
Epithelial Sodium Channel Inhibition by Amiloride Addressed with THz Spectroscopy and Molecular Modeling

 ,
,  ,
,  ,
,
Abstract
:1. Introduction
2. Results
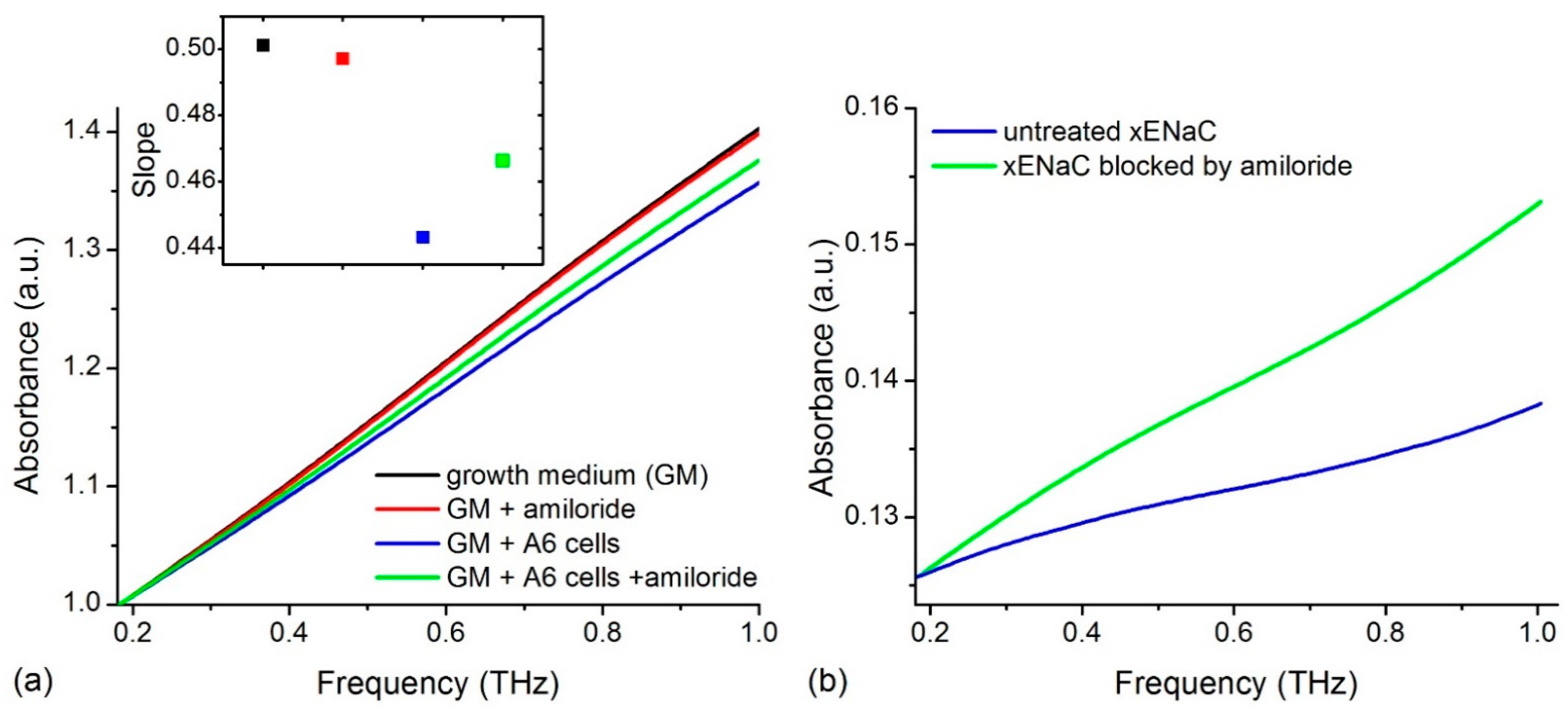
2.1. Experimental THz Spectra of A6 Cells
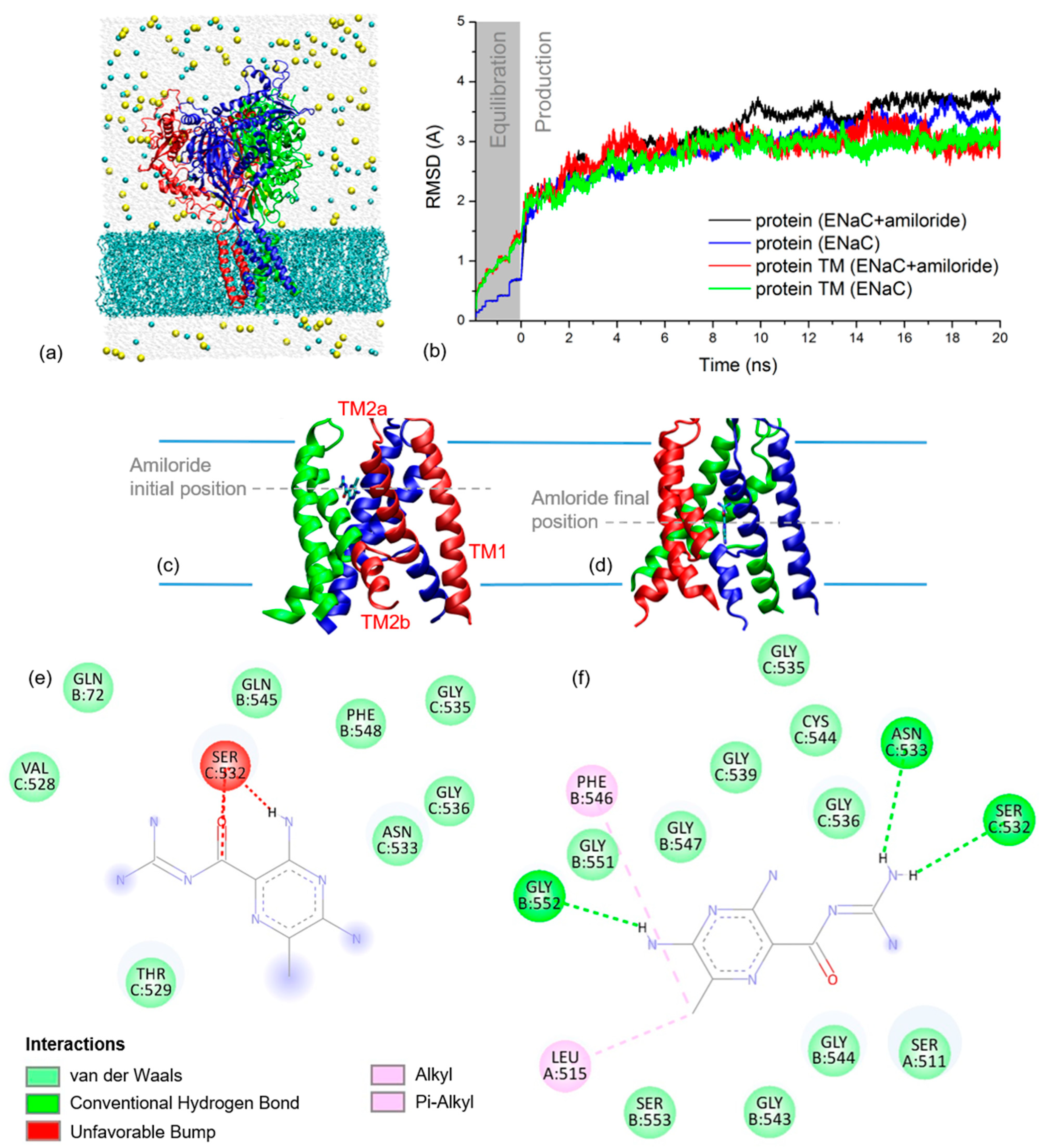
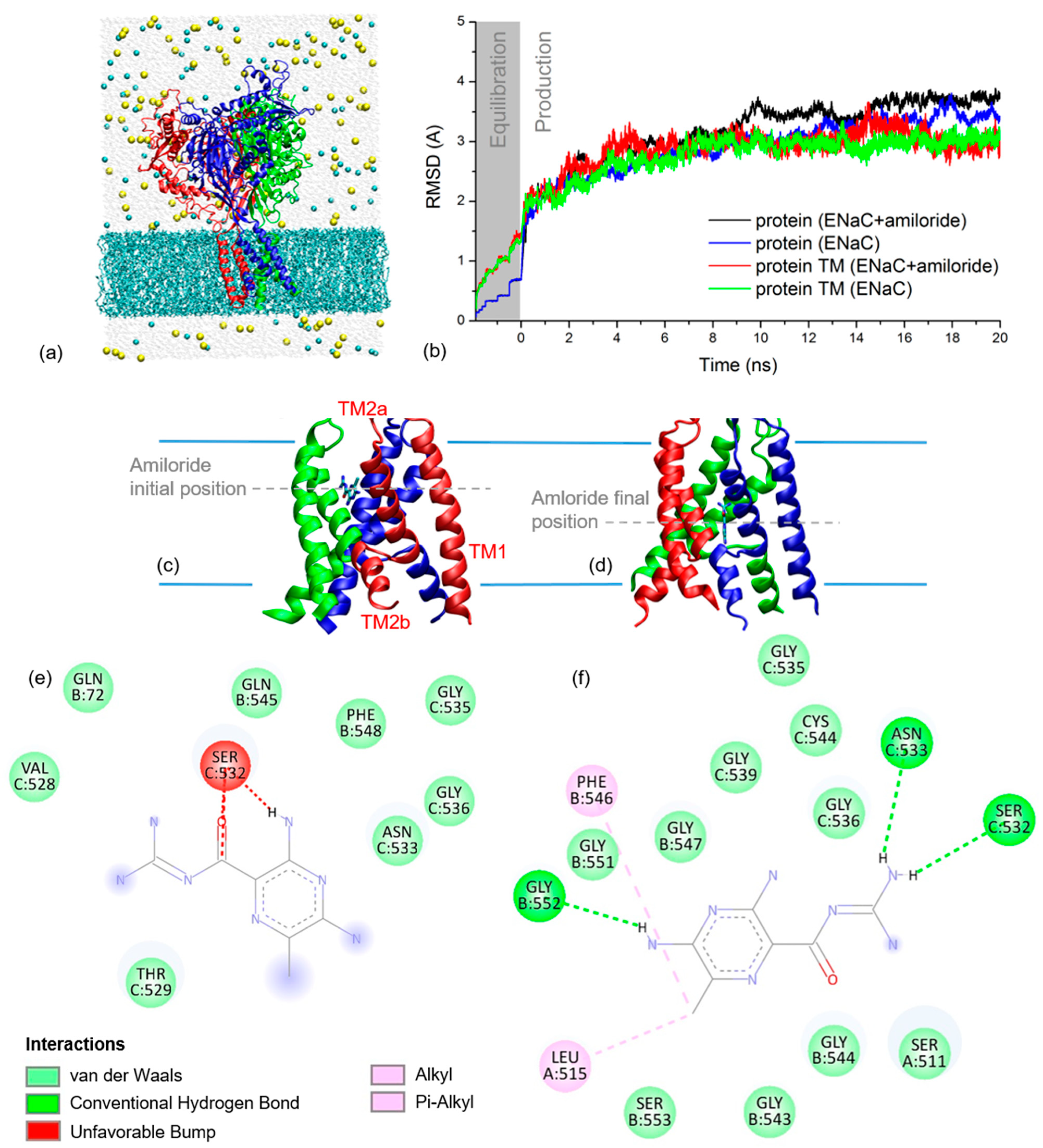
2.2. xENaC and xENaC—Amiloride Complex Models
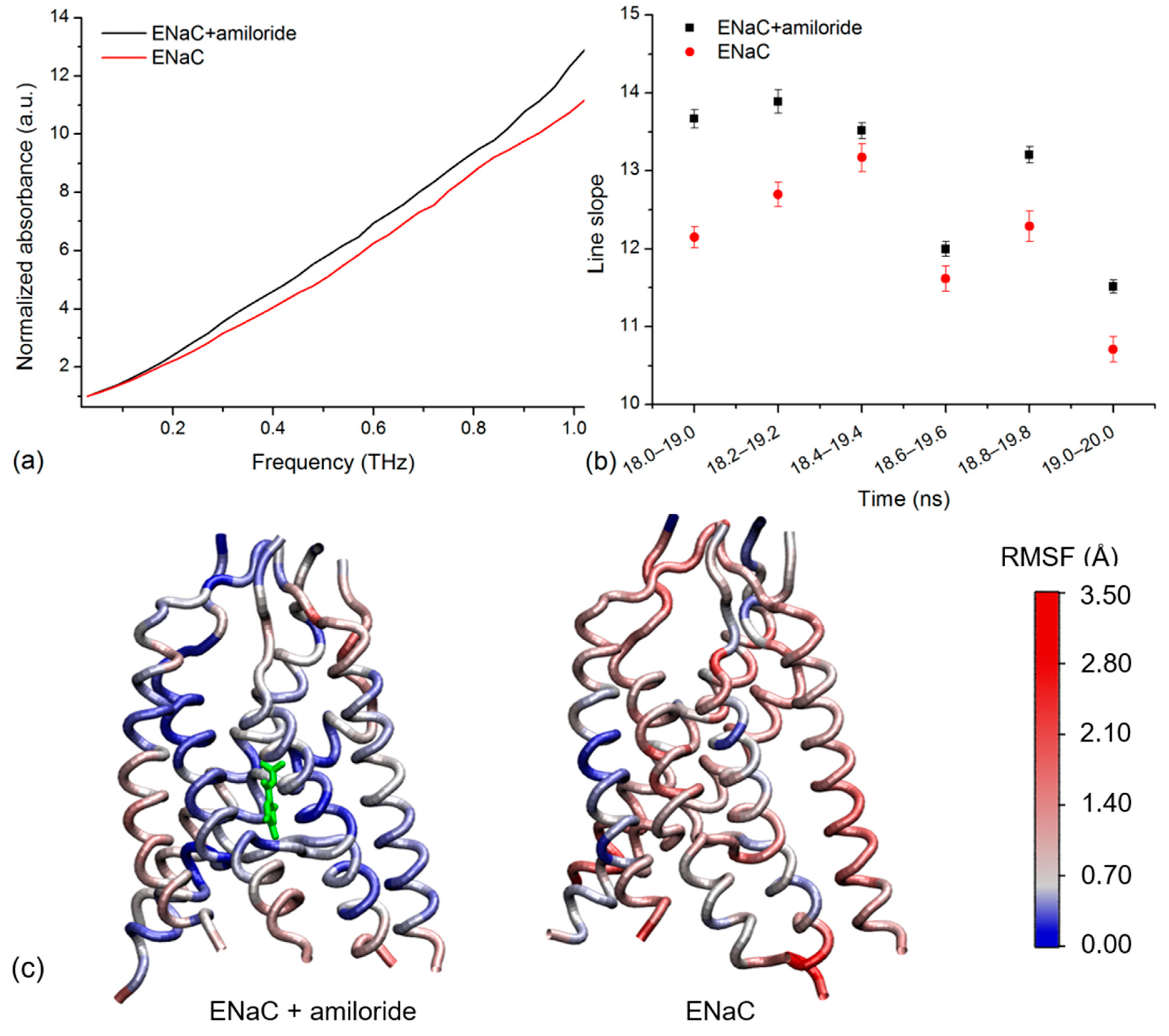
2.3. Simulated THz Spectra
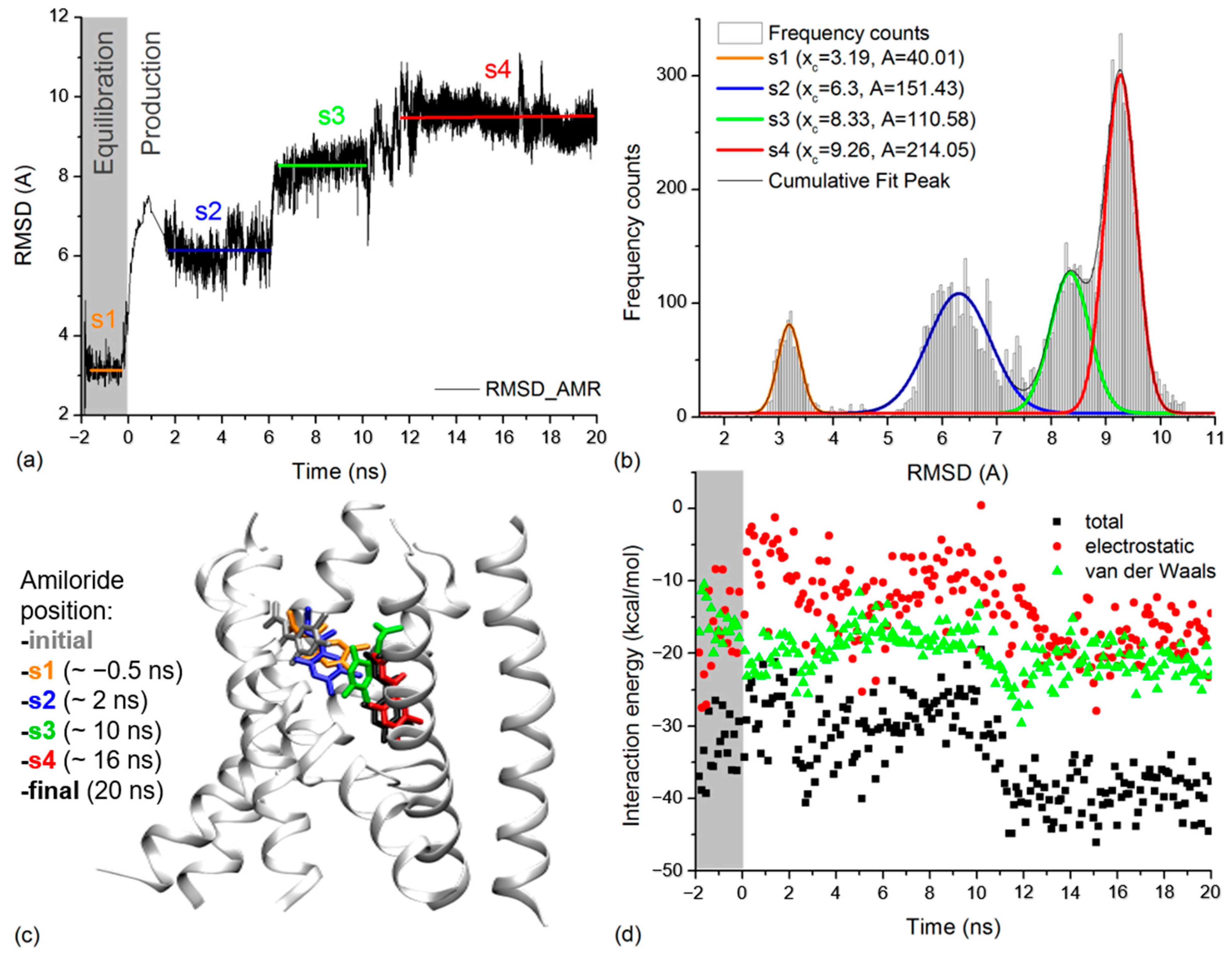
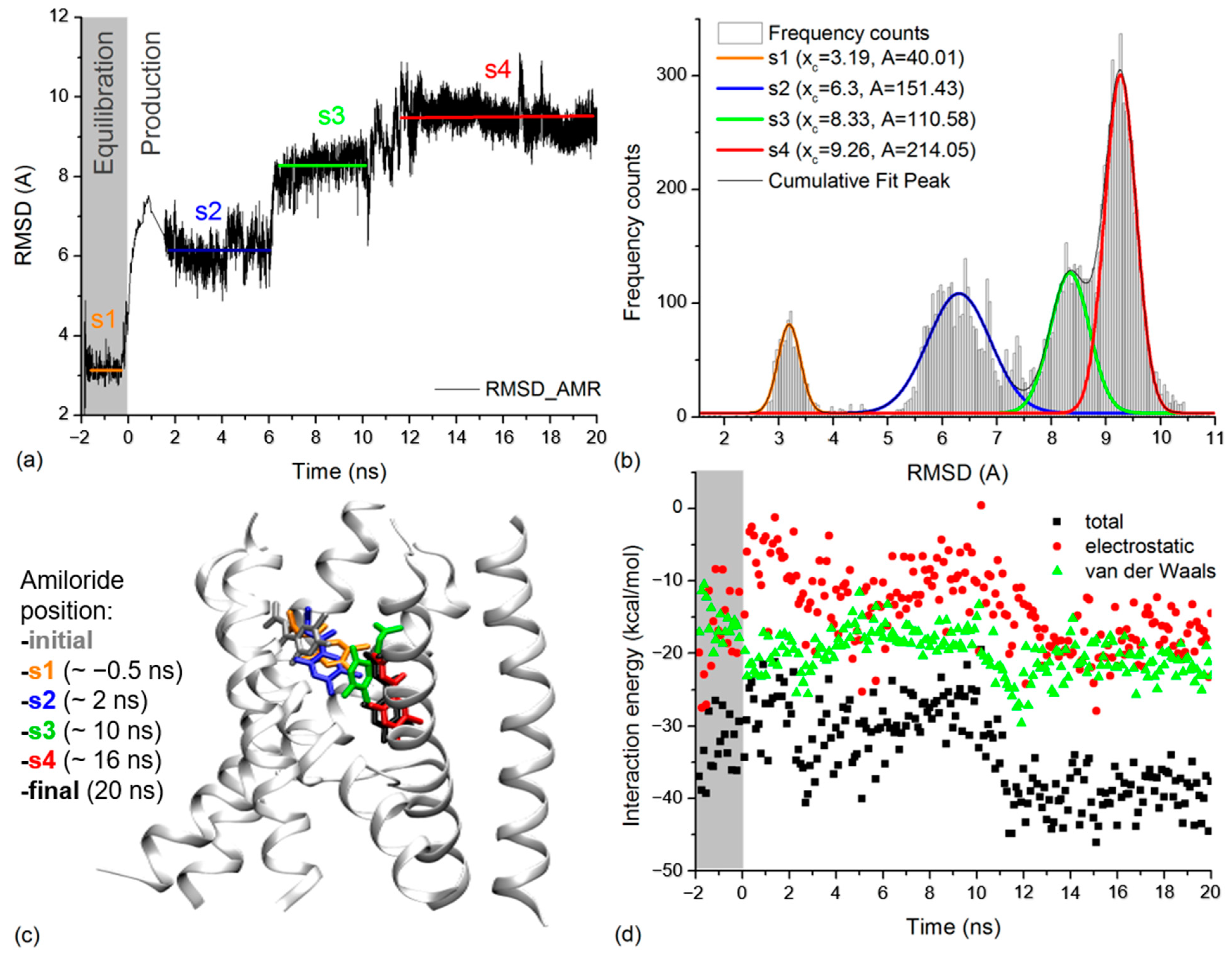
2.4. Amiloride Dynamics in xENaC Extracellular Vestibule
3. Discussion
4. Materials and Methods
4.1. Experimental Methods
4.1.1. A6 Cell Cultures and Sample Preparation
4.1.2. THz Spectroscopy Experiments
4.1.3. THz Absorption of A6 Cells in the Absence and Presence of Amiloride
4.2. Molecular Modeling Methods
4.2.1. xENaC Modelling and Amiloride Docking
4.2.2. Molecular Dynamics (MD) Simulations
4.2.3. THz Spectra Simulation
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Alexander, S.P.H.; Mathie, A.; Peters, J.A. Ion Channels. Br. J. Pharmacol. 2011, 164, S137–S174. [Google Scholar] [CrossRef]
- Brown, B.M.; Nguyen, H.M.; Wulff, H. Recent Advances in Our Understanding of the Structure and Function of More Unusual Cation Channels. F1000Research 2019, 8, 123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cordero-Morales, J.F.; Vásquez, V. How Lipids Contribute to Ion Channel Function, a Fat Perspective on Direct and Indirect Interactions. Curr. Opin. Struct. Biol. 2018, 51, 92–98. [Google Scholar] [CrossRef] [PubMed]
