
Influence of Multiple Binding Sites on the Supramolecular Assembly of N-[(3-pyridinylamino) Thioxomethyl] Carbamates


Abstract
:1. Introduction
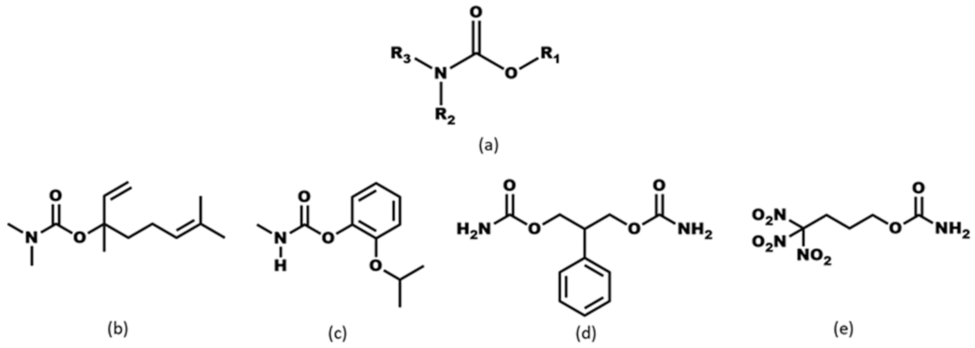
- ◾
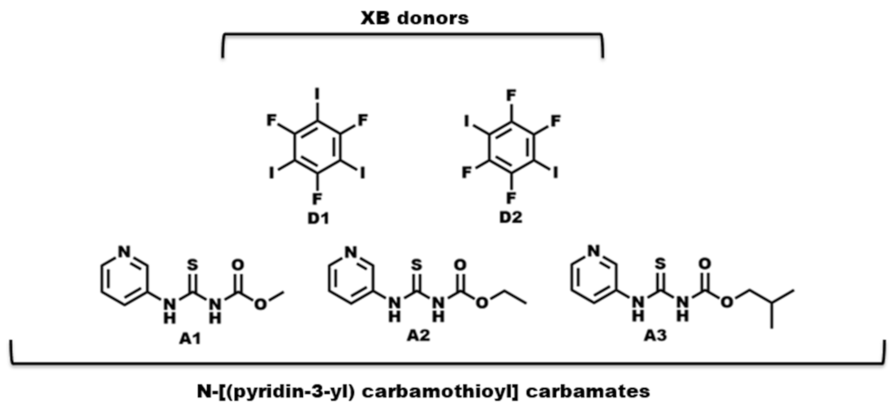
- What is the impact of varying chain length (R = methyl, ethyl, and isobutyl) on the crystal structures of the target molecules A1–A3 (Scheme 2)?
- ◾
- ◾
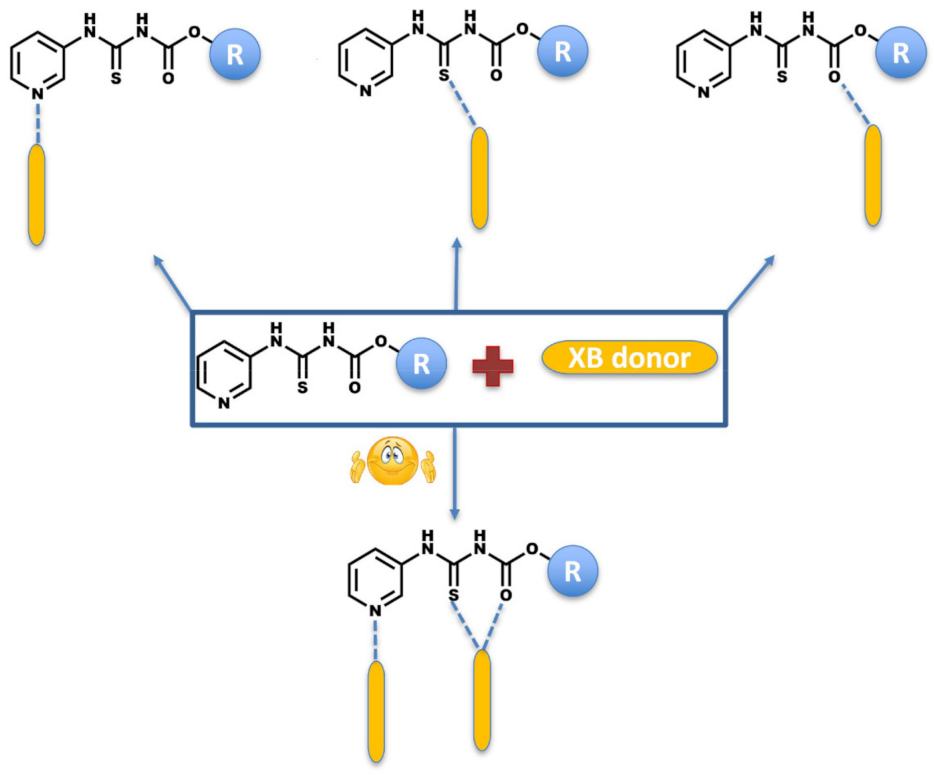
- What binding preference is observed in the solid state when hydrogen-bond and halogen-bond donors (D1 and D2) compete for three different acceptor sites (Scheme 2)?
