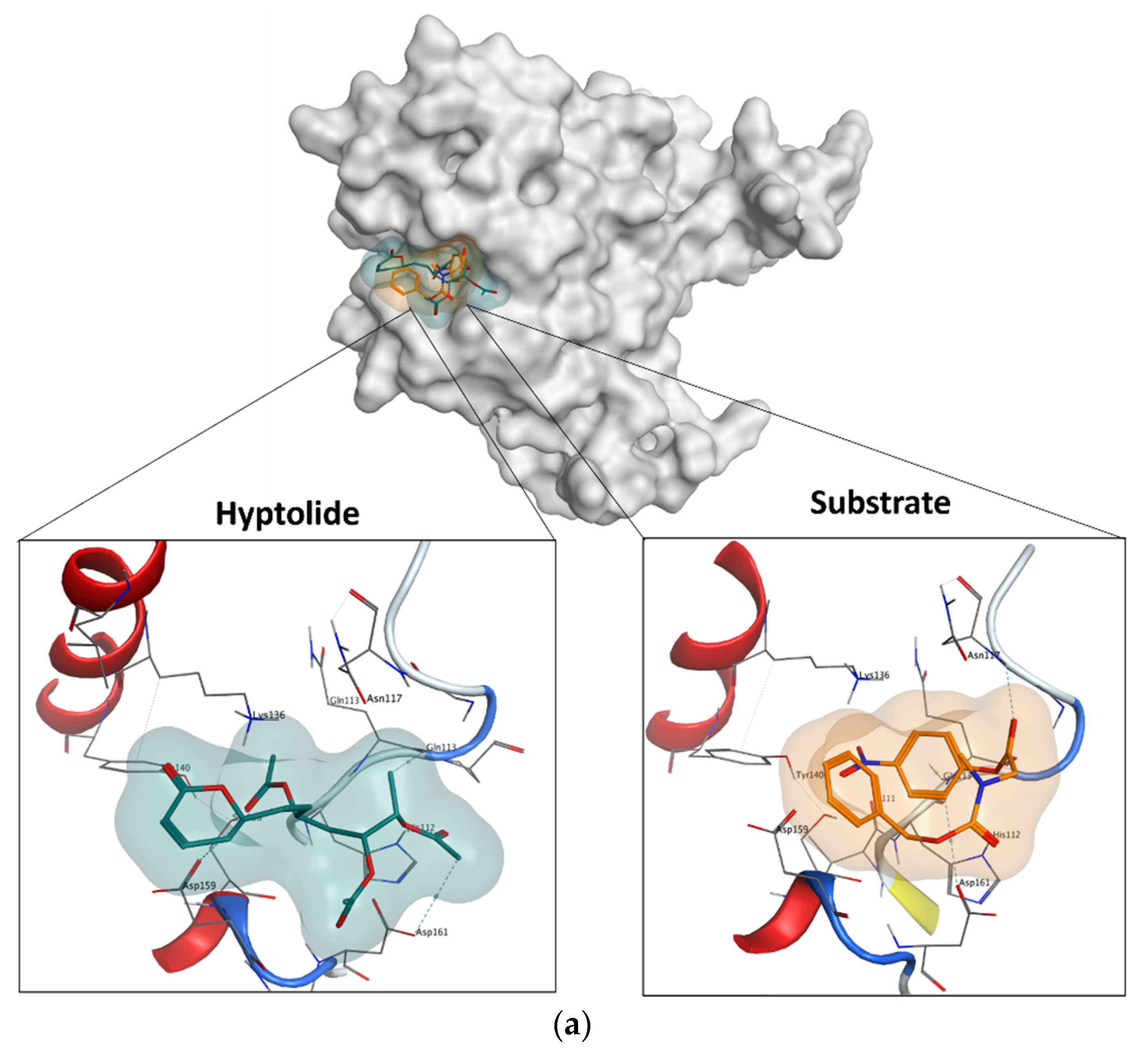
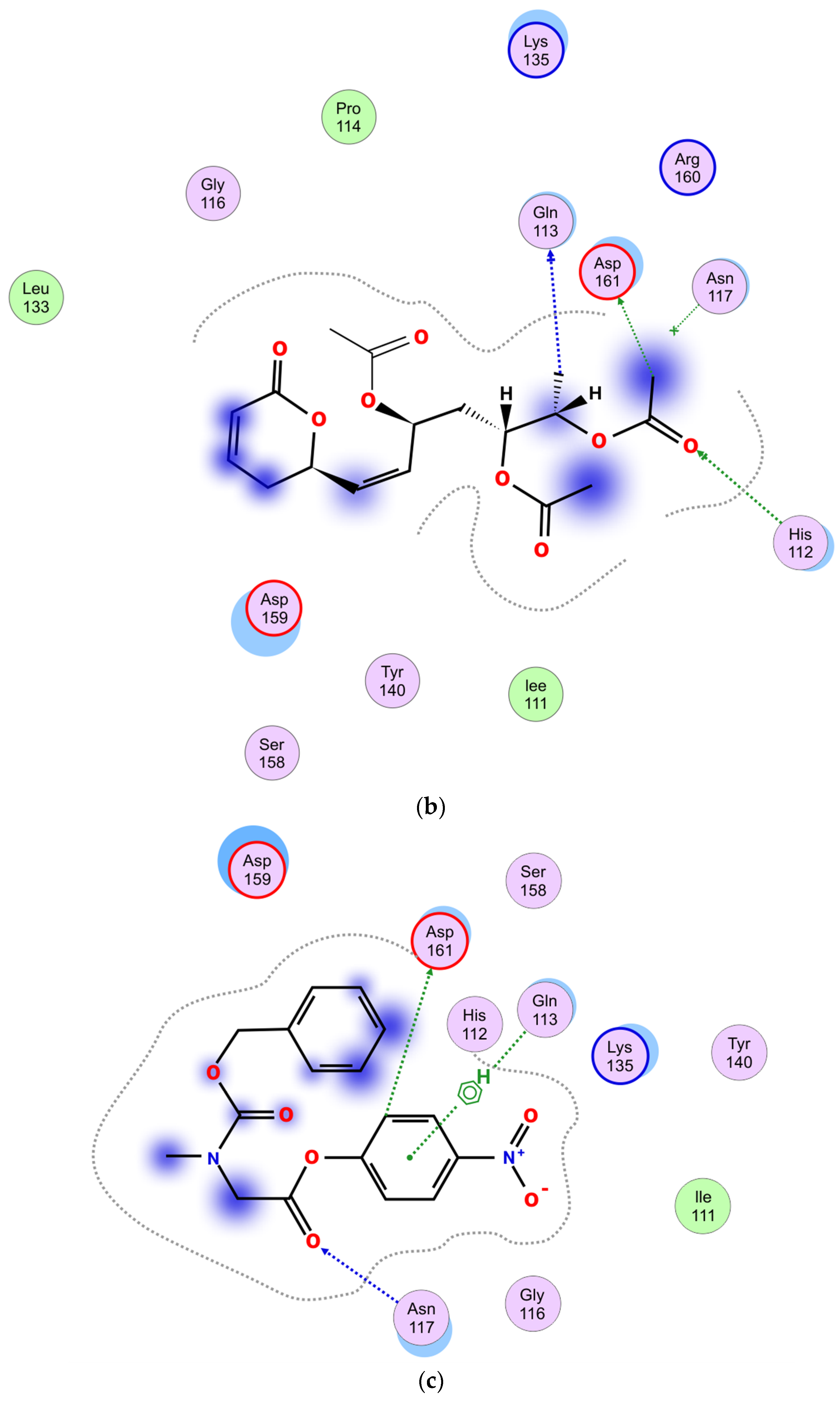
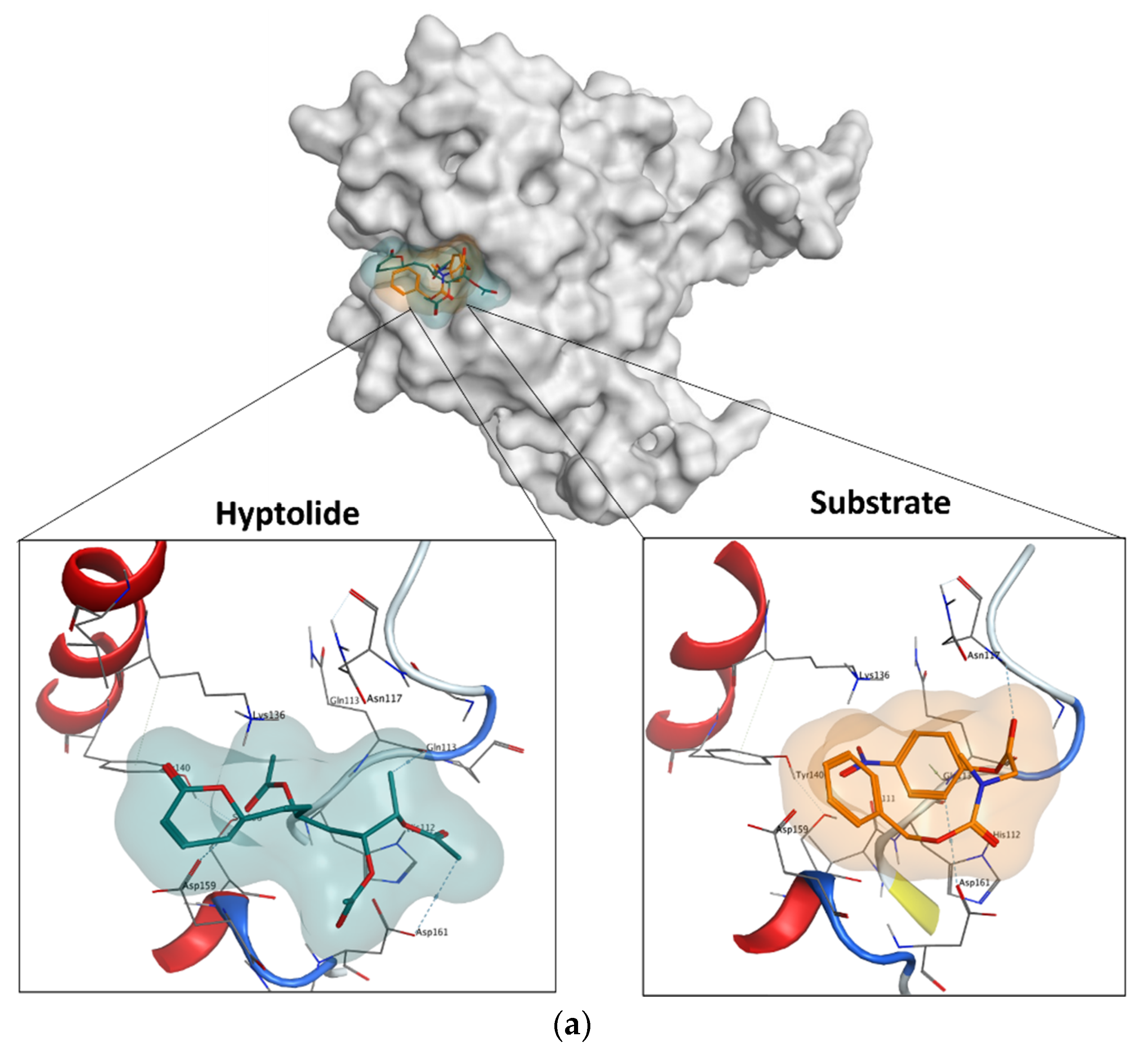
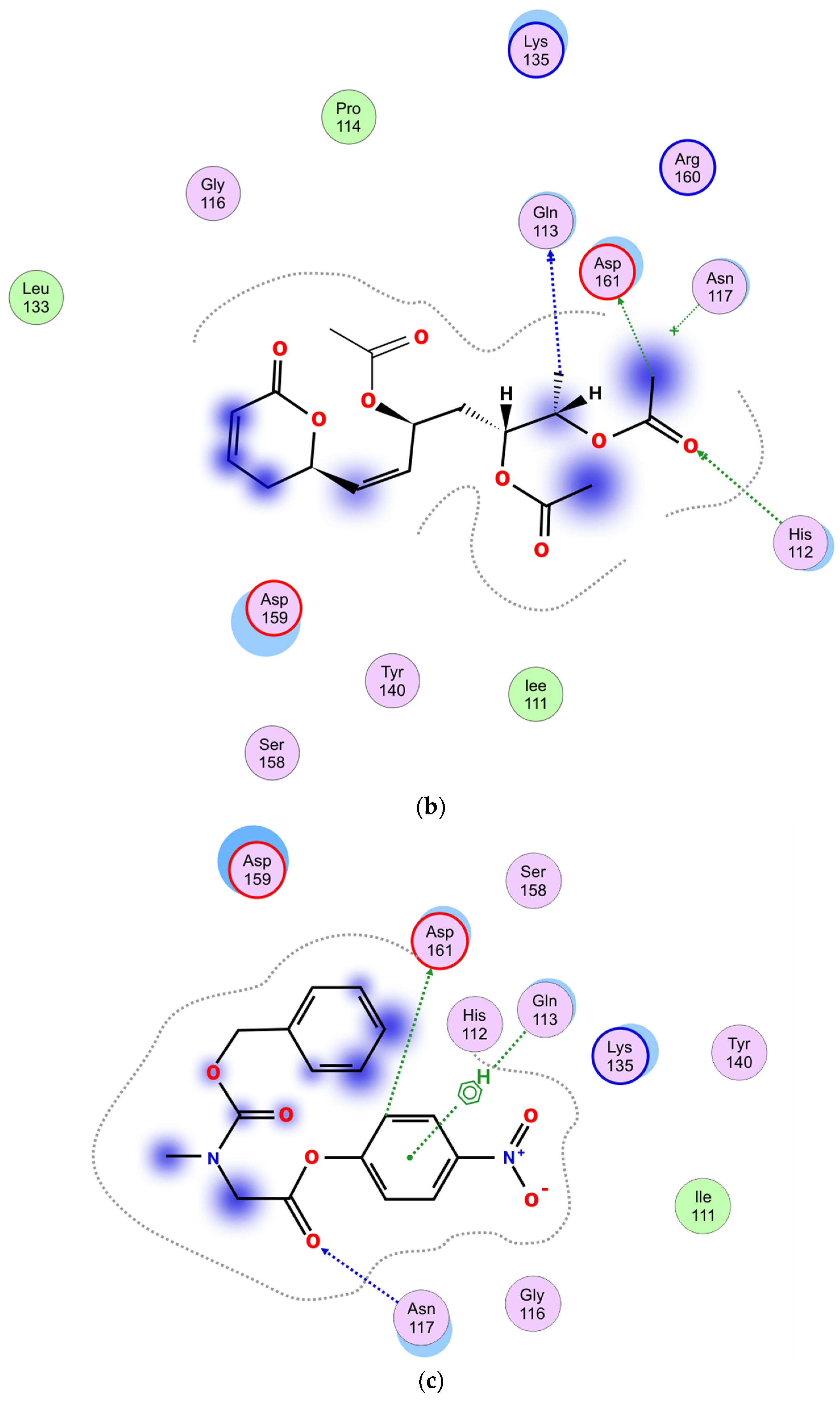
2.5.4. Molecular Docking
3. Discussion
4. Materials and Methods
4.1. Sequence Analysis and Structural Homology Modeling
4.2. Gene Synthesis and Expression System Construction
4.3. Over-Expression and Purification
4.4. Circular Dichroism (CD) Spectra
4.5. Oligomerization
4.6. Protease Activity
4.7. Effect of Temperature and pH on the Catalytic Activity
4.8. Catalytic Inhibition by Hyptolide
4.9. Molecular Docking Analysis
4.9.1. Ligand and Protein Structure Preparation
4.9.2. Molecular Docking Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Schalkwyk, D.A.; Moon, R.W.; Blasco, B.; Sutherland, C.J. Comparison of the susceptibility of Plasmodium knowlesi and Plasmodium falciparum to antimalarial agents. J. Antimicrob. Chemother. 2017, 72, 3051–3058. [Google Scholar] [CrossRef] [PubMed]
- Cox-Singh, J.; Davis, T.M.E.; Lee, K.S.; Shamsul, S.S.G.; Matusop, A.; Ratnam, S.; Rahman, H.A.; Conway, D.J.; Singh, B. Plasmodium knowlesi malaria in humans is widely distributed and potentially life threatening. Clinic. Infect. Dis. 2008, 46, 165–171. [Google Scholar] [CrossRef] [PubMed]
- Rajahram, G.S.; Barber, B.E.; William, T.; Grigg, M.J.; Menon, J.; Yeo, T.W.; Anstey, N.M. Falling Plasmodium knowlesi malaria death rate among adults despite rising incidence, Sabah, Malaysia, 2010–2014. Emerg. Infect. Dis. 2016, 22, 41–48. [Google Scholar] [CrossRef]
- Hussin, N.; Lim, Y.A.L.; Goh, P.P.; William, T.; Jelip, J.; Mudin, R.N. Updates on malaria incidence and profile in Malaysia from 2013 to 2017. Malar. J. 2020, 19, 55. [Google Scholar] [CrossRef] [PubMed]
- Kotepui, M.; Kotepui, K.U.; Milanez, G.D.J.; Masangkay, F.R. Prevalence of severe Plasmodium knowlesi infection and risk factors related to severe complications compared with non-severe P. knowlesi and severe P. falciparum malaria: A systematic review and meta-analysis. Infect. Dis. Poverty 2020, 9, 106. [Google Scholar] [CrossRef] [PubMed]
- Akter, R.; Vythilingam, I.; Khaw, L.T.; Qvist, R.; Lim, Y.A.; Sitam, F.T.; Venugopalan, B.; Sekaran, S.D. Simian malaria in wild macaques: First report from Hulu Selangor district, Selangor, Malaysia. Malar. J. 2015, 14, 386. [Google Scholar] [CrossRef]
