
Synthesis and Antiproliferative Activity of Novel Dehydroabietic Acid-Chalcone Hybrids

 , , , and
, , , and 
Abstract
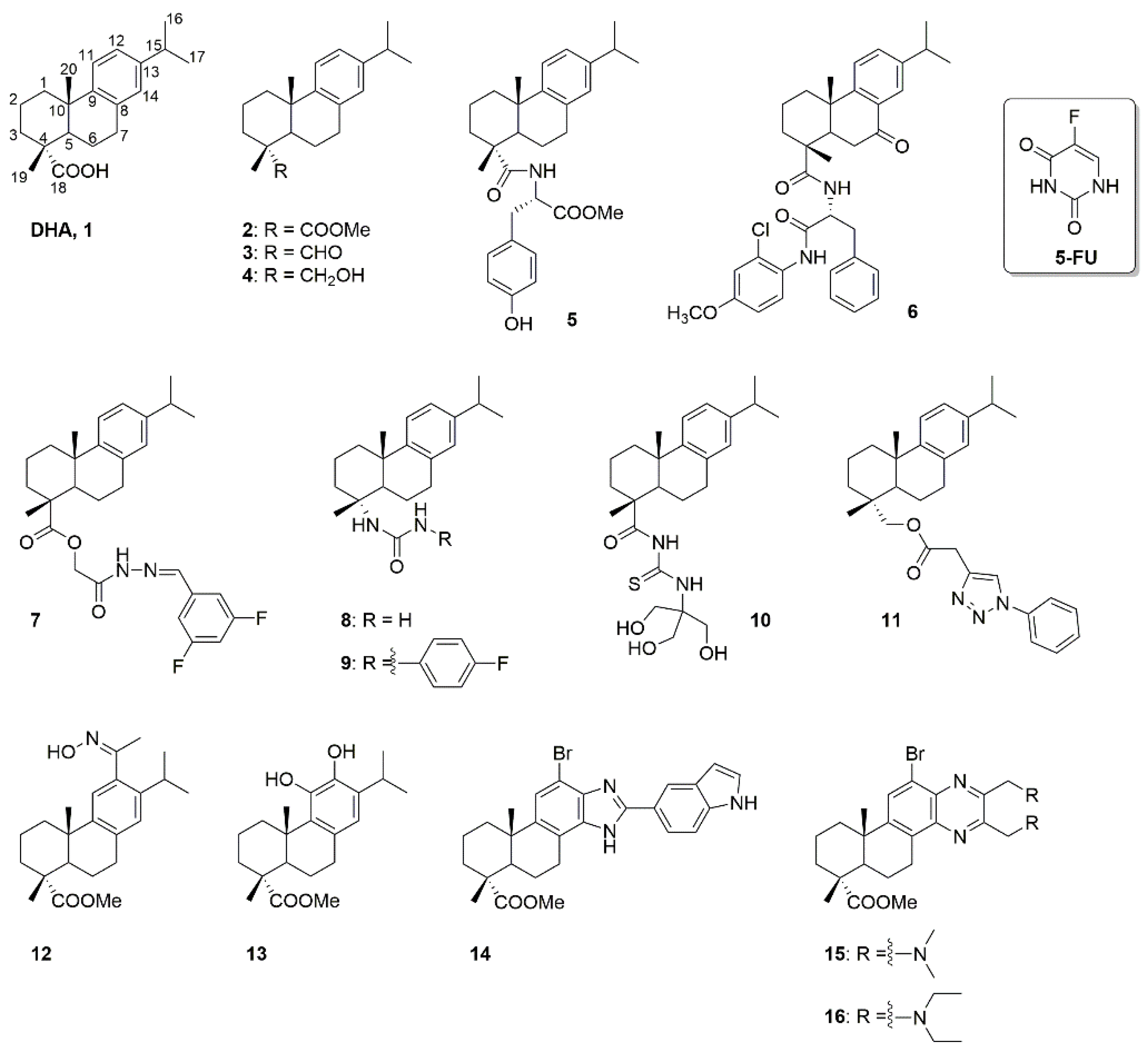
:1. Introduction
2. Results and Discussion
2.1. Synthesis of the DHA-Chalcone Hybrids 31–53
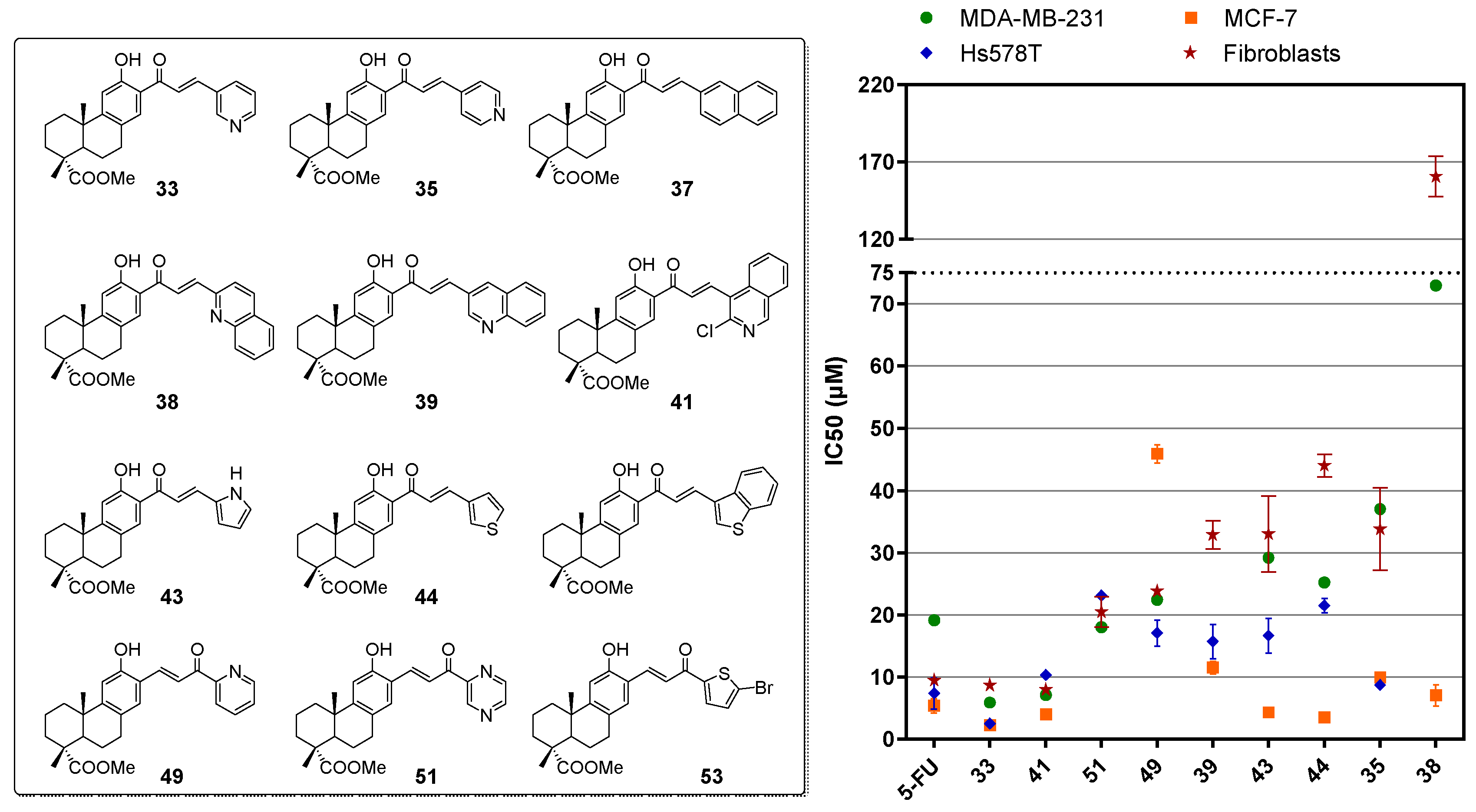
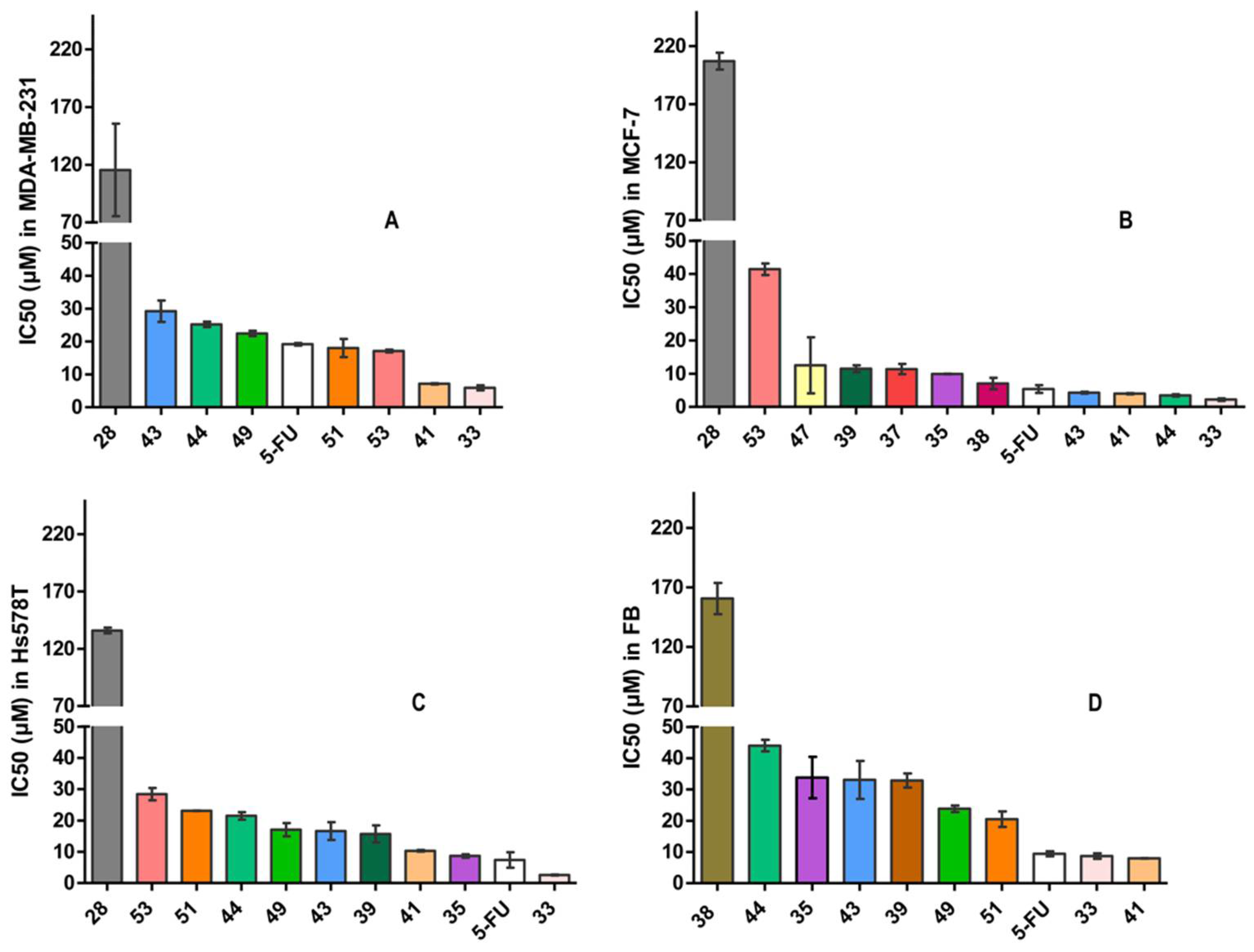
2.2. Biological Evaluation
2.3. Structure-Activity Relationships
3. Materials and Methods
3.1. General Methods
3.2. Experimental Procedures
General Procedure for the Synthesis of the DHA-Chalcone Hybrids
3.3. Biological Assays
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Wang, H.; Naghavi, M.; Allen, C.; Barber, R.M.; Bhutta, Z.A.; Carter, A.; Casey, D.C.; Charlson, F.J.; Chen, A.Z.; Coates, M.M.; et al. Global, regional, and national incidence, prevalence, and years lived with disability for 310 diseases and injuries, 1990–2015: A systematic analysis for the Global Burden of Disease Study 2015. Lancet 2016, 388, 1459–1544. [Google Scholar] [CrossRef] [Green Version]
- Newman, D.J.; Cragg, G.M.; Snader, K.M. Natural products as sources of new drugs over the period 1981–2002. J. Nat. Prod. 2003, 66, 1022–1037. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.-H. Current Developments in the Discovery and Design of New Drug Candidates from Plant Natural Product Leads. J. Nat. Prod. 2004, 67, 273–283. [Google Scholar] [CrossRef] [PubMed]
- You, X.; Zhu, D.; Lu, W.; Sun, Y.; Qiao, S.; Luo, B.; Du, Y.; Pi, R.; Hu, Y.; Huang, P.; et al. Design, synthesis and biological evaluation of N-arylsulfonyl carbazoles as novel anticancer agents. RSC Adv. 2018, 8, 17183–17190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, M.M.