Abstract
The crystal and molecular structures, intermolecular interactions, and energy of CL-20, HATO, and FOX-7 were comparatively predicted based on molecular dynamic (MD) simulations. By comparison, the 2D fingerprint plot, Hirshfeld surface, reduced density gradient isosurface, and electrostatic potential surface were studied to detect the intermolecular interactions. Meanwhile, the effects of vacuum and different solvents on the crystal habit of CL-20, HATO, and FOX-7 were studied by AE and MAE model, respectively. The energy calculation was also analysed based on the equilibrium structures of these crystal models by MD simulations. Our results would provide fundamental insights for the crystal engineering of energetic materials.
1. Introduction
Energetic materials (EMs), mainly including explosives, propellants, and pyrotechnics are a kind of important compounds that can store and release considerable energy, and have occupied an important place in mining, military equipment, space exploration, and fireworks [1,2,3,4,5,6].
The 2,4,6,8,10,12-Hexanitro-2,4,6,8,10,12-hexaazaisowurtzitane (CL-20) with a cage-shaped molecular structure is currently the most powerful and commercially available EM [7]. Though the CL-20 is the highest energy-density explosive than traditional EMS, it has failed to meet widespread application in many weapons due to its higher impact sensitivity and poorer security property [8].
Dihydroxylammonium 5,5′-bistetrazole-1,1′-diolate (HATO or TKX-50) is a newly synthesized explosive with excellent comprehensive properties, which possesses lower impact sensitivity and higher detonation performance than the common explosives. Apart from the higher energy level and lower sensitivity, low toxicity is another advantage of HATO. However, poor crystal habit, irregular crystal morphology, and smaller crystal particle make an extremely constraint in its application [9,10,11].
The 1,1-diamino-2,2-dinitroethene (FOX-7) is synthesized in 1998 and is a push–pull symmetrical structure. There is a nitro-enamine structure with two nitro group at one carbon and two amino group at other in its molecular structure. Due to the special chemistry structure, FOX-7 possesses an excellent detonation performances and safety property, and is honoured as outstanding low sensitivity and highly energetic materials. However, poor crystal quality, monotonous crystal particle, more crystal defect, and poor solubility make its widespread application enormous challenging [12,13,14,15].
To meet the high requirement, the study on Ems with high crystal quality has always been an ultimate goal that researchers strive for. The morphology of energetic materials is a vital component and plays an important role in the crystal quality. It can be affected by intermolecular interaction, solvent, additive, and temperature in the process of crystallization [16,17]. Nevertheless, the crystallization mechanism of CL-20, HATO, and FOX-7 remain unclear, which significantly motivates us to probe the crystal habit in three explosives.
Herein, CL-20, HATO, and FOX-7 were chosen for comparatively simulating their structures and predicting their properties by quantum chemistry calculation. By comparison, the intermolecular interactions of CL-20, HATO, and FOX-7 were studied by two-dimensional (2D) fingerprint plots, Hirshfeld surfaces, reduced density gradient (RDG) isosurface, and electrostatic potential surface analysis. Then, the effects of different solvents and additives on the crystal structures of CL-20, HATO, and FOX-7 were evaluated. This work aims to deepen the understanding of crystal growth and provide a better insight for the crystallization process of energetic material.
The chemical diagrams of CL-20, HATO, and FOX-7 are shown in Figure 1.
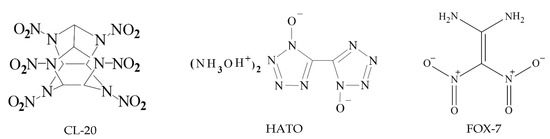
Figure 1.
The chemical diagrams of CL-20, HATO, and FOX-7, respectively.
2. Results and Discussion
2.1. Hirshfeld Surface Analysis
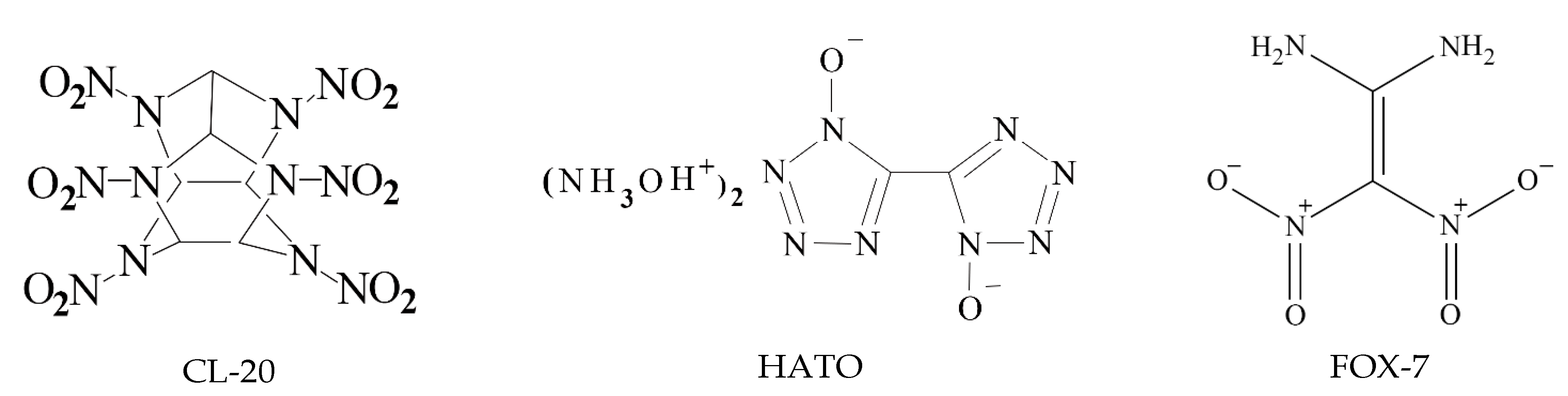
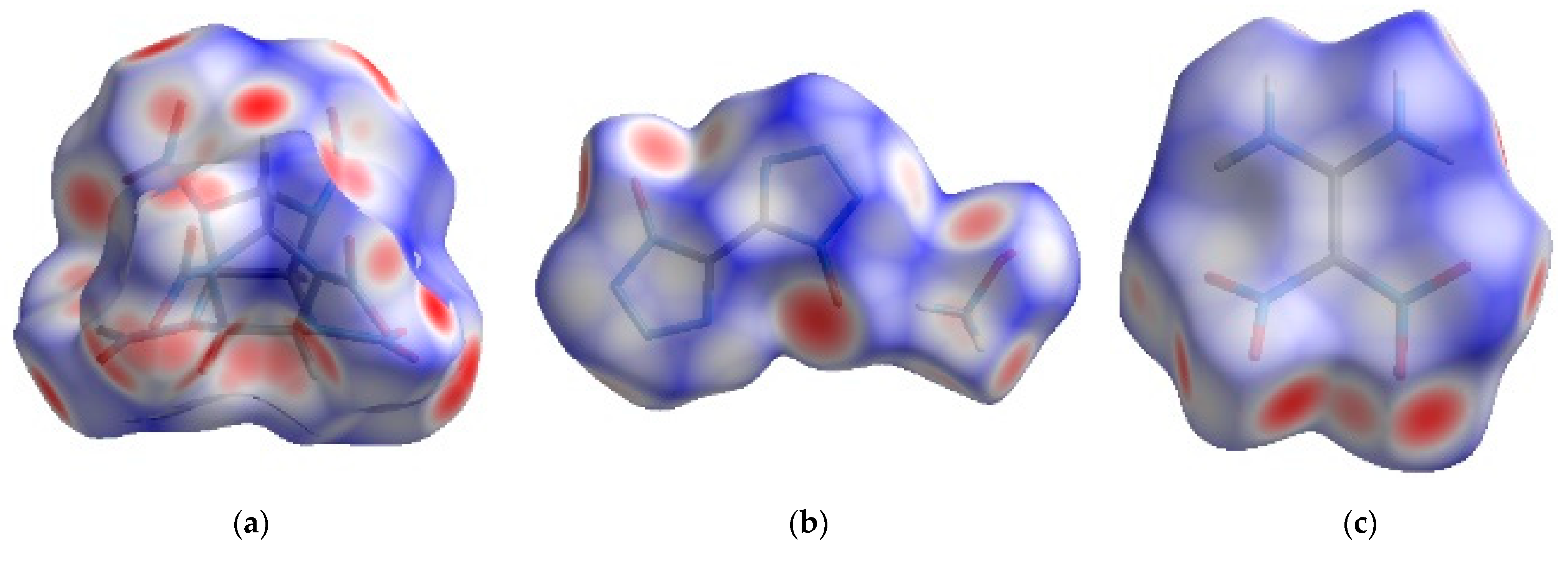
The surface and 2D fingerprint plot, which was formed by the location of (di de) point and their relative frequency, can confirm distance and intensity of intermolecular interaction [18,19]. The 2D fingerprints and the associated Hirshfeld surfaces of CL-20, HATO and FOX-7 were shown in Figure 2, in which the intermolecular interaction was visualized.


