
Synthesis and Activity of Triazole-Adenosine Analogs as Protein Arginine Methyltransferase 5 Inhibitors



Abstract
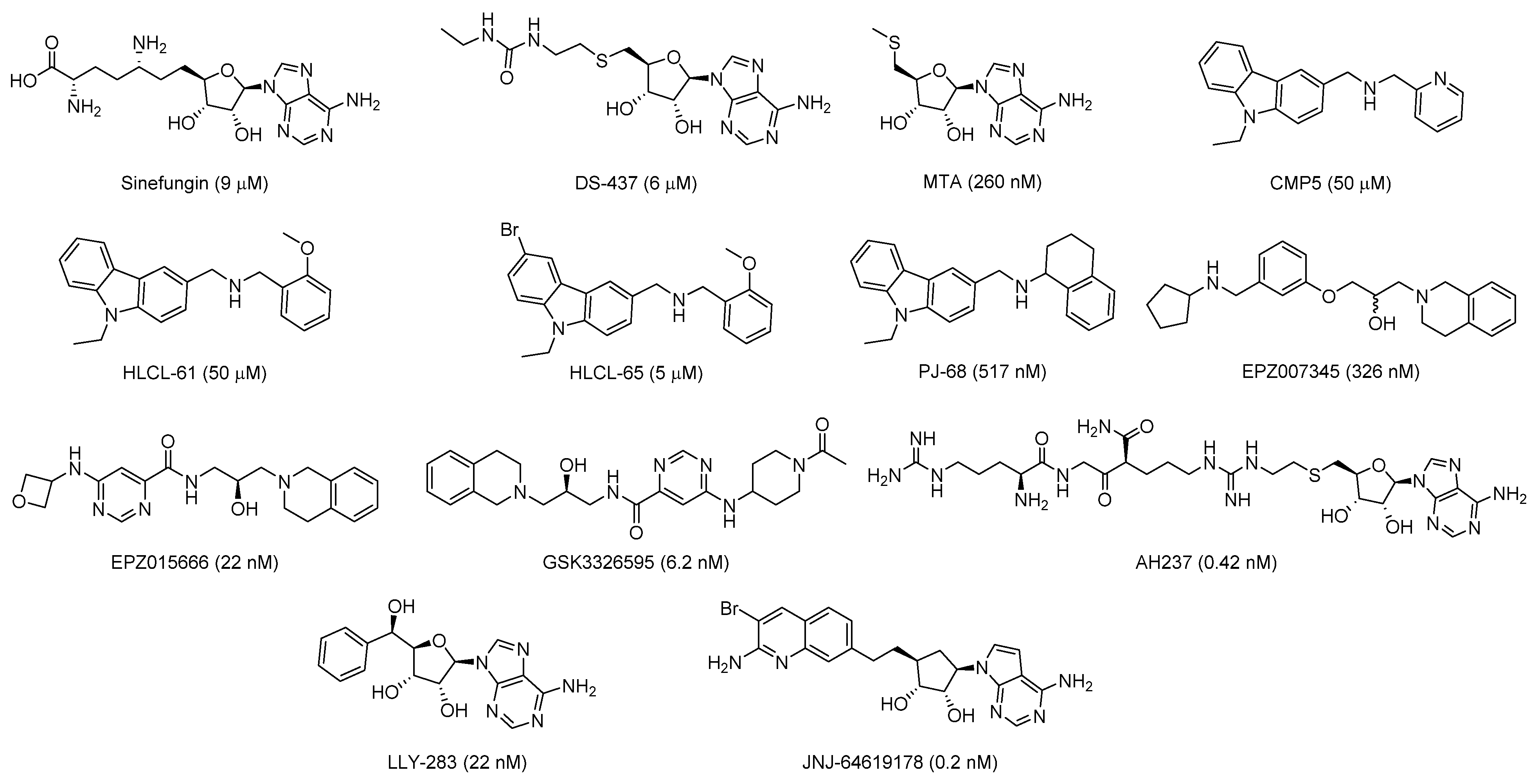
:1. Introduction
2. Results and Discussion
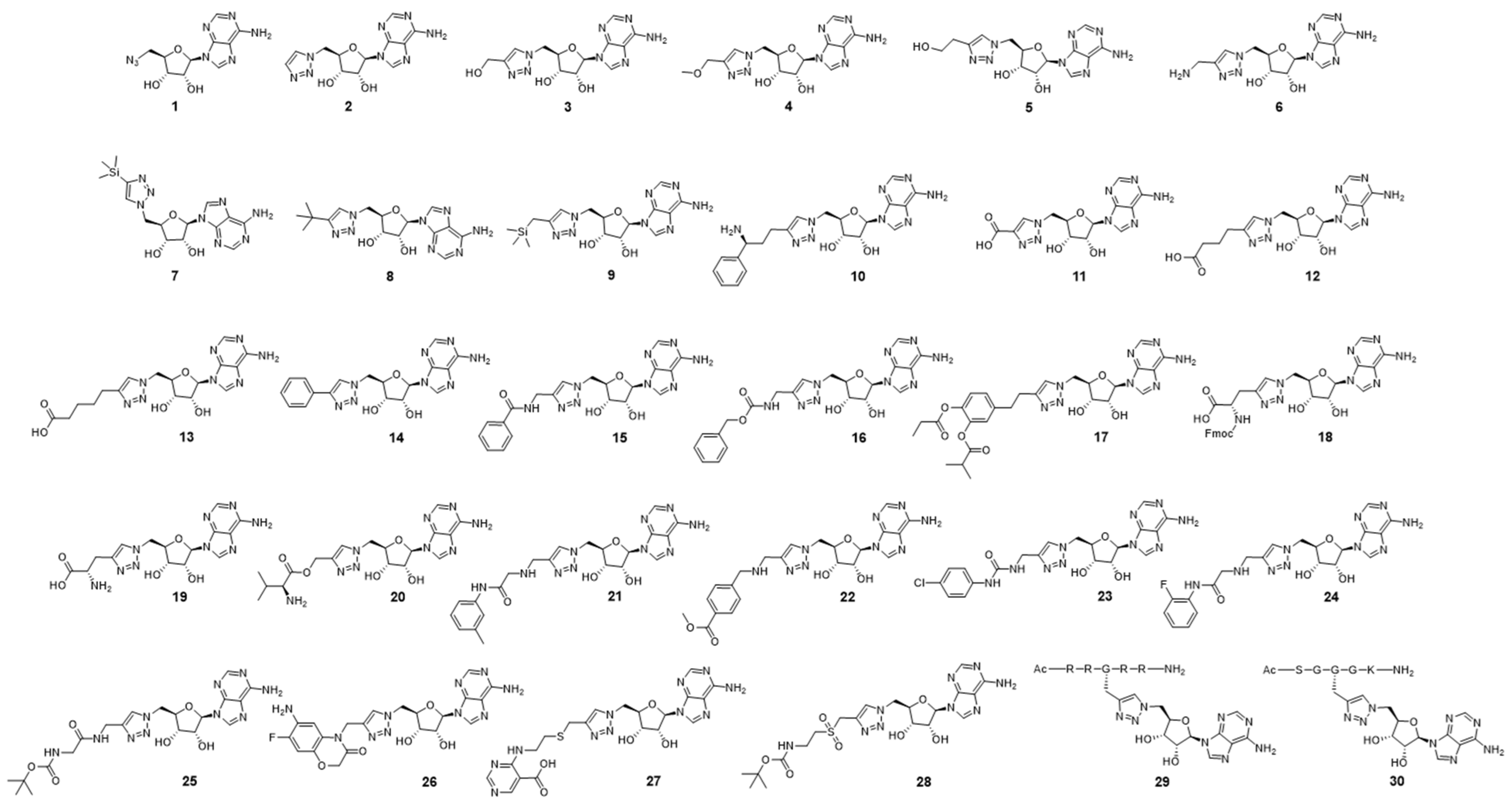
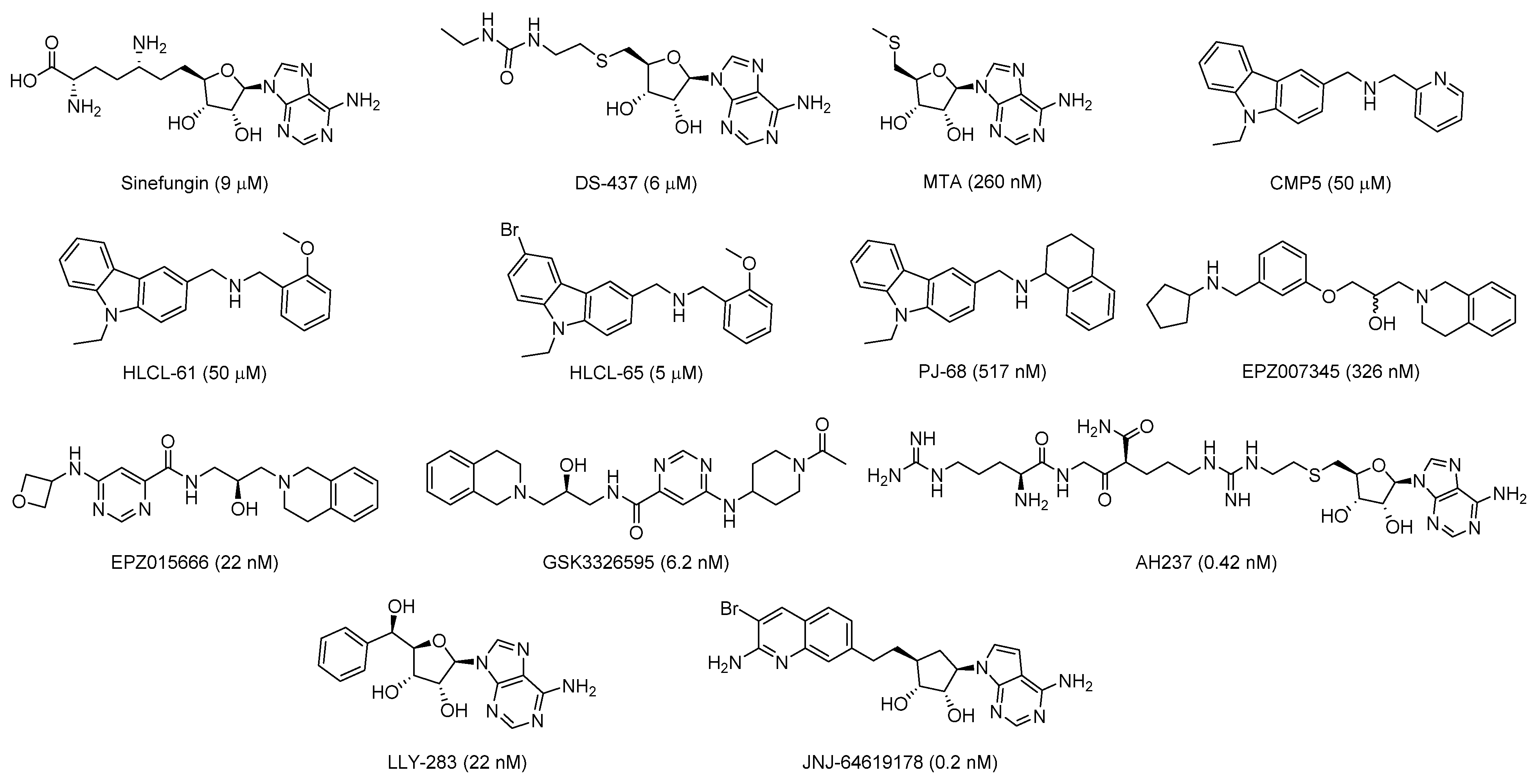
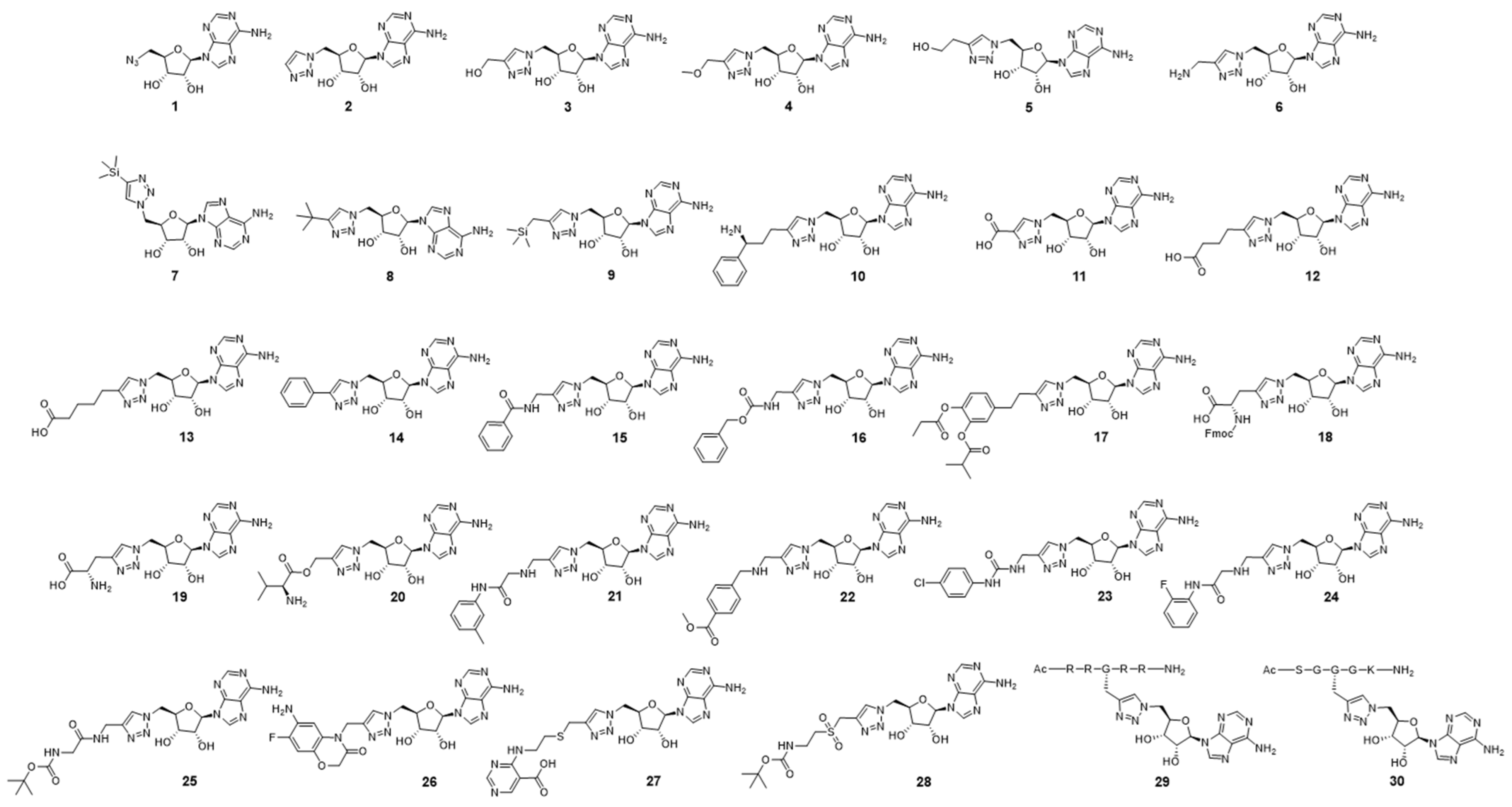
2.1. Design of Triazole-Adenosine Analog Inhibitors of PRMT5
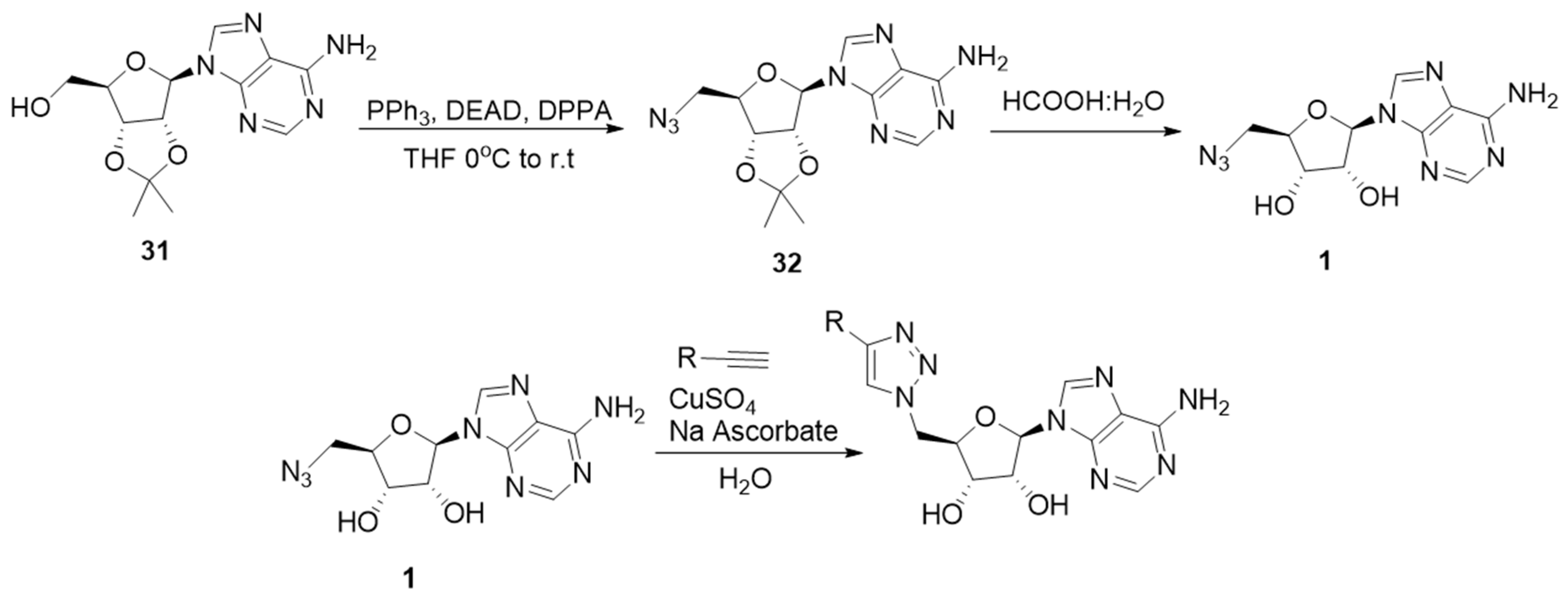
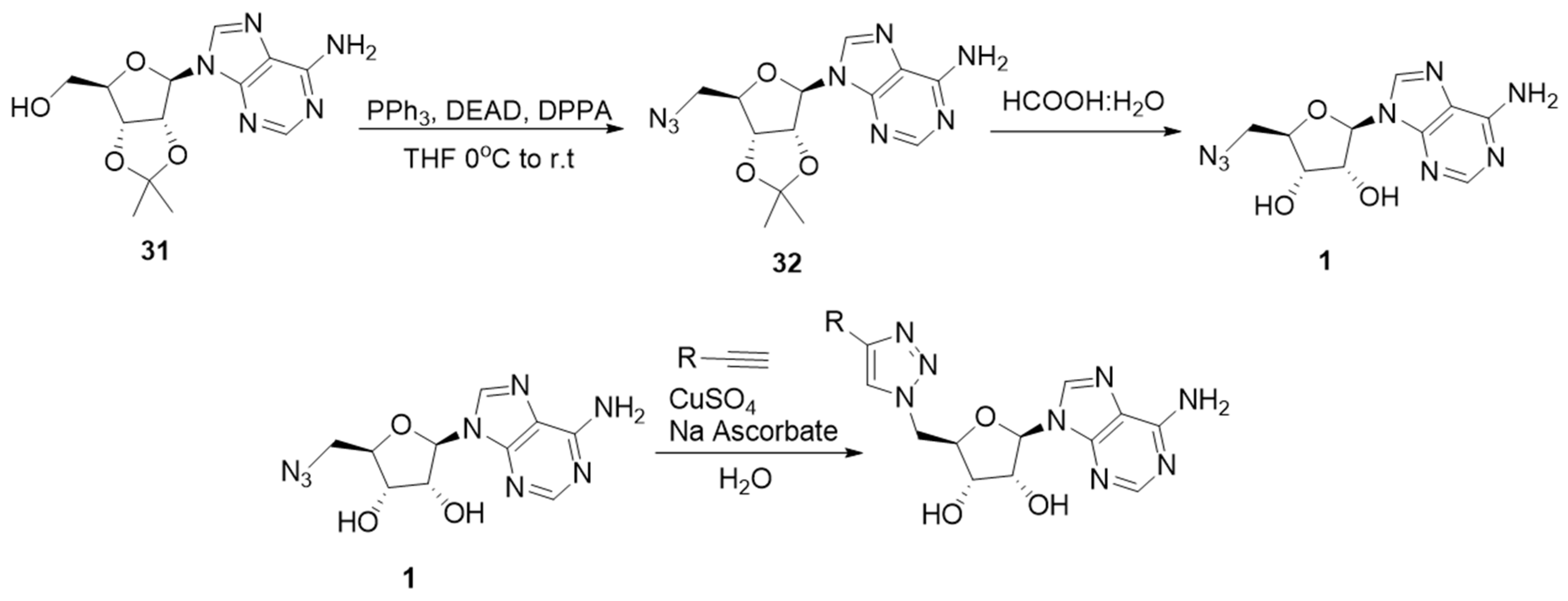
2.2. Synthesis of the Triazole-Adenosine Inhibitors
2.3. Biochemical Evaluation of the Triazole-Adenosine Analogs
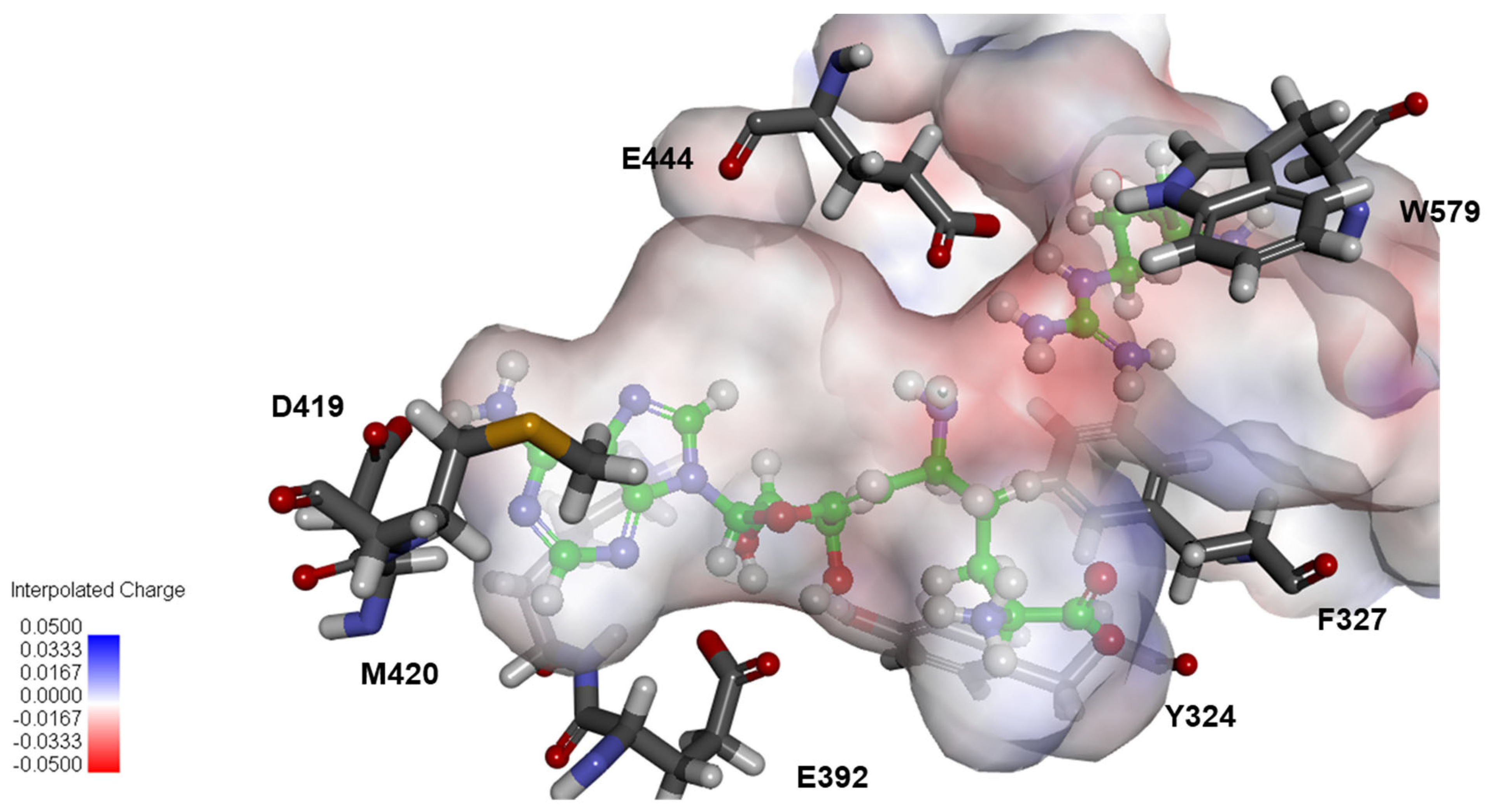
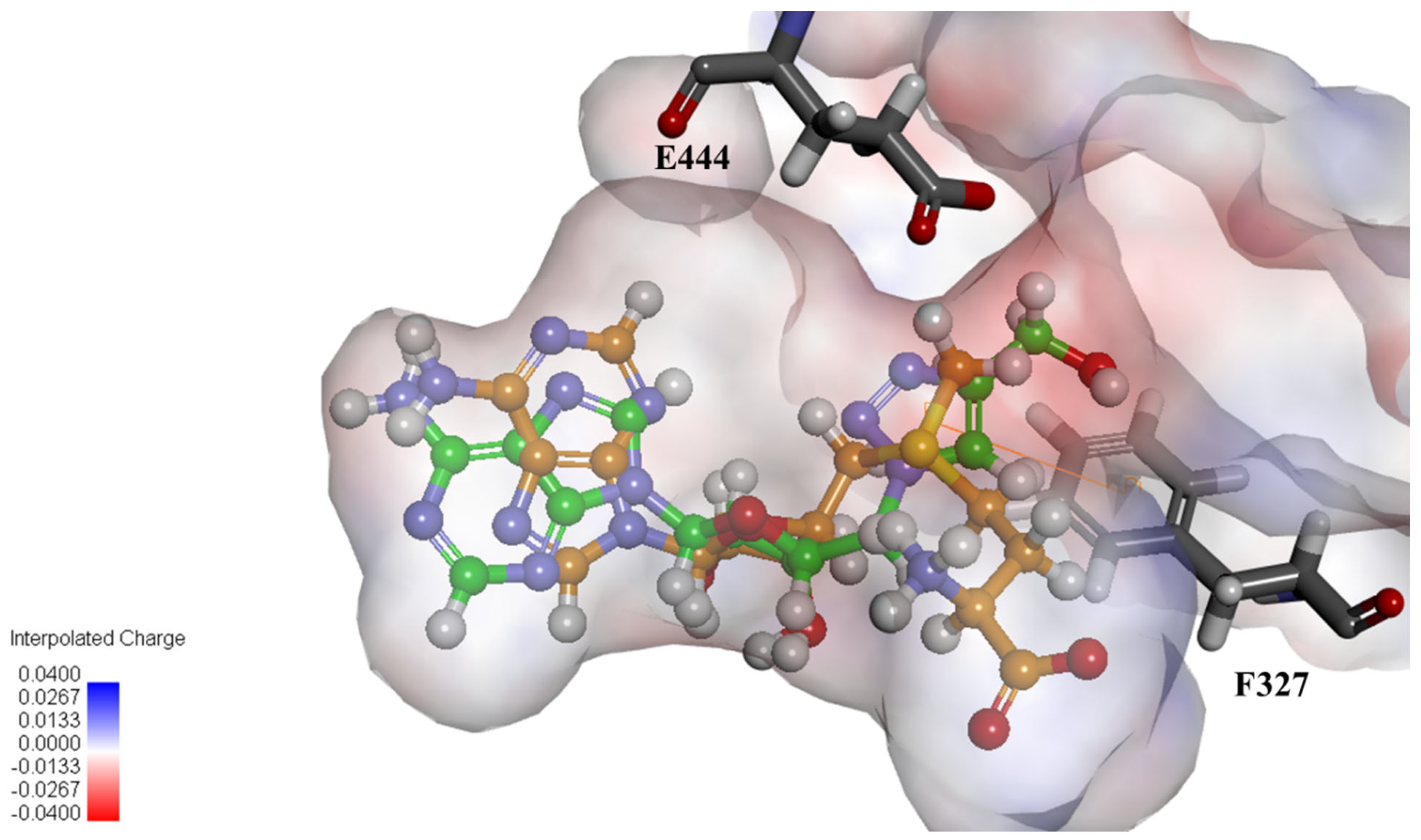
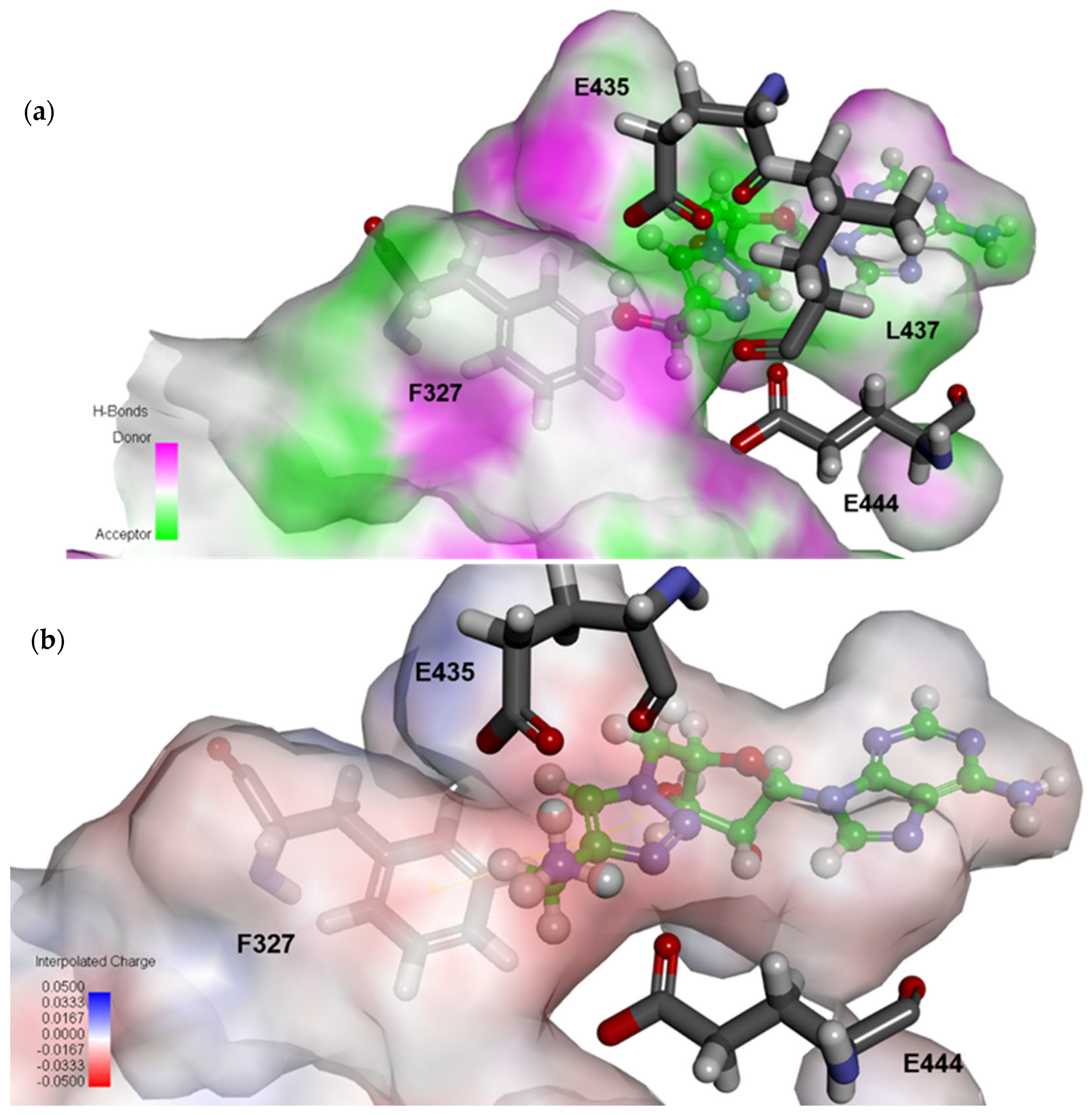
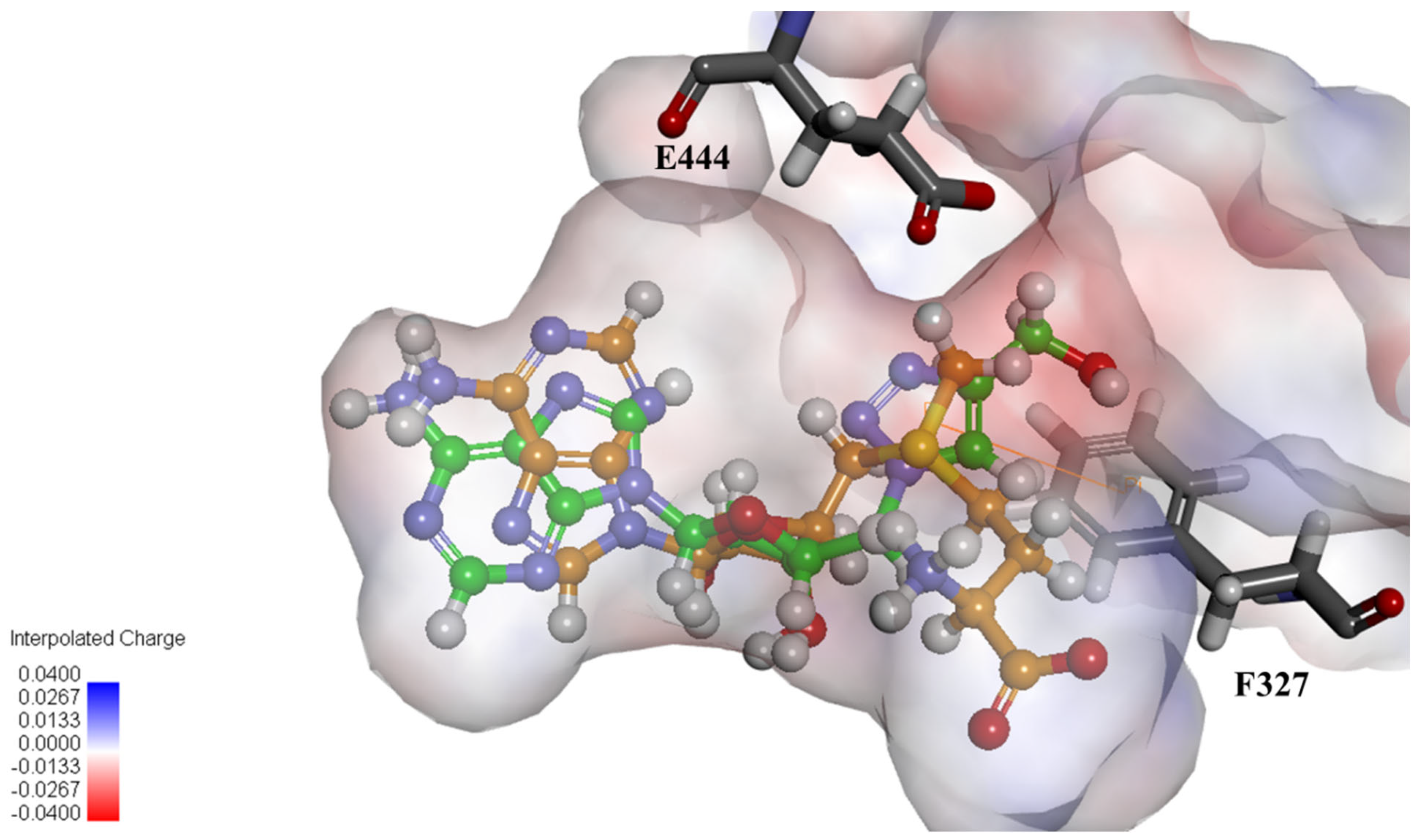
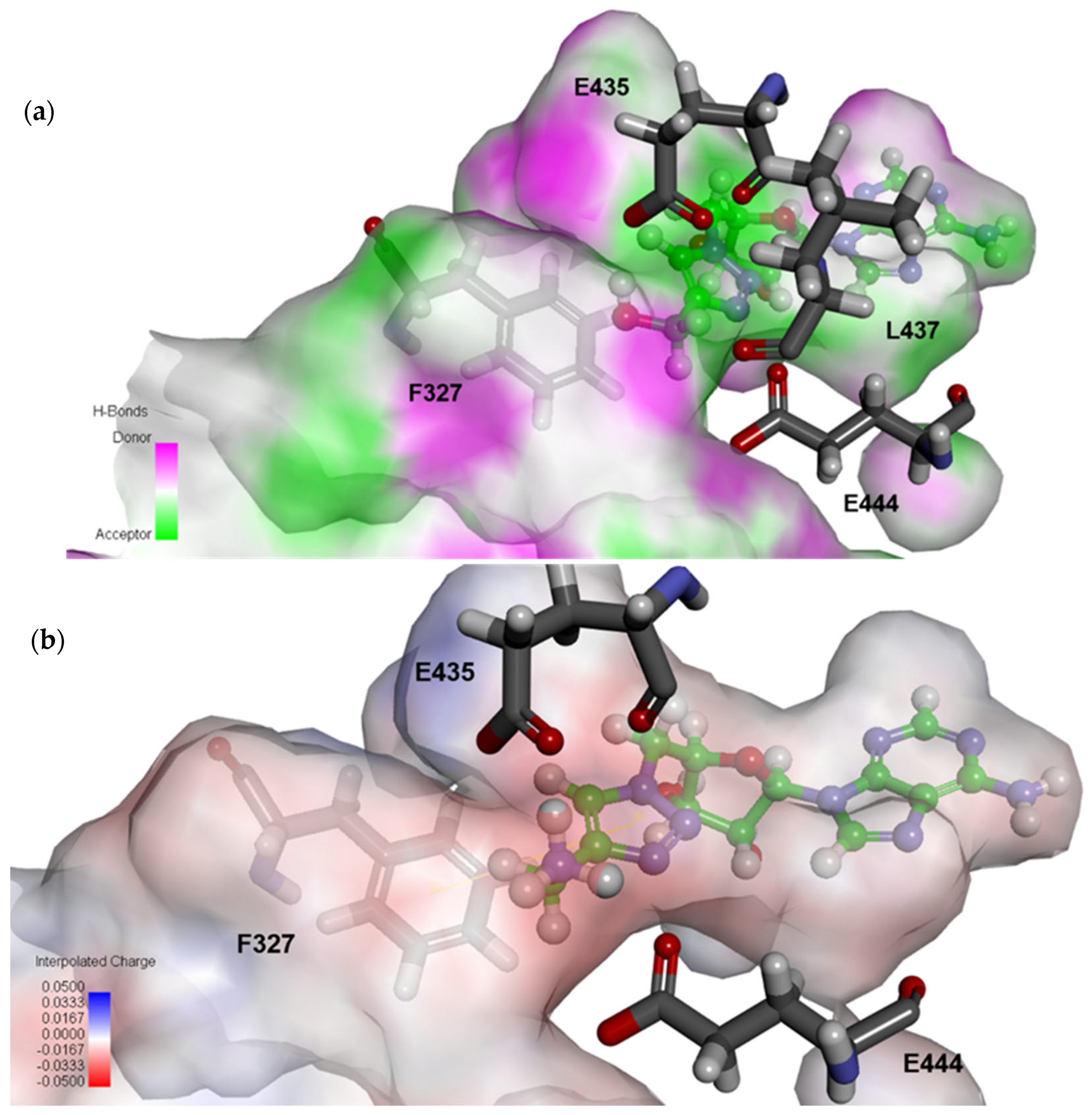
2.4. Structural Analysis for PRMT5 Inhibition
3. Materials and Methods
3.1. Synthesis
3.2. General Click Reaction for Hit Validation of Select Compounds
3.3. Solid-Phase Peptide Synthesis of 29 and 30
3.4. PRMT5 Expression and Purification
3.5. Biological Activity Assays
3.6. In Silico Docking of Ligands to PRMT5
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Migliori, V.; Phalke, S.; Bezzi, M.; Guccione, E. Arginine/lysine-methyl/methyl switches: Biochemical role of histone arginine methylation in transcriptional regulation. Epigenomics 2010, 2, 119–137. [Google Scholar] [CrossRef]