-L.; Chan, B.D.; Wong, W.-Y.; Leung, T.-W.; Qu, Z.; Huang, J.; Zhu, L.; Lee, C.-S.; Chen, S.; Tai, W.C.-S. Synthesis and Evaluation of Novel Anticancer Compounds Derived from the Natural Product Brevilin A. ACS Omega 2020, 5, 14586–14596. [Google Scholar] [CrossRef] [PubMed]
- Gazim, Z.C.; Rodrigues, F.; Amorin, A.C.L.; de Rezende, C.M.; Soković, M.; Tešević, V.; Vučković, I.; Krstić, G.; Cortez, L.E.R.; Colauto, N.B.; et al. New natural diterpene-type abietane from Tetradenia riparia essential oil with cytotoxic and antioxidant activities. Molecules 2014, 19, 514–524. [Google Scholar] [CrossRef] [Green Version]
- Neto, Í.; Faustino, C.; Rijo, P. Antimicrobial abietane diterpenoids against resistant bacteria and biofilms. In the Battle against Microbial Pathogens: Basic Science, Technological Advances and Educational Programs; Formatex Research Center: Badajoz, Spain, 2015; pp. 15–26. [Google Scholar]
- Johnson, J.J. Carnosol: A promising anti-cancer and anti-inflammatory agent. Cancer Lett. 2011, 305, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Burmistrova, O.; Simões, M.F.; Rijo, P.; Quintana, J.; Bermejo, J.; Estévez, F. Antiproliferative Activity of Abietane Diterpenoids against Human Tumor Cells. J. Nat. Prod. 2013, 76, 1413–1423. [Google Scholar] [CrossRef] [Green Version]
- Islam, M.T. Diterpenes and Their Derivatives as Potential Anticancer Agents. Phytother. Res. 2017, 31, 691–712. [Google Scholar] [CrossRef]
- Yang, N.-Y.; Liu, L.; Tao, W.-W.; Duan, J.-A.; Tian, L.-J. Diterpenoids from Pinus massoniana resin and their cytotoxicity against A431 and A549 cells. Phytochemistry 2010, 71, 1528–1533. [Google Scholar] [CrossRef]