- Lau, C.; Hunter, M.J.; Stewart, A.; Perozo, E.; Vandenberg, J.I. Never at Rest: Insights into the Conformational Dynamics of Ion Channels from Cryo-electron Microscopy. J. Physiol. 2018, 596, 1107–1119. [Google Scholar] [CrossRef] [Green Version]
- Kratochvil, H.T.; Carr, J.K.; Matulef, K.; Annen, A.W.; Li, H.; Maj, M.; Ostmeyer, J.; Serrano, A.L.; Raghuraman, H.; Moran, S.D.; et al. Instantaneous Ion Configurations in the K+ Ion Channel Selectivity Filter Revealed by 2D IR Spectroscopy. Science 2016, 353, 1040–1044. [Google Scholar] [CrossRef] [Green Version]
- Furutani, Y. Ion–Protein Interactions of a Potassium Ion Channel Studied by Attenuated Total Reflection Fourier Transform Infrared Spectroscopy. Biophys. Rev. 2018, 10, 235–239. [Google Scholar] [CrossRef] [Green Version]
- Mancini, T.; Mosetti, R.; Marcelli, A.; Petrarca, M.; Lupi, S.; D’Arco, A. Terahertz Spectroscopic Analysis in Protein Dynamics: Current Status. Radiation 2022, 2, 100–123. [Google Scholar] [CrossRef]
- Mernea, M.; Ulăreanu, R.; Călboreanu, O.; Chirițoiu, G.; Cucu, D.; Mihăilescu, D.F. N-Glycosylation State of TRPM8 Protein Revealed by Terahertz Spectroscopy and Molecular Modelling. Biochim. Biophys. Acta BBA Gen. Subj. 2020, 1864, 129580. [Google Scholar] [CrossRef]
- Hanukoglu, I.; Hanukoglu, A. Epithelial Sodium Channel (ENaC) Family: Phylogeny, Structure-Function, Tissue Distribution, and Associated Inherited Diseases. Gene 2016, 579, 95–132. [Google Scholar] [CrossRef] [Green Version]