- Don, E.; Wilson, D.M.R. Mammal Species of the World. A Taxonomic and Geographic Reference, 3rd ed.; Johns Hopkins University Press: Baltimore, MA, USA, 2005. [Google Scholar]
- Riley, C.M. Monkeys on the edge: Ecology and management of long-tailed macaques and their interface with humans. Int. J. Primatol. 2012, 33, 284–286. [Google Scholar] [CrossRef]
- Nath, M.J.; Bora, A.; Talukdar, P.K.; Das, N.G.; Dhiman, S.; Baruah, I.; Singh, L. A longitudinal study of malaria associated with deforestation in Sonitpur district of Assam. India Geocarto Int. 2012, 27, 79–88. [Google Scholar] [CrossRef]
- Ramdzan, A.R.; Ismail, A.; Zanib, M.Z.S. Prevalence of malaria and its risk factors in Sabah, Malaysia. Int. J. Infect. Dis. 2020, 91, 68–72. [Google Scholar] [CrossRef]
- William, T.; Menon, J. A review of malaria research in Malaysia. Med. J. Malays. 2014, 69, 82–87. [Google Scholar]
- Jenarun, J. Laporan Tahunan RKPBV Sabah 2014; Ibu Pejabat Jabatan Kesihatan Negeri Sabah: Kota Kinabalu, Malaysia, 2014. [Google Scholar]
- Cotter, C.; Sturrock, H.J.; Hsiang, M.S.; Liu, J.; Phillips, A.A.; Hwang, J.; Gueye, C.S.; Fullman, N.; Gosling, R.D.; Feachem, R.G.A. The changing epidemiology of malaria elimination: New strategies for new challenges. Lancet 2013, 382, 900–911. [Google Scholar] [CrossRef]
- Bojang, K.A.; Milligan, P.J.; Pinder, M.; Vigneron, L.; Alloueche, A.; Kester, K.E.; Ballou, W.R.; Conway, D.J.; Reece, W.H.; Gothard, P.; et al. Efficacy of RTS, S/AS02 malaria vaccine against Plasmodium falciparum infection in semi-immune adult men in The Gambia: A randomised trial. Lancet 2001, 358, 1927–1934. [Google Scholar] [CrossRef]
- Pousibet-Puerto, J.; Salas-Coronas, J.; Sánchez-Crespo, A.; Molina-Arrebola, M.A.; Soriano-Pérez, M.J.; Giménez-López, M.J.; Vázquez-Villegas, J.; Cabezas-Fernández, M.T. Impact of using artemisinin-based combination therapy (ACT) in the treatment of uncomplicated malaria from Plasmodium falciparum in a non-endemic zone. Malar. J. 2016, 15, 339. [Google Scholar] [CrossRef] [PubMed]
- Baragaña, B.; Hallyburton, I.; Lee, M.C.; Norcross, N.R.; Grimaldi, R.; Otto, T.D.; Proto, W.R.; Blagborough, A.M.; Meister, S.; Wirjanata, G.; et al. A novel multiple-stage antimalarial agent that inhibits protein synthesis. Nature 2015, 522, 315–320. [Google Scholar] [CrossRef] [PubMed]
- Cai, H.; Kuang, R.; Gu, J.; Wang, Y. Proteases in malaria parasites—A phylogenomic perspective. Curr. Genom. 2011, 12, 417–427. [Google Scholar] [CrossRef]