- Fronza, M.; Murillo, R.; Ślusarczyk, S.; Adams, M.; Hamburger, M.; Heinzmann, B.; Laufer, S.; Merfort, I. In vitro cytotoxic activity of abietane diterpenes from Peltodon longipes as well as Salvia miltiorrhiza and Salvia sahendica. Bioorg. Med. Chem. 2011, 19, 4876–4881. [Google Scholar] [CrossRef] [PubMed]
- González, M.A.; Pérez-Guaita, D.; Correa-Royero, J.; Zapata, B.; Agudelo, L.; Mesa-Arango, A.; Betancur-Galvis, L. Synthesis and biological evaluation of dehydroabietic acid derivatives. Eur. J. Med. Chem. 2010, 45, 811–816. [Google Scholar] [CrossRef] [PubMed]
- Ukiya, M.; Kawaguchi, T.; Ishii, K.; Ogihara, E.; Tachi, Y.; Kurita, M.; Ezaki, Y.; Fukatsu, M.; Kushi, Y.; Akihisa, T. Cytotoxic Activities of Amino Acid-Conjugate Derivatives of Abietane-Type Diterpenoids against Human Cancer Cell Lines. Chem. Biodivers. 2013, 10, 1260–1268. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.-C.; Wang, M.; Wang, H.-S.; Chen, Z.-F.; Zhang, Y.; Pan, Y.-M. Synthesis and antitumor activities of novel dipeptide derivatives derived from dehydroabietic acid. Bioorg. Med. Chem. Lett. 2014, 24, 1511–1518. [Google Scholar] [CrossRef]
- Li, F.-Y.; Wang, X.; Duan, W.-G.; Lin, G.-S. Synthesis and In Vitro Anticancer Activity of Novel Dehydroabietic Acid-Based Acylhydrazones. Molecules 2017, 22, 1087. [Google Scholar] [CrossRef] [Green Version]
- Rao, X.; Song, Z.; He, L.; Jia, W. Synthesis, structure analysis and cytotoxicity studies of novel unsymmetrically n,n’-substituted ureas from dehydroabietic Acid. Chem. Pharm. Bull. 2008, 56, 1575–1578. [Google Scholar] [CrossRef] [Green Version]