- Fuhrmann, J.; Clancy, K.W.; Thompson, P.R. Chemical biology of protein arginine modifications in epigenetic regulation. Chem. Rev. 2015, 115, 5413–5461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, A.S.M.; Li, F.; Eram, M.S.; Bolotokova, A.; Dela Sena, C.C.; Vedadi, M. Chemical probes for protein arginine methyltransferases. Methods 2020, 175, 30–43. [Google Scholar] [CrossRef] [PubMed]
- Fulton, M.D.; Brown, T.; Zheng, Y.G. Mechanisms and Inhibitors of Histone Arginine Methylation. Chem. Rec. 2018, 18, 1792–1807. [Google Scholar] [CrossRef] [PubMed]
- Pawlak, M.R.; Scherer, C.A.; Chen, J.; Roshon, M.J.; Ruley, H.E. Arginine N-methyltransferase 1 is required for early postimplantation mouse development, but cells deficient in the enzyme are viable. Mol. Cell. Biol. 2000, 20, 4859–4869. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hadjikyriacou, A.; Yang, Y.; Espejo, A.; Bedford, M.T.; Clarke, S.G. Unique Features of Human Protein Arginine Methyltransferase 9 (PRMT9) and Its Substrate RNA Splicing Factor SF3B2. J. Biol. Chem. 2015, 290, 16723–16743. [Google Scholar] [CrossRef] [Green Version]