Figure 2.
The 2D fingerprints plots and the associated Hirshfeld surfaces: Images (a–c) showing the 2D fingerprints of CL-20, HATO, and FOX-7, respectively; Images (d–f) showing the Hirshfeld surfaces of CL-20, HATO, and FOX-7, respectively.
As shown in Figure 2a–c, there are the Hirshfeld surfaces of CL-20, HATO and FOX-7, respectively. The shape of surface of HATO and FOX-7 is plate, which indicates an insensitivity energetic material than CL-20. Furthermore, there are red, white, and blue dots on the surface, which represents the close contact around molecules. The red dot represents the distance d less the Van der Waals, white dot represents equal and blue dot represents exceeds. Compared with HATO and FOX-7, more red dots, which represent the predominant intermolecular interaction, are located on the surface of CL-20, particularly at edges, indicating that high close contact around CL-20 molecule.
In Figure 2d–f, we can see the intermolecular interaction in 2D fingerprints plots of crystals of CL-20, HATO, and FOX-7, respectively. We can see that there are O···H, O···N, and O···O intermolecular interactions in both of CL-20 and FOX-7 crystal. Compared with CL-20 and FOX-7, intermolecular interactions are O···H, O···N, and O···O in HATO. In addition, we can know that there are O···H intermolecular interactions in all of CL-20, HATO, and FOX-7, and it is a pair of remarkable spikes on the bottom left in 2D fingerprints.
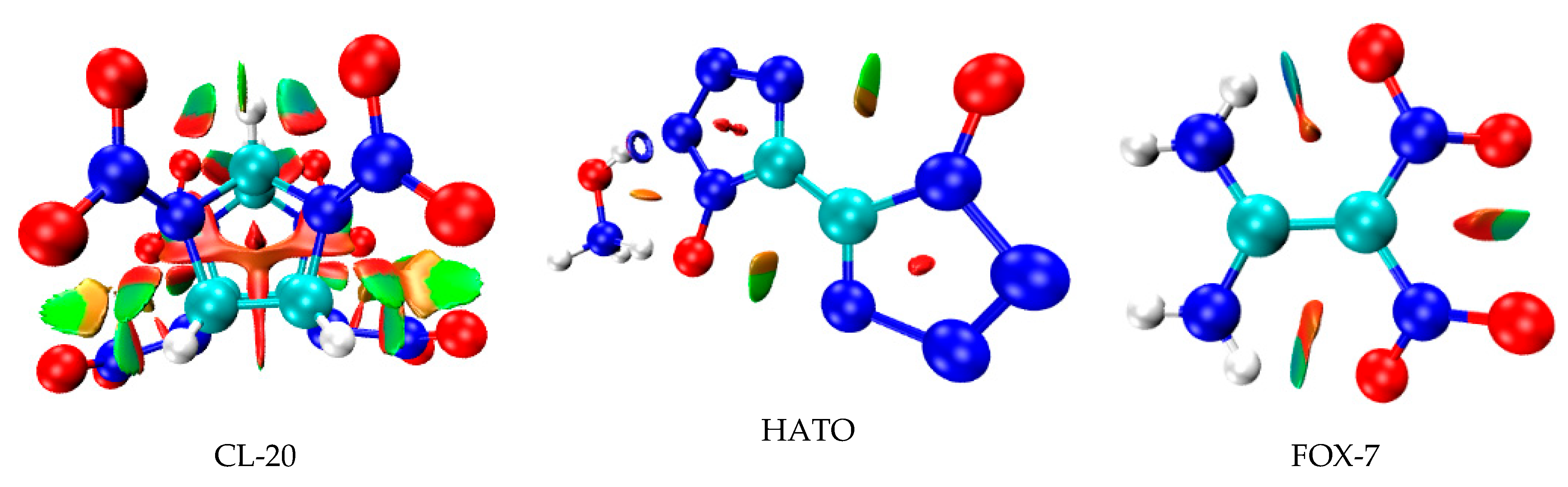
2.2. Reduced Density Gradient Analysis
To further reveal the information on inter- and intra-molecular effects and comprehensively study their influence on crystal structure, on the basis of the electron density and its RDG, the NCI plots of CL-20, HATO, and FOX-7 were determined in real space. In this method, the differences between hydrogen bonds, Van der Waals interactions, and repulsive steric clashes were observed by the study of the relationship between quantum-mechanical electron density and the reduced density gradient [20,21,22,23]. As shown in Figure 3, it is clear that there are abundant elliptical green slabs around the nitro group of each molecular edge, which denotes that weak hydrogen bonds and strong Van der Waals interactions exist in CL-20, HATO, and FOX-7. In addition to this, we can see that there are red slabs among five-membered ring of CL-20 and HATO, which indicates the steric effect in CL-20 and HATO. Due to more red slabs, CL-20 possess more greater steric effect than HATO. Due to red region only on the edge of elliptical slabs, FOX-7 possess the weakest steric effect in three molecule structure.

Figure 3.
NCI plots of gradient isosurfaces.
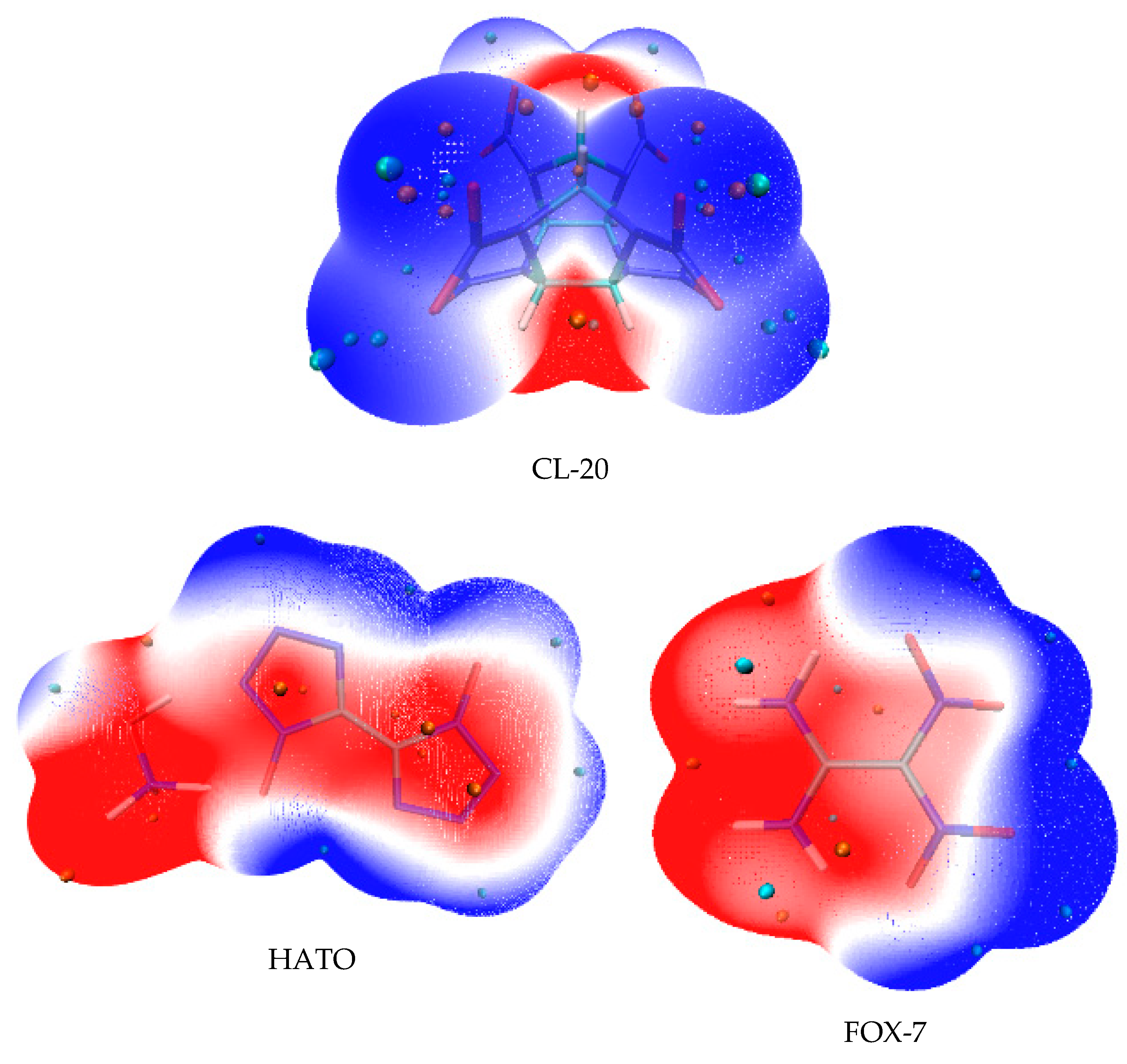
2.3. Electrostatic Potential Surface Analysis
Electrostatic potential surfaces (ESP) illustrate the necessary energy that a unit positive charge moves from infinity to a certain position in a chemical system [24]. It clearly shows the charge density distribution and the charge variation region of molecule, which can be used to determine the electrophilicity and nucleophilicity at different positions in the system, and also to reveal the properties of non-covalent molecular interaction [25,26]. Hence, the ESP was carried out in order to understand the electronic property and the bond strength variation in CL-20, HATO, and FOX-7. The ESP mapped surfaces of CL-20, HATO, and FOX-7 were presented in Figure 4.