- Rao, X.-P.; Wu, Y.; Song, Z.-Q.; Shang, S.-B.; Wang, Z.-D. Synthesis and antitumor activities of unsymmetrically disubstituted acylthioureas fused with hydrophenanthrene structure. Med. Chem. Res. 2011, 20, 333–338. [Google Scholar] [CrossRef]
- Pertino, M.W.; Verdugo, V.; Theoduloz, C.; Schmeda-Hirschmann, G. Synthesis and antiproliferative activity of some novel triazole derivatives from dehydroabietic acid. Molecules 2014, 19, 2523–2535. [Google Scholar] [CrossRef] [Green Version]
- Kolsi, L.E.; Leal, A.S.; Yli-Kauhaluoma, J.; Liby, K.T.; Moreira, V.M. Dehydroabietic oximes halt pancreatic cancer cell growth in the G1 phase through induction of p27 and downregulation of cyclin D1. Sci. Rep. 2018, 8, 15923. [Google Scholar] [CrossRef]
- Gigante, B.; Santos, C.; Silva, A.M.; Curto, M.J.M.; Nascimento, M.S.J.; Pinto, E.; Pedro, M.; Cerqueira, F.; Pinto, M.M.; Duarte, M.P.; et al. Catechols from abietic acid: Synthesis and evaluation as bioactive compounds. Bioorg. Med. Chem. 2003, 11, 1631–1638. [Google Scholar] [CrossRef]
- Miao, T.-T.; Tao, X.-B.; Li, D.-D.; Chen, H.; Jin, X.-Y.; Geng, Y.; Wang, S.-F.; Gu, W. Synthesis and biological evaluation of 2-aryl-benzimidazole derivatives of dehydroabietic acid as novel tubulin polymerization inhibitors. RSC Adv. 2018, 8, 17511–17526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gu, W.; Wang, S.; Jin, X.; Zhang, Y.; Hua, D.; Miao, T.; Tao, X.; Wang, S. Synthesis and Evaluation of New Quinoxaline Derivatives of Dehydroabietic Acid as Potential Antitumor Agents. Molecules 2017, 22, 1154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shalini; Kumar, V. Have molecular hybrids delivered effective anti-cancer treatments and what should future drug discovery focus on? Expert Opin. Drug Discov. 2021, 16, 335–363. [Google Scholar] [CrossRef] [PubMed]