- Canessa, C.M.; Schild, L.; Buell, G.; Thorens, B.; Gautschi, I.; Horisberger, J.D.; Rossier, B.C. Amiloride-Sensitive Epithelial Na+ Channel Is Made of Three Homologous Subunits. Nature 1994, 367, 463–467. [Google Scholar] [CrossRef]
- Mernea, M.; Calborean, O.; Grigore, O.; Dascalu, T.; Mihailescu, D.F. Validation of Protein Structural Models Using THz Spectroscopy: A Promising Approach to Solve Three-Dimensional Structures. Opt. Quantum Electron. 2014, 46, 505–514. [Google Scholar] [CrossRef]
- Balu, R.; Zhang, H.; Zukowski, E.; Chen, J.-Y.; Markelz, A.G.; Gregurick, S.K. Terahertz Spectroscopy of Bacteriorhodopsin and Rhodopsin: Similarities and Differences. Biophys. J. 2008, 94, 3217–3226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noreng, S.; Posert, R.; Bharadwaj, A.; Houser, A.; Baconguis, I. Molecular Principles of Assembly, Activation, and Inhibition in Epithelial Sodium Channel. eLife 2020, 9, e59038. [Google Scholar] [CrossRef] [PubMed]
- Baconguis, I.; Bohlen, C.J.; Goehring, A.; Julius, D.; Gouaux, E. X-ray Structure of Acid-Sensing Ion Channel 1–Snake Toxin Complex Reveals Open State of a Na+-Selective Channel. Cell 2014, 156, 717–729. [Google Scholar] [CrossRef] [Green Version]
- Handler, J.S.; Steele, R.E.; Sahib, M.K.; Wade, J.B.; Preston, A.S.; Lawson, N.L.; Johnson, J.P. Toad Urinary Bladder Epithelial Cells in Culture: Maintenance of Epithelial Structure, Sodium Transport, and Response to Hormones. Proc. Natl. Acad. Sci. USA 1979, 76, 4151–4155. [Google Scholar] [CrossRef] [Green Version]