Figure 4.
Electrostatic potential surfaces.
In Figure 4, it can be seen that the surface regions of ESP are coloured by different colours and the sites of maxima and minima are depicted by orange and cyan spheres, respectively. The blue region indicates negative ESP, which is the attraction of the proton by the concentrated electron density [27,28]. The blue region unexceptionally distributes on the edges of the molecules. Particularly, it situates on -NO2 group (in CL-20 and FOX-7) and -NO group (in HATO). The red region indicates positive ESP, which is the repulsion of the proton by the atomic nuclei in regions where low electron density exists and the nuclear charge is incompletely shielded [27,28]. The red region distributes on the edge of the -NH group (in HATO and FOX-7) and the central regions of the CL-20 molecule. When the polar of ESP is the same on the solvent and crystal face, they are manifested as a repulsive interaction, while the attractive interaction exists between the solvent and crystal face, when they are the opposite polar. The ESP qualitatively expresses the charged areas of molecules and the site of molecular reactively [29].
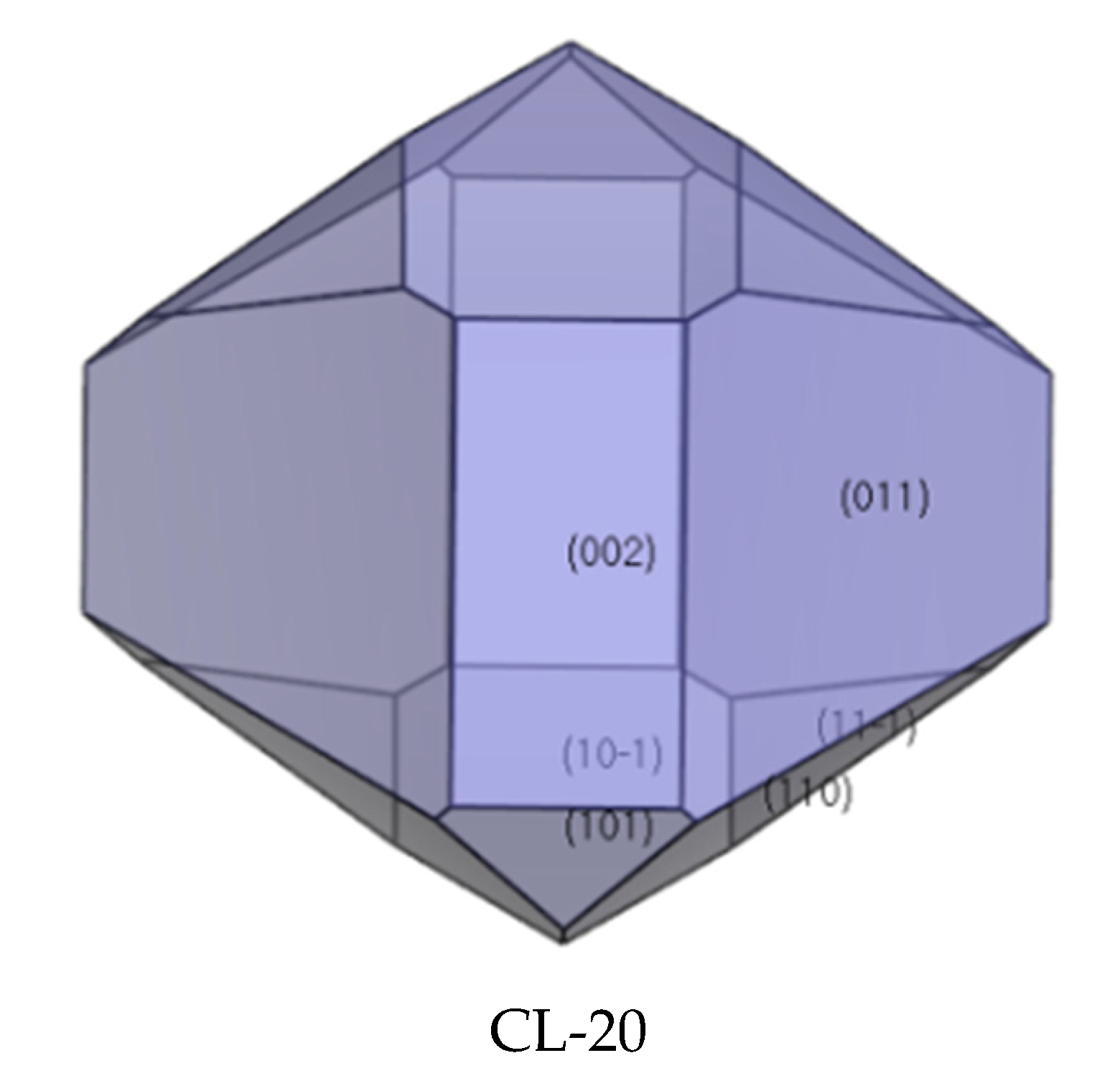
2.4. Crystal Morphology in Vacuum and Dominant Crystal Facets Analysis
The morphologically important facets of CL-20 crystal were simulated by the growth morphology algorithm (see Figure 5 and Table 1). This process was calculated based on the AE model, which was proposed by Hartman and Bennema according to the PBC theory in 1980. The attachment energy (Eatt) describes an amount of energy release when a crystal slice of thickness dhkl attached to the (hkl) facet, and this model does not consider the solvent media, temperature, pressure, and other factors during the simulation process. It can be seen from Figure 6 that the aspect ratio of CL-20 is 1.44 and there are six dominant crystal facets in CL-20, including {0 1 1}, {1 0 −1}, {1 1 0}, {1 1 −1}, {0 0 2}, and {1 0 1}. It can be seen from Table 1 that the largest area percentage is {0 1 1} facet, so this facet exhibits the most morphological important in CL-20 crystal morphology. Due to a larger interplanar spacing {dhkl}, {0 1 1} and {1 0 −1} facets are the slow-growing facets whereas other facets occupy smaller {dhkl} are the fast-growing facets.


Figure 5.
Crystal growth morphology in vacuum.

Table 1.
Dominant crystal facets of CL-20.

Figure 6.
Crystal growth morphology of CL-20 in solvent by MAE model and recrystallization experiment.