- Florentin, A.; Stephens, D.R.; Brooks, C.F.; Baptista, R.P.; Muralidharan, V. Plastid biogenesis in malaria parasites requires the interactions and catalytic activity of the Clp proteolytic system. Proc. Natl. Acad. Sci. USA 2020, 117, 13719–13729. [Google Scholar] [CrossRef]
- Frees, D.; Gerth, U.; Ingmer, H. Clp chaperones and proteases are central in stress survival, virulence and antibiotic resistance of Staphylococcus aureus. Int. J. Med. Microb. 2014, 304, 142–149. [Google Scholar] [CrossRef]
- Böttcher, T.; Sieber, S.A. β-Lactones as specific inhibitors of ClpP Attenuate the production of extracellular virulence factors of Staphylococcus aureus. J. Am. Chem. Soc. 2008, 130, 14400–14401. [Google Scholar] [CrossRef]
- Brötz-Oesterhelt, H.; Sass, P. Bacterial caseinolytic proteases as novel targets for antibacterial treatment. Int. J. Med. Microbiol. 2014, 304, 23–30. [Google Scholar] [CrossRef]
- Conlon, B.P.; Nakayasu, E.S.; Fleck, L.E.; LaFleur, M.D.; Isabella, V.M.; Coleman, K.; Leonard, S.N.; Smith, R.D.; Adkins, J.N.; Lewis, K. Activated ClpP kills persisters and eradicates a chronic biofilm infection. Nature 2013, 503, 365–370. [Google Scholar] [CrossRef]
- Gersch, M.; Famulla, K.; Dahmen, M.; Göbl, C.; Malik, I.; Richter, K.; Korotkov, V.S.; Sass, P.; Rübsamen-Schaeff, H.; Madl, T.; et al. AAA+ chaperones and acyldepsipeptides activate the ClpP protease via conformational control. Nat. Commun. 2015, 6, 6320. [Google Scholar] [CrossRef] [PubMed]
- Hackl, M.W.; Lakemeyer, M.; Dahmen, M.; Glaser, M.; Pahl, A.; Lorenz-Baath, K.; Menzel, T.; Sievers, S.; Böttcher, T.; Antes, I.; et al. Phenyl esters are potent inhibitors of caseinolytic Protease P and reveal a stereogenic switch for deoligomerization. J. Am. Chem. Soc. 2015, 137, 8475–8483. [Google Scholar] [CrossRef] [PubMed]
- Nagpal, J.; Paxman, J.J.; Zammit, J.E.; Alhuwaider, A.; Truscott, K.N.; Heras, B.; Dougan, D.A. Molecular and structural insights into an asymmetric proteolytic complex (ClpP1P2) from Mycobacterium smegmatis. Sci. Rep. 2019, 9, 18019. [Google Scholar] [CrossRef] [PubMed]
- El Bakkouri, M.; Pow, A.; Mulichak, A.M.; Cheung, K.L.Y.; Artz, J.D.; Amani, M.; Fell, S.; de Koning-Ward, T.F.; Goodman, C.D.; McFadden, G.I.; et al. The Clp chaperones and proteases of the human malaria parasite Plasmodium falciparum. J. Mol. Biol. 2010, 404, 456–477. [Google Scholar] [CrossRef] [PubMed]
