
Coumarinolignans with Reactive Oxygen Species (ROS) and NF-κB Inhibitory Activities from the Roots of Waltheria indica

 , ,
, ,  and
and
Abstract
1. Introduction
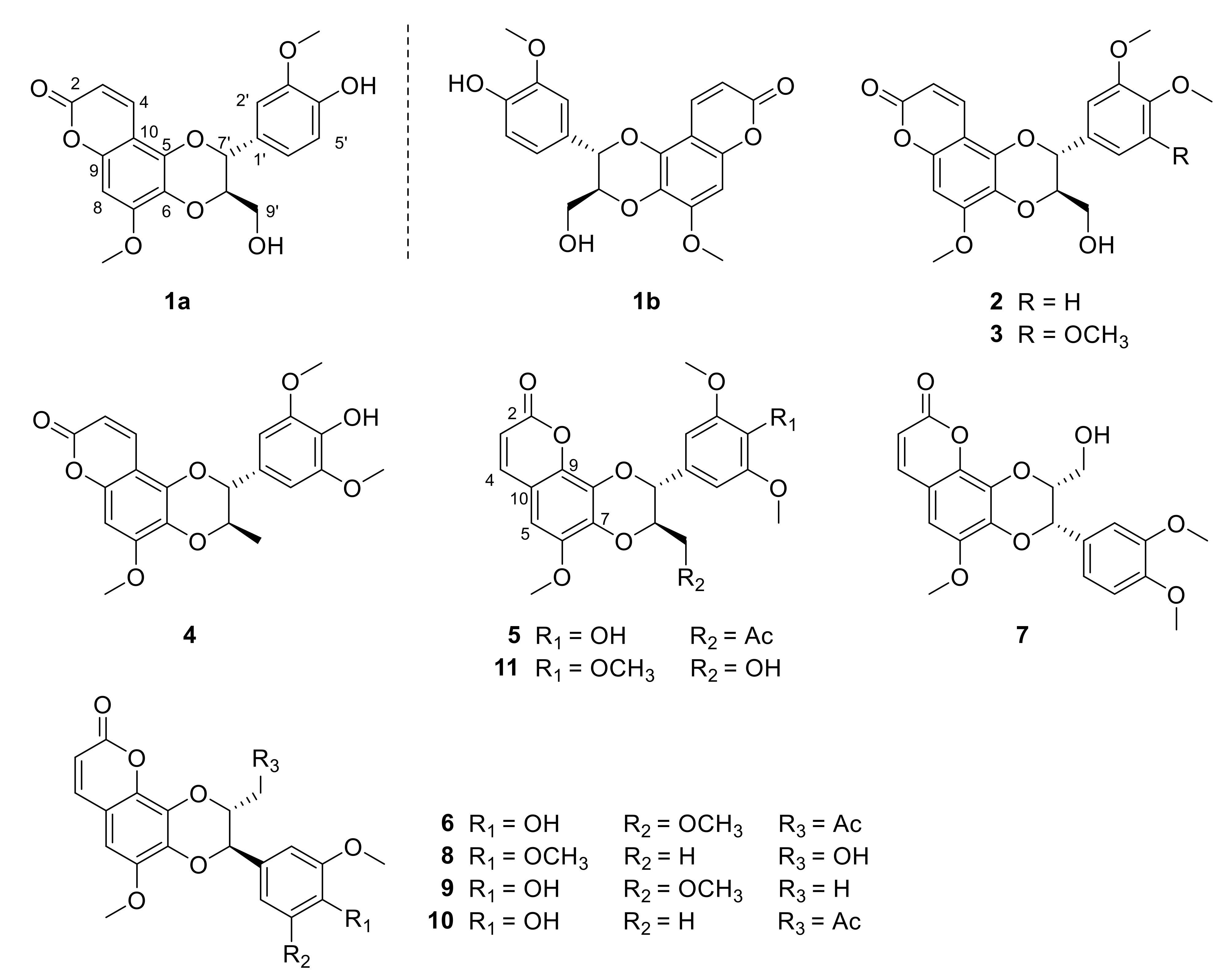
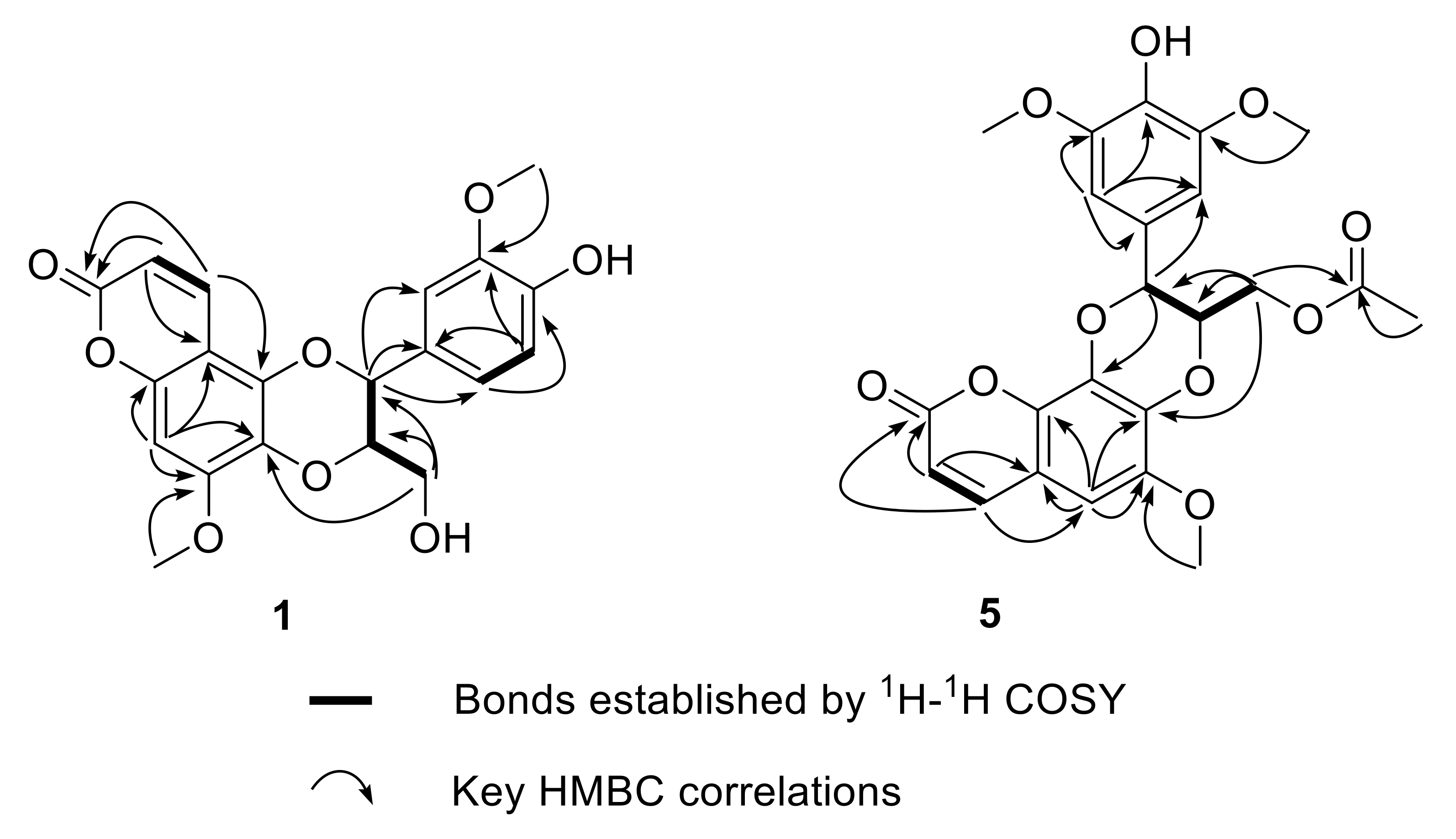
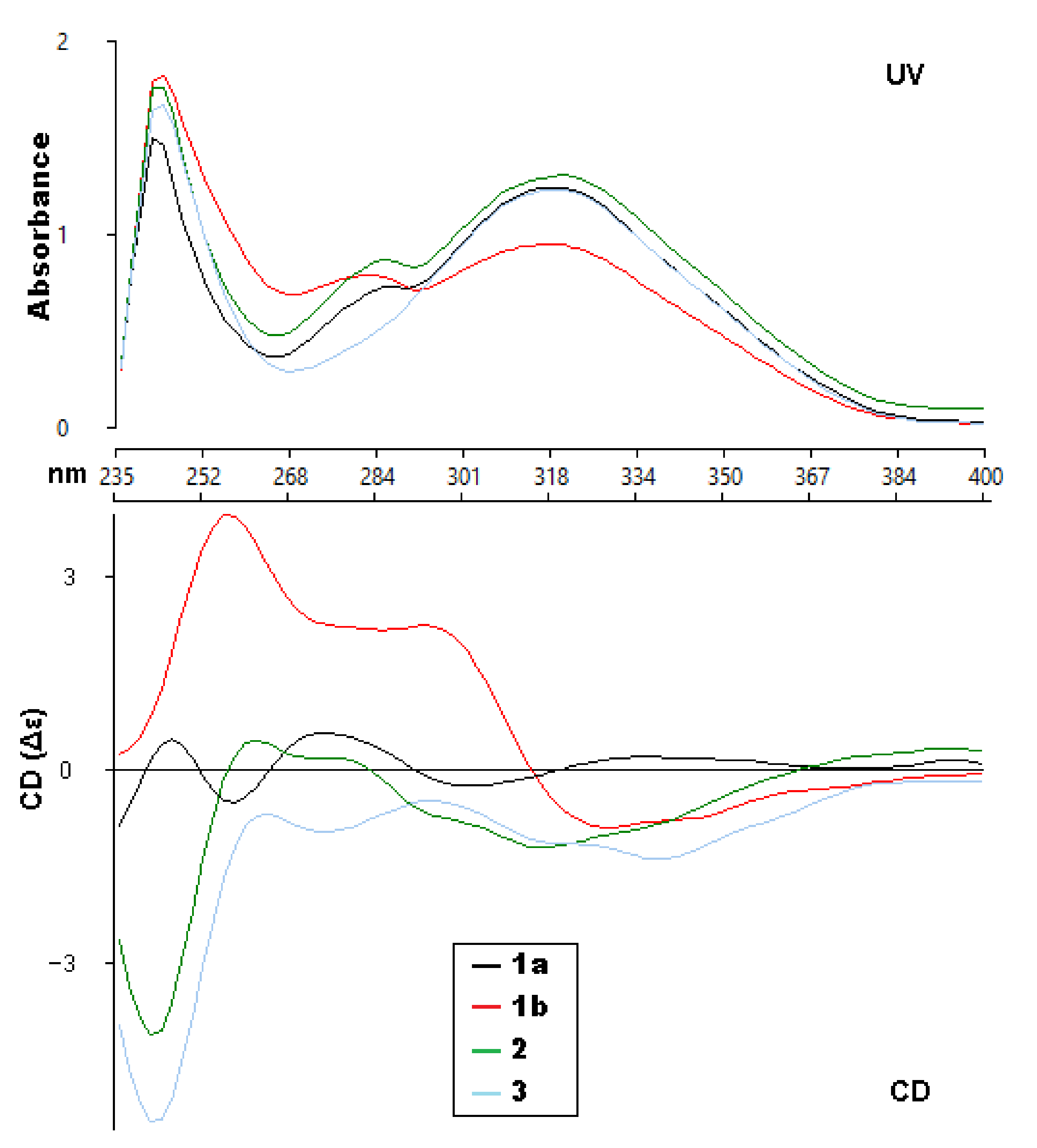
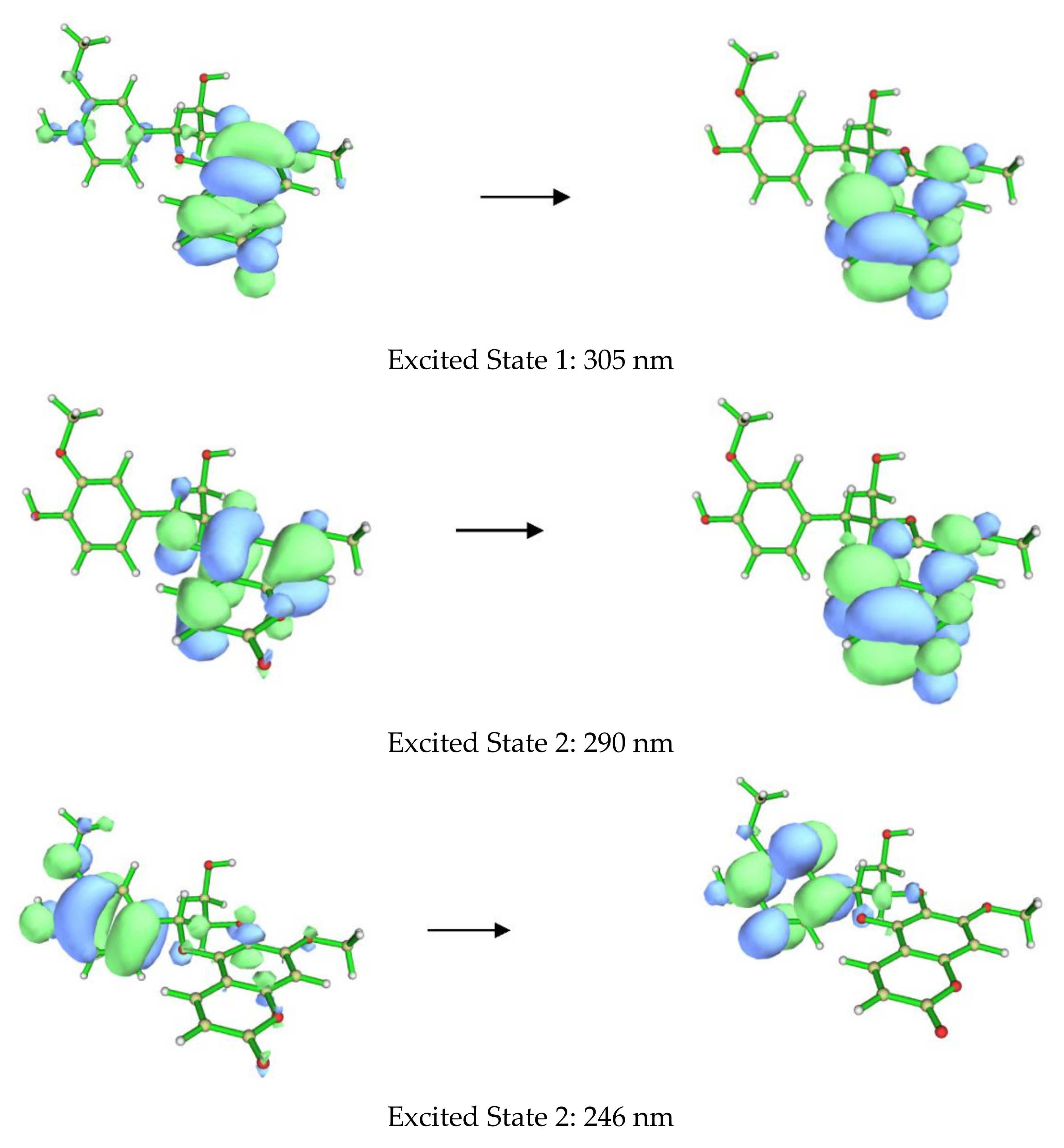
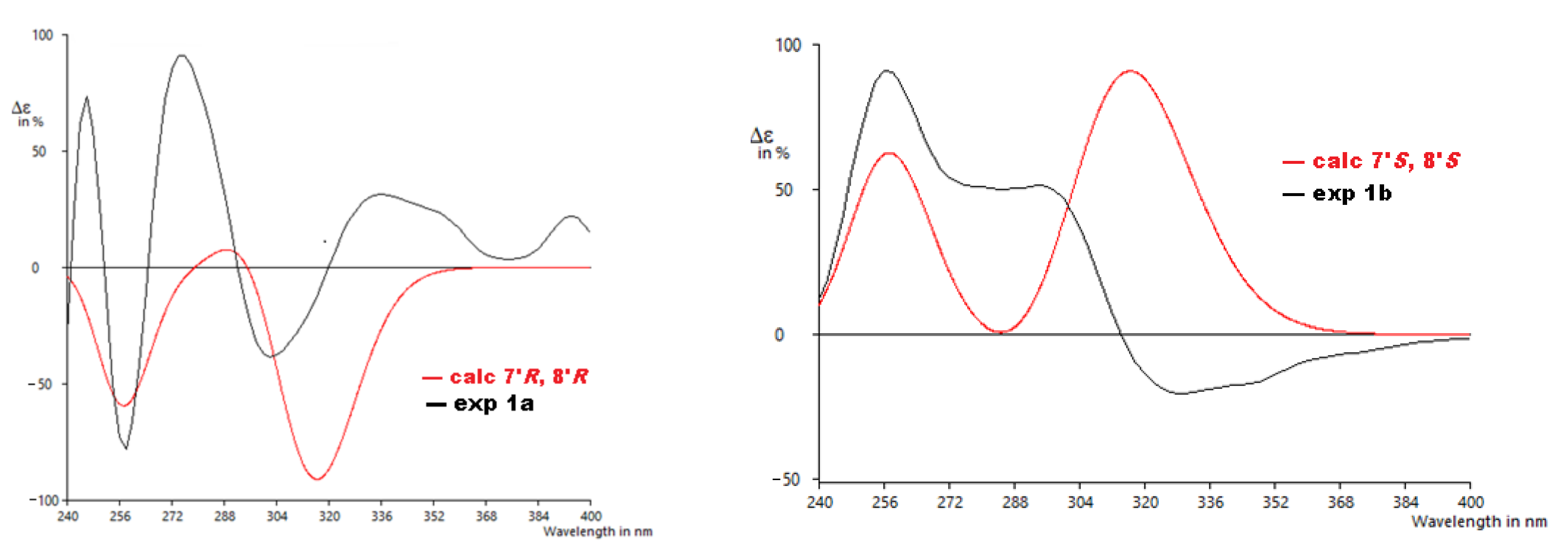
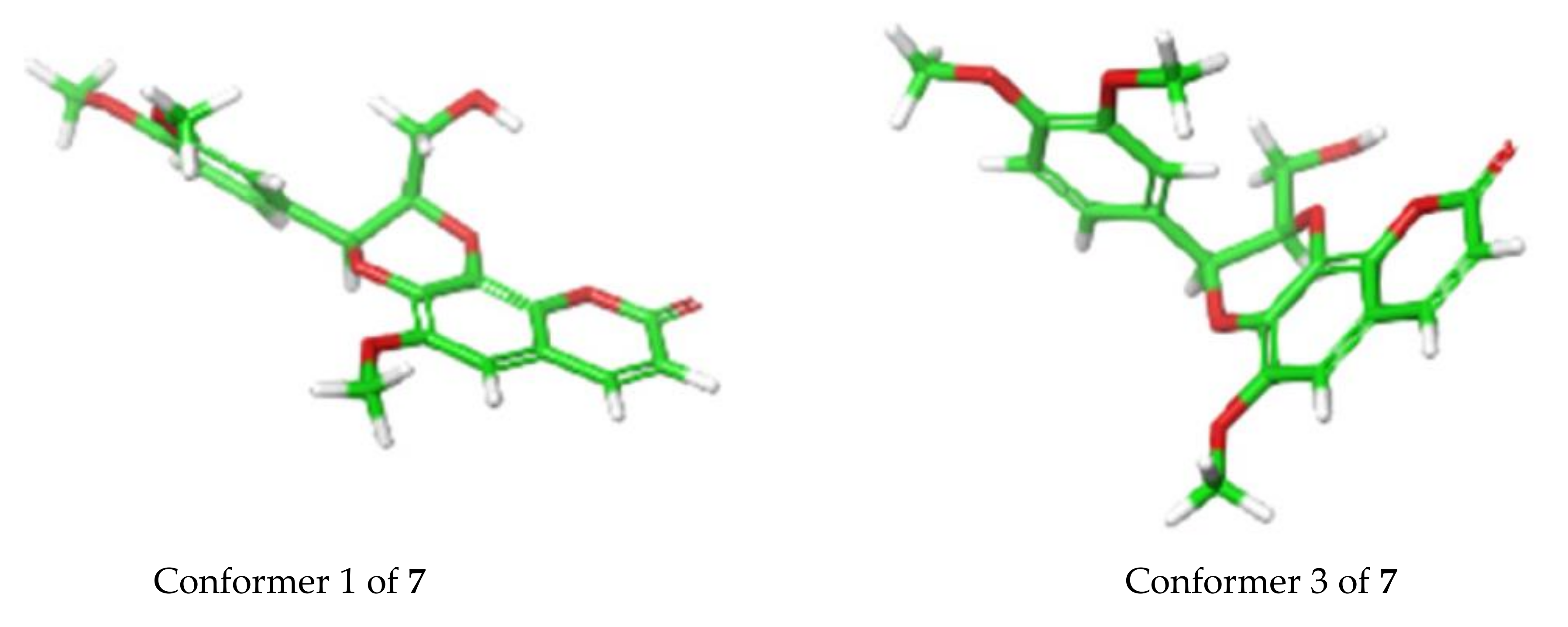
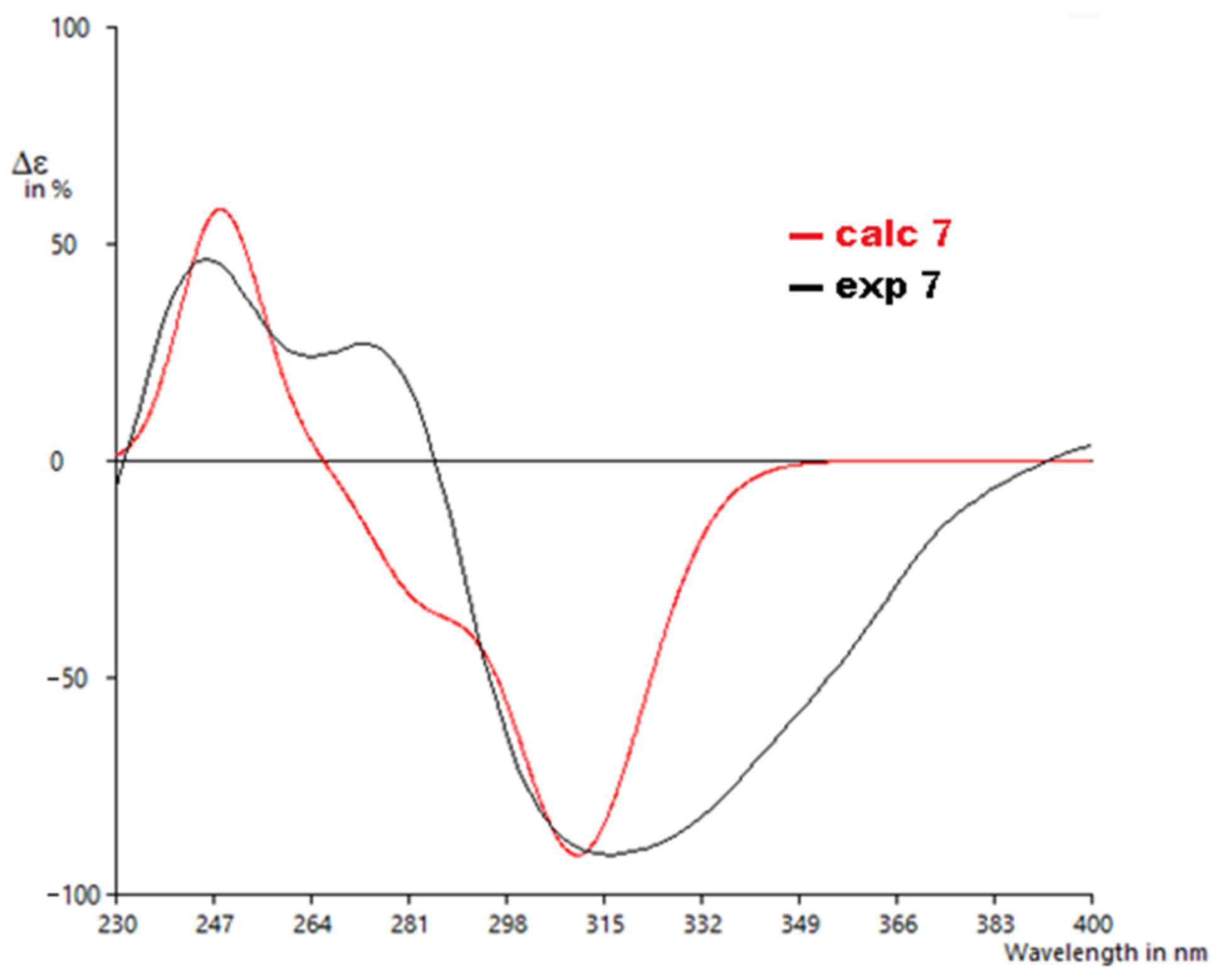
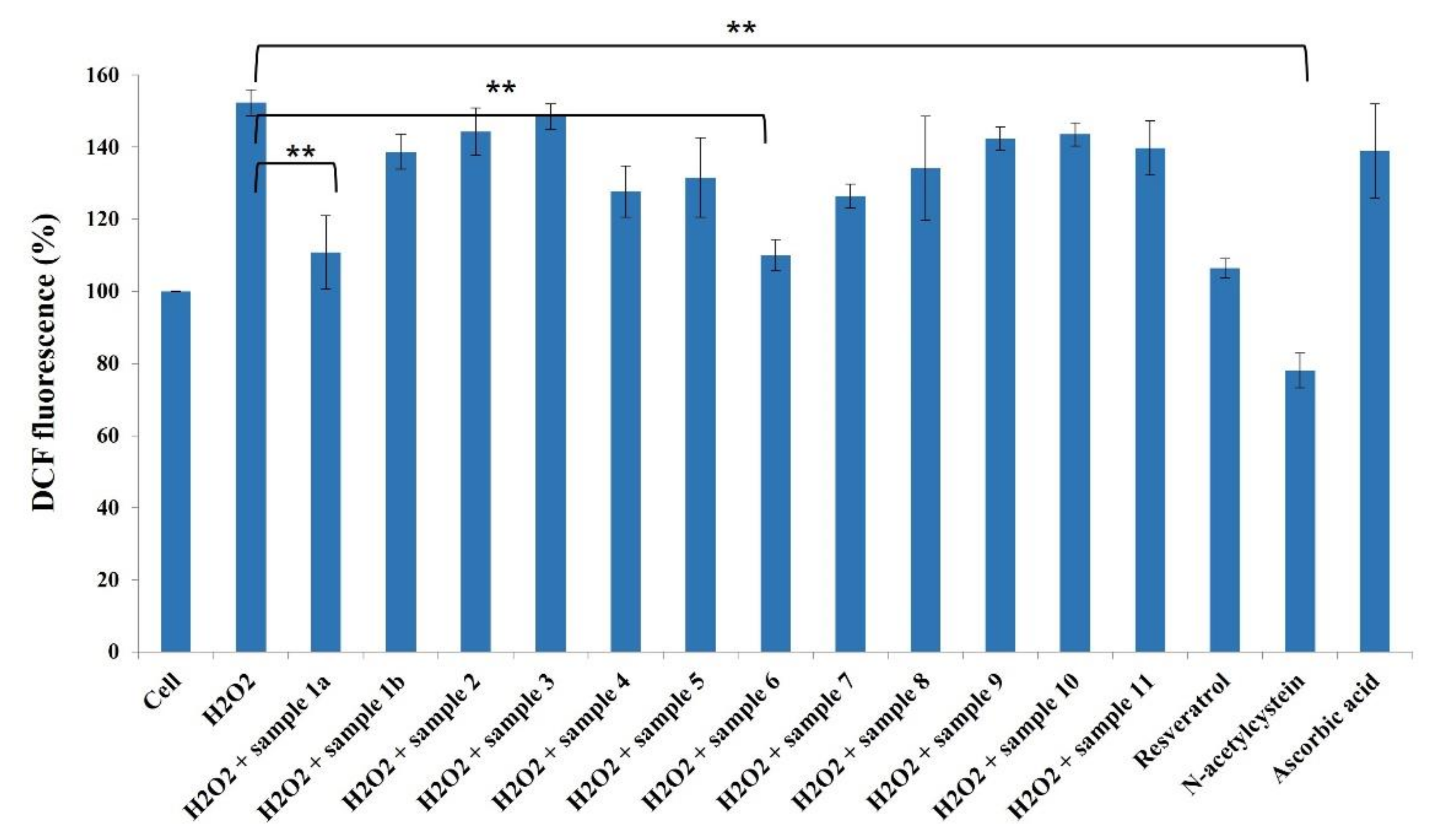
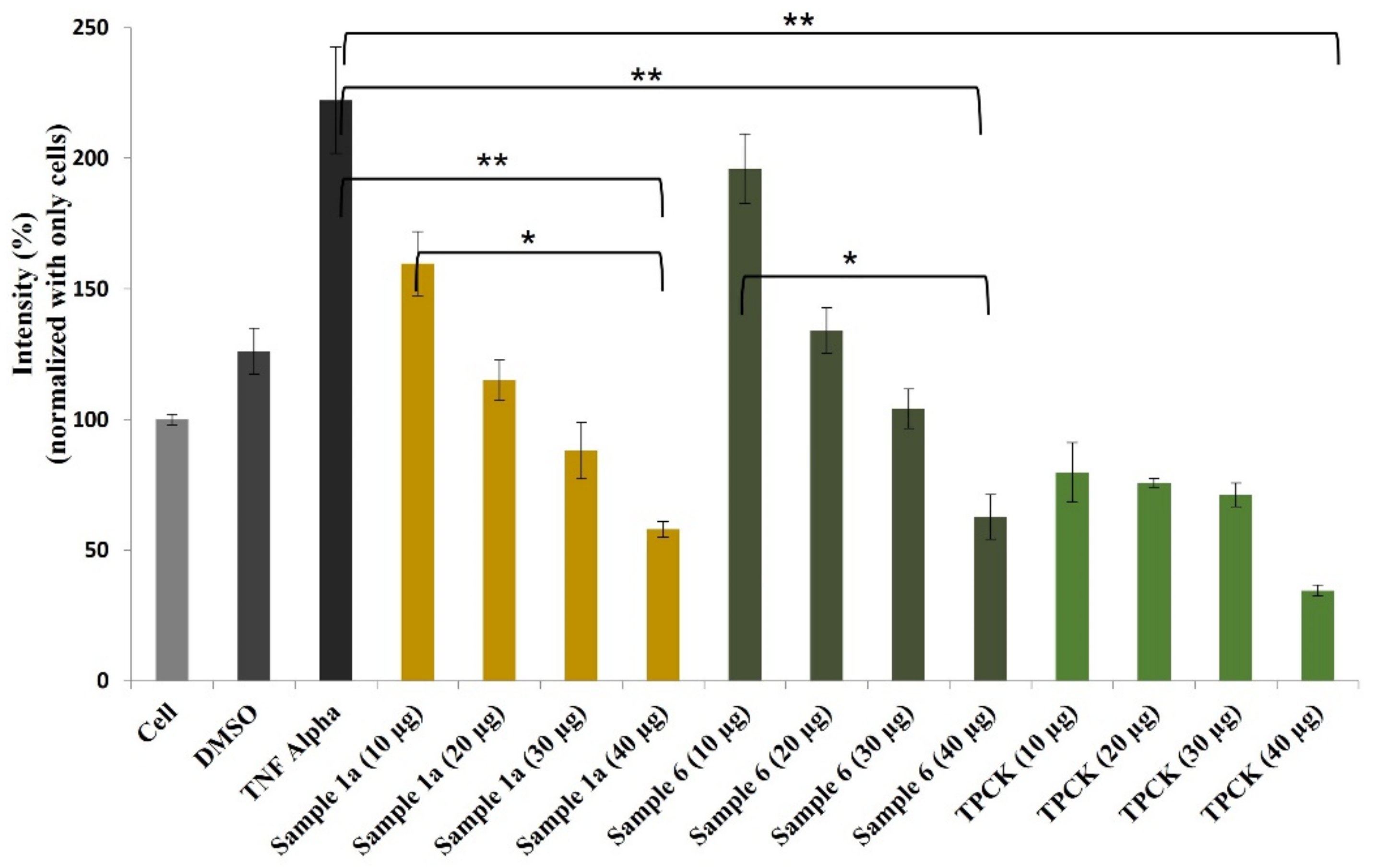
2. Results and Discussion
3. Materials and Methods
3.1. General Experimental Procedures
3.2. Plant Material
3.3. Extraction and Isolation
3.4. Computational Methods
3.5. Assessment of ROS Inhibition in HeLa Cells
3.6. TNF-α Activated NF-κB Assay
3.7. Statistical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Almeida, F.V.M.; Santos, J.C.; Silveira, F.A.O.; Fernandes, G.W. Distribution and frequency of galls induced by Anisodiplosis waltheriae Maia (Diptera: Cecidomyiidae) on the invasive plant Waltheria indica L. (Sterculiaceae). Neotrop. Entomol. 2006, 35, 435–439. [Google Scholar] [CrossRef] [PubMed]