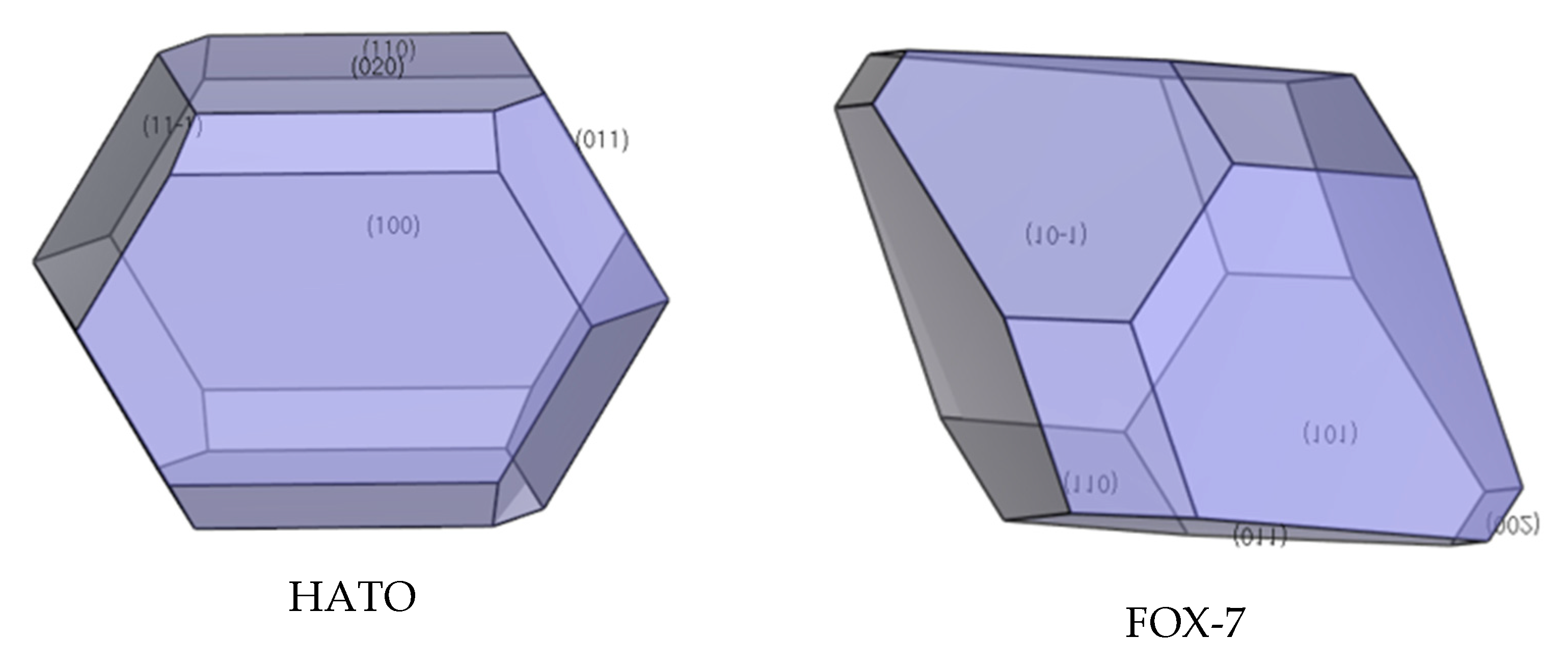
The morphologically important facets of HATO (see Figure 5, Table 2) and FOX-7 (see Figure 5, Table 3) crystals can be seen from the below figures and tables. In Figure 6, we can know that with an aspect ratio of 1.88, the predicted morphology of HATO crystal is mainly dominated by the {1 0 0}, {1 1 0}, {0 2 0}, {0 1 1}, and {1 1 −1} crystal facets. In Table 2, the largest area percentage of HATO is (1 0 0) facet, and this facet is the most morphological important in HATO crystal morphology. With a larger interplanar spacing {dhkl}, {1 0 0} facet is the slow-growing facet whereas other facets occupy smaller {dhkl} are the fast-growing facets. In the same way, the aspect ratio of FOX-7 is 1.83, and the dominant crystal facets of FOX-7 crystal are the {0 1 1}, {1 0 −1}, {0 0 2}, {1 0 1}, and {1 1 0} crystal facets (shown in Figure 6). In Table 3, the largest area percentage of FOX-7 is {0 1 1} facet, and this facet is the most morphological important in FOX-7 crystal morphology. With a larger interplanar spacing {dhkl}, {1 1 0} facet is the slow-growing facet whereas other facets occupy smaller {dhkl} are the fast-growing facets.
Table 2.
Dominant crystal facets of HATO.
Table 3.
Dominant crystal facets of FOX-7.
Apart from, there is Eatt mainly composed with electrostatic interaction, Van der Waals force and hydrogen bonding interaction in three crystals. In CL-20 and FOX-7, the Eatt is mainly involved Van der Waals force and electrostatic interaction, whereas, in HATO, the Eatt is mainly involved in Van der Waals force and hydrogen bonding interaction.
2.5. Effect of Ethyl Acetate, Acetonitrile, Acetone, DMF, DMSO, and NMP
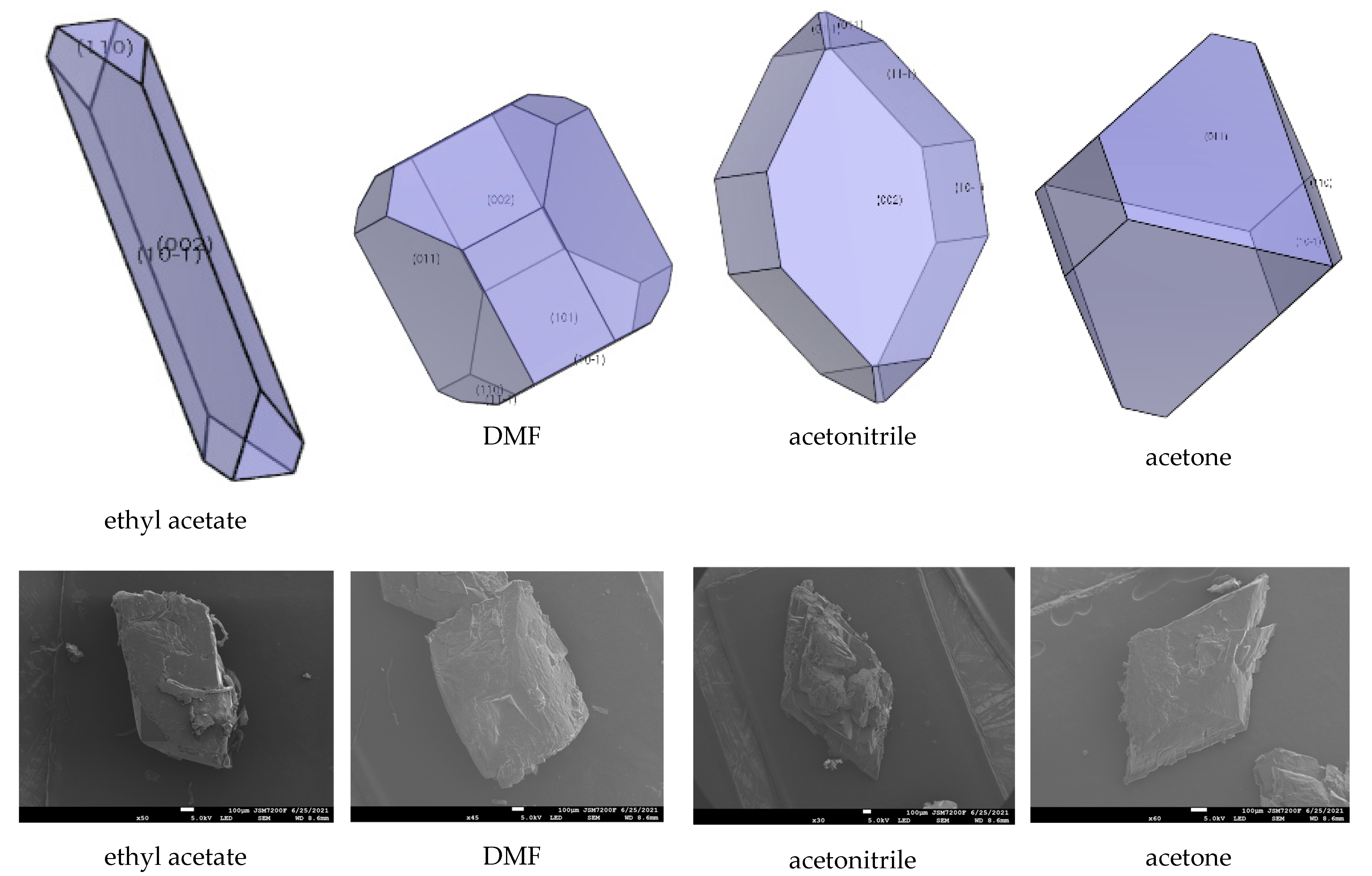
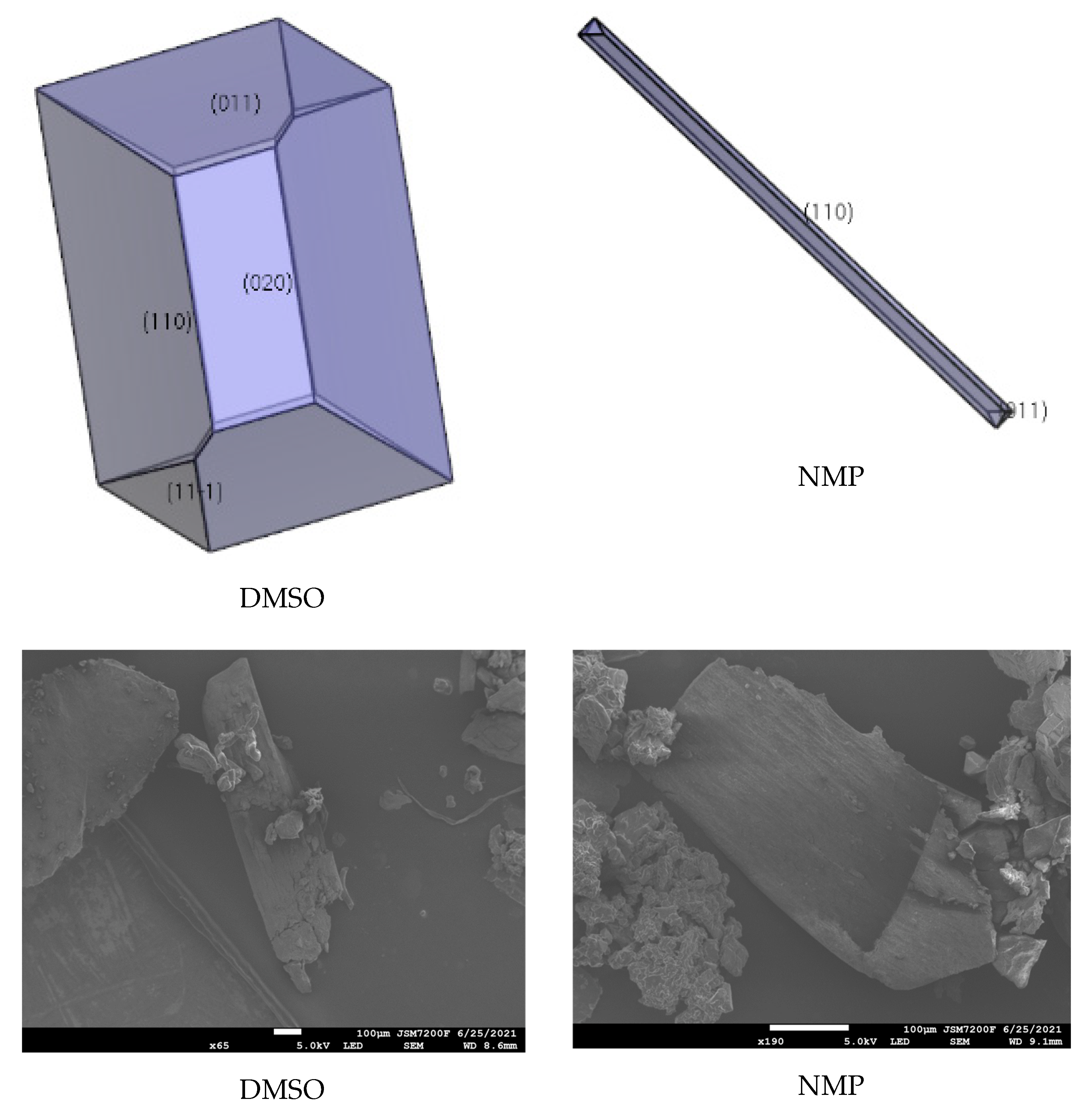
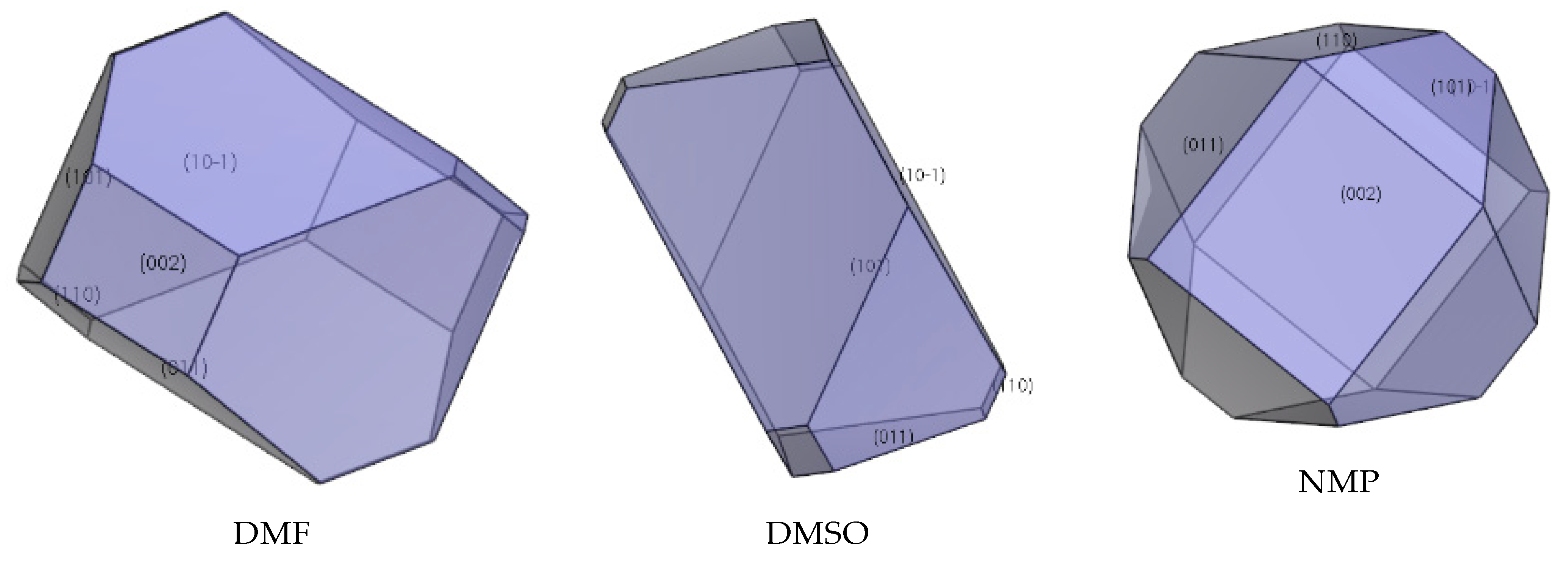
The interaction of solvents in the morphologically important crystal facets was simulated by the molecular dynamic method (shown in Table 4, Table 5 and Table 6). The interaction energy (Eint) is the energy between each crystal face and solvent molecule which is negative value in three crystals, indicating that the adsorption of solvent in the crystal