
TP-CSO: A Triptolide Prodrug for Pancreatic Cancer Treatment



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
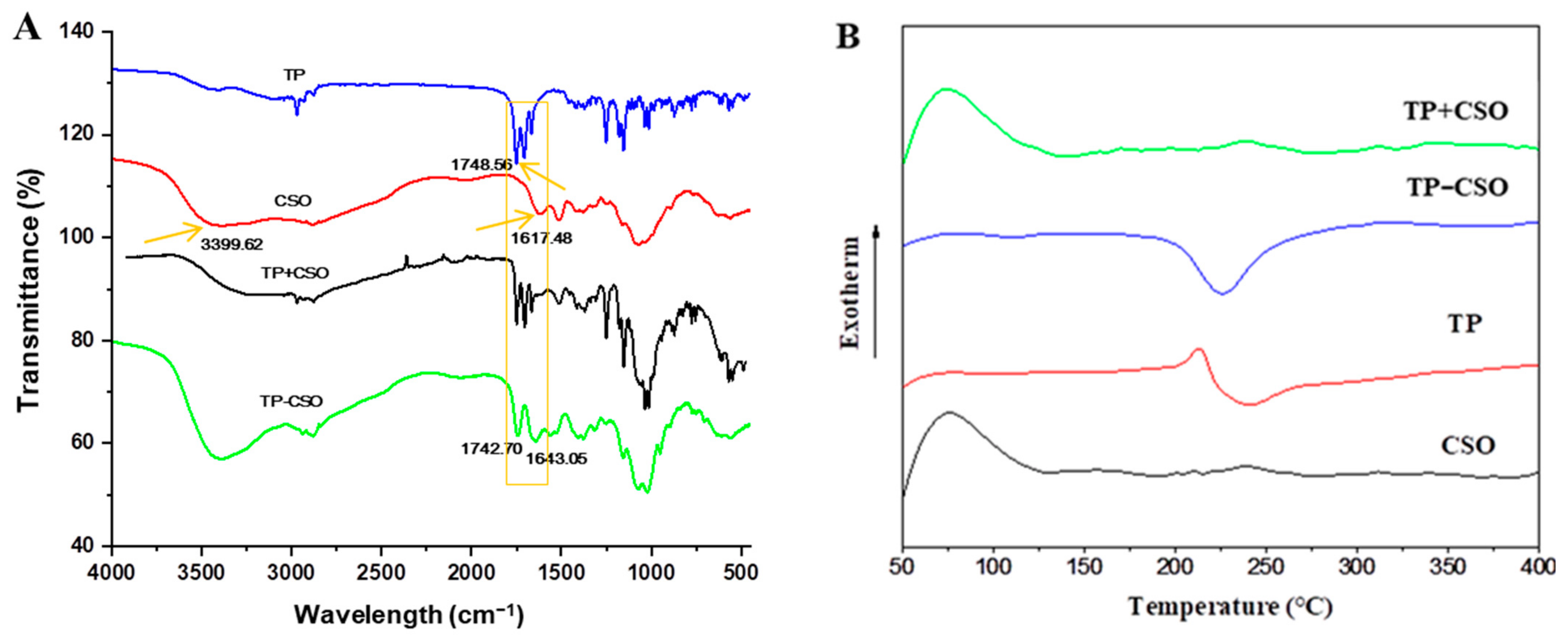
2.1. Preparation and Characterization of TP-CSO
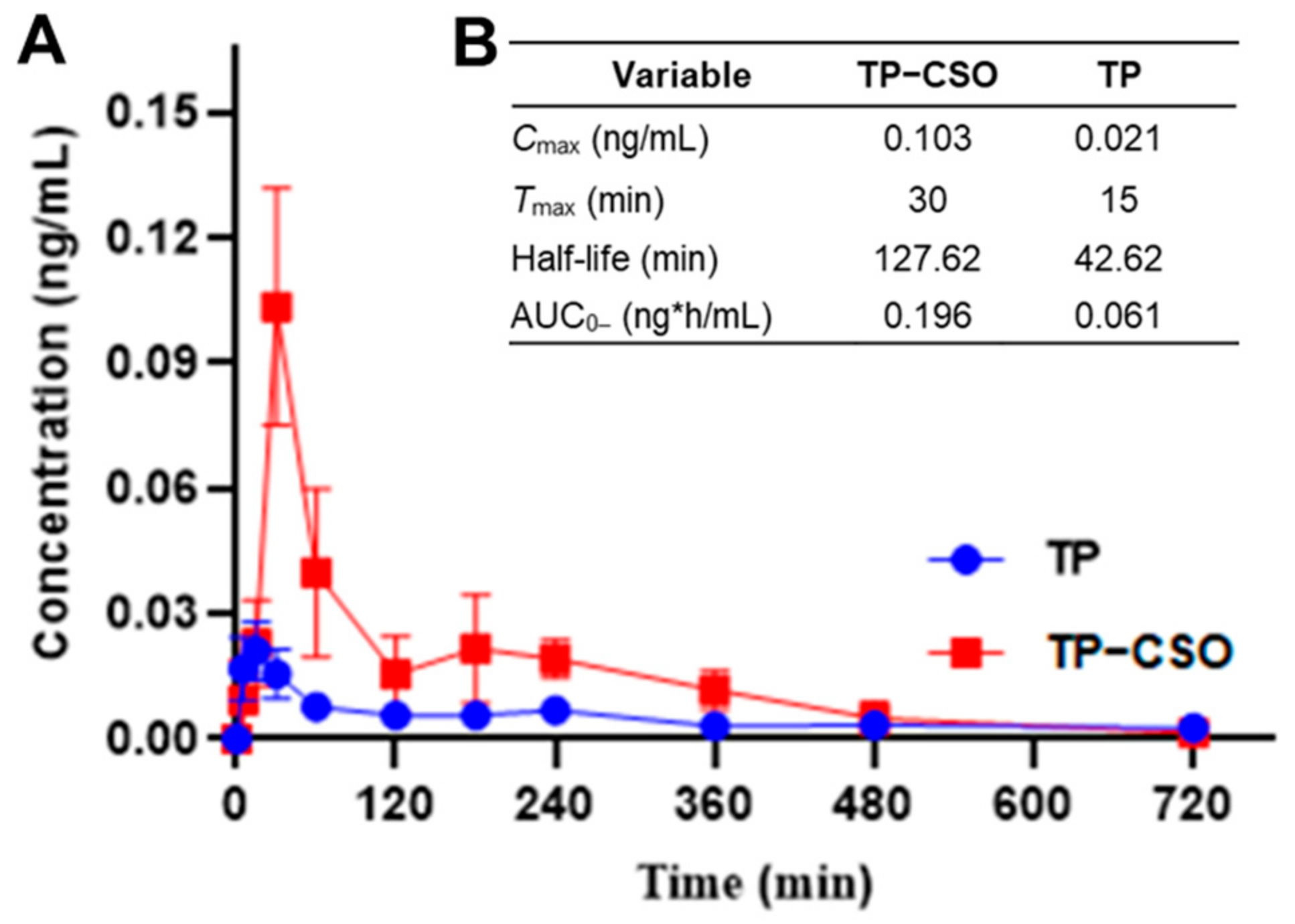
2.2. Pharmacokinetics after Gavage Administration of TP-CSO to Rats
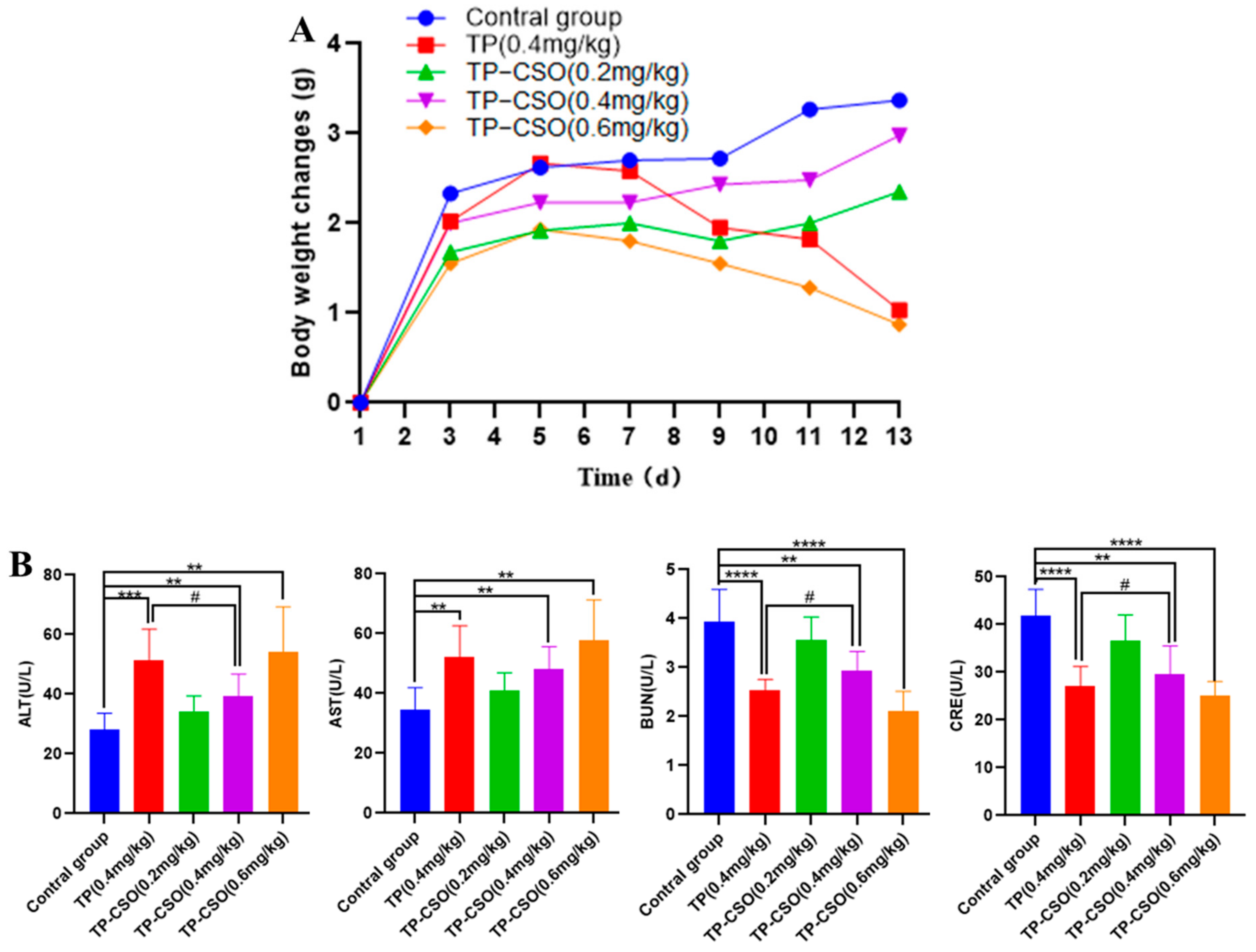
2.3. In Vivo Toxicity of TP-CSO
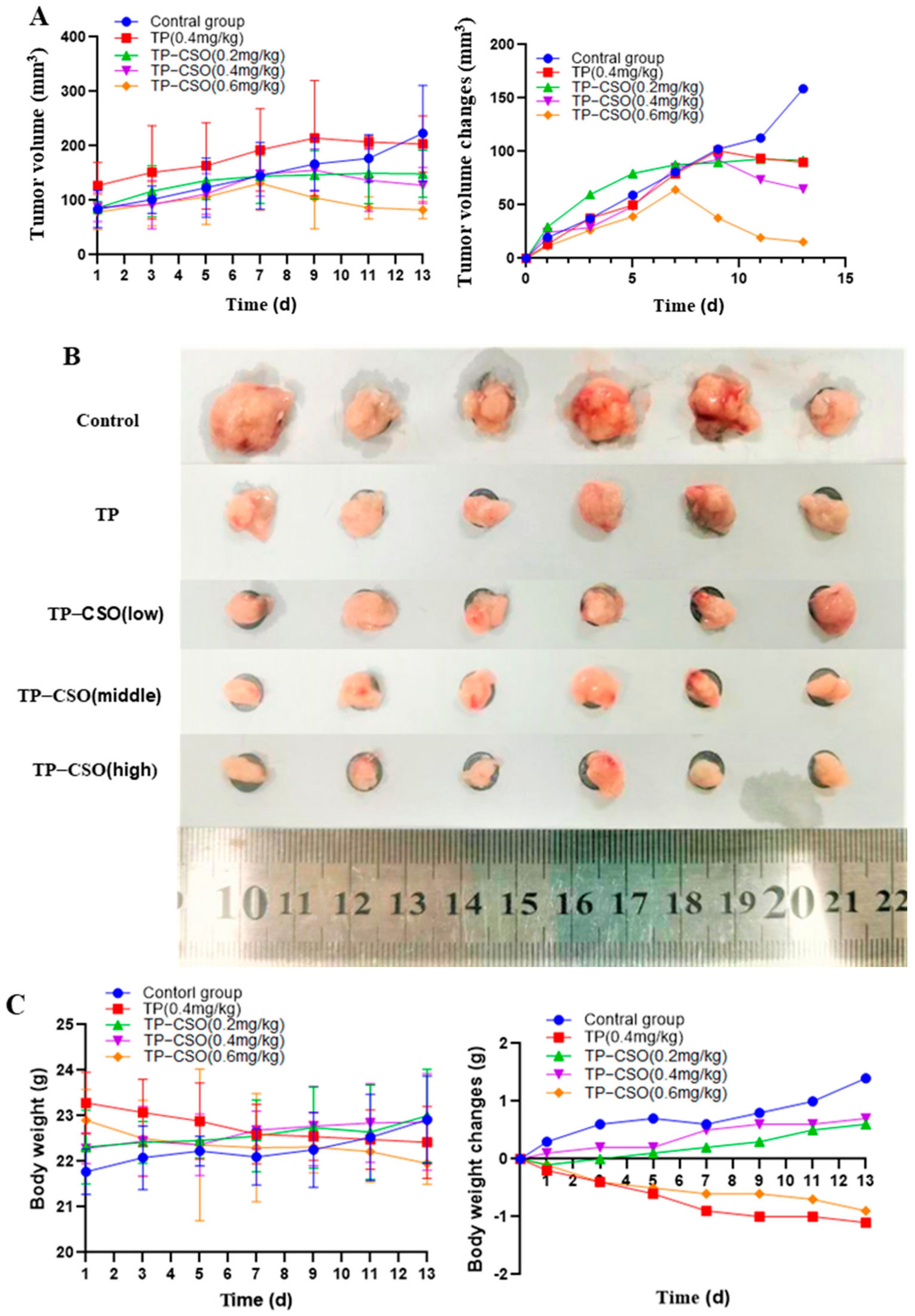
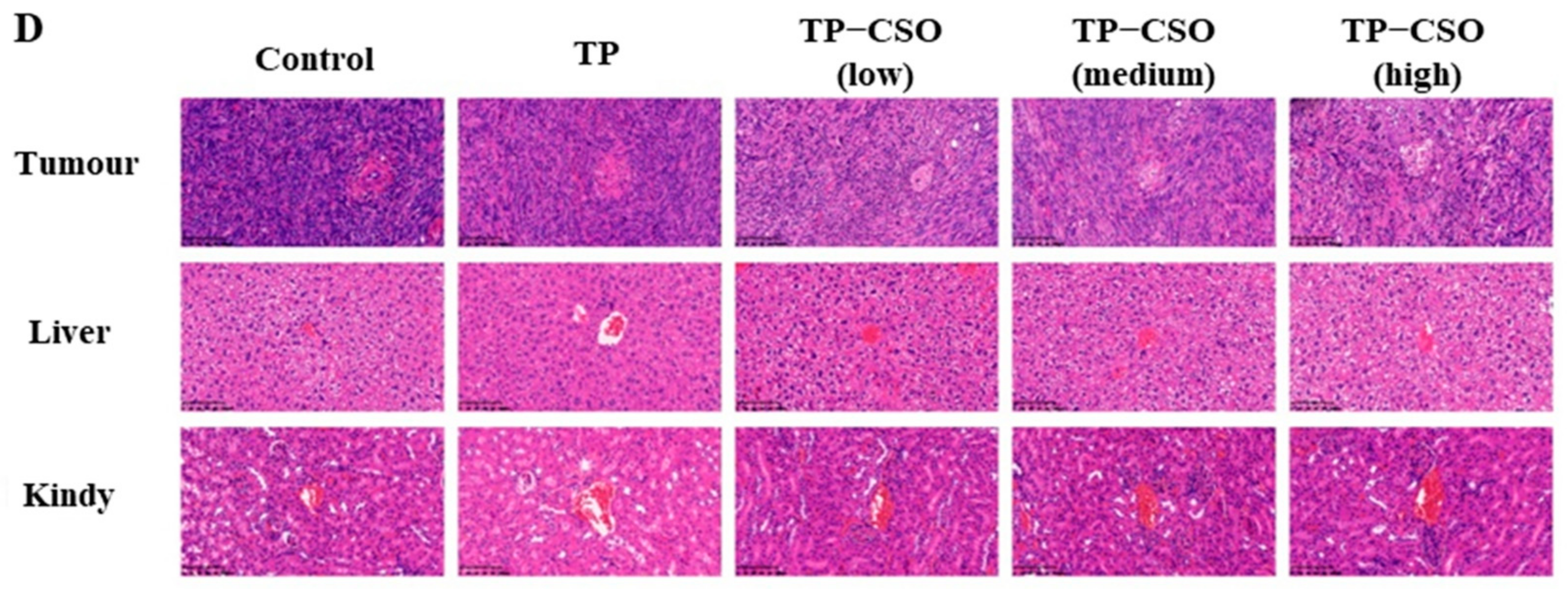
2.4. Antitumor Activity of TP-CSO
3. Materials and Methods
3.1. Materials
3.2. Preparation and Characterization of TP-CSO Conjugate
3.3. Differential Scanning Calorimetry (DSC)
3.4. X-ray Diffraction (XRD)
3.5. Determination of TP Loading Capacity
3.6. The Water Solubility Study
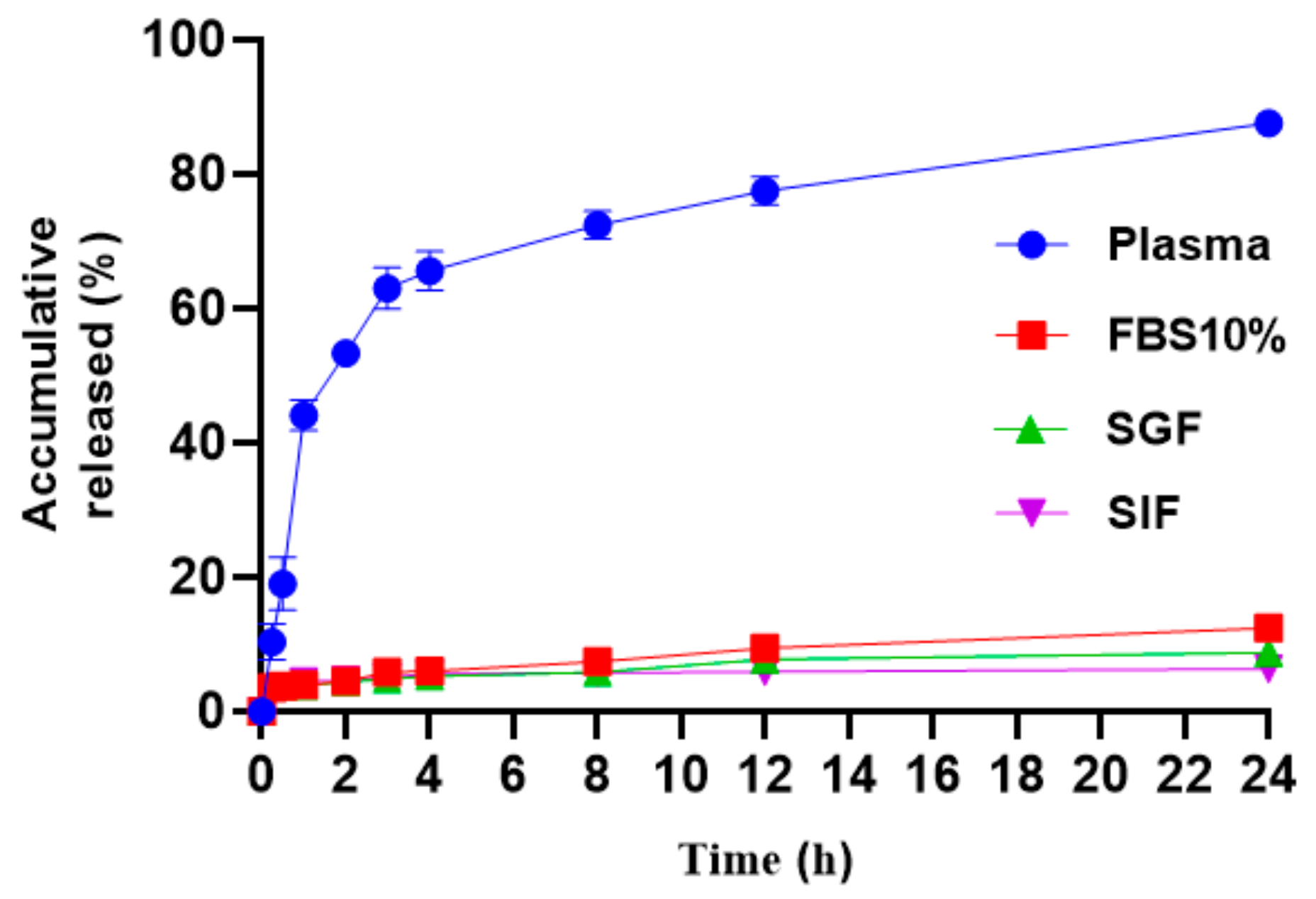
3.7. In Vitro Release Study of TP-CSO
3.8. Pharmacokinetic Study
3.9. In Vivo Toxicity Study
3.10. In Vivo Antitumor Study