- Jansen, O.; Angenot, L.; Tits, M.; Nicolas, J.P.; Mol, P.D.; Nikiéma, J.; Frédérich, M. Evaluation of 13 selected medicinal plants from Burkina Faso for their antiplasmodial properties. J. Ethnopharmacol. 2010, 130, 143–150. [Google Scholar] [CrossRef] [PubMed]
- Zongo, F.; Ribuot, C.; Boumendjel, A.; Guissou, I. Botany, traditional uses, phytochemistry and pharmacology of Waltheria indica L. (syn Waltheria americana): A review. J. Ethnopharmacol. 2013, 148, 14–26. [Google Scholar] [PubMed]
- Saunders, J.G. Sterculiaceae of Paraguay II. Waltheria. Bonplandia 2007, 16, 143–180. [Google Scholar] [CrossRef]
- Monteillier, A.; Cretton, S.; Ciclet, O.; Marcourt, L.; Ebrahimi, S.N.; Christen, P.; Cuendet, M. Cancer chemopreventive activity of compounds isolated from Waltheria indica. J. Ethnopharmacol. 2017, 203, 214–225. [Google Scholar] [CrossRef]
- Zongo, F.; Ribuot, C.; Boumendjel, A.; Guissou, I. Bioguidage search of active compounds from Waltheria indica L. (Malvaceae) used for asthma and inflammation treatment in Burkina Faso. Fundam. Clin. Pharmacol. 2014, 28, 323–330. [Google Scholar] [CrossRef]