face was exothermic, during which the solvent will spontaneously adhere to the crystal interface [30]. In Table 4, Table 5 and Table 6, we can see that different solvents have different effects on each crystal facet, which leads to the change of the importance of each crystal facet during the process of crystal growth. The crystal morphologies of CL-20, HATO, and FOX-7 in different solvents predicted and experimented were shown in Figure 6, Figure 7 and Figure 8, respectively. Compared with the morphology in vacuum, the crystal morphology predicted by MAE model in solvent is very obviously different.
Table 4.
Adsorption energies between the solvent and different CL-20 crystal faces.
Table 5.
Adsorption energies between the solvent and different HATO crystal faces.
Table 6.
Adsorption energies between the solvent and different FOX-7 crystal faces.

Figure 7.
Crystal growth morphology of HATO in solvent by MAE model and recrystallization experiment.


Figure 8.
Crystal growth morphology of FOX-7 in solvent by MAE model and recrystallization experiment.
In Table 4, we can see the change of the important crystal facet of CL-20. In ethyl acetate, the {0 0 2} facet is the most important crystal facet, which possesses the largest area percentage. The cohesive energy density (CED) of {0 0 2} is −3886.14 kcal/mol and the attachment energy of {0 0 2} varied from −92.86 kcal/mol to −47.13 kcal/mol. Furthermore, the crystal facets of {0 1 1}, {1 1 −1}, and {1 0 1} tend to disappear in ethyl acetate. In DMF and acetone, the most important crystal facet is still the {0 1 1} facet, which is in agreement with in-vacuum. In DMF and acetone, the CED of {0 1 1} are −8076.94 kcal/mol and −5979.91 kcal/mol, respectively. The attachment energy varied from −84.78 kcal/mol to 249.90 kcal/mol and from −84.78 kcal/mol to 6741.06 kcal/mol, respectively. However, in acetone, the crystal facets of {1 1 −1}, {0 0 2}, and {1 0 1} tend to disappear, which is different from in DMF solution. In acetonitrile, the most important crystal facet is {0 0 2}, which possesses the CED with −4189.15 kcal/mol. The crystal facets of {1 1 0} and {1 0 1} tend to disappear.