3.11. Statistical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- McGuigan, A.; Kelly, P.; Turkington, R.C.; Jones, C.; Coleman, H.G.; McCain, R.S. Pancreatic cancer: A review of clinical diagnosis, epidemiology, treatment and outcomes. World J. Gastroenterol. 2018, 24, 4846–4861. [Google Scholar] [CrossRef] [PubMed]
- Jentzsch, V.; Davis, J.; Djamgoz, M. Pancreatic Cancer (PDAC): Introduction of Evidence-Based Complementary Measures into Integrative Clinical Management. Cancers 2020, 12, 3096. [Google Scholar] [CrossRef] [PubMed]
- Mizrahi, J.D.; Surana, R.; Valle, J.W.; Shroff, R.T. Pancreatic cancer. Lancet 2020, 395, 2008–2020. [Google Scholar] [CrossRef]
- Conroy, T.; Hammel, P.; Hebbar, M.; Ben Abdelghani, M.; Wei, A.C.; Raoul, J.L.; Choné, L.; Francois, E.; Artru, P.; Biagi, J.J.; et al. FOLFIRINOX or Gemcitabine as Adjuvant Therapy for Pancreatic Cancer. N. Engl. J. Med. 2018, 379, 2395–2406. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.S.; Lee, J.C.; Kim, J.H.; Kim, J.; Hwang, J.H. Pharmacoethnicity of FOLFIRINOX versus gemcitabine plus nab-paclitaxel in metastatic pancreatic cancer: A systematic review and meta-analysis. Sci. Rep. 2021, 11, 20152. [Google Scholar] [CrossRef] [PubMed]
- Saung, M.T.; Zheng, L. Current Standards of Chemotherapy for Pancreatic Cancer. Clin. Ther. 2017, 39, 2125–2134. [Google Scholar] [CrossRef] [Green Version]
- Hackert, T.; Sachsenmaier, M.; Hinz, U.; Schneider, L.; Michalski, C.W.; Springfeld, C.; Strobel, O.; Jäger, D.; Ulrich, A.; Büchler, M.W. Locally Advanced Pancreatic Cancer: Neoadjuvant Therapy With Folfirinox Results in Resectability in 60% of the Patients. Ann. Surg. 2016, 264, 457–463. [Google Scholar] [CrossRef]
- Li, J.X.; Shi, J.F.; Wu, Y.H.; Xu, H.T.; Fu, C.M.; Zhang, J.M. Mechanisms and application of triptolide against breast cancer. Zhongguo Zhong Yao Za Zhi 2021, 46, 3249–3256. [Google Scholar]