- Xie, L.; Zhai, X.; Ren, L.; Meng, H.; Liu, C.; Zhu, W.; Zhao, Y. Design, Synthesis and Antitumor Activity of Novel Artemisinin Derivatives Using Hybrid Approach. Chem. Pharm. Bull. 2011, 59, 984–990. [Google Scholar] [CrossRef] [Green Version]
- Gaur, R.; Pathania, A.S.; Malik, F.A.; Bhakuni, R.S.; Verma, R.K. Synthesis of a series of novel dihydroartemisinin monomers and dimers containing chalcone as a linker and their anticancer activity. Eur. J. Med. Chem. 2016, 122, 232–246. [Google Scholar] [CrossRef]
- Palmieri, C.; Januszewski, A.; Stanway, S.; Coombes, R.C. Irosustat: A first-generation steroid sulfatase inhibitor in breast cancer. Expert Rev. Anticancer Ther. 2011, 11, 179–183. [Google Scholar] [CrossRef]
- Afzal, O.; Kumar, S.; Haider, M.R.; Ali, M.R.; Kumar, R.; Jaggi, M.; Bawa, S. A review on anticancer potential of bioactive heterocycle quinoline. Eur. J. Med. Chem. 2015, 97, 871–910. [Google Scholar] [CrossRef]
- Elshemy, H.A.H.; Zaki, M.A. Design and synthesis of new coumarin hybrids and insight into their mode of antiproliferative action. Bioorg. Med. Chem. 2017, 25, 1066–1075. [Google Scholar] [CrossRef]
- Shin, S.Y.; Yoon, H.; Ahn, S.; Kim, D.-W.; Kim, S.H.; Koh, D.; Lee, Y.H.; Lim, Y. Chromenylchalcones showing cytotoxicity on human colon cancer cell lines and in silico docking with aurora kinases. Bioorg. Med. Chem. 2013, 21, 4250–4258. [Google Scholar] [CrossRef]
- Tseng, C.-H.; Chen, Y.-L.; Hsu, C.-Y.; Chen, T.-C.; Cheng, C.-M.; Tso, H.-C.; Lu, Y.-J.; Tzeng, C.-C. Synthesis and antiproliferative evaluation of 3-phenylquinolinylchalcone derivatives against non-small cell lung cancers and breast cancers. Eur. J. Med. Chem. 2013, 59, 274–282. [Google Scholar] [CrossRef]
