
Prediction of Intact N-Glycopeptide Retention Time Windows in Hydrophilic Interaction Liquid Chromatography



Abstract
:1. Introduction
2. Results and Discussion
2.1. Relative Retention Time
2.2. Prediction of Retention Time Windows
3. Materials and Methods
3.1. Chemicals
3.2. Sample Processing
3.3. Instrumentation and Experimental Conditions
3.4. Prediction Model Construction
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Gutierrez Reyes, C.D.; Jiang, P.; Donohoo, K.; Atashi, M.; Mechref, Y.S. Glycomics and glycoproteomics: Approaches to address isomeric separation of glycans and glycopeptides. J. Sep. Sci. 2021, 44, 403–425. [Google Scholar] [CrossRef]
- Chang, D.; Zaia, J. Methods to improve quantitative glycoprotein coverage from bottom-up LC-MS data. Mass Spectrom. Rev. 2021. [Google Scholar] [CrossRef]
- Chernykh, A.; Kawahara, R.; Thaysen-Andersen, M. Towards structure-focused glycoproteomics. Biochem. Soc. Trans. 2021, 49, 161–186. [Google Scholar] [CrossRef]
- Ruhaak, L.R.; Xu, G.; Li, Q.; Goonatilleke, E.; Lebrilla, C.B. Mass Spectrometry Approaches to Glycomic and Glycoproteomic Analyses. Chem. Rev. 2018, 118, 7886–7930. [Google Scholar] [CrossRef] [PubMed]
- Choo, M.S.; Wan, C.; Rudd, P.M.; Nguyen-Khuong, T. GlycopeptideGraphMS: Improved Glycopeptide Detection and Identification by Exploiting Graph Theoretical Patterns in Mass and Retention Time. Anal. Chem. 2019, 91, 7236–7244. [Google Scholar] [CrossRef] [PubMed]
- Zeng, W.F.; Cao, W.Q.; Liu, M.Q.; He, S.M.; Yang, P.Y. Precise, fast and comprehensive analysis of intact glycopeptides and modified glycans with pGlyco3. Nat. Methods 2021, 18, 1515–1523. [Google Scholar] [CrossRef] [PubMed]
- Fang, Z.; Qin, H.; Mao, J.; Wang, Z.; Zhang, N.; Wang, Y.; Liu, L.; Nie, Y.; Dong, M.; Ye, M. Glyco-Decipher enables glycan database-independent peptide matching and in-depth characterization of site-specific N-glycosylation. Nat. Commun. 2022, 13, 1900. [Google Scholar] [CrossRef] [PubMed]
- Kawahara, R.; Chernykh, A.; Alagesan, K.; Bern, M.; Cao, W.; Chalkley, R.J.; Cheng, K.; Choo, M.S.; Edwards, N.; Goldman, R.; et al. Community evaluation of glycoproteomics informatics solutions reveals high-performance search strategies for serum glycopeptide analysis. Nat. Methods 2021, 18, 1304–1316. [Google Scholar] [CrossRef] [PubMed]
- Sanda, M.; Benicky, J.; Goldman, R. Low Collision Energy Fragmentation in Structure-Specific Glycoproteomics Analysis. Anal. Chem. 2020, 92, 8262–8267. [Google Scholar] [CrossRef]
- Bruderer, R.; Bernhardt, O.M.; Gandhi, T.; Reiter, L. High-precision iRT prediction in the targeted analysis of data-independent acquisition and its impact on identification and quantitation. Proteomics 2016, 16, 2246–2256. [Google Scholar] [CrossRef] [Green Version]
- Tarasova, I.A.; Masselon, C.D.; Gorshkov, A.V.; Gorshkov, M.V. Predictive chromatography of peptides and proteins as a complementary tool for proteomics. Analyst 2016, 141, 4816–4832. [Google Scholar] [CrossRef]
- Villacrés, C.; Spicer, V.; Krokhin, O.V. Confident Identification of Citrullination and Carbamylation Assisted by Peptide Retention Time Prediction. J. Proteome Res. 2021, 20, 1571–1581. [Google Scholar] [CrossRef]
- Ang, E.; Neustaeter, H.; Spicer, V.; Perreault, H.; Krokhin, O.V. Retention Time Prediction for Glycopeptides in Reversed Phase Chromatography for Glycoproteomic Applications. Anal. Chem. 2019, 91, 13360–13366. [Google Scholar] [CrossRef]
- Kozlik, P.; Goldman, R.; Sanda, M. Study of structure-dependent chromatographic behavior of glycopeptides using reversed phase nanoLC. Electrophoresis 2017, 38, 2193–2199. [Google Scholar] [CrossRef]
- Ozohanics, O.; Turiák, L.; Puerta, A.; Vékey, K.; Drahos, L. High-performance liquid chromatography coupled to mass spectrometry methodology for analyzing site-specific N-glycosylation patterns. J. Chromatogr. A 2012, 1259, 200–212. [Google Scholar] [CrossRef]