- Dhar, S.; Vemulapalli, V.; Patananan, A.N.; Huang, G.L.; Di Lorenzo, A.; Richard, S.; Comb, M.J.; Guo, A.; Clarke, S.G.; Bedford, M.T. Loss of the major Type I arginine methyltransferase PRMT1 causes substrate scavenging by other PRMTs. Sci. Rep. 2013, 3, 1311. [Google Scholar] [CrossRef]
- Di Lorenzo, A.; Bedford, M.T. Histone arginine methylation. FEBS Lett. 2011, 585, 2024–2031. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Litt, M.; Qiu, Y.; Huang, S. Histone arginine methylations: Their roles in chromatin dynamics and transcriptional regulation. Biosci. Rep. 2009, 29, 131–141. [Google Scholar] [CrossRef]
- Lorton, B.M.; Shechter, D. Cellular consequences of arginine methylation. Cell. Mol. Life Sci. 2019, 76, 2933–2956. [Google Scholar] [CrossRef]
- Schapira, M.; Ferreira de Freitas, R. Structural biology and chemistry of protein arginine methyltransferases. Medchemcomm 2014, 5, 1779–1788. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jansson, M.; Durant, S.T.; Cho, E.C.; Sheahan, S.; Edelmann, M.; Kessler, B.; La Thangue, N.B. Arginine methylation regulates the p53 response. Nat. Cell Biol. 2008, 10, 1431–1439. [Google Scholar] [CrossRef] [PubMed]
- Hsu, J.M.; Chen, C.T.; Chou, C.K.; Kuo, H.P.; Li, L.Y.; Lin, C.Y.; Lee, H.J.; Wang, Y.N.; Liu, M.; Liao, H.W.; et al. Crosstalk between Arg 1175 methylation and Tyr 1173 phosphorylation negatively modulates EGFR-mediated ERK activation. Nat. Cell Biol. 2011, 13, 174–181. [Google Scholar] [CrossRef] [Green Version]
