
Evaluation of Neurotropic Activity and Molecular Docking Study of New Derivatives of pyrano[4″,3″:4′,5′]pyrido[3′,2′:4,5]thieno[3,2-d]pyrimidines on the Basis of pyrano[3,4-c]pyridines

 ,
,  , , ,
, , ,
Abstract
:1. Introduction
2. Results and Discussion
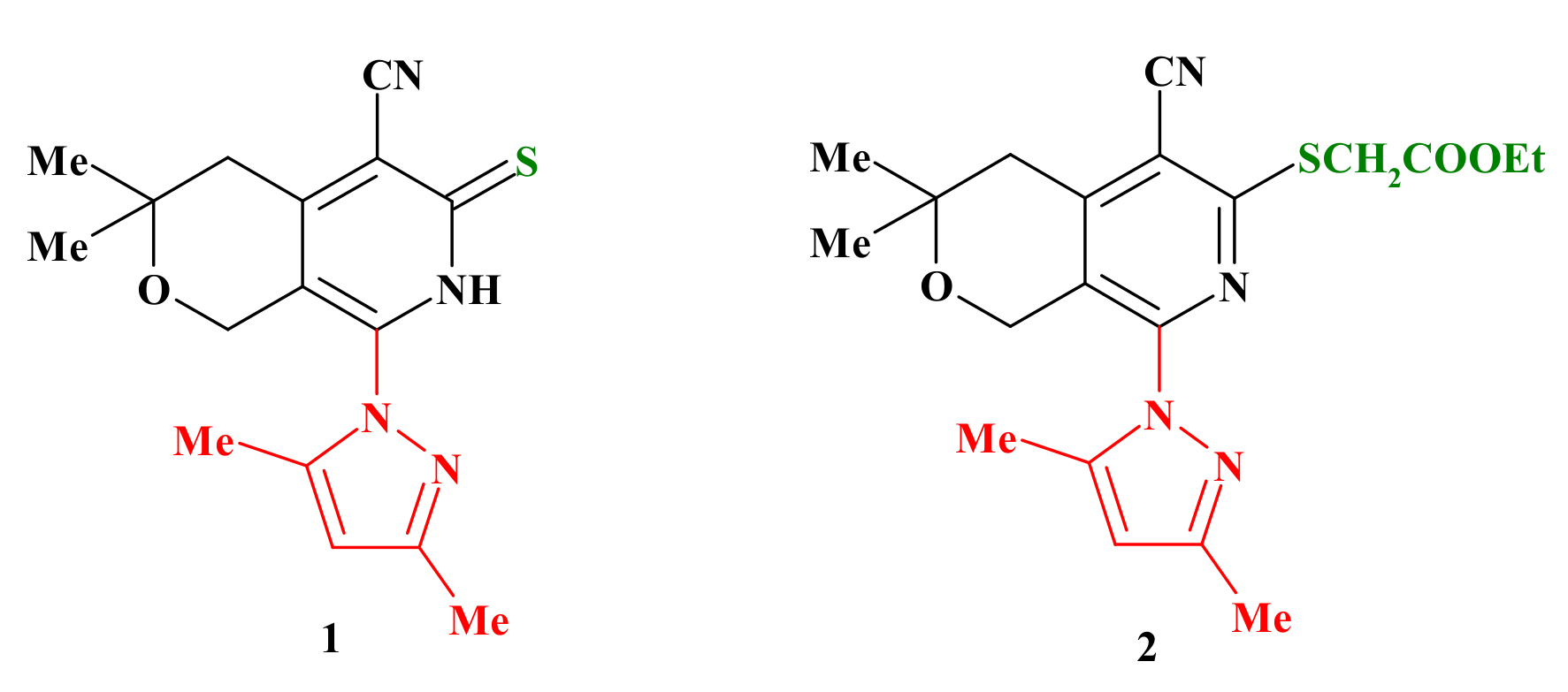
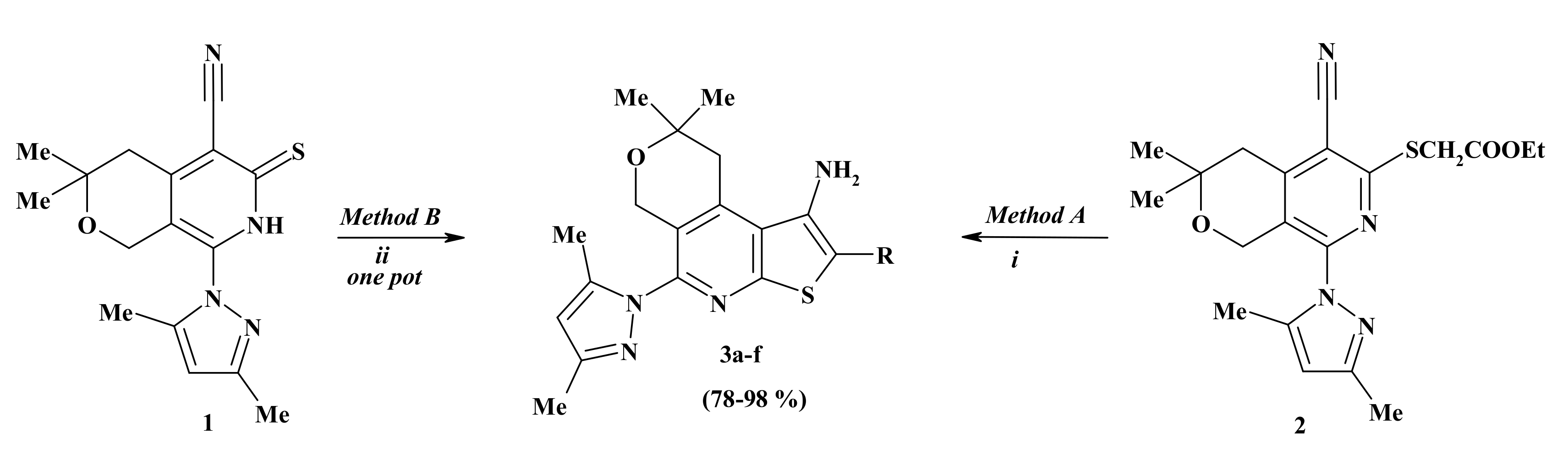
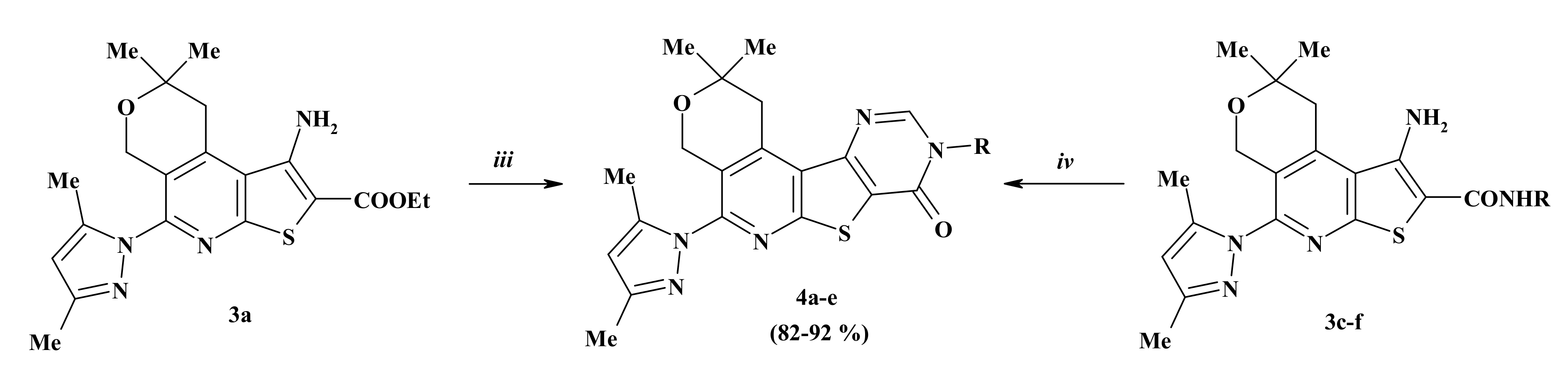
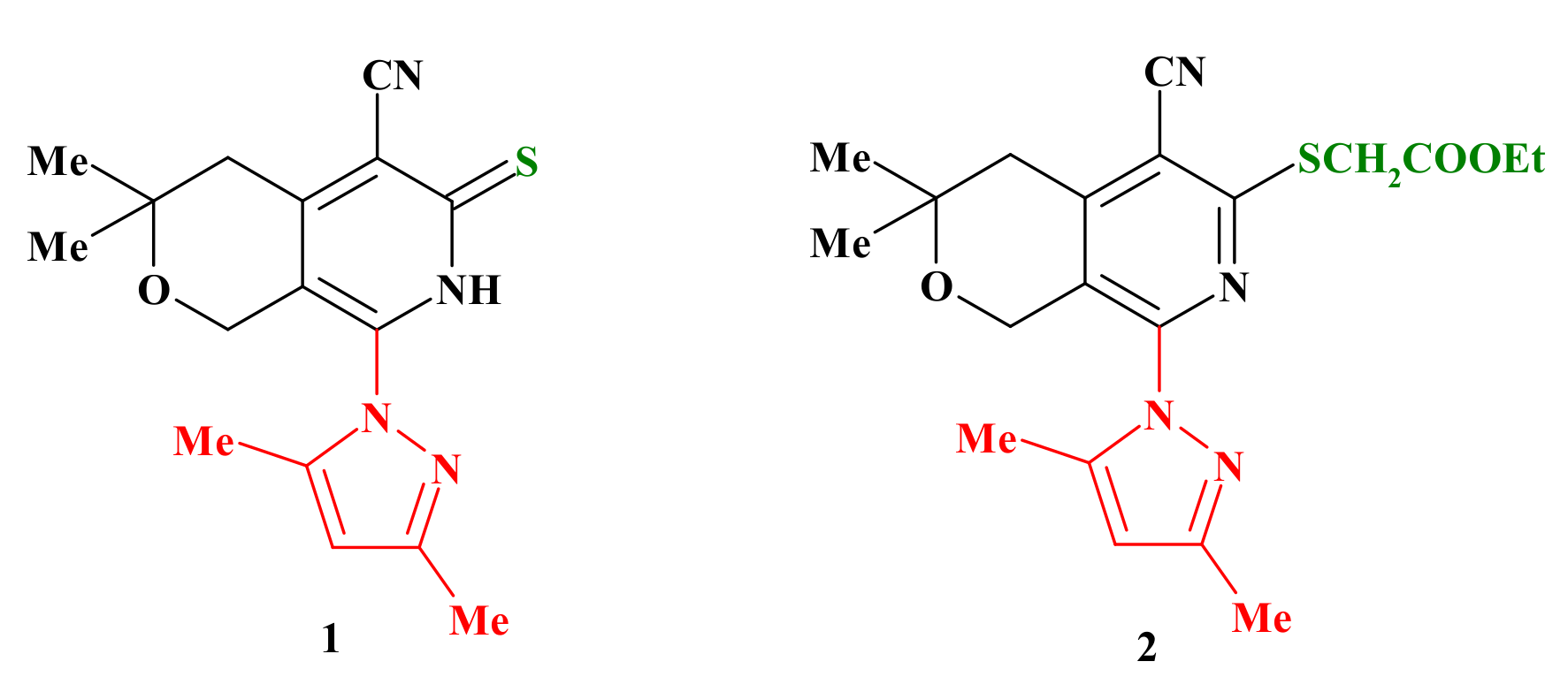
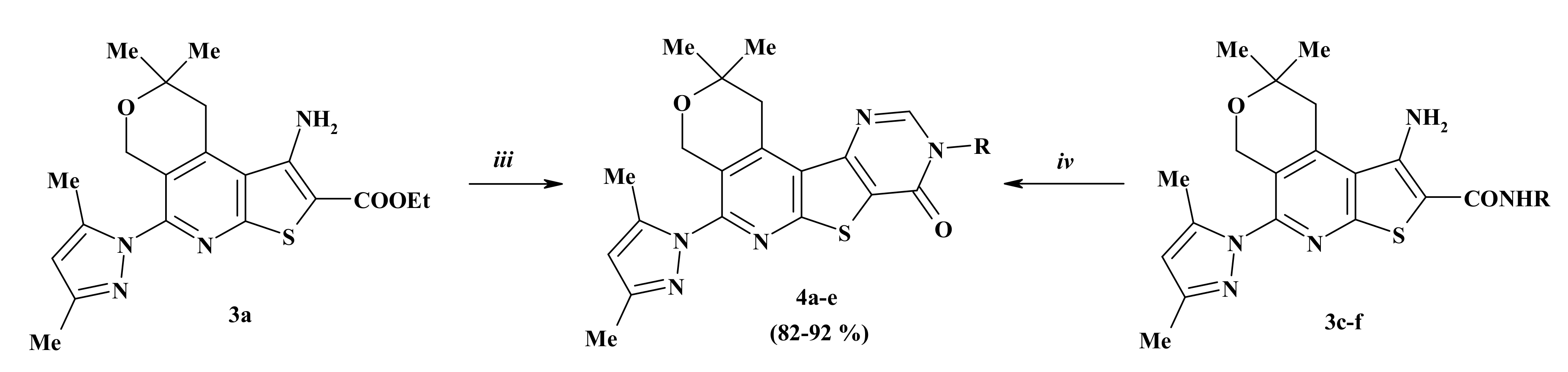
2.1. Chemistry
2.2. Biological Assays
2.3. Molecular Docking
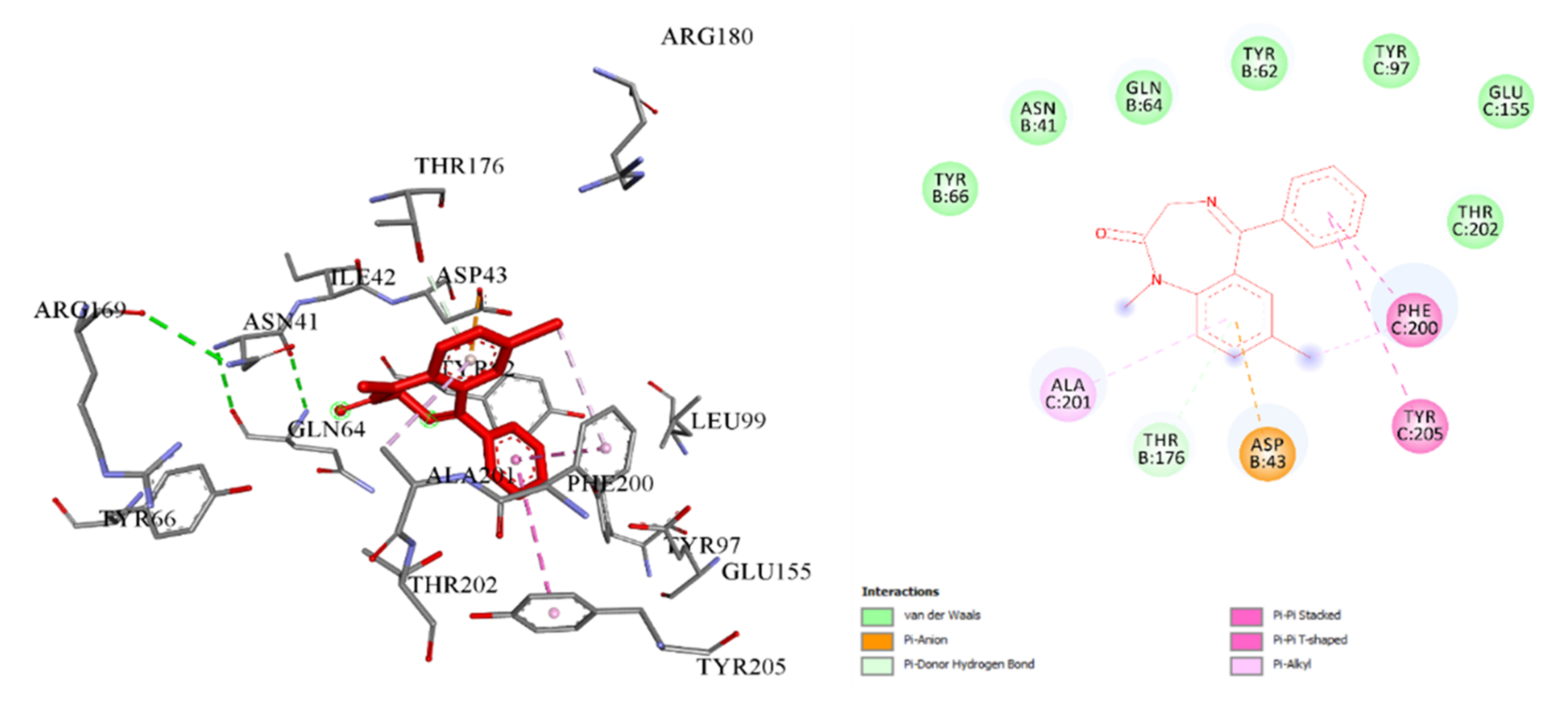
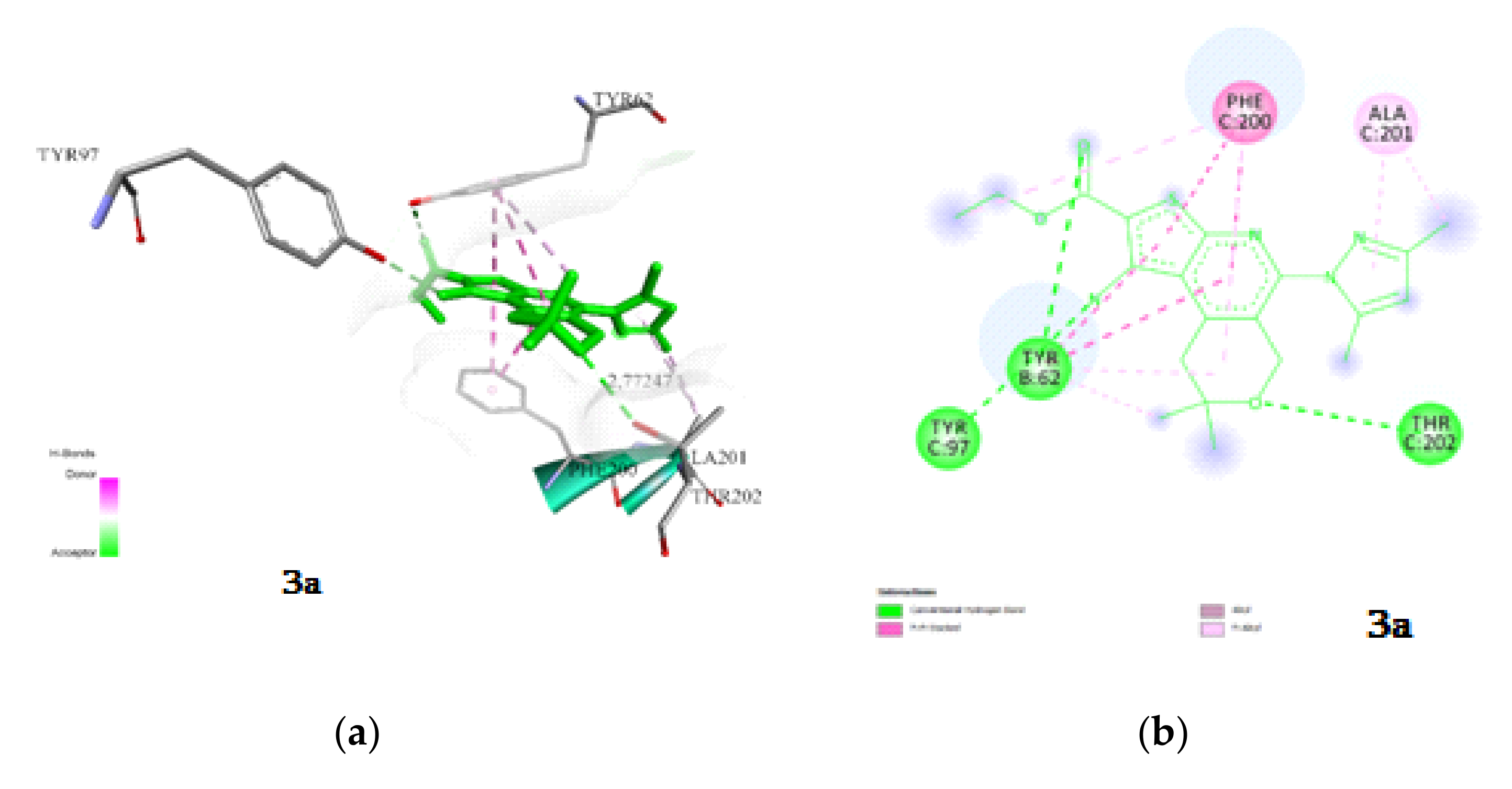
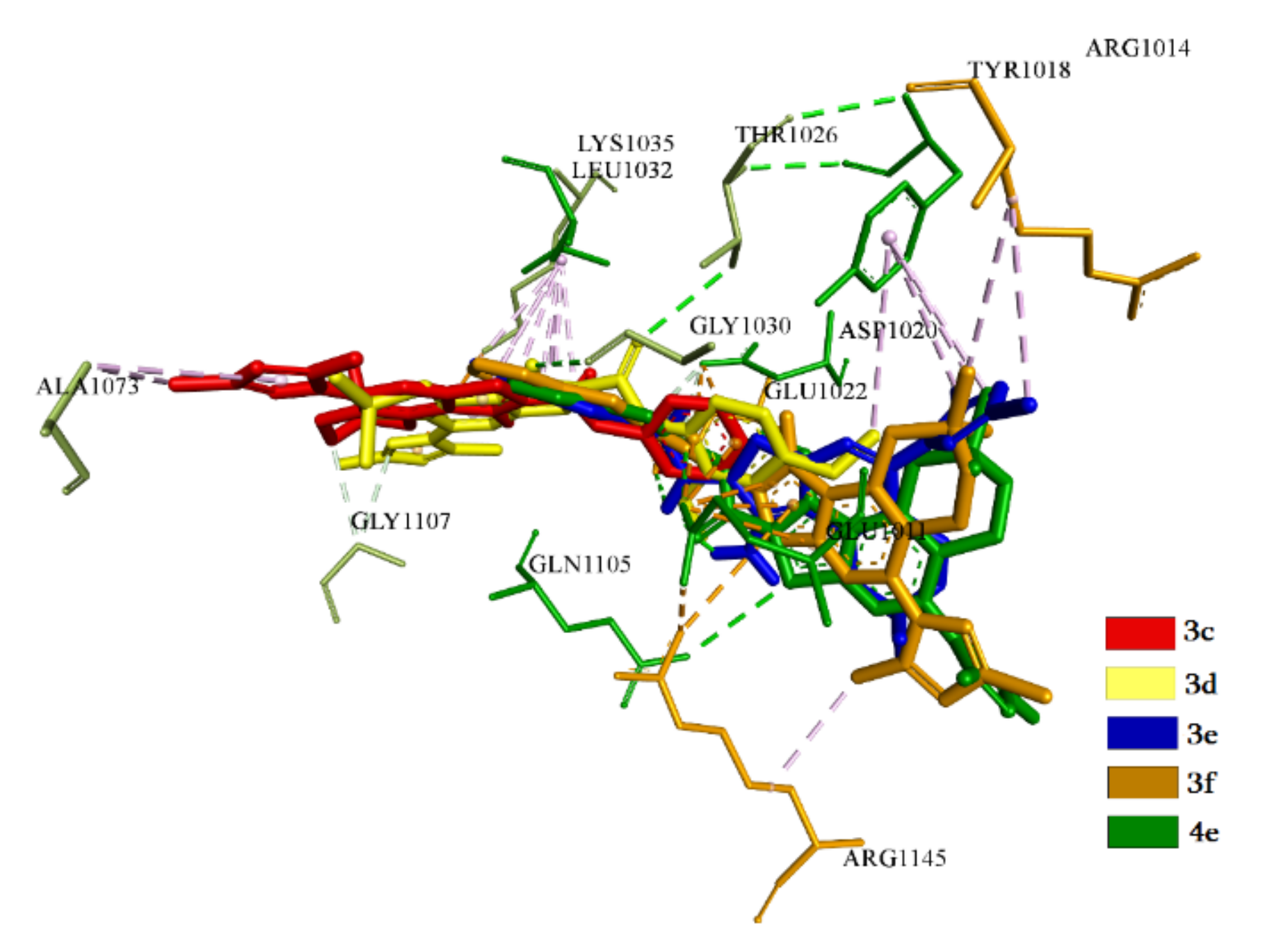
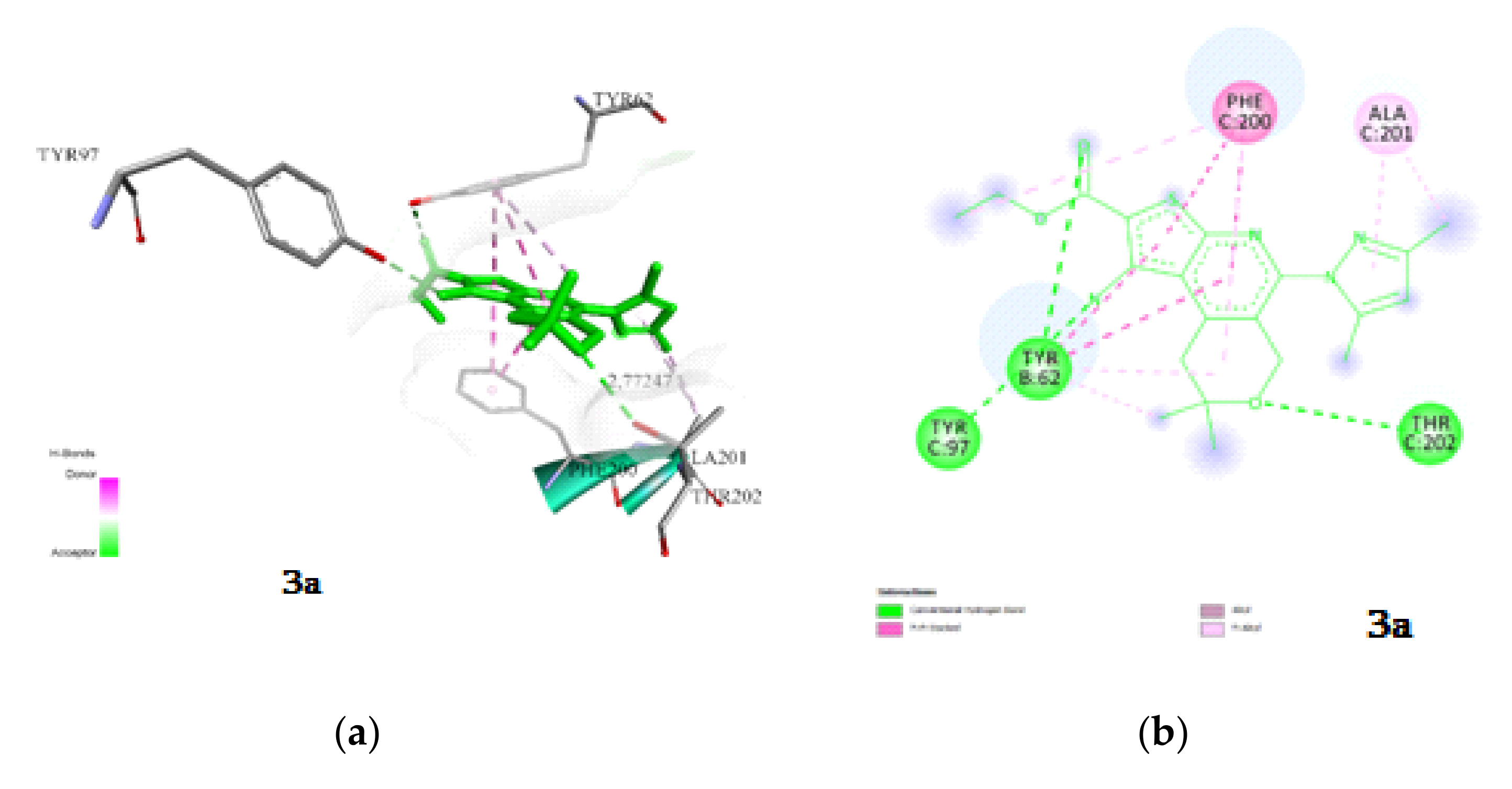
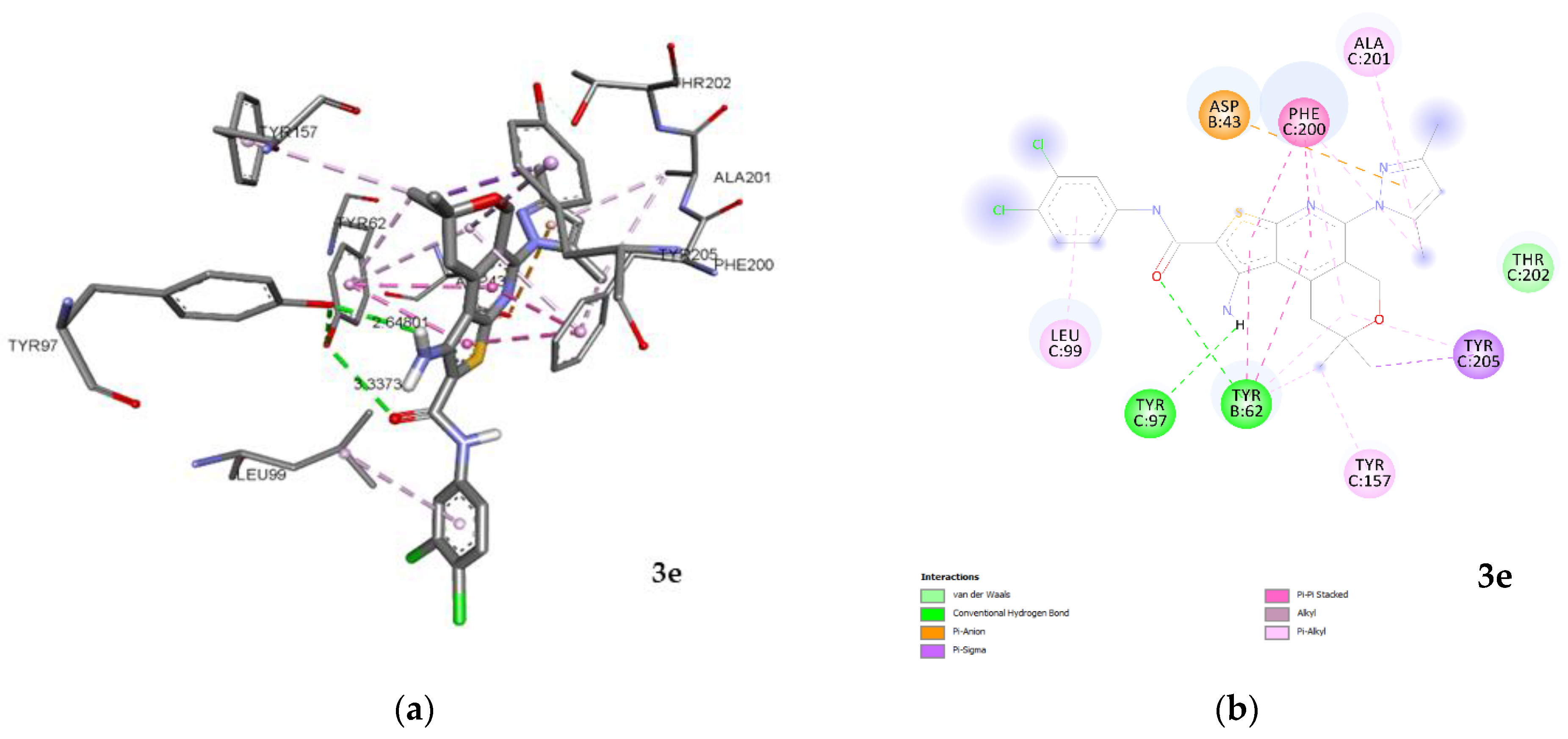
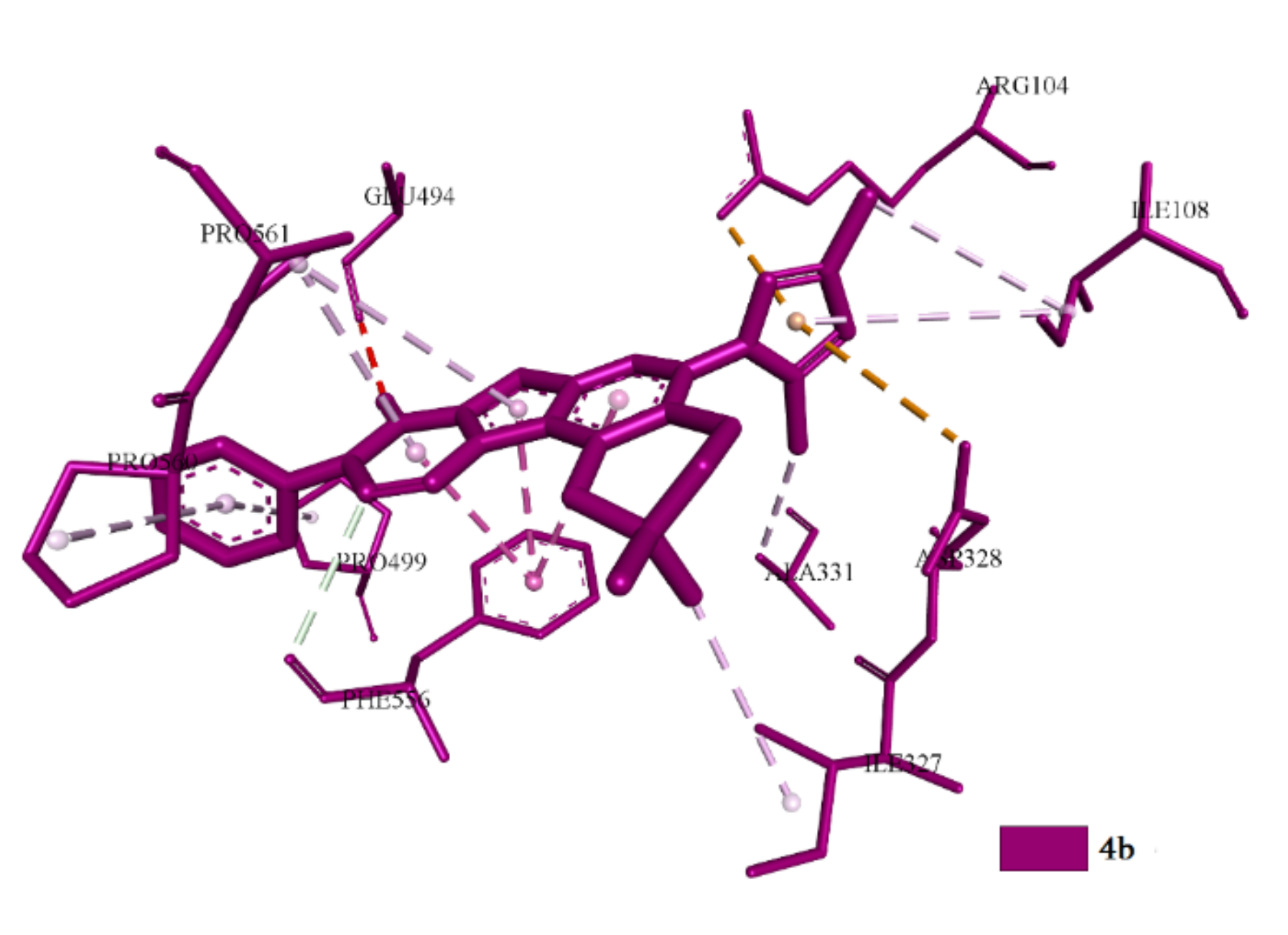
2.3.1. Docking and Conformational Analysis of GABAA Receptor Complexation
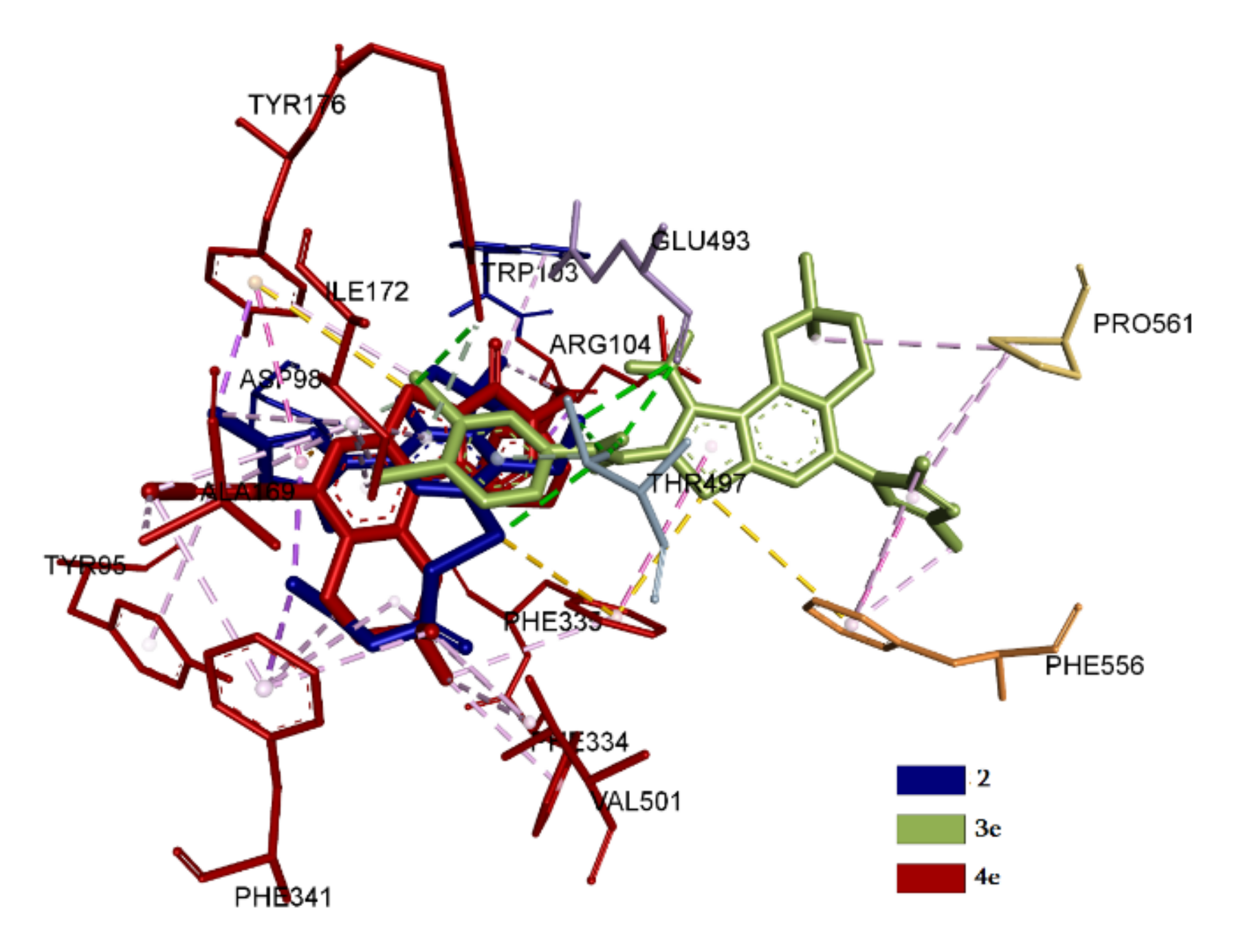
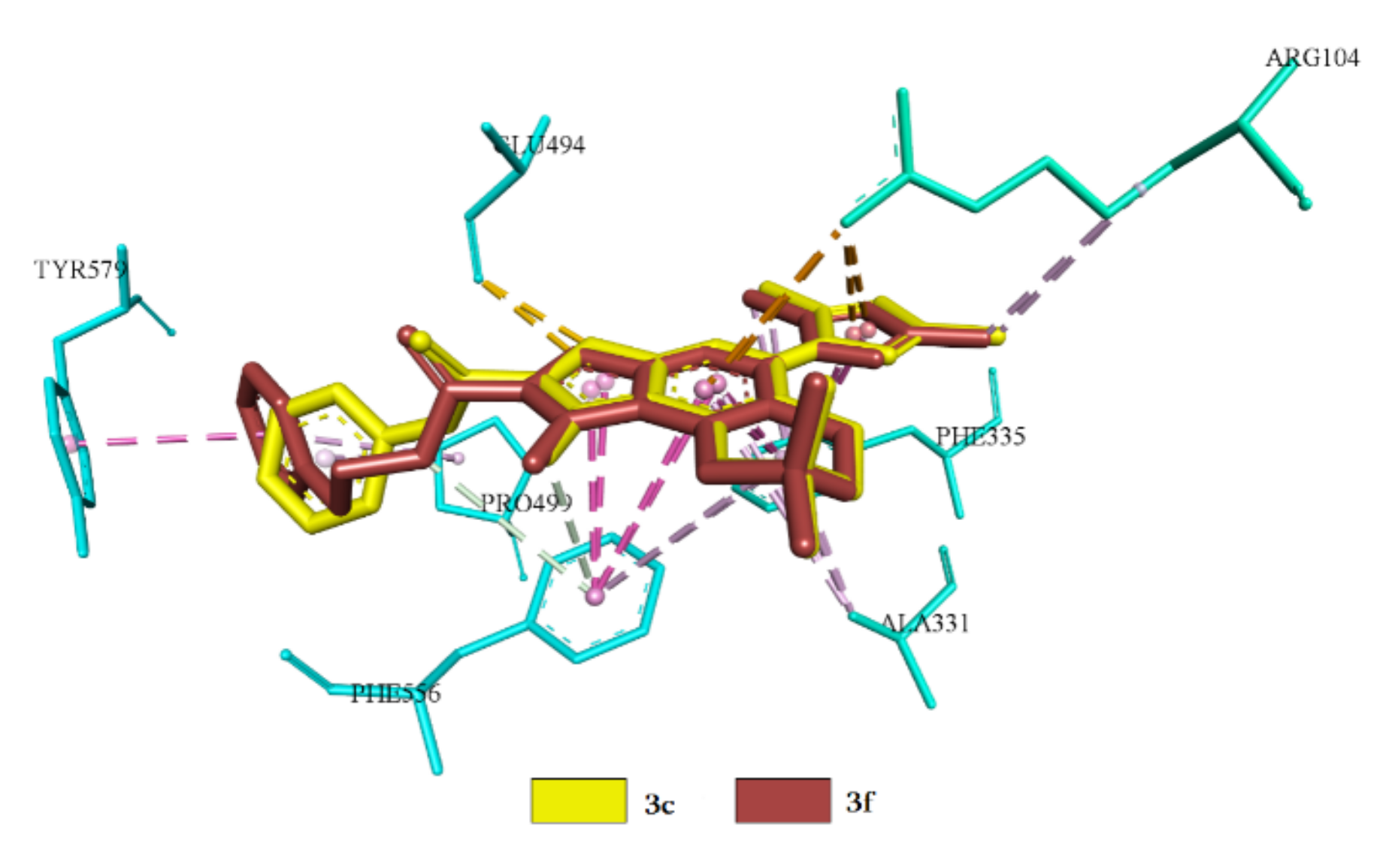
2.3.2. Docking and Conformational Analysis of SERT Complexation
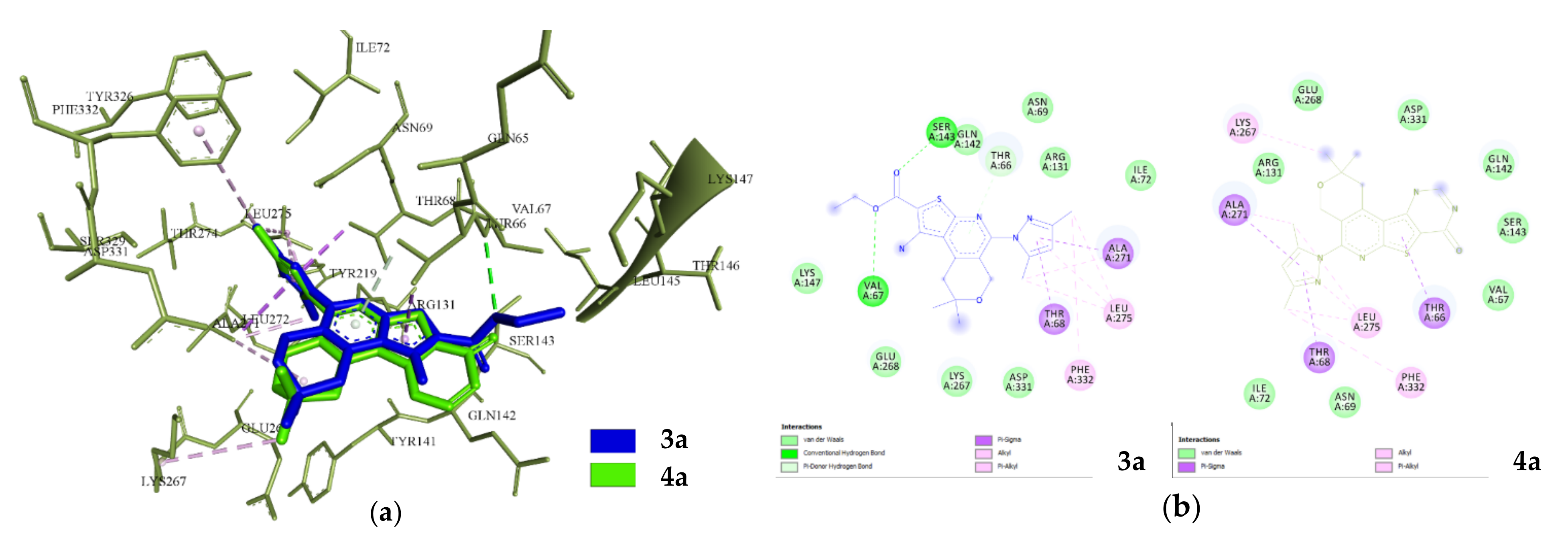
2.3.3. Docking and Conformational Analysis of 5HT_1A Complexation
3. Materials and Methods
3.1. Chemistry
3.1.1. General Information
3.1.2. Methods for the Synthesis of Ethyl 1-Amino-5-(3,5-dimethyl-1H-pyrazol-1-yl)-8,8-dimethyl-8,9-dihydro-6H-pyrano[4,3-d]thieno[2,3-b]pyridine-2-carboxylate (3a)