- Puoti, A.; May, A.; Canessa, C.M.; Horisberger, J.D.; Schild, L.; Rossier, B.C. The Highly Selective Low-Conductance Epithelial Na Channel of Xenopus Laevis A6 Kidney Cells. Am. J. Physiol. 1995, 269, C188–C197. [Google Scholar] [CrossRef]
- Bahar, I.; Lezon, T.R.; Bakan, A.; Shrivastava, I.H. Normal Mode Analysis of Biomolecular Structures: Functional Mechanisms of Membrane Proteins. Chem. Rev. 2010, 110, 1463–1497. [Google Scholar] [CrossRef] [Green Version]
- Guo, L.; Bo, W.; Wang, K.; Wang, S.; Gong, Y. Theoretical Investigation on the Effect of Terahertz Wave on Ca2+ Transport in the Calcium Channel. iScience 2022, 25, 103561. [Google Scholar] [CrossRef]
- Li, Y.; Chang, C.; Zhu, Z.; Sun, L.; Fan, C. Terahertz Wave Enhances Permeability of the Voltage-Gated Calcium Channel. J. Am. Chem. Soc. 2021, 143, 4311–4318. [Google Scholar] [CrossRef]
- Wang, K.; Wang, S.; Yang, L.; Wu, Z.; Zeng, B.; Gong, Y. THz Trapped Ion Model and THz Spectroscopy Detection of Potassium Channels. Nano Res. 2021, 15, 3825–3833. [Google Scholar] [CrossRef]
- Son, J.-H. (Ed.) Terahertz Biomedical Science and Technology; CRC Press: Boca Raton, FL, USA, 2014; ISBN 978-0-429-19428-3. [Google Scholar]
- Kawaguchi, S.; Kambara, O.; Ponseca, C.S., Jr.; Shibata, M.; Kandori, H.; Tominaga, K. Low-Frequency Dynamics of Biological Molecules Studied by Terahertz Time-Domain Spectroscopy. Spectroscopy 2010, 24, 153–158. [Google Scholar] [CrossRef]
