
An Easy Access to Furan-Fused Polyheterocyclic Systems

 ,
,  , ,
, ,
Abstract
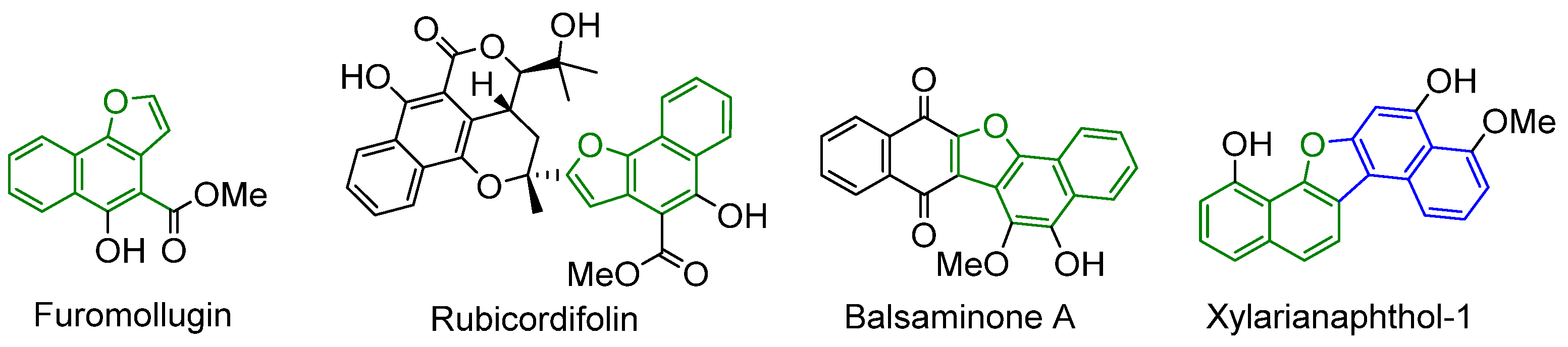
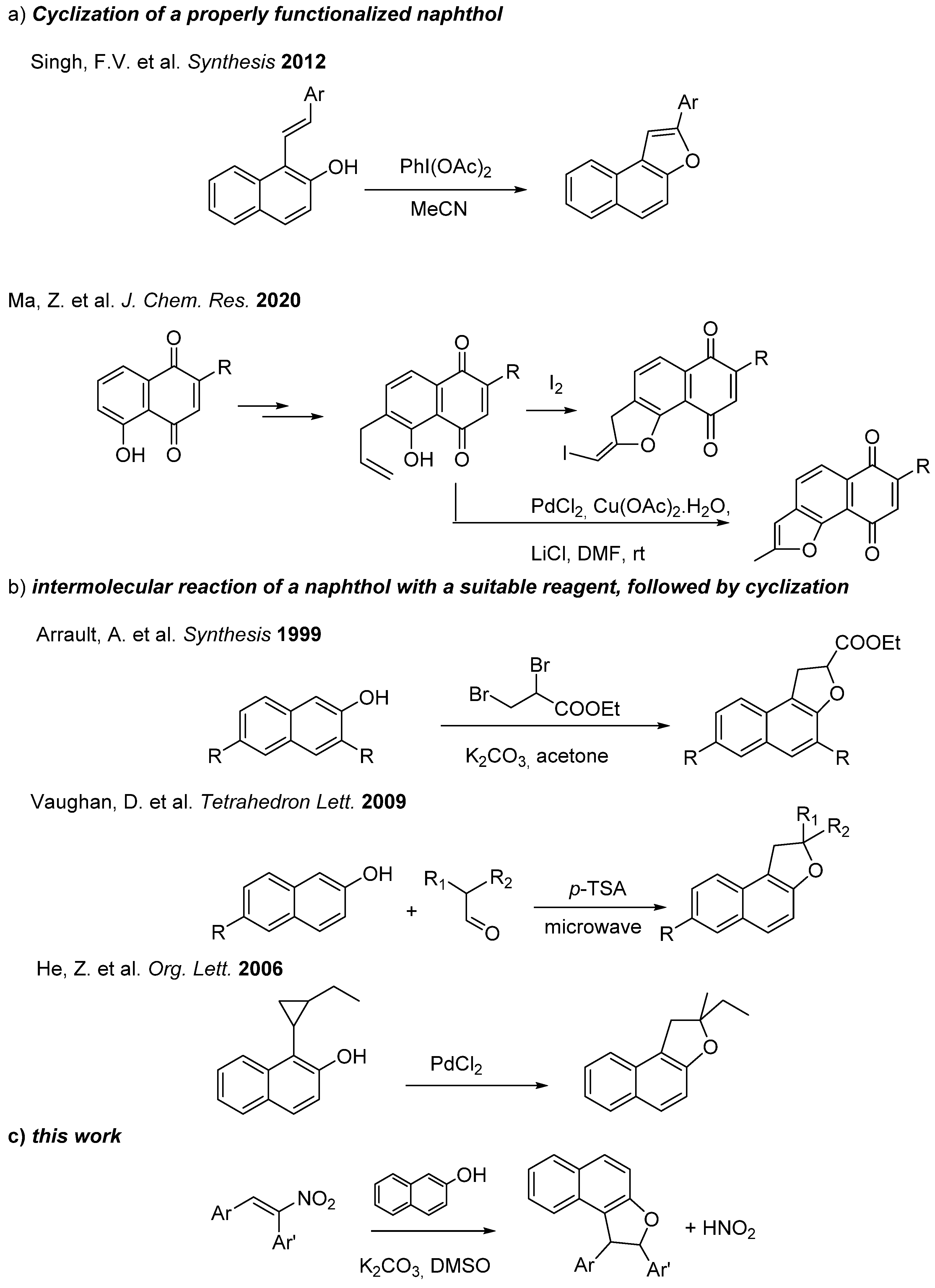
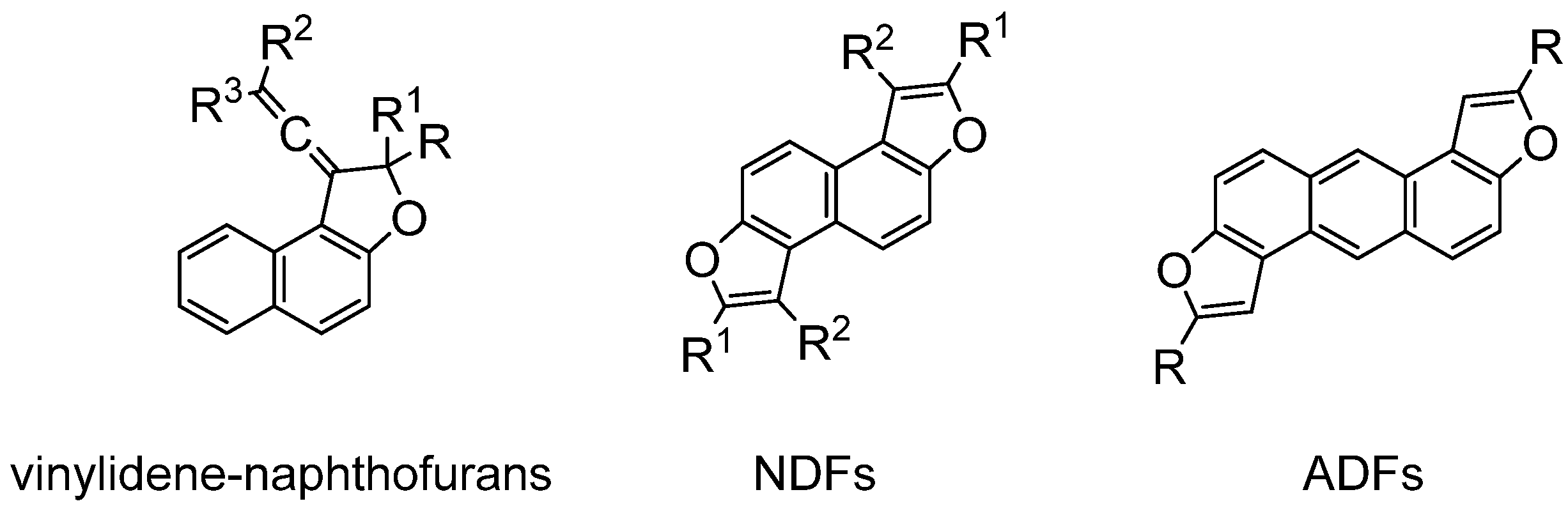
:1. Introduction