3.1.3. General Method for the Preparation of Compounds 3b–f
3.1.4. Method for the Synthesis of 5-(3,5-Dimethyl-1H-pyrazol-1-yl)-2,2-dimethyl-1,4-dihydro-2H-pyrano[4″,3″:4′,5′]pyrido[3′,2′:4,5]thieno[3,2-d]pyrimidin-8(9H)-one (4a)
3.1.5. General Method for the Preparation of Compounds 4b–f
3.2. Biological Evaluation
3.2.1. Evaluation of the Anticonvulsant Activity of the Synthesized Compounds
3.2.2. Evaluation of the Psychotropic Properties of the Synthesized Compounds
3.2.3. Evaluation of Incoordination of Movements in the Rotating Rod Test
3.3. Docking Studies
3.3.1. Design of Molecular Models
3.3.2. Molecular Docking
3.3.3. Binding Constant Calculation
3.3.4. Conformational Analysis and Visualization
3.3.5. Statistical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Kitchen, D.B.; Decornez, H.; Furr, J.R.; Bajorath, J. Docking and scoring in virtual screening for drug discovery: Methods and applications. Nat. Rev. Drug Discov. 2004, 3, 935–949. [Google Scholar] [CrossRef] [PubMed]
- Shiri, P. Novel Hybrid Molecules Based on triazole-β-lactam as Potential Biological Agents. Mini-Rev.Med. Chem. 2021, 21, 536–553. [Google Scholar] [CrossRef] [PubMed]
- Rayevsky, O.M. Modeling the Structure-Property Relationships; Poroikov, V.V., Ed.; Dobrosvet: Moscow, Russia, 2008; p. 288. (In Russian) [Google Scholar]