- Mitra, R.K.; Palit, D.K. Probing Biological Water Using Terahertz Absorption Spectroscopy. In Terahertz Technology [Working Title]; You, B., Lu, J., Eds.; IntechOpen: London, UK, 2021; ISBN 978-1-83962-684-5. [Google Scholar]
- Xu, J.; Plaxco, K.W.; Allen, S.J. Probing the Collective Vibrational Dynamics of a Protein in Liquid Water by Terahertz Absorption Spectroscopy. Protein Sci. Publ. Protein Soc. 2006, 15, 1175–1181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ebbinghaus, S.; Kim, S.J.; Heyden, M.; Yu, X.; Heugen, U.; Gruebele, M.; Leitner, D.M.; Havenith, M. An Extended Dynamical Hydration Shell around Proteins. Proc. Natl. Acad. Sci. USA 2007, 104, 20749–20752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Markelz, A.G.; Roitberg, A.; Heilweil, E.J. Pulsed Terahertz Spectroscopy of DNA, Bovine Serum Albumin and Collagen between 0.1 and 2.0 THz. Chem. Phys. Lett. 2000, 320, 42–48. [Google Scholar] [CrossRef]
- Yu, L.; Hao, L.; Meiqiong, T.; Jiaoqi, H.; Wei, L.; Jinying, D.; Xueping, C.; Weiling, F.; Yang, Z. The Medical Application of Terahertz Technology in Non-Invasive Detection of Cells and Tissues: Opportunities and Challenges. RSC Adv. 2019, 9, 9354–9363. [Google Scholar] [CrossRef] [Green Version]
- Moritsugu, K.; Njunda, B.M.; Smith, J.C. Theory and Normal-Mode Analysis of Change in Protein Vibrational Dynamics on Ligand Binding. J. Phys. Chem. B 2010, 114, 1479–1485. [Google Scholar] [CrossRef]
- Mernea, M.; Ulăreanu, R.; Călborean, O.; Chira, S.; Popescu, O.; Mihailescu, D.F.; Cucu, D. Effects of Cd2+ on the Epithelial Na+ Channel (ENaC) Investigated by Experimental and Modeling Studies. Gen. Physiol. Biophys. 2016, 35, 259–271. [Google Scholar] [CrossRef]