- Laczko, R.; Chang, A.; Watanabe, L.; Petelo, M.; Csiszar, K. Anti-inflammatory activities of Waltheria indica extracts by modulating expression of IL-1B, TNF-α, TNFRII and NF-κB in human macrophages. Inflammopharmacology 2020, 28, 525–540. [Google Scholar] [CrossRef]
- Petrus, A.J.A. Polyphenolic components of Waltheria indica. Fitoterapia 1990, 61, 371. [Google Scholar]
- Rao, Y.K.; Fang, S.H.; Tzeng, Y.M. Inhibitory effects of the flavonoids isolated from Waltheria indica on the production of NO, TNF-α and IL-12 in activated macrophages. Biol. Pharm. Bull. 2005, 28, 912–915. [Google Scholar] [CrossRef]
- Maheswara, M.; Rao, Y.K.; Rao, V.M.; Rao, C.V. Antibacterial activity of acylated flavonol glycoside from Waltheria indica. Asian J. Chem. 2006, 18, 2761–2765. [Google Scholar]
- Ragasa, C.Y.; Cruz, C.; Chiong, I.; Tada, M.; Rideout, J. Antifungal flavonoids from Waltheria americana. Philipp. J. Sci. 1997, 126, 243–250. [Google Scholar]
- Hua, Y.M.; Zhang, X.W.; Zeng, K.W.; Zhang, Q.Y.; Tu, P.F. Chemical constituents from the aerial parts of Waltheria indica Linn. J. Chin. Pharm. Sci. 2019, 27, 468–475. [Google Scholar]