- Shiri, P.; Ramezanpour, S.; Amani, A.M.; Dehaen, W. A patent review on efficient strategies for the total synthesis of pazopanib, regorafenib and lenvatinib as novel anti-angiogenesis receptor tyrosine kinase inhibitors for cancer therapy. Mol. Divers. 2022, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Tuccinardi, T.; Schenone, S.; Bondavalli, F.; Brullo, C.; Bruno, O.; Mosti, L.; Zizzari, A.T.; Tintori, C.; Manetti, F.; Ciampi, O.; et al. Substituted Pyrazolo[3,4-b]pyridines as Potent A1 Adenosine Antagonists: Synthesis, Biological Evaluation, and Development of an A1 Bovine Receptor Model. ChemMedChem 2008, 3, 898–913. [Google Scholar] [CrossRef]
- Salem, M.S.; Ali, M.A.M. Novel Pyrazolo[3,4-b]pyridine Derivatives: Synthesis, Characterization, Antimicrobial and Antiproliferative Profile. Biol. Pharm. Bull. 2016, 39, 473–483. [Google Scholar] [CrossRef] [Green Version]
- Lourenço, A.L.; Salvador, R.R.; Silva, L.A.; Saito, M.S.; Mello, J.F.; Cabral, L.M.; Rodrigues, C.R.; Vera, M.A.; Muri, E.M.; de Souza, A.M.; et al. Synthesis and mechanistic evaluation of novel N′-benzylidene-carbohydrazide-1 H -pyrazolo[3,4-b]pyridine derivatives as non-anionic antiplatelet agents. Eur. J. Med. Chem. 2017, 135, 213–229. [Google Scholar] [CrossRef]