- Shi, J.; Ren, Y.; Ma, J.; Luo, X.; Li, J.; Wu, Y.; Gu, H.; Fu, C.; Cao, Z.; Zhang, J. Novel CD44-targeting and pH/redox-dual-stimuli-responsive core-shell nanoparticles loading triptolide combats breast cancer growth and lung metastasis. J. Nanobiotech. 2021, 19, 188. [Google Scholar] [CrossRef]
- Hu, H.; Huang, G.; Wang, H.; Li, X.; Wang, X.; Feng, Y.; Tan, B.; Chen, T. Inhibition effect of triptolide on human epithelial ovarian cancer via adjusting cellular immunity and angiogenesis. Oncol. Rep. 2018, 39, 1191–1196. [Google Scholar] [CrossRef] [Green Version]
- Giri, B.; Gupta, V.K.; Yaffe, B.; Modi, S.; Roy, P.; Sethi, V.; Lavania, S.P.; Vickers, S.M.; Dudeja, V.; Banerjee, S.; et al. Pre-clinical evaluation of Minnelide as a therapy for acute myeloid leukemia. J. Transl. Med. 2019, 17, 163. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.L.; Yang, Y.X.; Ding, J.; Li, Y.C.; Miao, Z.H. Triptolide: Structural modifications, structure-activity relationships, bioactivities, clinical development and mechanisms. Nat. Prod. Rep. 2012, 29, 457–475. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.; Li, J.; Wu, S.; Li, S.; Le, L.; Su, X.; Qiu, P.; Hu, H.; Yan, G. Triptolide cooperates with Cisplatin to induce apoptosis in gemcitabine-resistant pancreatic cancer. Pancreas 2012, 41, 1029–1038. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Mao, Y.; Li, K.; Shi, T.; Yao, H.; Yao, J.; Wang, S. Pharmacokinetics and tissue distribution study in mice of triptolide-loaded lipid emulsion and accumulation effect on pancreas. Drug Deliv. 2016, 23, 1344–1354. [Google Scholar] [CrossRef] [Green Version]
- Patil, S.; Lis, L.G.; Schumacher, R.J.; Norris, B.J.; Morgan, M.L.; Cuellar, R.A.; Blazar, B.R.; Suryanarayanan, R.; Gurvich, V.J.; Georg, G.I. Phosphonooxymethyl Prodrug of Triptolide: Synthesis, Physicochemical Characterization, and Efficacy in Human Colon Adenocarcinoma and Ovarian Cancer Xenografts. J. Med. Chem. 2015, 58, 9334–9344. [Google Scholar] [CrossRef]
- Zhou, Y.; Xia, L.; Yao, W.; Han, J.; Wang, G. Arctiin Antagonizes Triptolide-Induced Hepatotoxicity via Activation of Nrf2 Pathway. Biomed. Res. Int. 2020, 2020, 2508952. [Google Scholar] [CrossRef]
- Xie, L.; Zhao, Y.; Duan, J.; Fan, S.; Shu, L.; Liu, H.; Wang, Y.; Xu, Y.; Li, Y. Integrated Proteomics and Metabolomics Reveal the Mechanism of Nephrotoxicity Induced by Triptolide. Chem. Res. Toxicol. 2020, 33, 1897–1906. [Google Scholar] [CrossRef]
- Xu, Y.; Fan, Y.F.; Zhao, Y.; Lin, N. Overview of reproductive toxicity studies on Tripterygium wilfordii in recent 40 years. Zhongguo Zhong Yao Za Zhi 2019, 44, 3406–3414. [Google Scholar]
- Lee, E.; Kim, H.; Lee, I.H.; Jon, S. In vivo antitumor effects of chitosan-conjugated docetaxel after oral administration. J. Control. Release 2009, 140, 79–85. [Google Scholar] [CrossRef]
- Lee, E.; Lee, J.; Lee, I.H.; Yu, M.; Kim, H.; Chae, S.Y.; Jon, S. Conjugated chitosan as a novel platform for oral delivery of paclitaxel. J. Med. Chem. 2008, 51, 6442–6449. [Google Scholar] [CrossRef]