- Li, W.; Xu, F.; Shuai, W.; Sun, H.; Yao, H.; Ma, C.; Xu, S.; Yao, H.; Zhu, Z.; Yang, D.-H.; et al. Discovery of Novel Quinoline–Chalcone Derivatives as Potent Antitumor Agents with Microtubule Polymerization Inhibitory Activity. J. Med. Chem. 2019, 62, 993–1013. [Google Scholar] [CrossRef] [PubMed]
- Lindamulage, I.K.; Vu, H.-Y.; Karthikeyan, C.; Knockleby, J.; Lee, Y.-F.; Trivedi, P.; Lee, H. Novel quinolone chalcones targeting colchicine-binding pocket kill multidrug-resistant cancer cells by inhibiting tubulin activity and MRP1 function. Sci. Rep. 2017, 7, 10298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Antoniou, A.; Chatzopoulou, M.; Bantzi, M.; Athanassopoulos, C.M.; Giannis, A.; Pitsinos, E.N. Identification of Gli-mediated transcription inhibitors through synthesis and evaluation of taepeenin D analogues. Med. Chem. Comm. 2016, 7, 2328–2331. [Google Scholar] [CrossRef]
- Chaban, M.F.; I Antoniou, A.; Karagianni, C.; Toumpa, D.; Joray, M.B.; Bocco, J.L.; Sola, C.; M Athanassopoulos, C.; Carpinella, M.C. Synthesis and structure-activity relationships of novel abietane diterpenoids with activity against Staphylococcus aureus. Future Med. Chem. 2019, 11, 3109–3124. [Google Scholar] [CrossRef]
- Mori, M.; Tottone, L.; Quaglio, D.; Zhdanovskaya, N.; Ingallina, C.; Fusto, M.; Ghirga, F.; Peruzzi, G.; Crestoni, M.E.; Simeoni, F.; et al. Identification of a novel chalcone derivative that inhibits Notch signaling in T-cell acute lymphoblastic leukemia. Sci. Rep. 2017, 7, 2213. [Google Scholar] [CrossRef]