- Kasabov, K.A.; Kudryashov, N.V.; Volkova, A.; Shimshirt, A.A.; Kalinina, T.S.; Zhmurenko, L.A.; Voronina, T.A. Psychotropic Effects of a New Pyrazolo[C]Pyridine Derivate GIZh-72 are Related to Functional Activity of Atp-Sensitive Potassium Channels. Bull. Exp. Biol. Med. 2020, 168, 449–452. [Google Scholar] [CrossRef]
- Samatov, A.; Akramov, S.T.; Yunusov, S.Y. Alkaloids of Gentiana. Structure of gentianadine and gentianamine. Chem. Nat. Compd. 1967, 3, 150–154. [Google Scholar] [CrossRef]
- Bhattacharya, S.; Ghosal, S.; Chaudhuri, R.; Singh, A.; Sharma, P. Chemical Constituents of Gentianaceae XI: Antipsychotic Activity of Gentianine. J. Pharm. Sci. 1974, 63, 1341–1342. [Google Scholar] [CrossRef]
- Yu, S.; Huang, Q.-Q.; Luo, Y.; Lu, W. Total Synthesis of Camptothecin and SN-38. J. Org. Chem. 2011, 77, 713–717. [Google Scholar] [CrossRef]
- Jubeen, F.; Iqbal, S.Z.; Shafiq, N.; Khan, M.; Parveen, S.; Iqbal, M.; Nazir, A. Eco-friendly synthesis of pyrimidines and its derivatives: A review on broad spectrum bioactive moiety with huge therapeutic profile. Synth. Commun. 2018, 48, 601–625. [Google Scholar] [CrossRef]
- Disch, J.S.; Evindar, G.; Chiu, C.H.; Blum, C.A.; Dai, H.; Jin, L.; Schuman, E.; Lind, K.E.; Belyanskaya, S.L.; Deng, J.; et al. Discovery of Thieno[3,2-d]pyrimidine-6-carboxamides as Potent Inhibitors of SIRT1, SIRT2, and SIRT3. J. Med. Chem. 2013, 56, 3666–3679. [Google Scholar] [CrossRef] [PubMed]