- Schröder, D.; Goldberg, N.; Zummack, W.; Schwarz, H.; Poutsma, J.C.; Squires, R.R. Generation of α-acetolactone and the acetoxyl diradical •CH2COO• in the gas phase. IMSPF8 1997, 165–166, 71–82. [Google Scholar] [CrossRef]
- Suzery, M.; Cahyono, B. Evaluation of cytotoxicity effect of Hyptis pectinata Poit (Lamiaceae) extracts using BSLT and MTT methods. Jurnal Sains Dan Matematika 2014, 22, 84–88. [Google Scholar]
- Suzery, M.; Cahyono, B.; Amalina, N.D.; Budiman, C.; Bayu, I.; Asy’ari, M.; Widayat, W. Antiplasmodial activity of Hyptis pectinata extract targeted ClpP inhibition. Thai. J. Pharm. Sci. 2021, 45, 527–531. [Google Scholar]
- Santana, F.R.; Lina-Dulcey, L.; Antunes, V.; Tormena, C.; Cominetti, M.R.; Duarte, M.C.; da Silva, J.A. Evaluation of the cytotoxicity on breast cancer cell of extracts and compounds isolated from Hyptis pectinata (L.) poit. Nat. Prod. Res. 2019, 34, 102–109. [Google Scholar] [CrossRef]
- Barbosa, C.V.; Aquino, P.G.V.; Ribeiro-Junior, K.A.L. Cytotoxic and antitumor activities of Hyptis pectinata (Sambacaitá) extract. Pharmacologyonline 2012, 3, 70–74. [Google Scholar]
- Wu, C.S.; Ikeda, K.; Yang, J.T. Ordered conformation of polypeptides and proteins in acidic dodecyl sulfate solution. Biochemistry 1981, 20, 566–570. [Google Scholar] [CrossRef]
- Kress, W.; Maglica, Z.; Weber-Ban, E. Clp chaperone-proteases: Structure and function. Res. Microbiol. 2009, 160, 618–628. [Google Scholar] [CrossRef] [PubMed]
- Zeiler, E.; List, A.; Alte, F.; Gersh, M.; Wachtel, R.; Poreba, M.; Drag, M.; Groll, M.; Sieber, S.A. Structural and functional insights into caseinolytic proteases reveal an unprecedented regulation principle of their catalytic triad. Proc. Natl. Acad. Sci. USA 2013, 110, 11302–11307. [Google Scholar] [CrossRef] [PubMed]
- El Bakkouri, M.; Rathore, S.; Calmettes, C. Structural insights into the inactive subunit of the apicoplast-localized caseinolytic protease complex of Plasmodium falciparum. J. Biol. Chem. 2013, 288, 1022–1031. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.E.; Baker, T.A.; Sauer, R.T. Control of substrate gating and translocation into ClpP by channel residues and ClpX binding. J. Mol. Biol. 2010, 399, 707–718. [Google Scholar] [CrossRef] [PubMed]