- Stavenhagen, K.; Hinneburg, H.; Kolarich, D.; Wuhrer, M. Site-specific N- and O-glycopeptide analysis using an integrated C18-PGC-LC-ESI-QTOF-MS/MS approach. Methods Mol. Biol. 2017, 1503, 109–119. [Google Scholar] [CrossRef]
- Kozlik, P.; Goldman, R.; Sanda, M. Hydrophilic interaction liquid chromatography in the separation of glycopeptides and their isomers. Anal. Bioanal. Chem. 2018, 410, 5001–5008. [Google Scholar] [CrossRef]
- Kozlik, P.; Sanda, M.; Goldman, R. Nano reversed phase versus nano hydrophilic interaction liquid chromatography on a chip in the analysis of hemopexin glycopeptides. J. Chromatogr. A 2017, 1519, 152–155. [Google Scholar] [CrossRef]
- Huang, Y.; Nie, Y.; Boyes, B.; Orlando, R. Resolving isomeric glycopeptide glycoforms with hydrophilic interaction chromatography (HILIC). J. Biomol. Tech. 2016, 27, 98–104. [Google Scholar] [CrossRef] [Green Version]
- Molnarova, K.; Kozlík, P. Comparison of different HILIC stationary phases in the separation of hemopexin and immunoglobulin G glycopeptides and their isomers. Molecules 2020, 25, 4655. [Google Scholar] [CrossRef]
- Badgett, M.J.; Mize, E.; Fletcher, T.; Boyes, B.; Orlando, R. Predicting the hilic retention behavior of the n-linked glycopeptides produced by trypsin digestion of immunoglobulin gs (Iggs). J. Biomol. Tech. 2018, 29, 98–104. [Google Scholar] [CrossRef]
- Molnarova, K.; Duris, A.; Jecmen, T.; Kozlik, P. Comparison of human IgG glycopeptides separation using mixed-mode hydrophilic interaction/ion-exchange liquid chromatography and reversed-phase mode. Anal. Bioanal. Chem. 2021, 413, 4321–4328. [Google Scholar] [CrossRef]
- Badgett, M.J.; Boyes, B.; Orlando, R. Predicting the retention behavior of specific O-Linked glycopeptides. J. Biomol. Tech. 2017, 28, 122–126. [Google Scholar] [CrossRef] [Green Version]
- Benicky, J.; Sanda, M.; Pompach, P.; Wu, J.; Goldman, R. Quantification of fucosylated hemopexin and complement factor H in plasma of patients with liver disease. Anal. Chem. 2014, 86, 10716–10723. [Google Scholar] [CrossRef] [Green Version]
- Pompach, P.; Brnakova, Z.; Sanda, M.; Wu, J.; Edwards, N.; Goldman, R. Site-specific glycoforms of haptoglobin in liver cirrhosis and hepatocellular carcinoma. Mol. Cell. Proteom. 2013, 12, 1281–1293. [Google Scholar] [CrossRef] [Green Version]
- Yuan, W.; Benicky, J.; Wei, R.; Goldman, R.; Sanda, M. Quantitative Analysis of Sex-Hormone-Binding Globulin Glycosylation in Liver Diseases by Liquid Chromatography-Mass Spectrometry Parallel Reaction Monitoring. J. Proteome Res. 2018, 17, 2755–2766. [Google Scholar] [CrossRef]
- Togayachi, A.; Tomioka, A.; Fujita, M.; Sukegawa, M.; Noro, E.; Takakura, D.; Miyazaki, M.; Shikanai, T.; Narimatsu, H.; Kaji, H. Identification of Poly-N-Acetyllactosamine-Carrying Glycoproteins from HL-60 Human Promyelocytic Leukemia Cells Using a Site-Specific Glycome Analysis Method, Glyco-RIDGE. J. Am. Soc. Mass Spectrom. 2018, 29, 1138–1152. [Google Scholar] [CrossRef]
- Badgett, M.J.; Boyes, B.; Orlando, R. Peptide retention prediction using hydrophilic interaction liquid chromatography coupled to mass spectrometry. J. Chromatogr. A 2018, 1537, 58–65. [Google Scholar] [CrossRef]
- Gilar, M.; Jaworski, A. Retention behavior of peptides in hydrophilic-interaction chromatography. J. Chromatogr. A 2011, 1218, 8890–8896. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
 , N-acetylglucosamine (GlcNAc);
, N-acetylglucosamine (GlcNAc);  , Mannose (Man);
, Mannose (Man);  , Galactose (Gal);
, Galactose (Gal);  , Fucose (Fuc);
, Fucose (Fuc);  , Sialic acid.
, N-acetylglucosamine (GlcNAc); , Mannose (Man); , Galactose (Gal); , Fucose (Fuc); , Sialic acid.
, Sialic acid.