- Clarke, T.L.; Sanchez-Bailon, M.P.; Chiang, K.; Reynolds, J.J.; Herrero-Ruiz, J.; Bandeiras, T.M.; Matias, P.M.; Maslen, S.L.; Skehel, J.M.; Stewart, G.S.; et al. PRMT5-Dependent Methylation of the TIP60 Coactivator RUVBL1 Is a Key Regulator of Homologous Recombination. Mol. Cell 2017, 65, 900–916.e907. [Google Scholar] [CrossRef] [PubMed]
- Hamard, P.J.; Santiago, G.E.; Liu, F.; Karl, D.L.; Martinez, C.; Man, N.; Mookhtiar, A.K.; Duffort, S.; Greenblatt, S.; Verdun, R.E.; et al. PRMT5 Regulates DNA Repair by Controlling the Alternative Splicing of Histone-Modifying Enzymes. Cell Rep. 2018, 24, 2643–2657. [Google Scholar] [CrossRef] [Green Version]
- Gerhart, S.V.; Kellner, W.A.; Thompson, C.; Pappalardi, M.B.; Zhang, X.P.; Montes de Oca, R.; Penebre, E.; Duncan, K.; Boriack-Sjodin, A.; Le, B.; et al. Activation of the p53-MDM4 regulatory axis defines the anti-tumour response to PRMT5 inhibition through its role in regulating cellular splicing. Sci. Rep. 2018, 8, 9711. [Google Scholar] [CrossRef] [PubMed]
- Lattouf, H.; Poulard, C.; Le Romancer, M. PRMT5 prognostic value in cancer. Oncotarget 2019, 10, 3151–3153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shailesh, H.; Zakaria, Z.Z.; Baiocchi, R.; Sif, S. Protein arginine methyltransferase 5 (PRMT5) dysregulation