- Baconguis, I.; Gouaux, E. Structural Plasticity and Dynamic Selectivity of Acid-Sensing Ion Channel–Spider Toxin Complexes. Nature 2012, 489, 400–405. [Google Scholar] [CrossRef] [Green Version]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [Green Version]
- Noreng, S.; Bharadwaj, A.; Posert, R.; Yoshioka, C.; Baconguis, I. Structure of the Human Epithelial Sodium Channel by Cryo-Electron Microscopy. eLife 2018, 7, e39340. [Google Scholar] [CrossRef]
- Kellenberger, S.; Gautschi, I.; Schild, L. Mutations in the Epithelial Na+ Channel ENaC Outer Pore Disrupt Amiloride Block by Increasing Its Dissociation Rate. Mol. Pharmacol. 2003, 64, 848–856. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhalla, V.; Hallows, K.R. Mechanisms of ENaC Regulation and Clinical Implications. J. Am. Soc. Nephrol. 2008, 19, 1845–1854. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sali, A.; Blundell, T.L. Comparative Protein Modelling by Satisfaction of Spatial Restraints. J. Mol. Biol. 1993, 234, 779–815. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.; Robertson, D.H.; Brooks, C.L., III; Vieth, M. Detailed Analysis of Grid-Based Molecular Docking: A Case Study of CDOCKER—A CHARMm-Based MD Docking Algorithm. J. Comput. Chem. 2003, 24, 1549–1562. [Google Scholar] [CrossRef]
- Jo, S.; Kim, T.; Iyer, V.G.; Im, W. CHARMM-GUI: A Web-Based Graphical User Interface for CHARMM. J. Comput. Chem. 2008, 29, 1859–1865. [Google Scholar] [CrossRef]