- ◾
- Does the supramolecular assembly change for different targets if we introduce the same XB donors as co-formers for co-crystallization?
- ◾
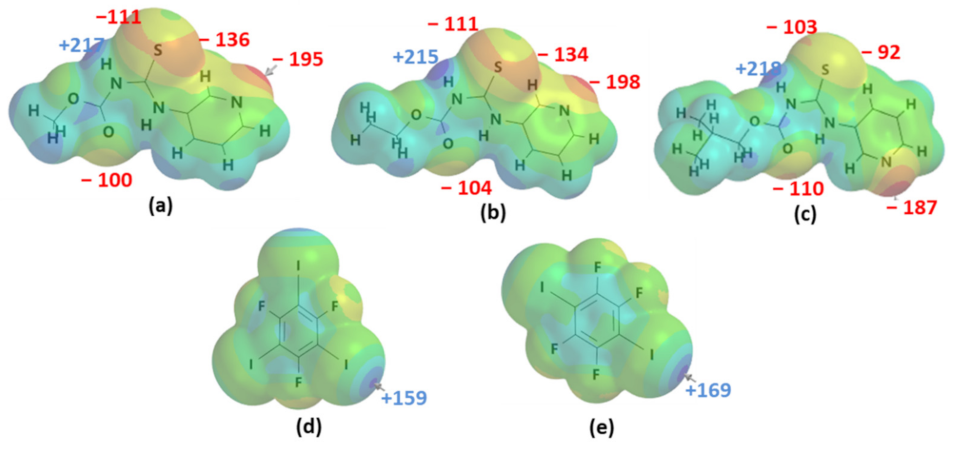
- How reliable are MEP rankings/predictions when multiple acceptors are present on the target molecules?
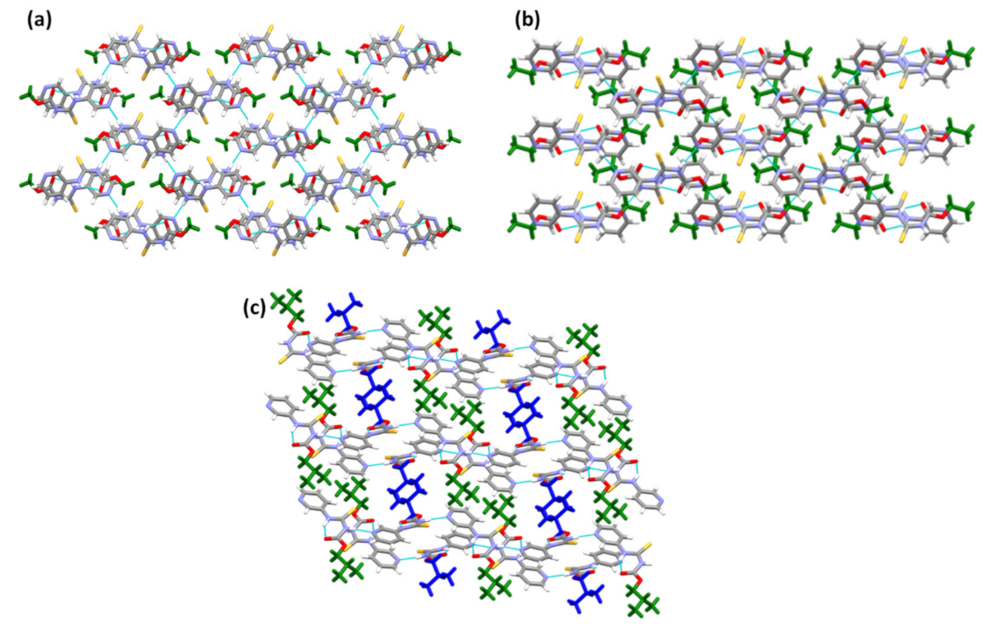
2. Results
3. Discussion
4. Materials and Methods
4.1. Reagents and General Methods
4.2. Synthesis of Methyl-N-[(3-pyridinylamino) Thioxomethyl] Carbamate (A1)
4.3. Synthesis of Ethyl-N-[(3-pyridinylamino) Thioxomethyl] Carbamate (A2)
4.4. Synthesis of Isobutyl-N-[(3-pyridinylamino) Thioxomethyl] Carbamate (A3)
4.5. Crystallization Experiments
4.5.1. Synthesis of Methyl-N-[(3-pyridinylamino) Thioxomethyl] Carbamate·1,3,5-trifluoro-2,4,6-triiodobenzene co-crystal (A1·D1)
4.5.2. Synthesis of di-(ethyl-N-[(3-pyridinylamino) Thioxomethyl] Carbamate)·1,3,5-trifluoro-2,4,6-triiodobenzene (A2)2·D1
4.5.3. Synthesis of Isobutyl-N-[(3-pyridinylamino) Thioxomethyl] Carbamate·1,3,5-trifluoro-2,4,6-triiodobenzene (A3·D1)
4.5.4. Synthesis of di-(methyl-N-[(3-pyridinylamino) Thioxomethyl] Carbamate)·1,2,4,5-tetrafluoro-3,6-diiodobenzene Chloroform Solvate (A1)2·D2
4.5.5. Synthesis of Ethyl-N-[(3-pyridinylamino) Thioxomethyl] Carbamate·1,2,4,5-tetrafluoro-3,6-diiodobenzene A2·D2
4.5.6. Synthesis of di-(isobutyl-N-[(3-pyridinylamino) Thioxomethyl] Carbamate)·1,2,4,5-tetrafluoro-3,6-diiodobenzene (A3)2·D2
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Ardila-Fierro, K.J.; André, V.; Tan, D.; Duarte, M.T.; Lancaster, R.W.; Karamertzanis, P.G.; Friščić, T. Molecular Recognition of Steroid Hormones in the Solid State: Stark Differences in Cocrystallization of β-Estradiol and Estrone. Cryst. Growth Des. 2015, 15, 1492–1501. [Google Scholar] [CrossRef]
- Pramanik, T.; Sarkar, S.; Guru Row, T.N. Halogen Bonded Network Modulating the Mechanical Property Elastic and Plastic Bending in Nonconventional Molecular Solid Solutions. Cryst. Growth Des. 2022, 22, 48–53. [Google Scholar] [CrossRef]
- Reddy, C.M.; Padmanabhan, K.A.; Desiraju, G.R. Structure−Property Correlations in Bending and Brittle Organic Crystals. Cryst. Growth Des. 2006, 6, 2720–2731. [Google Scholar] [CrossRef]
- Saha, S.; Desiraju, G.R. σ-Hole and π-Hole Synthon Mimicry in Third-Generation Crystal Engineering: Design of Elastic Crystals. Chem.—Eur. J. 2017, 23, 4936–4943. [Google Scholar] [CrossRef]
- Yadava, K.; Qin, X.; Liu, X.; Vittal, J.J. Straight, bendable and bent organic crystals. Chem. Commun. 2019, 55, 14749–14752. [Google Scholar] [CrossRef]
- Ghosh, S.; Mishra, M.K.; Kadambi, S.B.; Ramamurty, U.; Desiraju, G.R. Designing Elastic Organic Crystals: Highly Flexible Polyhalogenated N-Benzylideneanilines. Angew. Chem. Int. Ed. 2015, 54, 2674–2678. [Google Scholar] [CrossRef]