in cancer. Oncotarget 2018, 9, 36705–36718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, H.; Qian, K.; Ho, M.C.; Zheng, Y.G. Small Molecule Inhibitors of Protein Arginine Methyltransferases. Expert Opin. Investig. Drugs 2016, 25, 335–358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaniskan, H.U.; Konze, K.D.; Jin, J. Selective inhibitors of protein methyltransferases. J. Med. Chem. 2015, 58, 1596–1629. [Google Scholar] [CrossRef] [PubMed]
- Luo, M. Inhibitors of protein methyltransferases as chemical tools. Epigenomics 2015, 7, 1327–1338. [Google Scholar] [CrossRef] [Green Version]
- Schapira, M.; Arrowsmith, C.H. Methyltransferase inhibitors for modulation of the epigenome and beyond. Curr. Opin. Chem. Biol. 2016, 33, 81–87. [Google Scholar] [CrossRef] [PubMed]
- Pugh, C.S.; Borchardt, R.T.; Stone, H.O. Inhibition of Newcastle disease virion messenger RNA (guanine-7-)-methyltransferase by analogues of S-adenosylhomocysteine. Biochemistry 1977, 16, 3928–3932. [Google Scholar] [CrossRef] [PubMed]
- Borchardt, R.T.; Eiden, L.E.; Wu, B.; Rutledge, C.O. Sinefungin, a potent inhibitor or S-adenosylmethionine: Protein O-methyltransferase. Biochem. Biophys. Res. Commun. 1979, 89, 919–924. [Google Scholar] [CrossRef]