- Tan, Q.; Zhang, Z.; Hui, J.; Zhao, Y.; Zhu, L. Synthesis and anticancer activities of thieno[3,2-d]pyrimidines as novel HDAC inhibitors. Bioorganic Med. Chem. 2013, 22, 358–365. [Google Scholar] [CrossRef]
- Barthakur, M.G.; Gogoi, S.; Dutta, M.; Boruah, R.C. A facile three-component solid phase synthesis of steroidal A-ring fused pyrimidines under microwave irradiation. Steroids 2009, 74, 730–734. [Google Scholar] [CrossRef] [PubMed]
- Malhota, M.; Dhingra, R.; Sharma, R.; Bhadway, T.R. 17-Aza Steroids as 5α-Reductase Inhibitors: A Review. Int. J. Pharmacogn. Phytochem. Res. 2013, 5, 134–142. [Google Scholar]
- Kaur, S.; Kapoor, V.K.; Bhardwa, T.K.; Kaur, M. A facile route for the synthesis of bisquartenary azasteroids. Int. J. Pharmacogn. Phytochem. Res. 2013, 5, 728–732. [Google Scholar]
- Dashyan, S.; Paronikyan, E.G.; Noravyan, A.S. Synthesis and Anticonvulsive Activity of 5-Pyrrolidin-1-Ylpyrano[4″,3″:4′,5′]Pyrido-3′,2′:4,5]Thieno[3,2-D]Pyrimidine Derivatives. Pharm. Chem. J. 2016, 50, 221–225. [Google Scholar] [CrossRef]
- Babaev, E.V.; Koval, Y.I.; Rybakov, V.B.; Paronikyan, E.G.; Stepanyan, G.M.; Dashyan, S.S.; Rzhevskii, S.A.; Shadrin, I.A. 2-Allyloxy/propargyloxypyridines: Synthesis, structure, and biological activity. Bull. Acad. Sci. USSR Div. Chem. Sci. 2018, 67, 313–320. [Google Scholar] [CrossRef]