- Juneja, N.; Shapiro, N.M.; Unruh, D.K.; Bosch, E.; Groeneman, R.H.; Hutchins, K.M. Controlling Thermal Expansion in Supramolecular Halogen-Bonded Mixed Cocrystals through Synthetic Feed and Dynamic Motion. Angew. Chem. Int. Ed. 2022, e202202708. [Google Scholar] [CrossRef]
- Liu, Y.; Ward, M.D. Molecular Capsules in Modular Frameworks. Cryst. Growth Des. 2009, 9, 3859–3861. [Google Scholar] [CrossRef]
- Aakeröy, C.B.; Beatty, A.M.; Helfrich, B.A.; Nieuwenhuyzen, M. Do Polymorphic Compounds Make Good Cocrystallizing Agents? A Structural Case Study that Demonstrates the Importance of Synthon Flexibility. Cryst. Growth Des. 2003, 3, 159–165. [Google Scholar] [CrossRef]
- Sreekanth, B.R.; Vishweshwar, P.; Vyas, K. Supramolecular synthon polymorphism in 2:1 co-crystal of 4-hydroxybenzoic acid and 2,3,5,6-tetramethylpyrazine. Chem. Commun. 2007, 23, 2375–2377. [Google Scholar] [CrossRef]
- Mukherjee, A.; Desiraju, G.R. Synthon polymorphism and pseudopolymorphism in co-crystals. The 4,4′-bipyridine–4-hydroxybenzoic acid structural landscape. Chem. Commun. 2011, 47, 4090–4092. [Google Scholar] [CrossRef] [PubMed]
- Burrows, J.A.; Pauling, L. The nature of the chemical bond and the structure of molecules and crystals. Nature 1941, 148, 677. [Google Scholar]
- Aakeröy, C.B.; Spartz, C.L.; Dembowski, S.; Dwyre, S.; Desper, J. A systematic structural study of halogen bonding versus hydrogen bonding within competitive supramolecular systems. IUCrJ 2015, 2, 498–510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galek, P.T.A.; Allen, F.H.; Fábián, L.; Feeder, N. Knowledge-based H-bond prediction to aid experimental polymorph screening. CrystEngComm 2009, 11, 2634–2639. [Google Scholar] [CrossRef]
- Hunter, C.A. Quantifying Intermolecular Interactions: Guidelines for the Molecular Recognition Toolbox. Angew. Chem. Int. Ed. 2004, 43, 5310–5324. [Google Scholar] [CrossRef]
- Musumeci, D.; Hunter, C.A.; Prohens, R.; Scuderi, S.; McCabe, J.F. Virtual cocrystal screening. Chem. Sci. 2011, 2, 883–890. [Google Scholar] [CrossRef]
- DeHaven, B.A.; Chen, A.L.; Shimizu, E.A.; Salpage, S.R.; Smith, M.D.; Shimizu, L.S. Interplay between Hydrogen and Halogen Bonding in Cocrystals of Dipyridinylmethyl Oxalamides and Tetrafluorodiiodobenzenes. Cryst. Growth Des. 2019, 19, 5776–5783. [Google Scholar] [CrossRef]
- Martí-Rujas, J.; Colombo, L.; Lü, J.; Dey, A.; Terraneo, G.; Metrangolo, P.; Pilati, T.; Resnati, G. Hydrogen and halogen bonding drive the orthogonal self-assembly of an organic framework possessing 2D channels. Chem. Commun. 2012, 48, 8207–8209. [Google Scholar] [CrossRef] [Green Version]
- Aakeröy, C.B.; Wijethunga, T.K.; Haj, M.A.; Desper, J.; Moore, C. The structural landscape of heteroaryl-2-imidazoles: Competing halogen- and hydrogen-bond interactions. CrystEngComm 2014, 16, 7218–7225. [Google Scholar] [CrossRef]