- Budiman, C.; Bando, K.; Angkawidjaja, C.; Koga, Y.; Takano, K.; Kanaya, S. Engineering of monomeric FK506-binding protein 22 with peptidyl prolyl cis-trans isomerase: Importance of a V-shaped dimeric structure for binding to protein substrate. FEBS J. 2009, 276, 4091–4101. [Google Scholar] [CrossRef] [PubMed]
- Akopian, T.; Kandror, O.; Raju, R.M.; Unnikrishnan, M.; Rubin, E.J.; Goldberg, A.L. The active ClpP protease from M. tuberculosis is a complex composed of a heptameric ClpP1 and a ClpP2 ring. EMBO J. 2012, 31, 1529–1541. [Google Scholar] [CrossRef] [PubMed]
- Dhara, A.; Hussain, M.S.; Datta, D.; Kumar, M. Insights to the assembly of a functionally active leptospiral ClpP1P2 protease complex along with Its ATPase chaperone ClpX. ACS Omega 2019, 4, 12880–12895. [Google Scholar] [CrossRef]
- Kang, S.G.; Dimitrova, M.N.; Ortega, J.; Ginsburg, A.; Maurizi, M.R. Human mitochondrial ClpP is a stable heptamer that assembles into a tetradecamer in the presence of ClpX. J. Biol. Chem. 2005, 280, 35424–35432. [Google Scholar] [CrossRef]
- Budiman, C.; Angkawidjaja, C.; Motoike, H.; Koga, Y.; Takano, K.; Kanaya, S. Crystal structure of N-domain of FKBP22 from Shewanella sp. SIB1: Dimer dissociation by disruption of Val-Leu knot. Prot. Sci. 2011, 20, 1755–1764. [Google Scholar] [CrossRef]
- Obeng, E.M.; Brossette, T.; Ongkudon, C.M.; Budiman, C.; Maas, R.; Jose, J. The workability of Escherichia coli BL21 (DE3) and Pseudomonas putida KT2440 expression platforms with autodisplayed cellulases: A comparison. Appl. Microbiol. Biotechnol. 2018, 102, 4829–4841. [Google Scholar] [CrossRef]
- Goh, C.K.W.; Silvester, J.; Wan Mahadi, W.N.S.; Chin, L.P.; Ying, L.T.; Leow, T.C.; Kurahashi, R.; Takano, K.; Budiman, C. Expression and characterization of functional domains of FK506-binding protein 35 from Plasmodium knowlesi. Protein Eng. Des. Sel. 2018, 31, 489–498. [Google Scholar] [CrossRef] [PubMed]
- Razali, R.; Budiman, C.; Kamaruzaman, K.A. Soluble expression and catalytic properties of codon-optimized recombinant bromelain from MD2 pineapple in Escherichia coli. Protein 2021, 40, 406–418. [Google Scholar] [CrossRef] [PubMed]
- Van Noorden, C.J.F. Metabolic mapping by enzyme histochemistry in living animals, tissues and cells. In Proceedings of the XIIIth International Congress of Histochemistry and Cytochemistry, Gdansk, Poland, 23–27 August 2008. [Google Scholar]