- Paronikyan, E.G.; Dashyan, S.S.; Dzhagatspanyan, I.A.; Nazaryan, I.M.; Akopyan, A.G.; Minasyan, N.S. Synthesis and neurotropic activity of the derivatives of fused triazolo[4,3-c]- and triazolo[1,5-c]pyrimidines. Russ. J. Bioorganic Chem. 2017, 43, 595–603. [Google Scholar] [CrossRef]
- Dashyan, S.S.; Paronikyan, R.G.; Mamyan, S.S.; Paronikyan, E.G. Synthesis, neurotropic activity and SAR of new S-alkyl derivatives of 8- pyrazol-1-yl pyrano[3,4-c]pyridines. Arkivoc 2022, 2022, 43–53. [Google Scholar] [CrossRef]
- Vogel, H.G.; Vogel, V.H. Psychotropic and Neurotropic Activity. In Drug Discovery and Evaluation: Pharmacological Assays; Vogel, H.E., Ed.; Springer: Berlin, Germany, 2008; Volume 58, pp. 569–874. Available online: https://scholar.google.com/scholar_lookup?title=Drug+Discovery+and+Evaluation (accessed on 28 May 2021).
- Löscher, W.; Schmidt, D. Which animal models should be used in the search for new antiepileptic drugs? A proposal based on experimental and clinical considerations. Epilepsy Res. 1988, 2, 145–181. [Google Scholar] [CrossRef]
- Swinyard, E.A. Experimental Models of Epilepsy; Purpura, D.P., Penry, J.K., Tower, D., Woodbury, D.M., Walter, R., Eds.; Raven Press: New York, NY, USA, 1992; pp. 433–458. Available online: https://europepmc.org/article/med/4007184 (accessed on 28 May 2021).