- Gutierrez, P.; Altarejos, J.; Linares-Palomino, P.J.; Chahboun, R.; Alvarez-Manzaneda, E. Synthesis of cassane-type diterpenes from abietane compounds: The first synthesis of taepeenin F. Org. Chem. Front. 2018, 5, 2537–2541. [Google Scholar] [CrossRef]
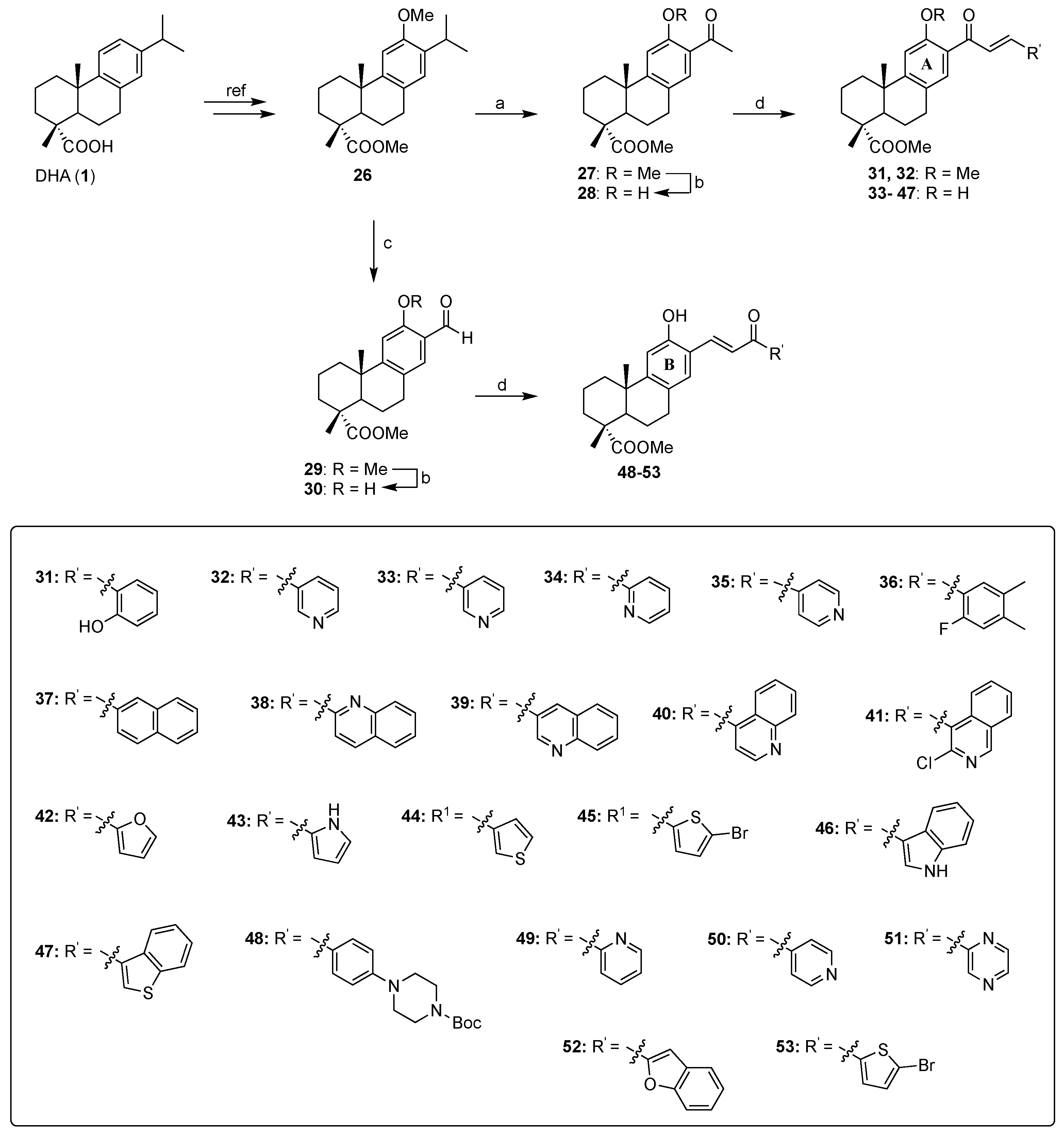

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | IC50 (μM) | Selectivity Index for | |||||
|---|---|---|---|---|---|---|---|
| MDA-MB-231 | MCF-7 | Hs578T | Fibroblasts | MDA-MB-231 | MCF-7 | Hs578T | |
| 31 | >75 | 23.90 ± 4.47 | >75 | n/a | n/a | n/a | n/a |
| 32 | >75 | >75 | >75 | n/a | n/a | n/a | n/a |
| 33 | 5.89 ± 0.77 | 2.21 ± 0.40 | 2.48 ± 0.13 | 8.68 ± 0.88 | 1.47 | 3.93 | 3.50 |
| 34 | >75 | 21.36 ± 0.60 | 28.09 ± 7.76 | n/a | n/a | n/a | n/a |
| 35 | 37.05 ± 1.82 | 9.90 ± 0.05 | 8.69 ± 0.55 | 33.83 ± 6.63 | 0.91 | 3.42 | 3.89 |
| 36 | >75 | >75 | >75 | n/a | n/a | n/a | n/a |
| 37 | >75 | 11.39 ± 1.55 | >75 | n/a | n/a | n/a | n/a |
| 38 | 73.00 ± 2.31 | 7.06 ± 1.71 | >75 | 160.65 ± 13.08 | 2.20 | 22.75 | - |
| 39 | >75 | 11.50 ± 1.02 | 15.71 ± 2.74 | 32.88 ± 2.27 | - | 2.86 | 2.09 |
| 40 | >75 | >75 | >75 | 92.11 ± 5.47 | - | - | - |
| 41 | 7.12 ± 0.20 | 3.99 ± 0.15 | 10.31 ± 0.25 | 7.96 ± 0.10 | 1.12 | 1.99 | 0.77 |
| 42 | >75 | 37.01 ± 6.89 | 43.33 ± 4.32 | n/a | n/a | n/a | n/a |
| 43 | 29.20 ± 3.26 | 4.29 ± 0.29 | 16.64 ± 2.83 | 33.05 ± 6.10 | 1.13 | 7.70 | 1.99 |
| 44 | 25.21 ± 0.84 | 3.45 ± 0.36 | 21.48 ± 1.20 | 44.03 ± 1.84 | 1.75 | 12.76 | 2.05 |
| 45 | >75 | 29.94 ± 5.26 | >75 | n/a | n/a | n/a | n/a |
| 46 | >75 | >75 | >75 | n/a | n/a | n/a | n/a |
| 47 | 40.37 ± 0.67 | 12.52 ± 8.43 | >75 | n/a | n/a | n/a | n/a |
| 48 | >75 | 25.62 ± 0.09 | 23.15 ± 2.50 | n/a | n/a | n/a | n/a |
| 49 | 22.43 ± 0.81 | 45.93 ± 1.52 | 17.07 ± 2.13 | 23.81 ± 1.07 | 1.06 | 0.52 | 1.39 |