The important crystal facet of HATO in solvent is shown in Table 5. we can see that in DMSO, the {1 1 0} facet is the most important crystal facet, which possesses the largest area percentage. The CED of {1 1 0} is −2633.03 kcal/mol and the attachment energy of {1 1 0} varied from −26.62 kcal/mol to 321.27 kcal/mol. Furthermore, the crystal facets of {1 0 0} and {1 1 −1} tend to disappear in DMSO. In NMP, the most important crystal facet is also the {1 1 0} facet, which is agree with in DMSO. The CED of {1 1 0} is −3031.07 kcal/mol and the attachment energy varied from −26.62 kcal/mol to −11.94 kcal/mol. In DMSO, the crystal facet of {1 0 0} tends to disappear, however, in NMP solution, there are three crystal facets tend to disappear, involving {1 0 0}, {0 2 0}, and {1 1 −1}.
The important crystal facet of FOX-7 in solvent is shown in Table 6. we can see that in DMF, the {0 1 1} facet is the most important crystal facet, which possesses the largest area percentage. The CED of {0 1 1} is −4643.31 kcal/mol and the attachment energy of {0 1 1} varied from −37.04 kcal/mol to 145.83 kcal/mol. In DMSO, the {1 0 −1} facet is the most important crystal facet. The CED of {1 0 −1} is −3842.51 kcal/mol and the attachment energy of {1 0 −1} varied from −36.61 kcal/mol to 125.28 kcal/mol. Furthermore, the crystal facets of {0 0 2} tend to disappear in DMSO. In NMP, the most important crystal facet is the {1 1 0} facet. The CED of {1 1 0} is −4630.11 kcal/mol and the attachment energy varied from −46.51 kcal/mol to 165.88 kcal/mol.
In Figure 6, we can see that the morphology of CL-20 in ethyl acetate and DMF is a column, whereas, in acetonitrile and acetone the morphology tends to a rhombus. Meanwhile, in Figure 7, the morphology of HATO is rodlike in DMSO and NMP. In Figure 8, the morphology of FOX-7 is irregular polyhedron in NMP, DMSO, and NMP. Moreover, we can know that the morphology predicted of three crystals in different solvents is quite consistent with the corresponding experimental results.
3. Experimental Computational Details
3.1. Hirshfeld Surface
The Hirshfeld surface (HS) and corresponding 2D fingerprints were calculated by CrystalExplorer [31]. In CrystalExplorer, the electron density, which comes from tabulations of atomic wavefunctions, was used to compute the HS. The pro-molecule electron density and the pro-crystal electron density were constructed based on the spherical atomic electron densities . Then, the weight function was defined based on and . The weight function is as follows:
In all space, this continuous scalar function has 0 < < 1, and the 0.5 isosurface of this function is the HS. The 2D fingerprint was displayed to its HS by CrystalExplorer.
3.2. Reduced Density Gradient
Reduced density gradient (RDG) is powerful to reveal the noncovalent interaction (NCI) [32,33,34]. RDG is defined as follows:
where is the electron density. The NCI plots were calculated by Multiwfn [35] and visualized using the VMD program [36].
3.3. Electrostatic Potential Surface
The calculation function of the electrostatic potential surface (ESP) is as follows:
The electrostatic interaction between a unit point charge placed at r and the system of interest can be measured by this function. ESP was calculated by Multiwfn [35] and visualized using the VMD program [36]. The cartesian atomic coordinates can be found in Supplementary Materials: Tables S1–S3.
3.4. Model and Computational Details
3.4.1. Construction of Models
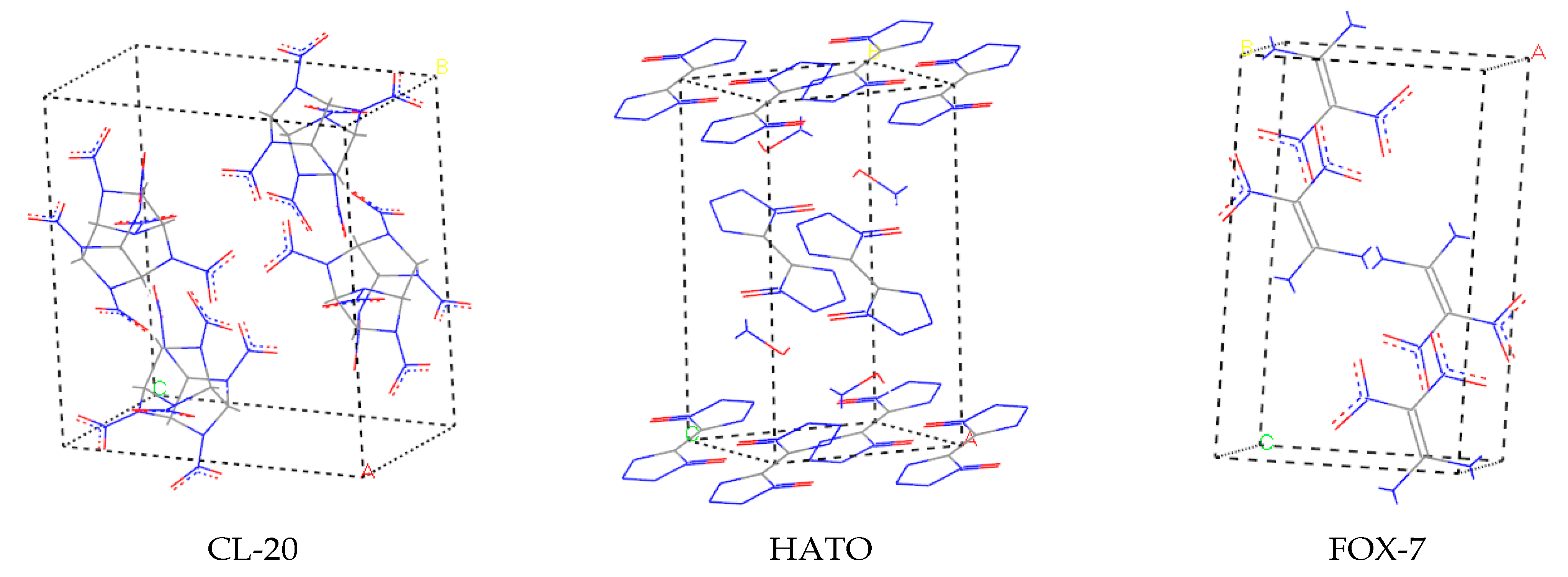
All computation simulations were performed using the Materials Studio software. We constructed the initial unit cell structure of CL-20, HATO, and FOX-7 based on the Cambridge Structural Database available at Cambridge Crystallographic Data Centre (CCDC) using Material Studio software. The crystal cell model of three crystals is shown in Figure 9.

Figure 9.
The crystal cell model of CL-20, HATO, and FOX-7, respectively.
3.4.2. Choice of Force Field
The force field plays a very important role to obtain reliable molecular dynamics simulation result. The popular COMPASS (condensed-phase optimized molecular potentials for atomistic molecular dynamics studies) force field has proved to be a universal all-atom force field, which has been calibrated from empirical parametrization techniques and state-of-the-art ab initio methods [8]. The COMPASS is suitable for molecular dynamic simulations involving conformation, vibration, structure, energy, and physical properties simulation of nitro (−NO2) containing compounds, such as RDX, HMX, and CL-20 [37,38]. So, CL-20 and FOX-7 were calculated by the COMPASS force field. However, there was no reasonable parameter for the N-oxides on the azole ring of HATO based on COMPASS force field in the simulation process. This result can be calibrated when replace COMPASS force field with Dreiding force field. Dreiding force field is a purely diagonal forcefield with harmonic valence terms and a cosine Fourier expansion torsion term and is more suitable for the N-oxides on the azole ring [39]. So, HATO cell structure was calculated by the Dreiding force field.