- Gunawardana, C.A.; Desper, J.; Sinha, A.S.; Ðaković, M.; Aakeröy, C.B. Competition and selectivity in supramolecular synthesis: Structural landscape around 1-(pyridylmethyl)-2,2′-biimidazoles. Faraday Discuss. 2017, 203, 371–388. [Google Scholar] [CrossRef]
- Abeysekera, A.M.; Averkiev, B.B.; Sinha, A.S.; Le Magueres, P.; Aakeröy, C.B. Establishing Halogen-Bond Preferences in Molecules with Multiple Acceptor Sites. ChemPlusChem 2021, 86, 1049–1057. [Google Scholar] [CrossRef] [PubMed]
- De Silva, V.; Averkiev, B.B.; Sinha, A.S.; Aakeröy, C.B. The Balance between Hydrogen Bonds, Halogen Bonds, and Chalcogen Bonds in the Crystal Structures of a Series of 1,3,4-Chalcogenadiazoles. Molecules 2021, 26, 4125. [Google Scholar] [CrossRef] [PubMed]
- Stanley, C.W.; Thornton, J.S.; Katague, D.B. Gas-chromatographic method for residues of Baygon and metabolites in plant tissues. J. Agric. Food Chem. 1972, 20, 1265–1269. [Google Scholar] [CrossRef] [PubMed]
- Teixeira, M.A.; Barrault, L.; Rodríguez, O.; Carvalho, C.C.; Rodrigues, A.E. Perfumery Radar 2.0: A Step toward Fragrance Design and Classification. Ind. Eng. Chem. Res. 2014, 53, 8890–8912. [Google Scholar] [CrossRef]
- Axthammer, Q.J.; Krumm, B.; Klapötke, T.M. Synthesis of Energetic Nitrocarbamates from Polynitro Alcohols and Their Potential as High Energetic Oxidizers. J. Org. Chem. 2015, 80, 6329–6335. [Google Scholar] [CrossRef]
- Popović, M.; Nierkens, S.; Pieters, R.; Uetrecht, J. Investigating the Role of 2-Phenylpropenal in Felbamate-Induced Idiosyncratic Drug Reactions. Chem. Res. Toxicol. 2004, 17, 1568–1576. [Google Scholar] [CrossRef]
- Parker, R.J.; Hartman, N.R.; Roecklein, B.A.; Mortko, H.; Kupferberg, H.J.; Stables, J.; Strong, J.M. Stability and Comparative Metabolism of Selected Felbamate Metabolites and Postulated Fluorofelbamate Metabolites by Postmitochondrial Suspensions. Chem. Res. Toxicol. 2005, 18, 1842–1848. [Google Scholar] [CrossRef]
- Tegeler, T.; El Rassi, Z. On-Column Trace Enrichment by Sequential Frontal and Elution Electrochromatography. 1. Application to Carbamate Insecticides. Anal. Chem. 2001, 73, 3365–3372. [Google Scholar] [CrossRef]
- Aly, O.M.; El-Dib, M.A. Studies of The Persistence of Some Carbamate Insecticides in The Aquatic Environment. In Fate of Organic Pesticides in the Aquatic Environment; American Chemical Society: Washington, DC, USA, 1972; Volume 111, pp. 210–243. [Google Scholar]
- Shamim, M.; Meléndez, J.; Sappington, K.; Ruhman, M. Conducting Ecological Risk Assessments of Urban Pesticide Uses. In Describing the Behavior and Effects of Pesticides in Urban and Agricultural Settings; American Chemical Society: Washington, DC, USA, 2014; Volume 1168, pp. 207–274. [Google Scholar]