- Gevorkyan, K.A.; Papayan, G.L.; Chshmarityan, S.G.; Paronikyan, R.G.; Akopyan, N.E.; Engoyan, A.P. Synthesis and anticonvulsant activity of oximes of 1-R-3-acetonyl-3-hydroxyoxindole and its derivatives. Pharm. Chem. J. 1987, 21, 95–98. [Google Scholar] [CrossRef]
- Katzung, B.G. Drugs Used in Generalized Seizures. In Basic and Clinical Pharmacology, 9th ed.; McGraw-Hill: New York, NY, USA, 2003; Large Medical Books. [Google Scholar]
- Patsalos, P.N. Properties of Antiepileptic Drugs in the Treatment of Idiopathic Generalized Epilepsies. Epilepsia 2005, 46, 140–148. [Google Scholar] [CrossRef]
- Rogawski, M.A.; Löscher, W. The neurobiology of antiepileptic drugs. Nat. Rev. Neurosci. 2004, 5, 553–564. [Google Scholar] [CrossRef] [Green Version]
- Dunham, N.W.; Miya, T.S.; Edwards, L.D. The Pharmacological Activity of a Series of Basic Esters of Mono- and Dialkylmalonic Acids. J. Am. Pharm. Assoc. 1957, 46, 64–66. [Google Scholar] [CrossRef]
- Litchfield, J.T., Jr.; Wilcoxon, F. A simplified method of evaluating dose-effect experiments. J. Pharmacol. Exp. Ther. 1949, 96, 99–113. [Google Scholar]
- Mironov, A.H. The 1th Part. In Manual for Preclinical Studies of Drugs; Medicine: Moscow, Russia, 2012; pp. 235–250. (In Russian) [Google Scholar]
- File, S.E. Factors controlling measures of anxiety and responses to novelty in the mouse. Behav. Brain Res. 2001, 125, 151–157. [Google Scholar] [CrossRef]
- Stanford, S.C. The Open Field Test: Reinventing the wheel. J. Psychopharmacol. 2007, 21, 134–135. [Google Scholar] [CrossRef]
- Prut, L.; Belzung, C. The open field as a paradigm to measure the effects of drugs on anxiety-like behaviors: A review. Eur. J. Pharmacol. 2003, 463, 3–33. [Google Scholar] [CrossRef]
- Pellow, S.; File, S.E. Anxiolytic and anxiogenic drug effects on exploratory activity in an elevated plus-maze: A novel test of anxiety in the rat. Pharmacol. Biochem. Behav. 1986, 24, 525–529. [Google Scholar] [CrossRef]
- Pellow, S.; Chopin, P.; File, S.E.; Briley, M. Validation of open:closed arm entries in an elevated plus-maze as a measure of anxiety in the rat. J. Neurosci. Methods 1985, 14, 149–167. [Google Scholar] [CrossRef]
- Jardim, M.C.; Nogueira, R.L.; Graeff, F.G.; Nunes-De-Souza, R.L. Evaluation of the elevated T-maze as an animal model of anxiety in the mouse. Brain Res. Bull. 1999, 48, 407–411. [Google Scholar] [CrossRef]
- Graeff, F.G.; Netto, C.F.; Zangrossi, H., Jr. The elevated T-maze as an experimental model of anxiety. Neurosci. Biobehav. Rev. 1998, 23, 237–246. [Google Scholar] [CrossRef]
- Porsolt, R.D.; Anton, G.; Blavet, N.; Jalfre, M. Behavioral despair in rats: A new model sensitive to antidepressant treatments. Eur. J. Pharmacol. 1978, 47, 379–391. [Google Scholar] [CrossRef]
- Puthenkalam, R.; Hieckel, M.; Simeone, X.; Suwattanasophon, C.; Feldbauer, R.; Ecker, G.F.; Ernst, M. Structural Studies of GABAA Receptor Binding Sites: Which Experimental Structure Tells us What? Front. Mol. Neurosci. 2016, 9, 44. [Google Scholar] [CrossRef] [Green Version]
- Miller, P.S.; Aricescu, A.R. Crystal structure of a human GABAA receptor. Nature 2014, 512, 270–275. [Google Scholar] [CrossRef] [Green Version]
- Sieghart, W. Allosteric Modulation of GABAA Receptors via Multiple Drug-Binding Sites. Adv. Pharmacol. 2014, 72, 53–96. [Google Scholar] [CrossRef]
- Coleman, J.; Green, E.M.; Gouaux, E. X-ray structures and mechanism of the human serotonin transporter. Nature 2016, 532, 334–339. [Google Scholar] [CrossRef] [Green Version]
- Jha, P.; Ragnarsson, L.; Lewis, R.J. Structure-Function of the High Affinity Substrate Binding Site (S1) of Human Norepinephrine Transporter. Front. Pharmacol. 2020, 11, 217. [Google Scholar] [CrossRef]
- Rannversson, H.; Andersen, J.; Bang-Andersen, B.; Strømgaard, K. Mapping the Binding Site for Escitalopram and Paroxetine in the Human Serotonin Transporter Using Genetically Encoded Photo-Cross-Linkers. ACS Chem. Biol. 2017, 12, 2558–2562. [Google Scholar] [CrossRef]
- Möller, I.R.; Slivacka, M.; Nielsen, A.K.; Rasmussen, S.G.F.; Gether, U.; Loland, C.J.; Rand, K.D. Conformational dynamics of the human serotonin transporter during substrate and drug binding. Nat. Commun. 2019, 10, 1687. [Google Scholar] [CrossRef]
- Chen, J.-G.; Sachpatzidis, A.; Rudnick, G. The Third Transmembrane Domain of the Serotonin Transporter Contains Residues Associated with Substrate and Cocaine Binding. J. Biol. Chem. 1997, 272, 28321–28327. [Google Scholar] [CrossRef] [Green Version]
- Freddolino, P.L.; Kalani, M.Y.S.; Vaidehi, N.; Floriano, W.B.; Hall, S.E.; Trabanino, R.J.; Kam, V.W.T.; Goddard, W.A. Predicted 3D structure for the human β2 adrenergic receptor and its binding site for agonists and antagonists. Proc. Natl. Acad. Sci. USA 2004, 101, 2736–2741. [Google Scholar] [CrossRef] [Green Version]
- Bang, I.; Choi, A.H.-J. Structural Features of β2 Adrenergic Receptor: Crystal Structures and Beyond. Mol. Cells 2014, 38, 105–111. [Google Scholar] [CrossRef]
- Thorsen, T.S.; Matt, R.; Weis, W.I.; Kobilka, B.K. Modified T4 Lysozyme Fusion Proteins Facilitate G Protein-Coupled Receptor Crystallogenesis. Structure 2014, 22, 1657–1664. [Google Scholar] [CrossRef] [Green Version]
- Zielesny, A. Chemistry Software Package ChemOffice Ultra 2005. J. Chem. Inf. Model. 2005, 45, 1474–1477. [Google Scholar] [CrossRef]
- Evans, M.J.; Moore, J.S. A Collaborative, Wiki-Based Organic Chemistry Project Incorporating Free Chemistry Software on the Web. J. Chem. Educ. 2011, 88, 764–768. [Google Scholar] [CrossRef]
- Rose, P.W.; Prlić, A.; Altunkaya, A.; Bi, C.; Bradley, A.R.; Christie, C.H.; Di Costanzo, L.; Duarte, J.M.; Dutta, S.; Feng, Z.; et al. The RCSB protein data bank: Integrative view of protein, gene and 3D structural information. Nucleic Acids Res. 2016, 45, D271–D281. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [Green Version]
- FFogolari, F.; Brigo, A.; Molinari, H. The Poisson-Boltzmann equation for biomolecular electrostatics: A tool for structural biology. J. Mol. Recognit. 2002, 15, 377–392. [Google Scholar] [CrossRef]
- Jejurikar, B.L.; Rohane, S.H. Drug designing in discovery studio. Asian J. Res. Chem. 2021, 14, 135–138. [Google Scholar] [CrossRef]
- Metsalu, T.; Vilo, J. ClustVis: A web tool for visualizing clustering of multivariate data using Principal Component Analysis and heatmap. Nucleic Acids Res. 2015, 43, W566–W570. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | R | Yield (%) 1 |
|---|---|---|
| 3a | COOEt | Method (A) 81/Method (B) 78 |
| 3b | CN | 89 |
| 3c | CONHC6H5 | 98 |
| 3d | CONH-2,4-(OMe)2C6H3 | 92 |
| 3e | CONH-3,4-Cl2C6H3 | 90 |
| 3f | CONH(CH2)2C6H5 | 89 |
| Compound | R | Yield (%) 1 |
|---|---|---|
| 4a | H | 82 |
| 4b | C6H5 | 92 |
| 4c | 2,4-(OMe)2C6H3 | 91 |
| 4d | 3,4-Cl2C6H3 | 89 |
| 4e | (CH2)2C6H5 | 84 |
| Compounds (50 mg/kg) | Antagonism with PTZ * | Myorelaxation (TD50, mg/kg) * | MTD, (mg/kg) | PI | |
|---|---|---|---|---|---|
| % | (ED50, mg/kg) * | ||||