- Budiman, C.; Tadokoro, T.; Angkwidjaja, C.; Koga, Y.; Kanaya, S. Role of polar and nonpolar residues at the active site for PPIase activity of FKBP22 from Shewanella sp. SIB1. FEBS J. 2012, 79, 976–986. [Google Scholar] [CrossRef] [PubMed]
- Akopian, T.; Kandror, O.; Tsu, C. Cleavage specificity of Mycobacterium tuberculosis ClpP1P2 protease and identification of novel peptide substrates and boronate inhibitors with anti-bacterial activity. Enzymology 2015, 290, 11008–11020. [Google Scholar] [CrossRef]
- Yu, A.Y.H.; Houry, W.A. ClpP: A distinctive family of cylindrical energy-dependent serine proteases. FEBS Lett. 2007, 581, 3749–3757. [Google Scholar] [CrossRef]
- Kuhn, Y.; Rohrbach, P.; Lanzer, M. Quantitative pH measurements in Plasmodium falciparum-infected erythrocytes using pHluorin. Cell Microbiol. 2007, 9, 1004–1013. [Google Scholar] [CrossRef]
- Saliba, K.J.; Kirk, K. pH regulation in the intracellular malaria parasite, Plasmodium falciparum. J. Biochem. 1999, 274, 33213–33219. [Google Scholar] [CrossRef]
- Bray, P.G.; Janneh, O.; Raynes, K.J.; Mungthin, M.; Ginsburg, H.; Ward, S.A. Cellular uptake of chloroquine is dependent on binding to ferriprotoporphyrin IX and is independent of NHE activity in Plasmodium falciparum. J. Cell Biol. 1999, 145, 363–376. [Google Scholar] [CrossRef]
- Rao, M.B.; Tanksale, A.M.; Ghatge, M.S.; Deshpande, V.V. Molecular and biotechnological aspects of microbial proteases. Microbiol. Mol. Biol. Rev. 1998, 62, 597–635. [Google Scholar] [CrossRef]
- Vieille, C.; Zeikus, G.J. Hyperthermophilic enzymes: Sources, uses, and molecular mechanisms for thermostability. Microbiol. Mol. Biol. Rev. 2001, 65, 1–43. [Google Scholar] [CrossRef]
- Crowther, G.J.; He, P.; Rodenbough, P.P.; Thomas, A.P.; Kovzun, K.V.; Leibly, D.J.; Bhandari, J.; Castaneda, L.J.; Hol, W.G.J.; Gelb, M.H.; et al. Use of thermal melt curves to assess the quality of enzyme preparations. Anal. Biochem. 2009, 399, 268–275. [Google Scholar] [CrossRef] [PubMed]
- Mathews, E.S.; Jezewski, A.J.; Audrey, R. Protein prenylation and Hsp40 in thermotolerance of Plasmodium falciparum malaria parasites. mBio 2021, 12, e00760-21. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, T.K.; Purkait, S. Total synthesis of hyptolide. Tetrahedron Lett. 2008, 49, 5502–5504. [Google Scholar] [CrossRef]
- Culp, E.; Wright, G.D. Bacterial proteases, untapped antimicrobial drug target. J. Antibiot. 2016, 70, 366–377. [Google Scholar] [CrossRef] [PubMed]