| 50 | >75 | >75 | >75 | 192.25 ± 51.41 | - | - | - |
| 51 | 18.01 ± 2.78 | >75 | 23.11 ± 0.04 | 20.47 ± 2.46 | 1.14 | - | 0.88 |
| 52 | >75 | >75 | 60.21 ± 6.94 | n/a | n/a | n/a | n/a |
| 53 | 17.12 ± 0.41 | 41.44 ± 1.74 | 28.44 ± 1.97 | n/a | n/a | n/a | n/a |
| 28 | 115.44 ± 40.11 | 206.95 ± 7.28 | 135.95 ± 2.47 | n/a | n/a | n/a | n/a |
| 5-FU | 19.17 ± 0.39 | 5.37 ± 1.16 | 7.38 ± 2.49 | 9.43 ± 0.80 | 0.49 | 1.76 | 1.28 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Grigoropoulou, S.; Manou, D.; Antoniou, A.I.; Tsirogianni, A.; Siciliano, C.; Theocharis, A.D.; Athanassopoulos, C.M. Synthesis and Antiproliferative Activity of Novel Dehydroabietic Acid-Chalcone Hybrids. Molecules 2022, 27, 3623. https://doi.org/10.3390/molecules27113623
Grigoropoulou S, Manou D, Antoniou AI, Tsirogianni A, Siciliano C, Theocharis AD, Athanassopoulos CM. Synthesis and Antiproliferative Activity of Novel Dehydroabietic Acid-Chalcone Hybrids. Molecules. 2022; 27(11):3623. https://doi.org/10.3390/molecules27113623
Chicago/Turabian StyleGrigoropoulou, Sophia, Dimitra Manou, Antonia I. Antoniou, Artemis Tsirogianni, Carlo Siciliano, Achilleas D. Theocharis, and Constantinos M. Athanassopoulos. 2022. "Synthesis and Antiproliferative Activity of Novel Dehydroabietic Acid-Chalcone Hybrids" Molecules 27, no. 11: 3623. https://doi.org/10.3390/molecules27113623
APA StyleGrigoropoulou, S., Manou, D., Antoniou, A. I., Tsirogianni, A., Siciliano, C., Theocharis, A. D., & Athanassopoulos, C. M. (2022). Synthesis and Antiproliferative Activity of Novel Dehydroabietic Acid-Chalcone Hybrids. Molecules, 27(11), 3623. https://doi.org/10.3390/molecules27113623