- Cretton, S.; Dorsaz, S.; Azzollini, A.; Favre-Godal, Q.; Marcourt, L.; Ebrahimi, S.N.; Voinesco, F.; Michellod, E.; Sanglard, D.; Gindro, K. Antifungal quinoline alkaloids from Waltheria indica. J. Nat. Prod. 2016, 79, 300–307. [Google Scholar] [CrossRef]
- Cretton, S.; Breant, L.; Pourrez, L.; Ambuehl, C.; Perozzo, R.; Marcourt, L.; Kaiser, M.; Cuendet, M.; Christen, P. Chemical constituents from Waltheria indica exert in vitro activity against Trypanosoma brucei and T. cruzi. Fitoterapia 2015, 105, 55–60. [Google Scholar] [CrossRef]
- Cretton, S.; Breant, L.; Pourrez, L.; Ambuehl, C.; Marcourt, L.; Ebrahimi, S.N.; Hamburger, M.; Perozzo, R.; Karimou, S.; Kaiser, M.; et al. Antitrypanosomal quinoline alkaloids from the roots of Waltheria indica. J. Nat. Prod. 2014, 77, 2304–2311. [Google Scholar] [CrossRef]
- Cheng, L.; Zhang, X.Y.; Zhang, M.; Zhang, P.; Song, Z.H.; Ma, Z.J.; Cheng, Y.Y.; Qu, H.B. Characterization of chemopreventive agents from the dichloromethane extract of Eurycorymbus cavaleriei by liquid chromatography–ion trap mass spectrometry. J. Chromatogr. A 2009, 1216, 4859–4867. [Google Scholar] [CrossRef]