| 3a | 40 | − | >500 | 1100 | − |
| 3b | 60 | 34.0 (22.6 ÷ 57.0) | 560.0 (455 ÷ 689) | 1050 | 16.5 |
| 3c | 60 | 36.0 (24.0 ÷ 54.0) | 525 (437.5 ÷ 630) | 1150 | 14.6 |
| 3d | 60 | 35.0 (21.8 ÷ 56.0) | 540.0 (439 ÷ 664) | 1300 | 15.4 |
| 3e | 80 | 26.0 (21.7 ÷ 31.2) | 580.0 (475 ÷ 708) | 1350 | 22.3 |
| 3f | 60 | 44.0 (33.8 ÷ 57.2) | 600.0 (500 ÷ 720) | 1500 | 13.6 |
| 4a | 40 | − | >500 | 1350 | - |
| 4b | 60 | 37.0 (30.8 ÷ 44.4) | 565.0 (463 ÷ 689) | 1200 | 15 |
| 4c | 60 | 44.0 (24.4 ÷ 65.6) | 528.0 (432.7÷644) | 1400 | 12 |
| 4d | 60 | 37.5 (20.8 ÷ 67.5) | 505.0 (417 ÷ 611) | 1250 | 13.5 |
| 4e | 60 | 40.0 (23.5 ÷ 68.0) | 545.0 (450.4÷660) | 1250 | 13.6 |
| Ethosuximide (200 mg/kg) | 60 | 155.0 (117.5 ÷ 205) | 520.0 (426 ÷ 634) | 1000 | 3.4 |
| Diazepam (2 mg/kg) | 80 | 0.5 (0.4 ÷ 0.7) | 2.7 (1.4 ÷ 5.5) | 200 | 5.4 |
| Compounds | Dose, mg/kg | Amount (Absolute Data over 5 min) * | ||
|---|---|---|---|---|
| Horizontal Displacement | Vertical Displacement | Cells | ||
| Control | – | 25.8 ± 3.5 | 6.1 ± 1.1 | 0.5 ± 0.02 |
| 3b | 50 | 26.4 ± 3.6 | 3.3 ± 0.6 ** | 2.6 ± 0.6 ** |
| 3c | 50 | 42.6 ± 6.3 ** | 6.8 ± 1.2 | 1.2 ± 0.06 ** |
| 3d | 50 | 44.8 ± 5.2 ** | 9.6 ± 1.9 ** | 2.8 ± 0.5 ** |
| 3e | 50 | 24.4 ± 3.7 | 2.8 ± 0.7 ** | 2.8 ± 0.53 ** |
| 3f | 50 | 58.2 ± 6.1 ** | 7.0 ± 2.2 | 2.4 ± 0.8 ** |
| 4b | 50 | 40.4 ± 7.5 ** | 4.5± 0.9 | 0.9 ± 0.05 ** |
| 4c | 50 | 43.8 ± 4.1 ** | 6.2 ± 1.8 | 2.2 ± 0.6 ** |
| 4d | 50 | 34.2 ± 4.8 ** | 5.2 ± 0.7 | 2.2 ± 0.3 ** |
| 4e | 50 | 50.4 ± 7.3 ** | 7.1 ± 3.0 | 2.5 ± 0.7 ** |
| Ethosuximide | 200 | 26.8 ± 3.8 | 5.8 ± 1.9 | 0.6 ± 0.08 |
| Diazepam | 2 | 33.6 ± 4.2 ** | 6.4 ± 1.0 | 3.2 ± 0.9 ** |
| Compound (50 mg/kg) | Time Spent in Closed Arms, /s/ * | Number of Entries into the Closed Arms * | Time Spent in the Center, /s/ * | Time Spent in the Open Arms, /s/ * |
|---|---|---|---|---|
| Control | 278.2 (262 ÷ 294.0) | 7.0 (5.83 ÷ 8.4) | 21.8 (11.0 ÷ 32.6) | – |
| 3b | 207.2 (180.1÷233.6) ** | 7.6 (5.2 ÷ 10.0) | 87.8 (73.2 ÷105.4) ** | 5.0 (4.34 ÷ 5.75) ** |
| 3c | 220.6 (186.9 ÷ 260.3) ** | 3.6 (2.9 ÷ 4.3) | 74.2 (61,3 ÷89.8) ** | 5.2 (4.3 ÷ 6.24) ** |
| 3d | 200.6 (171 ÷ 230.3) ** | 3.0 (2.2 ÷ 3.8) ** | 88.0 (70.1÷105.9) ** | 11.4 (6.3 ÷ 16.5) ** |
| 3e | 152.2 (112.4÷192) ** | 5.6 (4.66 ÷ 6.72) | 138.0 (115 ÷165.6) ** | 9.8 (8.3 ÷ 11.6) ** |
| 3f | 177.2 (137.8 ÷ 216.6) ** | 4.2 (3.5 ÷ 4.9) ** | 113.4 (94.5 ÷ 136) ** | 9.4 (7.9 ÷ 11.51) ** |
| 4b | 197.4 (143.0÷251.8) ** | 5.6 (3.5 ÷ 7.7) | 96.4 (79 ÷ 117.6) ** | 6.2 (3.0 ÷ 9.4) ** |
| 4c | 167.0 (144.4 ÷ 189.6) ** | 5.0 (3.2 ÷ 6.8) | 130 (107.5 ÷152.5) ** | 3.0 (2.6 ÷ 3.45) ** |
| 4d | 208.0 (179.2 ÷ 236.8) ** | 4.0 (3.1 ÷ 4.9) ** | 82.0 (53.9 ÷ 110.1) ** | 10.0 (8.3 ÷ 12) ** |
| 4e | 216.2 (183.2 ÷ 255.1) ** | 6.2 (5.1 ÷ 7.3) | 79.8 (66.2 ÷ 93.4) ** | 4.0 (3.48 ÷ 4.6) ** |
| Ethosux. 200 mg/kg | 245.2 (212.9 ÷ 277.5) | 8.1 (5.6 ÷ 10.6) | 52.8 (19.7 ÷ 85.9) | – |
| Diazep. 2 mg/kg | 257.5 (226.2÷ 288.8) | 5.5 (4.58 ÷ 6.6) | 42.5 (34.8 ÷ 51.9) ** | 57 (47.5 ÷ 68.4) ** |
| Compound | Dose, mg/kg | Latent Period I Immobilization, /s/ * | Total Time of Immobilization /s/ * | Total Time of Active Swimming, /s/ * |
|---|---|---|---|---|
| Control | – | 92 (80.0 ÷ 105.8) | 81 (59.3 ÷ 102.7) | 282 (245.2 ÷ 324.3) |
| 3b | 50 | 71.0 (60.6 ÷ 81.4) | 195.0 (141.0 ÷ 249.0) | 165.0 (132 ÷ 206.3) |
| 3c | 50 | 97.0 (70.8 ÷ 123.2) | 136.0 (92.2 ÷ 179.8) | 224.0 (180.2 ÷ 267.8) |
| 3d | 50 | 91.0 (66.0 ÷ 116.0) | 142.0 (108.4 ÷ 175.6) | 218.0 (184.4 ÷ 251.6) |
| 3e | 50 | 172.0 (143.0 ÷206.4) ** | 48.0 (40.0 ÷ 57.6) ** | 312.0 (276.1 ÷ 352.6) |
| 3f | 50 | 121.0 (108.0 ÷ 135.5) ** | 20.0 (16.5 ÷ 24.2) ** | 340.0 (283.3 ÷ 408.0) |
| 4b | 50 | 78.0 (58.0 ÷ 100.0) | 141.0 (116.0 ÷ 166.0) | 219.0 (193.2 ÷ 244.8) |
| 4c | 50 | 70.0(58.1 ÷ 81.9) | 159.0 (140.9 ÷ 177.1) | 201.0 (150.2 ÷ 251.8) |
| 4d | 50 | 124.0 (110.7 ÷ 138.9) ** | 177.0 (149.9 ÷ 204.1) | 183.0 (155.9 ÷210.1) |
| 4e | 50 | 104.0 (67.2 ÷ 140.8) | 96.0 (73.7÷ 118.3) | 264.0 (202.0 ÷ 326.0) |
| Ethosux. | 200 | 105 (87.5 ÷ 104.4) | 98 (75.3 ÷ 127.4) | 262 (199.9 ÷ 324.1) |
| Diazep. | 2 | 174 (144 ÷ 204) ** | 24 (13.9 ÷ 34.1) ** | 336 (282.6 ÷ 389.4) |
| Structure ID: | Complexation Energy kcal/mol | Root-Mean-Square Deviation (RMSD) | Binding Constant, Kb |
|---|---|---|---|
| 3a | −8.3 ± 0.41 | 1.35 | 1.1 × 106 |
| 3c | −10.0 ± 0.5 | 1.92 | 2.2 × 107 |
| 3d | −9.3 ± 0.46 | 1.92 | 6.0 × 106 |
| 3e | −9.8 ± 0.49 | 1.68 | 1.5 × 107 |
| 4b | −10.0 ± 0.5 | 0.83 | 2.2 × 107 |
| 4c | −8.8 ± 0.44 | 1.08 | 2.9 × 106 |
| 4d | −9.8 ± 0.49 | 1.81 | 1.5 × 107 |
| Diazepam | −7.5 ± 0.37 | 1.20 | 2.9 × 105 |
| Structure ID | Complexation Energy kcal/mol | Root-Mean-Square Deviation (RMSD) | Binding Constant, Kb |
|---|---|---|---|
| 2 | −7.8 ± 0.39 | 1.73 | 5.2 × 105 |
| 3c | −10.3 ± 0.51 | 1.86 | 3.2 × 107 |
| 3d | −9.1 ± 0.45 | 1.87 | 4.2 × 106 |
| 3e | −10.3 ± 0.51 | 0.92 | 3.0 × 107 |
| 3f | −10.8 ± 0.54 | 1.08 | 7.8 × 107 |
| 4b | −10.2 ± 0.51 | 0.62 | 2.7 × 107 |
| 4d | −9.8 ± 0.49 | 1.36 | 1.4 × 107 |
| 4e | −11.0 ± 0.54 | 1.41 | 9.7 × 107 |
| Structure ID: | Complexation Energy kcal/mol | RMSD | Binding Constant, Kb |
|---|---|---|---|
| 3a | −6.8 ± 0.34 | 1.98 | 9.8 × 104 |
| 3c | −8.9 ± 0.44 | 1.40 | 3.1 × 106 |
| 3d | −9.1 ± 0.45 | 1.95 | 4.2 × 106 |
| 3e | −8.9 ± 0.44 | 1.86 | 3.3 × 106 |
| 3f | −9.3 ± 0.46 | 0.92 | 6.0 × 106 |
| 4a | −8.1 ± 0.4 | 1.49 | 8.0 × 105 |
| 4e | −8.7 ± 0.43 | 1.17 | 2.5 × 106 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dashyan, S.S.; Babaev, E.V.; Paronikyan, E.G.; Ayvazyan, A.G.; Paronikyan, R.G.; Hunanyan, L.S. Evaluation of Neurotropic Activity and Molecular Docking Study of New Derivatives of pyrano[4″,3″:4′,5′]pyrido[3′,2′:4,5]thieno[3,2-d]pyrimidines on the Basis of pyrano[3,4-c]pyridines. Molecules 2022, 27, 3380. https://doi.org/10.3390/molecules27113380
Dashyan SS, Babaev EV, Paronikyan EG, Ayvazyan AG, Paronikyan RG, Hunanyan LS. Evaluation of Neurotropic Activity and Molecular Docking Study of New Derivatives of pyrano[4″,3″:4′,5′]pyrido[3′,2′:4,5]thieno[3,2-d]pyrimidines on the Basis of pyrano[3,4-c]pyridines. Molecules. 2022; 27(11):3380. https://doi.org/10.3390/molecules27113380
Chicago/Turabian StyleDashyan, Shushanik Sh., Eugene V. Babaev, Ervand G. Paronikyan, Armen G. Ayvazyan, Ruzanna G. Paronikyan, and Lernik S. Hunanyan. 2022. "Evaluation of Neurotropic Activity and Molecular Docking Study of New Derivatives of pyrano[4″,3″:4′,5′]pyrido[3′,2′:4,5]thieno[3,2-d]pyrimidines on the Basis of pyrano[3,4-c]pyridines" Molecules 27, no. 11: 3380. https://doi.org/10.3390/molecules27113380
APA StyleDashyan, S. S., Babaev, E. V., Paronikyan, E. G., Ayvazyan, A. G., Paronikyan, R. G., & Hunanyan, L. S. (2022). Evaluation of Neurotropic Activity and Molecular Docking Study of New Derivatives of pyrano[4″,3″:4′,5′]pyrido[3′,2′:4,5]thieno[3,2-d]pyrimidines on the Basis of pyrano[3,4-c]pyridines. Molecules, 27(11), 3380. https://doi.org/10.3390/molecules27113380