- Zeng, H.; Zhu, X.; Tian, Q.; Yan, Y.; Zhang, L.; Yan, M.; Li, R.; Li, X.; Wang, G.; Ma, J.; et al. In vivo antitumor effects of carboxymethyl chitosan-conjugated triptolide after oral administration. Drug Deliv. 2020, 27, 848–854. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.Y.; Termsarasab, U.; Lee, M.Y.; Kim, D.H.; Lee, S.Y.; Kim, J.S.; Cho, H.J.; Kim, D.D. Chemosensitizing indomethacin-conjugated chitosan oligosaccharide nanoparticles for tumor-targeted drug delivery. Acta Biomater. 2017, 57, 262–273. [Google Scholar] [CrossRef] [PubMed]
- Kittur, F.S.; Prashanth, K.V.H.; Sankar, K.U.; Tharanathan, R.N. Characterization of chitin, chitosan and their carboxylmethyl derivatives by differential scanning calorimetry. Carbohydr. Polym. 2002, 49, 185–193. [Google Scholar] [CrossRef]
- Xi, C.; Peng, S.; Wu, Z.; Zhou, Q.; Zhou, J. Toxicity of triptolide and the molecular mechanisms involved. Biomed. Pharmacother. 2017, 90, 531–541. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, X.; Zeng, H.; Zhu, X.; Xu, D.; Tian, Q.; Wang, C.; Zhao, L.; Zhao, J.; Miao, M.; Wu, X. TP-CSO: A Triptolide Prodrug for Pancreatic Cancer Treatment. Molecules 2022, 27, 3686. https://doi.org/10.3390/molecules27123686
Wang X, Zeng H, Zhu X, Xu D, Tian Q, Wang C, Zhao L, Zhao J, Miao M, Wu X. TP-CSO: A Triptolide Prodrug for Pancreatic Cancer Treatment. Molecules. 2022; 27(12):3686. https://doi.org/10.3390/molecules27123686
Chicago/Turabian StyleWang, Xinlong, Huahui Zeng, Xin Zhu, Duanjie Xu, Qikang Tian, Can Wang, Lingzhou Zhao, Junwei Zhao, Mingsan Miao, and Xiangxiang Wu. 2022. "TP-CSO: A Triptolide Prodrug for Pancreatic Cancer Treatment" Molecules 27, no. 12: 3686. https://doi.org/10.3390/molecules27123686
APA StyleWang, X., Zeng, H., Zhu, X., Xu, D., Tian, Q., Wang, C., Zhao, L., Zhao, J., Miao, M., & Wu, X. (2022). TP-CSO: A Triptolide Prodrug for Pancreatic Cancer Treatment. Molecules, 27(12), 3686. https://doi.org/10.3390/molecules27123686