- Zhang, J.J.; Yang, H. Advance in Methodology and Strategies To Unveil Metabolic Mechanisms of Pesticide Residues in Food Crops. J. Agric. Food Chem. 2021, 69, 2658–2667. [Google Scholar] [CrossRef]
- Ghosh, A.K.; Brindisi, M. Organic Carbamates in Drug Design and Medicinal Chemistry. J. Med. Chem. 2015, 58, 2895–2940. [Google Scholar] [CrossRef] [Green Version]
- Matošević, A.; Bosak, A. Carbamate group as structural motif in drugs: A review of carbamate derivatives used as therapeutic agents. Arch. Ind. Hyg. Toxicol. 2020, 71, 285–299. [Google Scholar] [CrossRef] [PubMed]
- Bennion, J.C.; Matzger, A.J. Development and Evolution of Energetic Cocrystals. Acc. Chem. Res. 2021, 54, 1699–1710. [Google Scholar] [CrossRef] [PubMed]
- Axthammer, Q.J.; Klapötke, T.M.; Krumm, B.; Reith, T. Energetic Sila-Nitrocarbamates: Silicon Analogues of Neo-Pentane Derivatives. Inorg. Chem. 2016, 55, 4683–4692. [Google Scholar] [CrossRef] [PubMed]
- Klapötke, T.M.; Krumm, B.; Unger, C.C. Energetic Metal and Nitrogen-Rich Salts of the Pentaerythritol Tetranitrate Analogue Pentaerythritol Tetranitrocarbamate. Inorg. Chem. 2019, 58, 2881–2887. [Google Scholar] [CrossRef] [PubMed]
- Aitipamula, S.; Banerjee, R.; Bansal, A.K.; Biradha, K.; Cheney, M.L.; Choudhury, A.R.; Desiraju, G.R.; Dikundwar, A.G.; Dubey, R.; Duggirala, N.; et al. Polymorphs, Salts, and Cocrystals: What’s in a Name? Cryst. Growth Des. 2012, 12, 2147–2152. [Google Scholar] [CrossRef]
- Aakeröy, C.B.; Fasulo, M.E.; Desper, J. Cocrystal or Salt: Does It Really Matter? Mol. Pharm. 2007, 4, 317–322. [Google Scholar] [CrossRef] [PubMed]
- Desiraju, G.R. Crystal Engineering: From Molecule to Crystal. J. Am. Chem. Soc. 2013, 135, 9952–9967. [Google Scholar] [CrossRef]
- Zhang, Y.-M.; Yang, L.-Z.; Lin, Q.; Wei, T.-B.; Wang, D.-Q. Synthesis and the crystal structure of N-ethoxycarbonyl-N′-(3-pyridyl)thiourea. Youji Huaxue 2006, 26, 134–138. [Google Scholar]
- Spek, A. Structure validation in chemical crystallography. Acta Crystallogr. Sect. D 2009, 65, 148–155. [Google Scholar] [CrossRef]
- Bondi, A. van der Waals Volumes and Radii. J. Phys. Chem. 1964, 68, 441–451. [Google Scholar] [CrossRef]
- Groom, C.R.; Bruno, I.J.; Lightfoot, M.P.; Ward, S.C. The Cambridge Structural Database. Acta Crystallogr. Sect. B 2016, 72, 171–179. [Google Scholar] [CrossRef] [PubMed]
- Macrae, C.F.; Sovago, I.; Cottrell, S.J.; Galek, P.T.A.; McCabe, P.; Pidcock, E.; Platings, M.; Shields, G.P.; Stevens, J.S.; Towler, M.; et al. Mercury 4.0: From visualization to analysis, design and prediction. J. Appl. Crystallogr. 2020, 53, 226–235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wavefunction, Inc. Irvine, CA 92612, USA. Available online: https://www.wavefun.com/products/spartan.html (accessed on 5 March 2022).