- Razak, A.R.; Suzery, M.; Razali, R.; Amin, Z.; Mokhtar, R.A.M.; Lee, P.C.; Budiman, C. Technical data on the inhibition properties of some medicinal plant extracts towards caseinolytic protease proteolytic subunit of Plasmodium knowlesi. Data Br. 2021, 39, 107588. [Google Scholar] [CrossRef] [PubMed]
- Faraday, C.J.; Craik, C.S. Mechanisms of macromolecular protease inhibitors. ChemBioChem 2010, 11, 2341–2346. [Google Scholar] [CrossRef] [PubMed]
- Du, X.; Li, Y.; Xia, Y.L.; Ai, S.; Liang, J.; Sang, P.; Ji, X.; Liu, S. Insights into protein–ligand interactions: Mechanisms, models, and methods. Int. J. Mol. Sci. 2016, 17, 144. [Google Scholar] [CrossRef]
- Fong, C.W. The effect of desolvation on the binding of inhibitors to HIV-1 protease and cyclin-dependent kinases: Causes of resistance. Bioorg. Med. Chem. Lett. 2016, 26, 3705–3713. [Google Scholar] [CrossRef]
- Razali, R.; Kumar, V.; Budiman, C. Structural insights into the enzymatic activity of cysteine protease bromelain of MD2 pineapple. Pak. J. Biol. Sci. 2020, 23, 829–838. [Google Scholar] [CrossRef]
- Geourjon, C.; Deleage, G. SOPMA: Significant improvements in protein secondary structure prediction by consensus prediction from multiple alignments. Comput. Appl. Biosci. 1995, 11, 681–684. [Google Scholar] [CrossRef]
- Benkert, P.; Biasini, M.; Schwede, T. Toward the estimation of absolute quality of individual protein structure models. Bioinformatics 2011, 27, 343–350. [Google Scholar] [CrossRef] [PubMed]
- Benkert, P.; Künzli, M.; Schwede, T. QMEAN server for protein model quality estimation. Nucleic Acids Res. 2009, 37, W510–W514. [Google Scholar] [CrossRef] [PubMed]
- Lovell, S.C.; Davis, I.W.; Arendall, W.B.; de Bakker, P.I.W.; Word, J.M.; Prisant, M.G.; Richardson, J.S.; Richardson, D.C. Structure validation by Calpha geometry: Phi, psi and Cbeta deviation. Proteins 2003, 50, 437–450. [Google Scholar] [CrossRef] [PubMed]
- Eisenberg, D.; Luthy, R.; Bowie, J.U. VERIFY3D: Assessment of protein models with three-dimensional profiles. Methods Enzymol. 1997, 277, 396–404. [Google Scholar]
- Laemmli, U. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 1970, 227, 680–685. [Google Scholar] [CrossRef]
- Goodwin, T.W.; Morton, R.A. The spectrophotometric determination of tyrosine and tryptophan in proteins. Biochem. J. 1946, 40, 628–632. [Google Scholar] [CrossRef]