- Wu, E.L.; Cheng, X.; Jo, S.; Rui, H.; Song, K.C.; Dávila-Contreras, E.M.; Qi, Y.; Lee, J.; Monje-Galvan, V.; Venable, R.M.; et al. CHARMM-GUI Membrane Builder toward Realistic Biological Membrane Simulations. J. Comput. Chem. 2014, 35, 1997–2004. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.; Cheng, X.; Swails, J.M.; Yeom, M.S.; Eastman, P.K.; Lemkul, J.A.; Wei, S.; Buckner, J.; Jeong, J.C.; Qi, Y.; et al. CHARMM-GUI Input Generator for NAMD, GROMACS, AMBER, OpenMM, and CHARMM/OpenMM Simulations Using the CHARMM36 Additive Force Field. J. Chem. Theory Comput. 2016, 12, 405–413. [Google Scholar] [CrossRef]
- Kim, S.; Lee, J.; Jo, S.; Brooks, C.L.; Lee, H.S.; Im, W. CHARMM-GUI Ligand Reader & Modeler for CHARMM Force Field Generation of Small Molecules. J. Comput. Chem. 2017, 38, 1879–1886. [Google Scholar] [CrossRef]
- Phillips, J.C.; Braun, R.; Wang, W.; Gumbart, J.; Tajkhorshid, E.; Villa, E.; Chipot, C.; Skeel, R.D.; Kalé, L.; Schulten, K. Scalable Molecular Dynamics with NAMD. J. Comput. Chem. 2005, 26, 1781–1802. [Google Scholar] [CrossRef] [Green Version]
- Brooks, B.R.; Brooks, C.L.; MacKerell, A.D.; Nilsson, L.; Petrella, R.J.; Roux, B.; Won, Y.; Archontis, G.; Bartels, C.; Boresch, S.; et al. CHARMM: The Biomolecular Simulation Program. J. Comput. Chem. 2009, 30, 1545–1614. [Google Scholar] [CrossRef]
- IR Spectral Density Calculator Plugin, Version 1.3. Available online: https://www.ks.uiuc.edu/Research/vmd/plugins/irspecgui/ (accessed on 12 March 2022).