- Wei, T.B.; Lin, Q.; Zhang, Y.M.; Wang, H. Efficient and Novel Synthesis of N-Aryl-N′-ethoxycarbonylthiourea and Arene-bis-ethoxycarbonylthiourea Derivatives Catalyzed by TMEDA. Synth. Commun. 2004, 34, 2205–2213. [Google Scholar] [CrossRef]
- Ren, Y.-H.; Song, J.-R.; Xu, K.-Z.; Ma, H.-X.; Huang, J.; Fu, D.-W.; Hu, H.-M. Preparation, Crystal Structure and Theoretical Calculation of N-(Pyrimidin-2-yl)-N′-methoxycarbonyl-thiourea. Chin. J. Chem. 2007, 25, 510–514. [Google Scholar] [CrossRef]
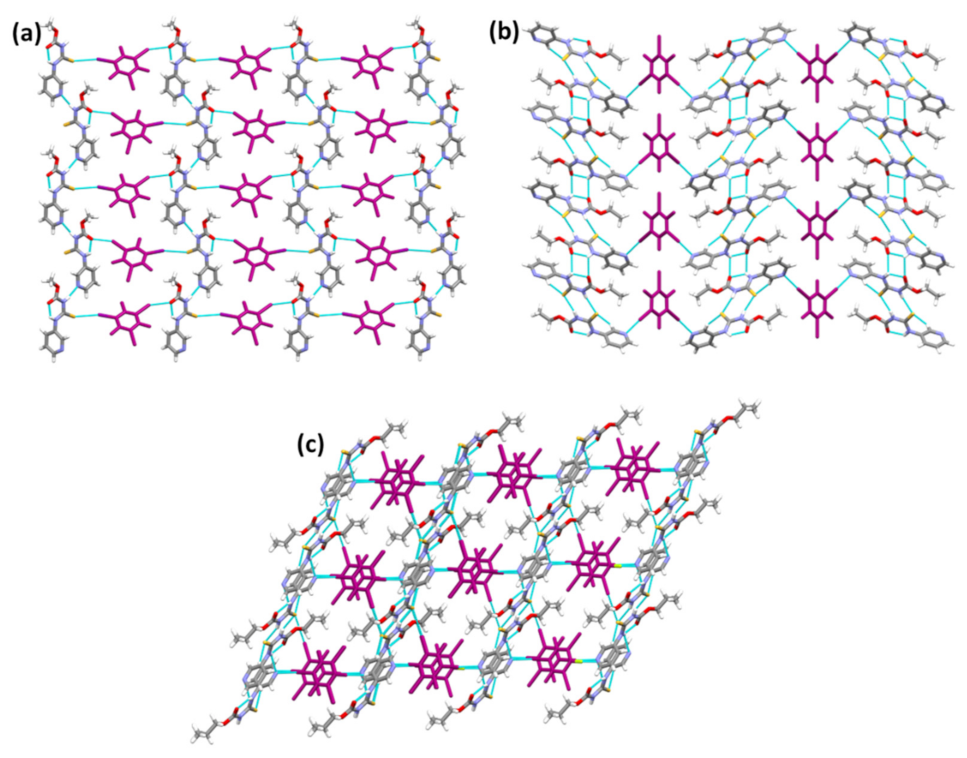
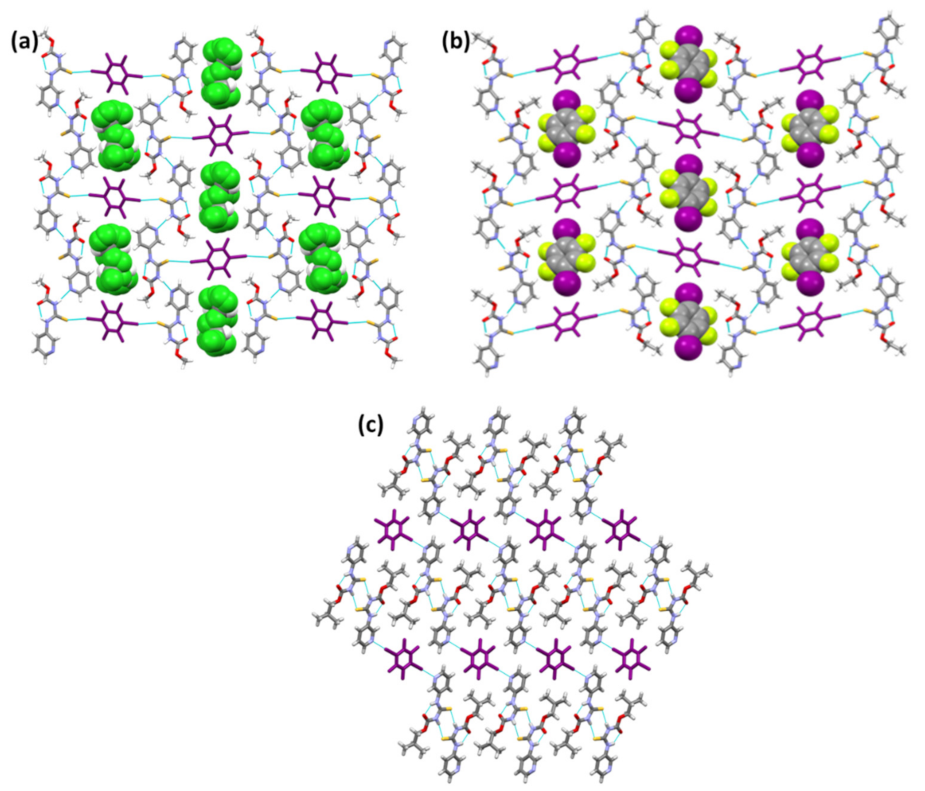

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
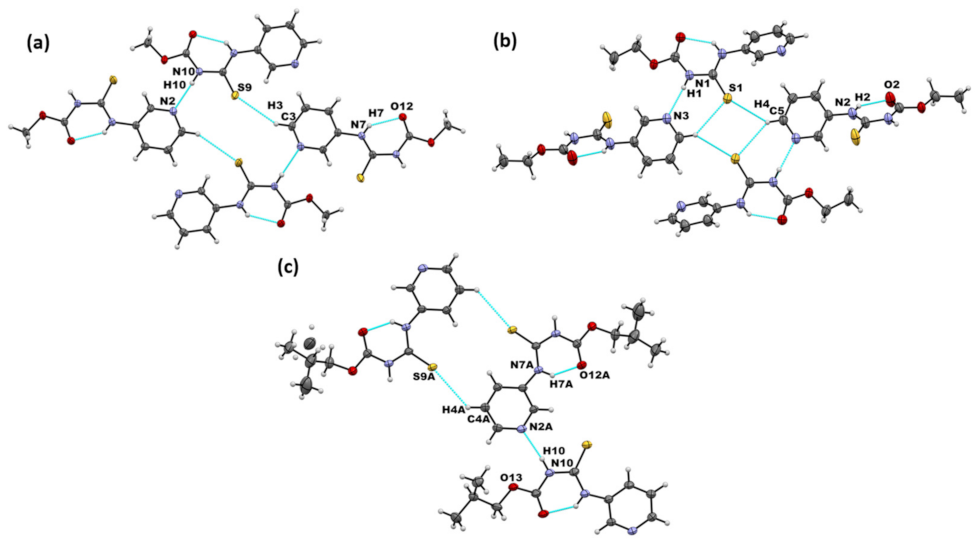
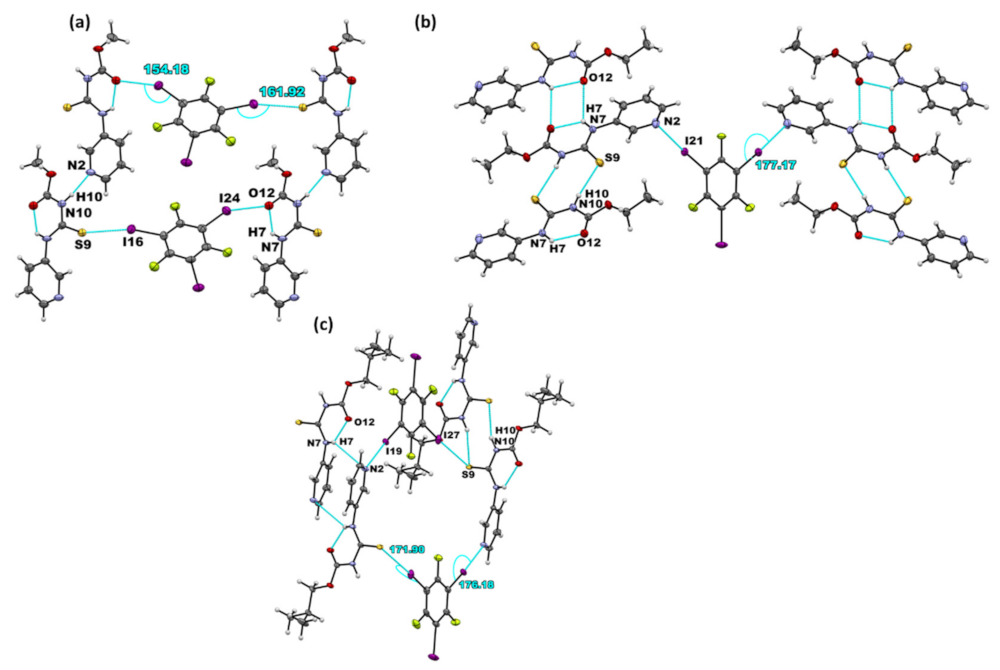
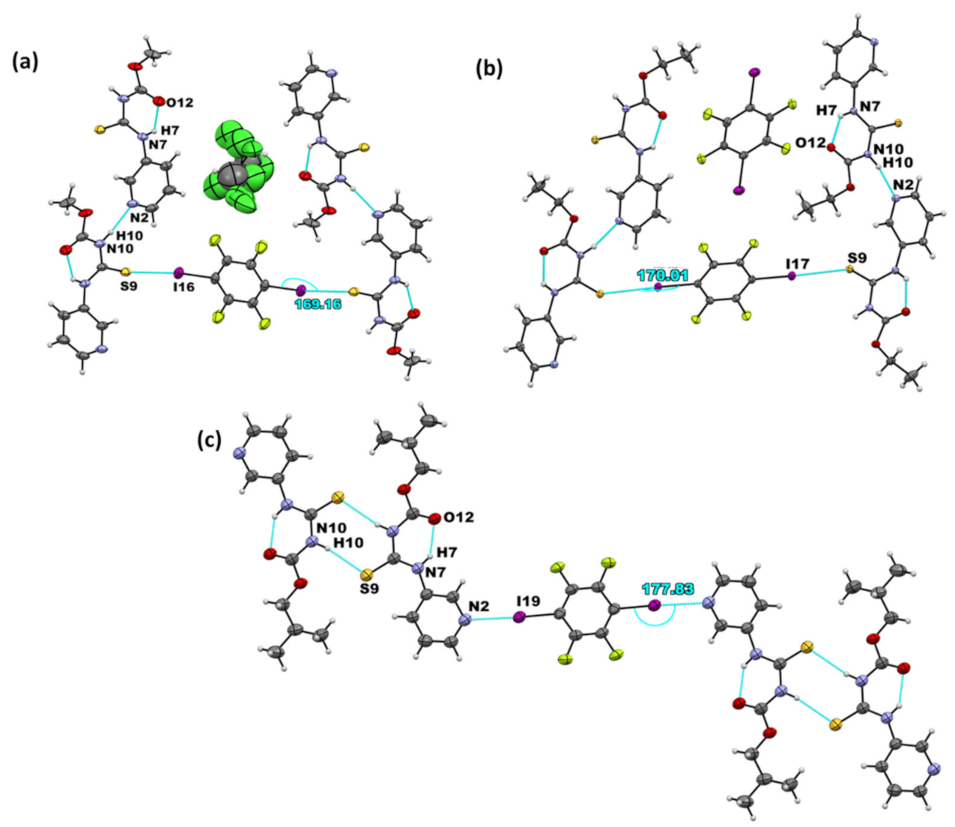
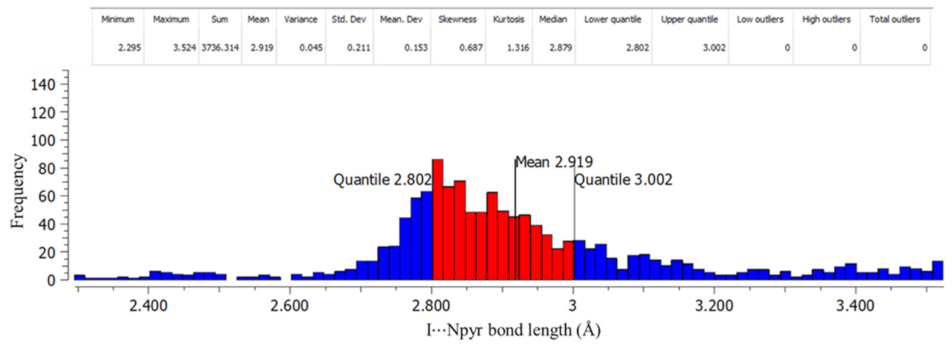