3.4.3. Structure Optimization and Morphology in Vacuum
The atomic positions, lattice parameters and the geometry structure of CL-20, HATO, and FOX-7 were firstly optimized to minimize the total energy and obtain an optimized molecule geometry by the selected force field. Then, the crystal morphology of CL-20, HATO, and FOX-7 was simulated under vacuum with an attachment energy (AE) model, in which the growth morphology method was employed to identify the morphologically important facets of these crystals.
3.4.4. Construction of Interface Models
The morphologically important growth crystal facets of CL-20, HATO, and FOX-7 crystals were cut to obtain the 3D periodic structure. Next, the periodic supercell was constructed. In the Amorphous Cell module, a solvent box was built with 200 molecules randomly distributed solvent molecules and it was consistent with the 3D periodic structure sizes of the important facets. Due to a favourable solubleness of CL-20 in ethyl acetate, N,N-Dimethylformamide(DMF), acetonitrile and acetone, these solvents were choice during the computation [40]. In the same way, the solvent molecules of HATO were dimethyl sulfoxide (DMSO) and N-methylpyrrolidone (NMP), and the solvent molecules of FOX-7 were DMF, DMSO, and NMP. Then, optimizing the solvent layer by the selected force field. The two-layer structure model containing solvent layer and main facet was constructed, which has a 50 Å thickness of the vacuum layer to eliminate the effect of additional free boundaries with a periodic boundary.
3.4.5. Molecular Dynamic Simulation
The ensemble with constant temperature and constant volume (NVT)-MD simulation was carried out on CL-20, HATO, and FOX-7 two-layer structure models at room temperature (298 K) to obtain their equilibrium structure. The Berendsen thermostat was used to control the temperature. The total simulation time was set as 300 ps with a time step of 1 fs and every 1000 time steps output a frame. The calculations of the Van der Waals (vdW) and electrostatic force were performed by an atom-based method and the Ewald method, respectively. The cut-off distance of the atom-based method was set to 12.5 Å, and the Ewald accuracy of the Ewald method was set to 0.001 kcal/mol. Due to a reduction in the attachment energy in a vacuum, a modified attachment energy (MAE) model is more suitable for solvent screening. So, CL-20, HATO, and FOX-7 two-layer structure models were calculated with the MAE model. Eventually, the crystal morphologies of the CL-20, HATO, and FOX-7 were predicted by the MAE model in different solvent medium.
4. Conclusions
In this work, we investigated the intermolecular interaction, crystal morphology change, and energy of CL-20, HATO, and FOX-7 with quantum chemical computation. Compared with HATO and FOX-7, more red dots in Hirshfeld surfaces of CL-20 indicated more intermolecular interactions, especially hydrogen bonds. Noncovalent interaction analysis showed that apart from hydrogen bond, there were Van der Waals interactions exist in CL-20, HATO, and FOX-7. The ESP analysis detected the reactive site between solvent and crystal face. MD simulation manifested the crystal habit in vacuum by AE model have apparently change compared with in the solvent system. Furthermore, different solvents also have different effects on crystal morphology. These results indicate that solvent and environmental condition can enormously affect crystal morphology. Our findings give us a better understanding of the crystal engineering of energetic materials.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/molecules27123755/s1, Table S1: Cartesian atomic coordinates for CL-20 used for reduced density gradient analysis; Table S2: Cartesian atomic coordinates for HATO used for reduced density gradient analysis; Table S3. Cartesian atomic coordinates for FOX-7 used for reduced density gradient analysis.
Author Contributions
Conceptualization, C.A.; methodology, B.Y.; software, D.L.; analysis, investigation, N.L.; investigation, M.W.; software, writing—review and editing, Y.L.; Funding acquisition, Y.L. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by scientific and technological innovation programs of higher education institutions in Shanxi. (Funder: Yan Liu. Grant No. 2021L539). This work was also supported by Taiyuan institute of technology scientific research initial funding. (Funder: Yan Liu. Grant No. 20020111 and 21010110).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
Sample Availability
Samples of the compounds are not available from the authors.
References
- Duan, B.-H.; Shu, Y.-J.; Liu, N.; Lu, Y.-Y.; Wang, B.-Z.; Lu, X.-M.; Zhang, J.-Q. Comparative studies on structure, sensitivity and mechanical properties of CL-20/DNDAP cocrystal and composite by molecular dynamics simulation. RSC Adv. 2018, 8, 34690–34698. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.-B.; Gong, J.; Hu, X.-Y.; Ju, X. Comparative investigation on the thermostability, sensitivity, and mechanical performance of RDX/HMX energetic cocrystal and its mixture. J. Mol. Model. 2020, 26, 176–186. [Google Scholar] [CrossRef] [PubMed]
- Sun, T.; Xiao, J.-J.; Liu, Q.; Zhao, F.; Xiao, H.-M. Comparative study on structure, energetic and mechanical properties of a 3-CL-20/HMX cocrystal and its composite with molecular dynamics simulation. J. Mater. Chem. A 2014, 2, 13898–13904. [Google Scholar] [CrossRef]
- Gamekkanda, J.-C.; Sinha, A.-S.; Aakeroy, C.-B. Cocrystals and salts of tetrazole-based energetic materials. Cryst. Growth Des. 2020, 20, 2432–2439. [Google Scholar] [CrossRef]
- Zhang, C. Origins of the energy and safety of energetic materials and of the energy & safety contradiction. Propellants Explos. Pyrotech. 2018, 43, 855–856. [Google Scholar]
- Bolton, O.; Matzger, A.-J. Improved stability and smart-material functionality realized in an energetic cocrystal. Angew. Chem. Int. Ed. 2011, 50, 8960–8963. [Google Scholar] [CrossRef] [PubMed]
- Pang, W.-Q.; Wang, K.; Zhang, W.; Luca, L.-T.-D.; Fan, X.-Z.; Li, J.-Q. CL-20-based cocrystal energetic materials: Simulation, preparation and performance. Molecules 2020, 25, 4311–4330. [Google Scholar] [CrossRef]
- Hang, G.-Y.; Yu, W.-L.; Wang, W.-T.; Wang, J.-T. Theoretical investigations into effects of adulteration crystal defect on properties of CL-20/TNT cocrystal explosive. Comput. Mater. Sci. 2019, 156, 77–83. [Google Scholar] [CrossRef]
- Fischer, N.; Fischer, D.; Klapotke, T.-M.; Piercey, D.-G.; Stierstorfer, J. Pushing the limits of energetic materials—The synthesis and characterization of dihydroxylammonium 5,5′-bistetrazole-1,1′-diolate. J. Mater. Chem. 2012, 22, 20418–20422. [Google Scholar] [CrossRef]
- Fischer, N.; Klapotke, T.-M.; Reymann, M.; Stierstorfer, J. Nitrogen-rich salts of 1 h,1′h-5,5′-Bitetrazole-1,1′-diol: Energetic materials with high thermal stability. Eur. J. Inorg. Chem. 2013, 12, 2167–2180. [Google Scholar] [CrossRef]
- An, Q.; Cheng, T.; Goddard, W.-A.; Zybin, S.-V. Anisotropic impact sensitivity and shock induced plasticity of TKX-50 (dihydroxylammonium 5,5′-bis(tetrazole)-1,1′-diolate) single crystals: From large-scale molecular dynamics simulations. J. Phys. Chem. C 2015, 119, 2196–2207. [Google Scholar] [CrossRef]