- Smil, D.; Eram, M.S.; Li, F.; Kennedy, S.; Szewczyk, M.M.; Brown, P.J.; Barsyte-Lovejoy, D.; Arrowsmith, C.H.; Vedadi, M.; Schapira, M. Discovery of a Dual PRMT5-PRMT7 Inhibitor. ACS Med. Chem. Lett. 2015, 6, 408–412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marjon, K.; Cameron, M.J.; Quang, P.; Clasquin, M.F.; Mandley, E.; Kunii, K.; McVay, M.; Choe, S.; Kernytsky, A.; Gross, S.; et al. MTAP Deletions in Cancer Create Vulnerability to Targeting of the MAT2A/PRMT5/RIOK1 Axis. Cell Rep. 2016, 15, 574–587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alinari, L.; Mahasenan, K.V.; Yan, F.; Karkhanis, V.; Chung, J.H.; Smith, E.M.; Quinion, C.; Smith, P.L.; Kim, L.; Patton, J.T.; et al. Selective inhibition of protein arginine methyltransferase 5 blocks initiation and maintenance of B-cell transformation. Blood 2015, 125, 2530–2543. [Google Scholar] [CrossRef]
- Tarighat, S.S.; Santhanam, R.; Frankhouser, D.; Radomska, H.S.; Lai, H.; Anghelina, M.; Wang, H.; Huang, X.; Alinari, L.; Walker, A.; et al. The dual epigenetic role of PRMT5 in acute myeloid leukemia: Gene activation and repression via histone arginine methylation. Leukemia 2016, 30, 789–799. [Google Scholar] [CrossRef]