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual Molecular Dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Keogh, E.; Ratanamahatana, C.A. Exact Indexing of Dynamic Time Warping. Knowl. Inf. Syst. 2005, 7, 358–386. [Google Scholar] [CrossRef]
- Keogh, E.; Chakrabarti, K.; Pazzani, M.; Mehrotra, S. Dimensionality Reduction for Fast Similarity Search in Large Time Series Databases. Knowl. Inf. Syst. 2001, 3, 263–286. [Google Scholar] [CrossRef]
- Dau, H.A.; Silva, D.F.; Petitjean, F.; Forestier, G.; Bagnall, A.; Mueen, A.; Keogh, E. Optimizing Dynamic Time Warping’s Window Width for Time Series Data Mining Applications. Data Min. Knowl. Discov. 2018, 32, 1074–1120. [Google Scholar] [CrossRef] [Green Version]
- Salvador, S.; Chan, P. Toward Accurate Dynamic Time Warping in Linear Time and Space. Intell. Data Anal. 2007, 11, 561–580. [Google Scholar] [CrossRef] [Green Version]
- Python Implementation of FastDTW. Available online: https://github.com/slaypni/fastdtw (accessed on 23 March 2022).
- Pandas—Python Data Analysis Library. Available online: https://pandas.pydata.org/ (accessed on 23 March 2022).
- NumPy. Available online: https://numpy.org/ (accessed on 23 March 2022).
- SciPy. Available online: https://scipy.org/ (accessed on 23 March 2022).



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Amiloride Pose | CDOCKER Energy (kcal/mol) | CDOCKER Interaction Energy (kcal/mol) |
|---|---|---|
| 1 | −6.34 | −18.58 |
| 2 | −4.47 | −17.10 |
| 3 | −4.11 | −16.58 |
| 4 | −4.05 | −16.61 |
| 5 | −3.75 | −16.40 |
| 6 | −3.55 | −16.02 |
| 7 | −3.55 | −16.03 |
| 8 | −3.35 | −15.89 |
| 9 | −3.19 | −17.57 |
| 10 | −3.05 | −15.63 |
| Simulation System | cDTW | FastDTW | |||||
|---|---|---|---|---|---|---|---|
| CI | Euclidean Distance | CI | Euclidean Distance | ||||
| min | max | min | max | ||||
| ENaC | 95% | 0.0002 | 0.0004 | 95% | 0.066 | 0.080 | |
| ENaC + amiloride | 95% | 0.00016 | 0.00034 | 95% | 0.066 | 0.078 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mernea, M.; Ulăreanu, R.Ș.; Cucu, D.; Al-Saedi, J.H.; Pop, C.-E.; Fendrihan, S.; Anghelescu, G.D.C.; Mihăilescu, D.F. Epithelial Sodium Channel Inhibition by Amiloride Addressed with THz Spectroscopy and Molecular Modeling. Molecules 2022, 27, 3271. https://doi.org/10.3390/molecules27103271
Mernea M, Ulăreanu RȘ, Cucu D, Al-Saedi JH, Pop C-E, Fendrihan S, Anghelescu GDC, Mihăilescu DF. Epithelial Sodium Channel Inhibition by Amiloride Addressed with THz Spectroscopy and Molecular Modeling. Molecules. 2022; 27(10):3271. https://doi.org/10.3390/molecules27103271
Chicago/Turabian StyleMernea, Maria, Roxana Ștefania Ulăreanu, Dana Cucu, Jasim Hafedh Al-Saedi, Cristian-Emilian Pop, Sergiu Fendrihan, Giorgiana Diana Carmen Anghelescu, and Dan Florin Mihăilescu. 2022. "Epithelial Sodium Channel Inhibition by Amiloride Addressed with THz Spectroscopy and Molecular Modeling" Molecules 27, no. 10: 3271. https://doi.org/10.3390/molecules27103271
APA StyleMernea, M., Ulăreanu, R. Ș., Cucu, D., Al-Saedi, J. H., Pop, C.-E., Fendrihan, S., Anghelescu, G. D. C., & Mihăilescu, D. F. (2022). Epithelial Sodium Channel Inhibition by Amiloride Addressed with THz Spectroscopy and Molecular Modeling. Molecules, 27(10), 3271. https://doi.org/10.3390/molecules27103271