- Yan, C.; Qi, X.-J.; Wang, K.-C.; Jin, J.-H.; Cheng, G.-B.; Liu, T.-L.; Yang, H.-W.; Zhang, Q.-H. Revisiting the reactive chemistry of FOX-7: Cyclization of FOX-7 affords the fused-ring polynitro compounds. Chem. Commun. 2019, 19, 3497–3500. [Google Scholar] [CrossRef] [PubMed]
- Bemm, U.; Ostmark, H. 1,1-diamino-2,2-dinitroethylene:t a novel energetic material with infinite layers in two dimensions. Acta Crystallogr. Sect. C Cryst. Struct. Commun. 1998, 54, 1997–1999. [Google Scholar] [CrossRef]
- Evers, J.; Klapotke, T.-M.; Mayer, P.; Oehlinger, G.; Welch, J. α- and β-FOX-7, polymorphs of a high energy density material, studied by X-ray single crystal and powder investigations in the temperature range from 200 to 423 K. Inorg. Chem. 2006, 45, 4996–5007. [Google Scholar] [CrossRef] [PubMed]
- Crawford, M.-J.; Evers, J.; Gobel, M.; Klapotke, T.-M.; Mayer, P.; Oehlinger, G.; Welch, J.-M. γ-FOX-7: Structure of a high energy density material immediately prior to decomposition. Propellants Explos. Pyrotech. 2007, 32, 478–495. [Google Scholar] [CrossRef]
- Xiong, S.-L.; Chen, S.-S.; Jin, S.-H.; Zhang, C.-Y. Molecular dynamics simulations on dihydroxylammonium 5,5′-bistetrazole-1,1′-diolate/hexanitrohexaazaisowurtzitane cocrystal. RSC Adv. 2016, 6, 4221–4226. [Google Scholar] [CrossRef]
- Zhou, T.; Chen, F.; Li, J.; He, L.; Ren, Y.-Y.; Wang, X.-L.; Cao, D.-L. Morphology prediction of 5,5′-bistetrazole-1,1′-diolate (BTO) crystal in solvents with different models using molecular dynamics simulation. J. Cryst. Growth 2020, 548, 125843–125854. [Google Scholar] [CrossRef]
- Spackman, M.-A.; McKinnon, J.-J. Fingerprinting intermolecular interactions in molecular crystals. CrystEngComm 2002, 4, 378–392. [Google Scholar] [CrossRef]
- McKinnon, J.-J.; Jayatilaka, D.; Spackman, M.-A. Towards quantitative analysis of intermolecular interactions with Hirshfeld surfaces. Chem. Commun. 2007, 37, 3814–3816. [Google Scholar] [CrossRef]
- Lin, H.; Zhu, S.-G.; Li, H.-Z.; Peng, X.-H. Structure and detonation performance of a novel HMX/LLM-105 cocrystal explosive. J. Phys. Org. Chem. 2013, 26, 898–907. [Google Scholar] [CrossRef]
- Gao, H.-F.; Zhang, S.-H.; Ren, F.-D.; Liu, F.; Gou, R.-J.; Ding, X. Theoretical insight into the co-crystal explosive of 2,4,6,8,10,12-hexanitrohexaazaisowurtzitane (CL-20)/1,1-diamino-2,2-dinitroethylene (FOX-7). Comput. Mater. Sci. 2015, 107, 33–41. [Google Scholar] [CrossRef]
- Xu, Y.-G.; Tian, L.-L.; Li, D.-X.; Wang, P.-C.; Lu, M. A series of energetic cyclo-pentazolate salts: Rapid synthesis, characterization, and promising performance. J. Mater. Chem. A. 2019, 7, 12468–12479. [Google Scholar] [CrossRef]
- Johnson, E.-R.; Keinan, S.; Mori-Sanchez, P.; Contreras-Garcia, J.; Cohen, A.-J.; Yang, W.-T. Revealing Noncovalent Interactions. J. Am. Chem. Soc. 2010, 132, 6498–6506. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.-R.; Li, H.-Z.; Gou, R.-J.; Zhang, C.-Y. Packing structures of CL-20-based cocrystals. Cryst. Growth Des. 2018, 18, 7065–7078. [Google Scholar] [CrossRef]
- Ren, J.-R.; Chen, D.; Liu, G.-R.; Wang, K.-C.; Fan, G.-J.; Yu, Y.-W.; Zhang, C.-Y.; Li, H.-Z. Crystal structure evolution of the high energetic compound carbonic dihydrazidinium bis[3-(5-nitroimino-1,2,4-triazolate)] induced by solvents. CrystEngComm 2020, 22, 593–601. [Google Scholar] [CrossRef]
- Ma, Y.; Meng, L.-Y.; Li, H.-Z.; Zhang, C.-Y. Enhancing intermolecular interactions and their anisotropy to build low-impact-sensitivity energetic crystals. CrystEngComm 2017, 19, 3145–3155. [Google Scholar] [CrossRef]
- Zhang, J.-H.; Zhang, Q.-H.; Vo, T.-T.; Parrish, D.-A.; Shreeve, J.-M. Energetic salts with π-stacking and hydrogen-bonding interactions lead the way to future energetic materials. J. Am. Chem. Soc. 2015, 137, 1697–1704. [Google Scholar] [CrossRef]
- Bu, R.-P.; Liu, G.-R.; Zhong, K.; Wang, R.; Xiong, Y.; Zhang, C.-Y. Relationship between the molecular structure and stacking mode: Characteristics of the D2h and D3h molecules in planar layer-stacked crystals. Cryst. Growth Des. 2021, 21, 6847–6861. [Google Scholar] [CrossRef]
- Xie, Z.-B.; Hu, S.-Q.; Cao, X. Theoretical insight into the influence of molecular ratio on the binding energy and mechanical property of HMX/2-picoline-N-oxide cocrystal, cooperativity effect and surface electrostatic potential. Mol. Phys. 2016, 114, 2164–2176. [Google Scholar] [CrossRef]
- Chen, L.-Z.; She, C.-C.; Pan, H.-X.; Wang, J.-L.; Cao, D.-L. Habit prediction of 3,4,5-trinitro-1H-pyrazole in four solvent mediums using a molecular dynamics simulation. J. Cryst. Growth 2019, 507, 58–64. [Google Scholar] [CrossRef]
- Spackman, P.-R.; Turner, M.-J.; McKinnon, J.-J.; Wolff, S.-K.; Grimwood, D.-J.; Jayatilaka, D.; Spackman, M.-A. CrystalExplorer: A program for Hirshfeld surface analysis, visualization and quantitative analysis of molecular crystals. J. Appl. Cryst. 2021, 54, 1006–1011. [Google Scholar] [CrossRef] [PubMed]
- Khrustalev, V.-N.; Grishina, M.-M.; Matsulevich, Z.-V.; Lukiyanova, J.-M.; Borisova, G.-N.; Osmanov, V.-K.; Novikov, A.-S.; Kirichuk, A.-A.; Borisov, A.-V.; Solari, E.; et al. Novel cationic 1,2,4-selenadiazoles: Synthesis via addition of 2-pyridylselenyl halides to unactivated nitriles, structures and four-center Se⋯N contacts. Dalton Trans. 2021, 50, 10689–10691. [Google Scholar] [CrossRef] [PubMed]
- Grudova, M.-V.; Kubasov, A.-S.; Khrustalev, V.-N.; Novikov, A.-S.; Kritchenkov, A.-S.; Nenajdenko, V.-G.; Borisov, A.-V.; Tskhovrebov, A.-G. Exploring supramolecular assembly space of cationic 1,2,4-selenodiazoles: Effect of the substituent at the carbon atom and anions. Molecules 2022, 27, 1029–1041. [Google Scholar] [CrossRef] [PubMed]
- Artemjev, A.-A.; Astafiev, A.-A.; Vologzhanina, A.-V.; Kubasov, A.-S.; Burkin, G.-M.; Novikov, A.-S.; Kritchenkov, A.-S.; Kirichuk, A.-A.; Tskhovrebov, A.-G. Triarylazoimidazole-ZnII, CdII, and HgII complexes: Structures, photophysics, and antibacterial properties. Crystals 2022, 12, 680–693. [Google Scholar] [CrossRef]
- Lu, T.; Chen, F.-W. Multiwfn: A Multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef] [PubMed]
- Pant, S.; Zhang, J.; Kim, E.-C.; Lam, K.; Chung, H.-J.; Tajkhorshid, E. PIP2-dependent coupling of voltage sensor and pore domains in Kv7.2 channel. Commun. Biol. 2021, 4, 1189. [Google Scholar] [CrossRef] [PubMed]
- Zhai, P.-F.; Shi, C.-Y.; Zhao, S.-X.; Mei, Z.-S.; Pan, Y.-G. Molecular dynamics simulations of a cyclotetramethylene tetra-nitramine/hydrazine 5,5’-bitetrazole-1,1’-diolate cocrystal. RSC Adv. 2019, 9, 19390–19396. [Google Scholar] [CrossRef] [PubMed]
- Bunte, S.-W.; Sun, H. Molecular modeling of energetic materials: The parameterization and validation of nitrate esters in the COMPASS force field. J. Phys. Chem. B 2000, 104, 2477–2489. [Google Scholar] [CrossRef]
- Wang, F. Study on the Interaction and Thermal Stability of TKX-50/Additives System; Beijing Institute of Technology: Beijing, China, 2017. [Google Scholar]
- Liu, Y. Research on Design and Controlled Assembly of Nitroamine Explosive Cocrystal; North University of China: Taiyuan, China, 2020. [Google Scholar]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).