- Webb, L.M.; Amici, S.A.; Jablonski, K.A.; Savardekar, H.; Panfil, A.R.; Li, L.; Zhou, W.; Peine, K.; Karkhanis, V.; Bachelder, E.M.; et al. PRMT5-Selective Inhibitors Suppress Inflammatory T Cell Responses and Experimental Autoimmune Encephalomyelitis. J. Immunol. 2017, 198, 1439–1451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, Y.; Zhou, J.; Xu, F.; Jin, B.; Cui, L.; Wang, Y.; Du, X.; Li, J.; Li, P.; Ren, R.; et al. Targeting methyltransferase PRMT5 eliminates leukemia stem cells in chronic myelogenous leukemia. J. Clin. Investig. 2016, 126, 3961–3980. [Google Scholar] [CrossRef] [Green Version]
- Chan-Penebre, E.; Kuplast, K.G.; Majer, C.R.; Boriack-Sjodin, P.A.; Wigle, T.J.; Johnston, L.D.; Rioux, N.; Munchhof, M.J.; Jin, L.; Jacques, S.L.; et al. A selective inhibitor of PRMT5 with in vivo and in vitro potency in MCL models. Nat. Chem. Biol. 2015, 11, 432–437. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Gao, G.; Yu, X.; Kim, H.; Wang, L.; Xie, L.; Schwarz, M.; Chen, X.; Guccione, E.; Liu, J.; et al. Discovery of First-in-Class Protein Arginine Methyltransferase 5 (PRMT5) Degraders. J. Med. Chem. 2020, 63, 9977–9989. [Google Scholar] [CrossRef]
- Al-Hamashi, A.A.; Chen, D.; Deng, Y.; Dong, G.; Huang, R. Discovery of a potent and dual-selective bisubstrate inhibitor for protein arginine methyltransferase 4/5. Acta Pharm. Sin. B 2021, 11, 2709–2718. [Google Scholar] [CrossRef] [PubMed]
- ClincialTrials.gov. Available online: https://clinicaltrials.gov (accessed on 25 March 2022).