- Begum, S.; Saxena, B.; Goyal, M.; Ranjan, R.; Joshi, V.B.; Rao, C.V.; Krishnamurthy, S.; Sahai, M. Study of anti-inflammatory, analgesic and antipyretic activities of seeds of Hyoscyamus niger and isolation of a new coumarinolignan. Fitoterapia 2010, 81, 178–184. [Google Scholar] [CrossRef]
- Chang, H.S.; Lin, C.H.; Hsiao, P.Y.; Peng, H.T.; Lee, S.J.; Cheng, M.J.; Chen, I.S. Bioactive composition of Reevesia formosana root and stem with cytotoxic activity potential. RSC Adv. 2017, 7, 27040–27047. [Google Scholar] [CrossRef]
- Ma, C.; Zhang, H.J.; Tan, G.T.; Hung, N.V.; Fong, H.H.S. Antimalarial compounds from Grewia bilamellata. J. Nat. Prod. 2006, 69, 346–350. [Google Scholar] [CrossRef]
- Sajeli, B.; Sahai, M.; Suessmuth, R.; Asai, T.; Hara, N.; Fujimoto, Y. Hyosgerin, a new optically active coumarinolignan, from the seeds of Hyoscyamus niger. Chem. Pharm. Bull. 2006, 54, 538–541. [Google Scholar] [CrossRef][Green Version]
- Mándi, A.; Kurtán, T. Applications of OR/ECD/VCD to the structure elucidation of natural products. Nat. Prod. Rep. 2019, 36, 889–918. [Google Scholar] [CrossRef]
- Iqbal, K.; Anis, I.; Muhktar, N.; Malik, A.; Fatima, N.; Chaudhary, M.I. Phosphodiesterase inhibitory coumarinolignoids from Duranta repens. Heterocycles 2003, 60, 151–157. [Google Scholar]