- Tripathi, P. Calculation of thermodynamic parameters of protein unfolding using far-ultraviolet circular dichroism. J. Protein Proteomics. 2013, 4, 85–91. [Google Scholar]
- Foukis, A.; Stergiou, P.-Y.; Theodorou, L.G.; Papagianni, M.; Papamichael, E.M. Purification, kinetic characterization and properties of a novel thermo-tolerant extracellular protease from Kluyveromyces marxianus IFO 0288 with potential biotechnological interest. Bioresour. Technol. 2012, 123, 214–220. [Google Scholar] [CrossRef]
- Suzery, M.; Cahyono, B.; Amalina, N.D. Antiproliferative and apoptosis effect of hyptolide from Hyptis pectinata (L.) Poit on human breast cancer cells. J. Appl. Pharm. Sci. 2020, 10, 001–006. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Step | Total Activity (U) | Total Protein (mg) | Specific Activity (U mg−1) | Purification Fold | Yield (%) |
|---|---|---|---|---|---|
| Cell lysate | 1822.03 | 64.38 | 28.30 | 1 | 100 |
| Ni2+-NTA affinity chromatography | 1607.21 | 2.2 | 730.51 | 25.81 | 88.21 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Budiman, C.; Razak, R.A.; Unggit, A.R.A.; Razali, R.; Suzery, M.; Mokhtar, R.A.M.; Lee, P.-C.; Utomo, D.H. Catalytic Properties of Caseinolytic Protease Subunit of Plasmodium knowlesi and Its Inhibition by a Member of δ-Lactone, Hyptolide. Molecules 2022, 27, 3787. https://doi.org/10.3390/molecules27123787
Budiman C, Razak RA, Unggit ARA, Razali R, Suzery M, Mokhtar RAM, Lee P-C, Utomo DH. Catalytic Properties of Caseinolytic Protease Subunit of Plasmodium knowlesi and Its Inhibition by a Member of δ-Lactone, Hyptolide. Molecules. 2022; 27(12):3787. https://doi.org/10.3390/molecules27123787
Chicago/Turabian StyleBudiman, Cahyo, Raimalynah Abd Razak, Angelesa Runin Anak Unggit, Rafida Razali, Meiny Suzery, Ruzaidi Azli Mohd Mokhtar, Ping-Chin Lee, and Didik Huswo Utomo. 2022. "Catalytic Properties of Caseinolytic Protease Subunit of Plasmodium knowlesi and Its Inhibition by a Member of δ-Lactone, Hyptolide" Molecules 27, no. 12: 3787. https://doi.org/10.3390/molecules27123787
APA StyleBudiman, C., Razak, R. A., Unggit, A. R. A., Razali, R., Suzery, M., Mokhtar, R. A. M., Lee, P.-C., & Utomo, D. H. (2022). Catalytic Properties of Caseinolytic Protease Subunit of Plasmodium knowlesi and Its Inhibition by a Member of δ-Lactone, Hyptolide. Molecules, 27(12), 3787. https://doi.org/10.3390/molecules27123787