, N-acetylglucosamine (GlcNAc); , Mannose (Man); , Galactose (Gal); , Fucose (Fuc); , Sialic acid.| Sex-Hormone Binding Globulin | |||||
|---|---|---|---|---|---|
| SHEIWTHSCPQSPGNGTDASH LDVDQALNR |  A2G2 |  A2G2F1 |  A2G2S1 | 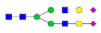 A2G2S2 | 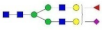 A2G2S1F1 |
 A2G2S2F1 | 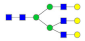 A3G3 | 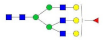 A3G3F1 | 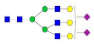 A3G3S2 | 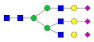 A3G3S3 | |
| Haptoglobin | |||||
| VVLHPNYSQVDIGLIK | A2G2 | A2G2F1 | A2G2S1 | A2G2S2 | A2G2S1F1 |
| A2G2S2F1 | A3G3 | A3G3F1 | 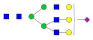 A3G3S1 | A3G3S2 | |
| A3G3S3 | |||||
| MVSHHNLTTGATLINEQWLLTTAK | A2G2 | A2G2F1 | A2G2S1 | A2G2S2 | A2G2S1F1 |
| A3G3 | A3G3F1 | A3G3S1 | A3G3S2 | A3G3S3 | |
| Hemopexin | |||||
| SWPAVGNCSSALR | A2G2 | A2G2F1 | A2G2S1 | A2G2S2 | A2G2S2F1 |
| A3G3 | A3G3F1 | A3G3S1 | A3G3S2 | A3G3S3 | |
| ALPQPQNVTSLLGCTH | A2G2 | A2G2F1 | A2G2S1 | A2G2S2 | A2G2S1F1 |
| A2G2S2F1 | A3G3 | A3G3F1 | |||
| A2G2F1 | A3G3 | A3G3F1 | A2G2S1 | A2G2S2 | |
| Median | 1.032 | 1.088 | 1.121 | 1.387 | 1.879 |
| Lower limit | 1.027 | 1.074 | 1.102 | 1.347 | 1.846 |
| Upper limit | 1.034 | 1.101 | 1.140 | 1.427 | 1.912 |
| A2G2S1F1 | A2G2S2F1 | A3G3S1 | A3G3S2 | A3G3S3 | |
| Median | 1.410 | 1.892 | 1.447 | 1.953 | 2.307 |
| Lower limit | 1.388 | 1.813 | 1.372 | 1.886 | 2.232 |
| Upper limit | 1.433 | 1.971 | 1.522 | 2.019 | 2.381 |
| Identified Glycoforms | Predicted Retention Window (min) | Measured Retention Time (min) |
|---|---|---|
| LCPDCPLLAPLNDSR | ||
| A2G2 | 30.2 | |
| A2G2S2 | 54.8–59.6 | 55.8 |
| A3G3 | 32.5–33.3 | 32.9 |
| A3G3S2 | 57.0–61.0 | 59.4 |
| A3G3S3 | 67.5–71.9 | 68.1 |
| RPTGEVYDIEIDTLETTCHVLDPTPLANCSVR | ||
| A2G2 | 36.5 | |
| A2G2S1 | 49.2–52.1 | 49.8 |
| A2G2S2 | 66.2–72.0 | 67.1 |
| A3G3 | 39.2–40.2 | 39.6 |
| A3G3S1 | 50.7–52.3 | 52.1 |
| A3G3S3 | 81.5–86.8 | 84.1 |
| VVHAVEVALATFNAESNGSYLQLVEISR | ||
| A2G2 | 33.1 | |
| A3G3 | 35.6–36.5 | 35.8 |
| A3G3S3 | 73.9–78.7 | 75.7 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kozlik, P.; Molnarova, K.; Jecmen, T.; Krizek, T.; Bosakova, Z. Prediction of Intact N-Glycopeptide Retention Time Windows in Hydrophilic Interaction Liquid Chromatography. Molecules 2022, 27, 3723. https://doi.org/10.3390/molecules27123723
Kozlik P, Molnarova K, Jecmen T, Krizek T, Bosakova Z. Prediction of Intact N-Glycopeptide Retention Time Windows in Hydrophilic Interaction Liquid Chromatography. Molecules. 2022; 27(12):3723. https://doi.org/10.3390/molecules27123723
Chicago/Turabian StyleKozlik, Petr, Katarina Molnarova, Tomas Jecmen, Tomas Krizek, and Zuzana Bosakova. 2022. "Prediction of Intact N-Glycopeptide Retention Time Windows in Hydrophilic Interaction Liquid Chromatography" Molecules 27, no. 12: 3723. https://doi.org/10.3390/molecules27123723
APA StyleKozlik, P., Molnarova, K., Jecmen, T., Krizek, T., & Bosakova, Z. (2022). Prediction of Intact N-Glycopeptide Retention Time Windows in Hydrophilic Interaction Liquid Chromatography. Molecules, 27(12), 3723. https://doi.org/10.3390/molecules27123723