- Arnoldi, A.; Arnone, A.; Merlini, L. Synthesis of the natural coumarinolignoids propacin and cleomiscosin A and B. An empirical spectroscopic method to distinguish regioisomers of natural benzodioxane lignoids. Heterocycles 1984, 22, 1537–1544. [Google Scholar]
- Wu, H.C.; Cheng, M.J.; Yen, C.H.; Chen, Y.; Chang, H.S. Chemical Constituents with GNMT-Promoter-Enhancing and NRF2-Reduction Activities from Taiwan Agarwood Excoecaria formosana. Molecules 2020, 25, 1746. [Google Scholar] [CrossRef]
- Begum, S.A.; Sahai, M.; Ray, A.B. Non-conventional lignans: Coumarinolignans, flavonolignans, and stilbenolignans. Prog. Chem. Org. Nat. Prod. 2010, 93, 1–70. [Google Scholar]
- Yin, H.L.; Li, J.H.; Li, J.; Li, B.; Chen, L.; Tian, Y.; Liu, S.J.; Zhang, T.; Dong, J.X. Four new coumarinolignoids from seeds of Solanum indicum. Fitoterapia 2013, 84, 360–365. [Google Scholar] [CrossRef]
- Arnoldi, A.; Camarda, L.; Merlini, L. Synthesis and sweet taste of some 2-phenylbenzodioxanes. J. Agric. Food Chem. 1986, 34, 339–344. [Google Scholar] [CrossRef]
- Bruhn, T.; Schaumlöffel, A.; Hemberger, Y. SpecDis, Version 1.63; University of Wuerzburg: Wurzburg, Germany, 2015.