| Co-Crystal | Halogen Bond | Experimental XA Distance | XA Angle | |
|---|---|---|---|---|
| (Å) | % vdW Reduction | (°) | ||
| A1·D1 | I···S=C | 3.310(3) | 12.2 | 161.9(3) |
| I···O=C | 3.168(9) | 9.1 | 154.2(4) | |
| (A2)2·D1 | I···Npyr | 2.877(2) | 18.2 | 177.2(8) |
| A3·D1 | I···S=C | 3.363(6) | 10.9 | 171.9(7) |
| I···Npyr | 2.927(3) | 16 | 176.2(1) | |
| (A1)2·D2 | I···S=C | 3.222(1) | 14.6 | 169.2(1) |
| A2·D2 | I···S=C | 3.248(7) | 13.8 | 170.0(6) |
| (A3)2·D2 | I···Npyr | 2.802(6) | 20.5 | 177.8(2) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shunje, K.N.; Averkiev, B.B.; Aakeröy, C.B. Influence of Multiple Binding Sites on the Supramolecular Assembly of N-[(3-pyridinylamino) Thioxomethyl] Carbamates. Molecules 2022, 27, 3685. https://doi.org/10.3390/molecules27123685
Shunje KN, Averkiev BB, Aakeröy CB. Influence of Multiple Binding Sites on the Supramolecular Assembly of N-[(3-pyridinylamino) Thioxomethyl] Carbamates. Molecules. 2022; 27(12):3685. https://doi.org/10.3390/molecules27123685
Chicago/Turabian StyleShunje, Kelly N., Boris B. Averkiev, and Christer B. Aakeröy. 2022. "Influence of Multiple Binding Sites on the Supramolecular Assembly of N-[(3-pyridinylamino) Thioxomethyl] Carbamates" Molecules 27, no. 12: 3685. https://doi.org/10.3390/molecules27123685
APA StyleShunje, K. N., Averkiev, B. B., & Aakeröy, C. B. (2022). Influence of Multiple Binding Sites on the Supramolecular Assembly of N-[(3-pyridinylamino) Thioxomethyl] Carbamates. Molecules, 27(12), 3685. https://doi.org/10.3390/molecules27123685