- Bonday, Z.Q.; Cortez, G.S.; Grogan, M.J.; Antonysamy, S.; Weichert, K.; Bocchinfuso, W.P.; Li, F.L.; Kennedy, S.; Li, B.H.; Mader, M.M.; et al. LLY-283, a Potent and Selective Inhibitor of Arginine Methyltransferase 5, PRMT5, with Antitumor Activity. ACS Med. Chem. Lett. 2018, 9, 612–617. [Google Scholar] [CrossRef]
- Brehmer, D.; Beke, L.; Wu, T.; Millar, H.J.; Moy, C.; Sun, W.; Mannens, G.; Pande, V.; Boeckx, A.; van Heerde, E.; et al. Discovery and Pharmacological Characterization of JNJ-64619178, a Novel Small-Molecule Inhibitor of PRMT5 with Potent Antitumor Activity. Mol. Cancer Ther. 2021, 20, 2317–2328. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Wang, C.; Jiang, H.; Luo, C. A patent review of arginine methyltransferase inhibitors (2010-2018). Expert Opin. Ther. Pat. 2019, 29, 97–114. [Google Scholar] [CrossRef]
- Sun, L.; Wang, M.; Lv, Z.; Yang, N.; Liu, Y.; Bao, S.; Gong, W.; Xu, R.M. Structural insights into protein arginine symmetric dimethylation by PRMT5. Proc. Natl. Acad. Sci. USA 2011, 108, 20538–20543. [Google Scholar] [CrossRef] [Green Version]
- Lee, L.; Chang, K.-H.; Valiyev, F.; Liu, H.-J.; Li, W.-S. Synthesis and Biological Evaluation of 5′-Triazole Nucleosides. J. Chin. Chem. Soc. 2006, 53, 1547–1555. [Google Scholar] [CrossRef]
- Zhang, G.; Richardson, S.L.; Mao, Y.; Huang, R. Design, synthesis, and kinetic analysis of potent protein N-terminal methyltransferase 1 inhibitors. Org. Biomol. Chem. 2015, 13, 4149–4154. [Google Scholar] [CrossRef]
- Wu, G.; Robertson, D.H.; Brooks, C.L., 3rd; Vieth, M. Detailed analysis of grid-based molecular docking: A case study of CDOCKER-A CHARMm-based MD docking algorithm. J. Comput. Chem. 2003, 24, 1549–1562. [Google Scholar] [CrossRef]
- Schulze, B.; Schubert, U.S. Beyond click chemistry - supramolecular interactions of 1,2,3-triazoles. Chem. Soc. Rev. 2014, 43, 2522–2571. [Google Scholar] [CrossRef] [PubMed]
- Luan, Y.; Blazer, L.L.; Hu, H.; Hajian, T.; Zhang, J.; Wu, H.; Houliston, S.; Arrowsmith, C.H.; Vedadi, M.; Zheng, Y.G. Design of a fluorescent ligand targeting the S-adenosylmethionine binding site of the histone methyltransferase MLL1. Org. Biomol. Chem. 2016, 14, 631–638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turner, R.A.; Oliver, A.G.; Lokey, R.S. Click chemistry as a macrocyclization tool in the solid-phase synthesis of small cyclic peptides. Org. Lett. 2007, 9, 5011–5014. [Google Scholar] [CrossRef]
- Wu, J.; Xie, N.; Feng, Y.; Zheng, Y.G. Scintillation proximity assay of arginine methylation. J. Biomol. Screen 2012, 17, 237–244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- 4.0.100.13345; Discovery Studio Client. Dassault Systemes BIOVIA: San Diego, CA, USA, 2005.
- Fulton, M.D.; Dang, T.; Brown, T.; Zheng, Y.G. Effects of substrate modifications on the arginine dimethylation activities of PRMT1 and PRMT5. Epigenetics 2022, 17, 1–18. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Relative Activity of PRMT5 at 10 μM (IC50) | Relative Activity of PRMT1 at 10 μM (IC50) |
|---|---|---|
| 1 | 48.0 ± 9.7% (3.0 ± 0.3 μM) | 97.5 ± 7.6% (>1000 μM) |
| 2 | (27.0 ± 4.5 μM) | (>1000 μM) |
| 3 | (4.4 ± 0.3 μM) | (>1000 μM) |
| 4 | (143.9 ± 8.4 μM) | (>1000 μM) |
| 5 | (194.9 ± 26.2 μM) | (>1000 μM) |
| 6 | 61.0 ± 8.0% (1.2 ± 0.2 µM) | 96.3 ± 8.0% (7.83 ± 8.4 μM) |
| 7 | (8.3 ± 2.2 μM) | (>1000 μM) |
| 8 | (7.7 ± 3.9 μM) | (>1000 μM) |
| 9 | (>200 μM) | (>1000 μM) |
| 10 | 71.0 ± 17.0% | 102.6 ± 5.6% |
| 11 | 74.0 ± 11.0%(3.15 ± 0.11 µM) | 96.9 ± 2.2% |
| 12 | 73 ± 12.0% (>400 µM) | 99.3 ± 6.7% |
| 13 | 57.0 ± 4.4% (>40 µM) | 92.7 ± 10.0% |
| 14 | 51.0 ± 5.7% (2.6 ± 0.1 µM) | 90.1 ± 5.7% |
| 15 | 94.2 ± 12.0% (>40 µM) | 123.0 ± 4.4% |
| 16 | 80.0 ± 9.5% | 101.4 ± 10.0% |
| 17 | 56.0 ± 10.0% | 91.0 ± 11.0% |
| 18 | 56.0 ± 6.4% | 91.2 ± 4.7% |
| 19 | (18.9 ± 3.0 μM) | (>200 μM) |
| 20 | 26.0 ± 0.45% (0.88 ± 0.09 µM) | 111.0 ± 5.5% (43.5 ± 11.0 μM) |
| 21 | 32.0 ± 3.3% | 84 ± 55.0% |
| 22 | 30.0 ± 1.7% (1.3 ± 0.2 µM) | 94.2 ± 2.2% (129.6 ± 38.7 μM) |
| 23 | 70.0 ± 5.6% | 91.8 ± 16.0% |
| 24 | 40.0 ± 3.6% (13.2 ± 3.0 µM) | 93.0 ± 1.3% |
| 25 | 72.0 ± 9.6% | 99.1 ± 13.0% |
| 26 | 83.0 ± 2.5% | 100.1 ± 4.6% |
| 27 | 40.0 ± 3.4% | 50.0 ± 9.9% |
| 28 | 76.0 ± 11.0% | 77.0 ± 23.0% |
| 29 | (53.7 ± 6.5 μM) | (>400 μM) |
| 30 | (45.6 ± 8.7 μM) | (46.1 ± 18.0 μM) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brown, T.; Cao, M.; Zheng, Y.G. Synthesis and Activity of Triazole-Adenosine Analogs as Protein Arginine Methyltransferase 5 Inhibitors. Molecules 2022, 27, 3779. https://doi.org/10.3390/molecules27123779
Brown T, Cao M, Zheng YG. Synthesis and Activity of Triazole-Adenosine Analogs as Protein Arginine Methyltransferase 5 Inhibitors. Molecules. 2022; 27(12):3779. https://doi.org/10.3390/molecules27123779
Chicago/Turabian StyleBrown, Tyler, Mengtong Cao, and Y. George Zheng. 2022. "Synthesis and Activity of Triazole-Adenosine Analogs as Protein Arginine Methyltransferase 5 Inhibitors" Molecules 27, no. 12: 3779. https://doi.org/10.3390/molecules27123779
APA StyleBrown, T., Cao, M., & Zheng, Y. G. (2022). Synthesis and Activity of Triazole-Adenosine Analogs as Protein Arginine Methyltransferase 5 Inhibitors. Molecules, 27(12), 3779. https://doi.org/10.3390/molecules27123779