- Patnam, R.; Kadali, S.S.; Koumaglo, K.H.; Roy, R. A chlorinated coumarinolignan from the African medicinal plant, Mondia whitei. Phytochemistry 2005, 66, 683–686. [Google Scholar] [CrossRef]
- Bai, B.; Gu, X.W.; Chen, Y.; Guan, F.Q.; Feng, X. Virginicin, a new naphthalene from Kosteletzkya virginica (Malvaceae). J. Braz. Chem. Soc. 2015, 26, 723–728. [Google Scholar]
- Bhandari, P.; Pant, P.; Rastogi, R.P. Aquillochin, a coumarinolignan from Aquilaria agallocha. Phytochemistry 1982, 21, 2147–2149. [Google Scholar] [CrossRef]
- Baker, R.G.; Hayden, M.S.; Ghosh, S. NF-kappaB, inflammation, and metabolic disease. Cell Metab. 2011, 13, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Karin, M.; Greten, F.R. FRNF-B: Linking inflammation and immunity to cancer development and progression. Nat. Rev. Immunol. 2005, 5, 749–759. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, T. The nuclear factor NF-κB pathway in Inflammation. Cold Spring Harb. Perspect. Biol. 2009, 1, a001651. [Google Scholar] [CrossRef] [PubMed]
- Marcu, K.B.; Otero, M.; Olivotto, E.; Borzi, R.M.; Goldring, M.B. NF-kappaB signaling: Multiple angles to target OA. Curr. Drug Targets 2010, 11, 599–613. [Google Scholar] [CrossRef]
- Higuchi, Y.; Otsu, K.; Nishida, K.; Hirotani, S.; Nakayama, H.; Yamaguchi, O.; Matsumura, Y.; Ueno, H.; Tada, M.; Hori, M. Involvement of reactive oxygen species-mediated NF-κB activation in TNF-α-induced cardiomyocyte hypertrophy. J. Mol. Cell. Cardiol. 2002, 34, 233–240. [Google Scholar] [CrossRef]
- Li, Z.Q.; Scheraga, H.A. Monte carlo-minimization approach to the multiple-minima problem in protein folding. Proc. Natl. Acad. Sci. USA 1987, 84, 6611–6615. [Google Scholar] [CrossRef]
- Kaminski, G.A.; Friesner, R.A.; Tirado-Rives, J.; Jorgensen, W.L. Evaluation and reparametrization of the OPLS-AA force field for proteins via comparison with accurate quantum chemical calculations on peptides. J. Phys. Chem. B 2001, 105, 6474–6487. [Google Scholar] [CrossRef]
- Schrödinger Release 2020-4; MacroModel. Schrödinger, LLC: New York, NY, USA, 2020.
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision, B.01; Gaussian Inc.: Wallingford, CT, USA, 2009.
- Yanai, T.; Tew, D.P.; Handy, N.C. A new hybrid exchange-correlation functional using the Coulomb-attenuating method (CAM-B3LYP). Chem. Phys. Lett. 2004, 393, 51–57. [Google Scholar] [CrossRef]
- Clark, T.; Chandrasekhar, J.; Spitznagel, G.W.; Schleyer, P.V.R. Efficient diffuse function-augmented basis sets for anion calculations. III. The 3-21+G basis set for first-row elements, Li–F. J. Comput. Chem. 1983, 4, 294–301. [Google Scholar] [CrossRef]
- Ditchfield, R.; Hehre, W.J.; Pople, J.A. Self-consistent molecular-orbital methods. IX. An extended gaussian-type basis for molecular-orbital studies of organic molecules. J. Chem. Phys. 1971, 54, 724–728. [Google Scholar] [CrossRef]
- Hehre, W.J.; Ditchfield, R.; Pople, J.A. Self-consistent molecular orbital methods. XII. Further extensions of gaussian—Type basis sets for use in molecular orbital studies of organic molecules. J. Chem. Phys. 1972, 56, 2257–2261. [Google Scholar] [CrossRef]
- Hariharan, P.C.; Pople, J.A. The influence of polarization functions on molecular orbital hydrogenation energies. Theor. Chim. Acta 1973, 28, 213–222. [Google Scholar] [CrossRef]
- Tomasi, J.; Mennucci, B.; Cammi, R. Quantum mechanical continuum solvation models. Chem. Rev. 2005, 105, 2999–3094. [Google Scholar] [CrossRef]
- Runge, E.; Gross, E.K.U. Density-functional theory for time-dependent systems. Phys. Rev. Lett. 1984, 52, 997–1000. [Google Scholar] [CrossRef]
- Gross, E.K.U.; Dobson, J.F.; Petersilka, M. Density functional theory of time-dependent phenomena. Top. Curr. Chem. 1996, 181, 81–159. [Google Scholar]
- Grauso, L.; Teta, R.; Esposito, G.; Menna, M.; Mangoni, A. Computational prediction of chiroptical properties in structure elucidation of natural products. Nat. Prod. Rep. 2019, 36, 1005–1030. [Google Scholar] [CrossRef]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef]
- Tian, L.; Chen, F. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar]
- Youn, U.J.; Park, E.J.; Kondratyuk, T.P.; Sang-Ngern, M.; Wall, M.M.; Wei, Y.Z.; Pezzuto, J.M.; Chang, L.C. Anti-inflammatory and quinone reductase inducing compounds from fermented noni (Morinda citrifolia) juice exudates. J. Nat. Prod. 2016, 79, 1508–1513. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Position | 1a a | 1b a | 2 b | 3 a | 4 b | 5 a | 7 a |
|---|---|---|---|---|---|---|---|
| 3 | 6.17, d (9.6) | 6.18, d (9.6) | 6.16, d (9.6) | 6.18, d (9.6) | 6.16, d (9.6) | 6.28, d (9.5) | 6.29, d (9.5) |
| 4 | 7.90, d (9.6) | 7.90, d (9.6) | 7.96, d (9.6) | 7.92, d (9.6) | 7.98, d (9.6) | 7.58, d (9.5) | 7.59, d (9.5) |
| 5 | 6.53, (s) | 6.53, (s) | |||||
| 8 | 6.50, (s) | 6.51, (s) | 6.62, (s) | 6.52, (s) | 6.63, (s) | ||
| 2′ | 6.89, d (1.5) | 6.90, (brs) | 7.06, d (1.4) | 6.65, (s) | 6.74, (s) | 6.58, (s) | 6.86, d (2.0) |
| 5′ | 6.95, d (8.0) | 6.96, d (8.2) | 7.01, d (8.2) | 6.81, d (8.3) | |||
| 6′ | 6.94, dd (1.5, 8.0) | 6.95, d (8.2) | 7.03, dd (1.4, 8.2) | 6.65, (s) | 6.74, (s) | 6.58, (s) | 6.92, dd (2.0, 8.3) |
| 7′ | 5.03, d (8.0) | 5.04, d (8.0) | 5.07, d (8.0) | 5.05, d (8.0) | 4.70, d (8.0) | 4.91, d (7.4) | 5.34, d (2.9) |
| 8′ | 3.98, ddd (2.9, 3.5, 8.0) | 3.99, (m) | 4.08, ddd (2.4, 4.0, 8.0) | 4.00, (m) | 4.19, dq (8.0, 6.4) | 4.36, (m) | 4.63, ddd (2.9, 4.0, 8.3) |
| 9′ | 3.55, dd (3.5, 12.8) 3.88, dd (2.9, 12.8) | 3.56, dd (3.4, 12.5) 3.89, (m) | 3.49, dd (4.0, 12.5) 3.78, dd (2.4, 12.5) | 3.58, dd (3.0, 12.5) 3.97, (m) | 1.22, d (6.4) | 4.07, dd (5.4, 13.3) 4.35, (m) | 3.57, dd (4.0, 12.3) 3.79, dd (8.3, 12.3) |
| Ac-9′ | 2.05 (s) | ||||||
| OCH3-5 | 3.90 (s) | 3.87 (s) | |||||
| OCH3-7 | 3.92 (s) | 3.93 (s) | 3.93 (s) | 3.93 (s) | 3.93 (s) | ||
| OCH3-3′ | 3.90 (s) | 3.91 (s) | 3.85 (s) | 3.87 (s) | 3.87 (s) | 3.87 (s) | 3.82 (s) |
| OCH3-4′ | 3.86 (s) | 3.86 (s) | 3.83 (s) | ||||
| OCH3-5′ | 3.87 (s) | 3.87 (s) | 3.87 (s) |
| Position | 1a a | 1b a | 2 b | 3 a | 4 b | 5 a | 7 a |
|---|---|---|---|---|---|---|---|
| 2 | 161.7, C | 161.7, C | 163.8, C | 161.6, C | 163.9, C | 160.8, C | 160.9, C |
| 3 | 112.3, CH | 112.3, CH | 112.4, CH | 112.4, CH | 112.3, CH | 114.6, CH | 114.5, CH |
| 4 | 138.3 CH | 138.3 CH | 140.0, CH | 138.1, CH | 140.2, CH | 143.8, CH | 144.0, CH |
| 5 | 140.0, C | 140.0, C | 141.4, C | 139.8, C | 141.5, C | 100.9, CH | 101.0, CH |
| 6 | 129.6, C | 129.6, C | 131.4, C | 129.6, C | 131.2, C | 146.0, C | 146.1, C |
| 7 | 152.4, C | 152.4, C | 154.4, C | 152.4, C | 154.3, C | 136.8, C | 137.0, C |
| 8 | 93.1, CH | 93.1, CH | 94.0, CH | 93.2, CH | 93.9, CH | 132.7, C | 130.9, C |
| 9 | 149.9, C | 149.9, C | 150.9, C | 149.9, C | 150.8, C | 139.2, C | 139.3, C |
| 10 | 103.7, C | 103.7, C | 104.7, C | 103.7, C | 104.7, C | 112.0, C | 112.0, C |
| 1′ | 127.4, C | 127.4, C | 130.1, C | 131.1, C | 128.4, C | 125.8, C | 127.6, C |
| 2′ | 109.9, CH | 109.8, CH | 112.5, CH | 104.6, CH | 106.3, CH | 104.6, CH | 110.2, CH |
| 3′ | 147.1, C | 147.1, C | 150.9, C | 153.9, C | 149.7, C | 147.6, C | 149.4, C |
| 4′ | 146.8, C | 146.8, C | 151.4, C | 138.9, C | 137.8, C | 136.1, C | 149.7, C |
| 5′ | 115.0, CH | 115.0, CH | 113.1, CH | 153.9, C | 149.7, C | 147.6, C | 111.6, CH |
| 6′ | 121.0, CH | 121.0, CH | 121.8, CH | 104.6, CH | 106.3, CH | 104.6, CH | 119.4, CH |
| 7′ | 77.3, CH | 77.3, CH | 78.6, CH | 77.3, CH | 83.4, CH | 76.7, CH | 76.4, CH |
| 8′ | 78.5, CH | 78.5, CH | 79.8, CH | 78.3, CH | 75.4, CH | 76.2, CH | 77.6, CH |
| 9′ | 61.6, CH2 | 61.6, CH2 | 62.0, CH2 | 61.5, CH2 | 17.5, CH3 | 63.0, CH2 | 60.0, CH2 |
| Ac-9′ | 170.6, C | ||||||
| Ac-9′ | 21.0, CH3 | ||||||
| OCH3-5 | 56.7, CH3 | 56.6, CH3 | |||||
| OCH3-7 | 56.7, CH3 | 56.5, CH3 | 57.2, CH3 | 56.7, CH3 | 57.1, CH3 | ||
| OCH3-3′ | 56.3, CH3 | 56.3, CH3 | 56.7, CH3 | 56.5, CH3 | 57.1, CH3 | 56.7, CH3 | 56.3, CH3 |
| OCH3-4′ | 56.6, CH3 | 61.1, CH3 | 56.1, CH3 | ||||
| OCH3-5′ | 56.5, CH3 | 57.1, CH3 | 56.7, CH3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, F.; Mallick, S.; O’Donnell, T.J.; Rouzimaimaiti, R.; Luo, Y.; Sun, R.; Wall, M.; Wongwiwatthananukit, S.; Date, A.; Silva, D.K.; et al. Coumarinolignans with Reactive Oxygen Species (ROS) and NF-κB Inhibitory Activities from the Roots of Waltheria indica. Molecules 2022, 27, 3270. https://doi.org/10.3390/molecules27103270
Liu F, Mallick S, O’Donnell TJ, Rouzimaimaiti R, Luo Y, Sun R, Wall M, Wongwiwatthananukit S, Date A, Silva DK, et al. Coumarinolignans with Reactive Oxygen Species (ROS) and NF-κB Inhibitory Activities from the Roots of Waltheria indica. Molecules. 2022; 27(10):3270. https://doi.org/10.3390/molecules27103270
Chicago/Turabian StyleLiu, Feifei, Sudipta Mallick, Timothy J. O’Donnell, Ruxianguli Rouzimaimaiti, Yuheng Luo, Rui Sun, Marisa Wall, Supakit Wongwiwatthananukit, Abhijit Date, Dane Kaohelani Silva, and et al. 2022. "Coumarinolignans with Reactive Oxygen Species (ROS) and NF-κB Inhibitory Activities from the Roots of Waltheria indica" Molecules 27, no. 10: 3270. https://doi.org/10.3390/molecules27103270
APA StyleLiu, F., Mallick, S., O’Donnell, T. J., Rouzimaimaiti, R., Luo, Y., Sun, R., Wall, M., Wongwiwatthananukit, S., Date, A., Silva, D. K., Williams, P. G., & Chang, L. C. (2022). Coumarinolignans with Reactive Oxygen Species (ROS) and NF-κB Inhibitory Activities from the Roots of Waltheria indica. Molecules, 27(10), 3270. https://doi.org/10.3390/molecules27103270