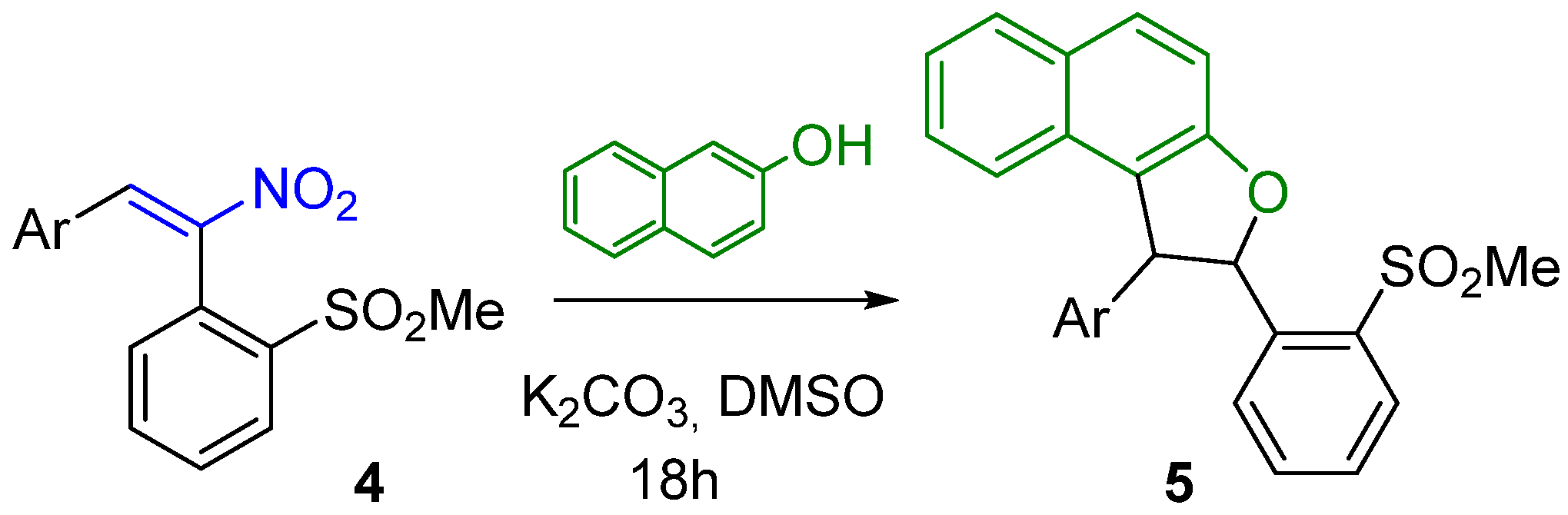
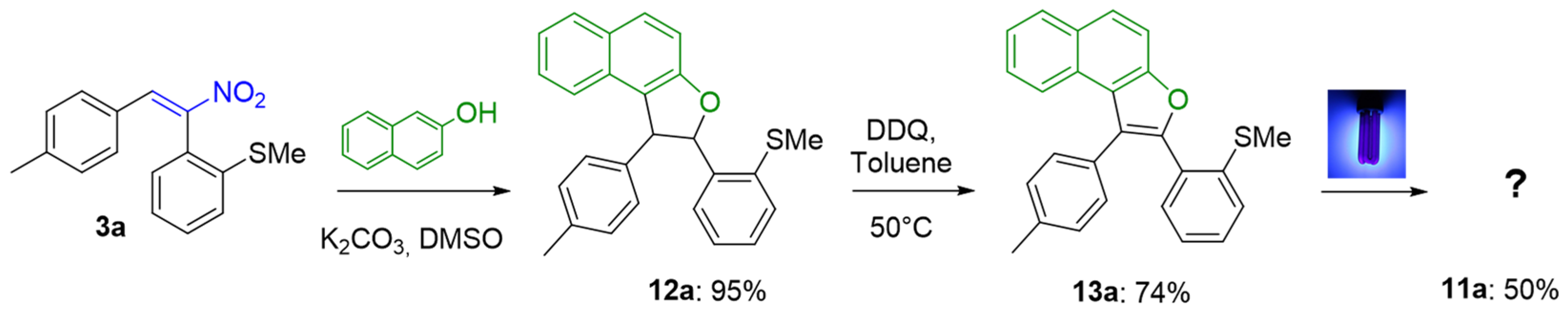
2. Results
3. Materials and Methods
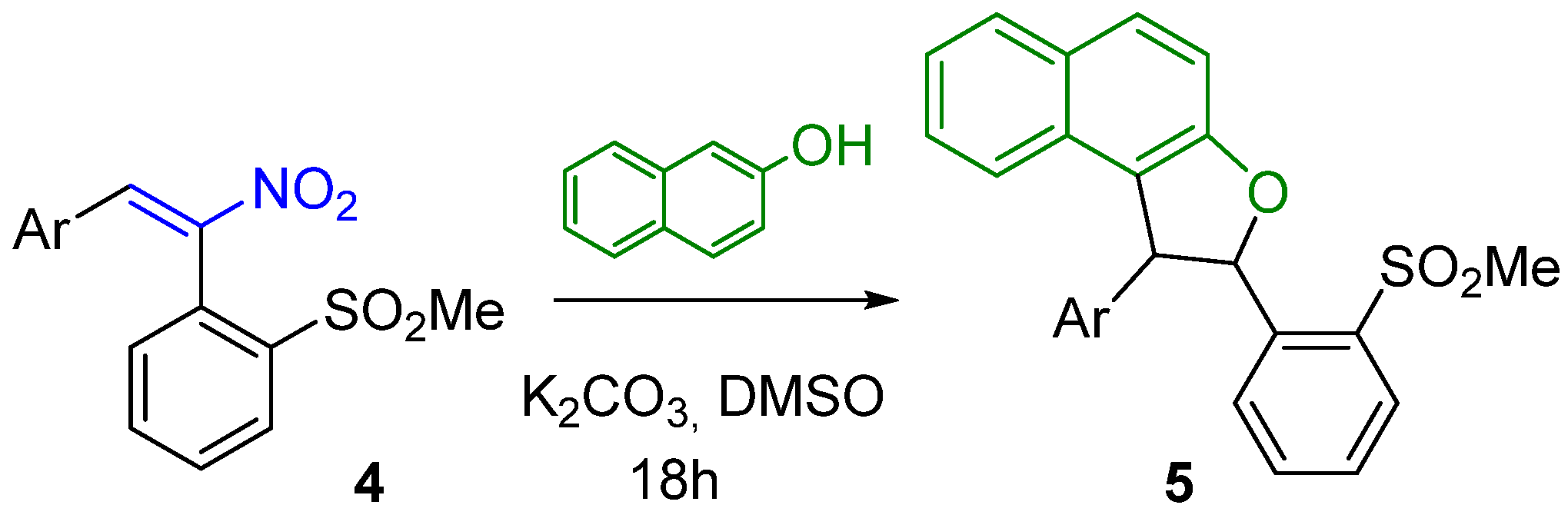
3.1. General Procedure for the Reactions of Substrates 4 with β-Naphthol
2-(2-(Methylsulfonyl)phenyl)-1-(p-tolyl)-1,2-dihydronaphtho[2,1-b]furan (5a)
2-(2-(Methylsulfonyl)phenyl)-1-(o-tolyl)-1,2-dihydronaphtho[2,1-b]furan (5b)
1-(4-Methoxyphenyl)-2-(2-(methylsulfonyl)phenyl)-1,2-dihydronaphtho[2,1-b]furan (5c)
1-(4-Chlorophenyl)-2-(2-(methylsulfonyl)phenyl)-1,2-dihydronaphtho[2,1-b]furan (5d)
2-(2-(Methylsulfonyl)phenyl)-1-(naphthalen-1-yl)-1,2-dihydronaphtho[2,1-b]furan (5e)
2-(2-(Methylsulfonyl)phenyl)-1-(thiophen-2-yl)-1,2-dihydronaphtho[2,1-b]furan (5f)
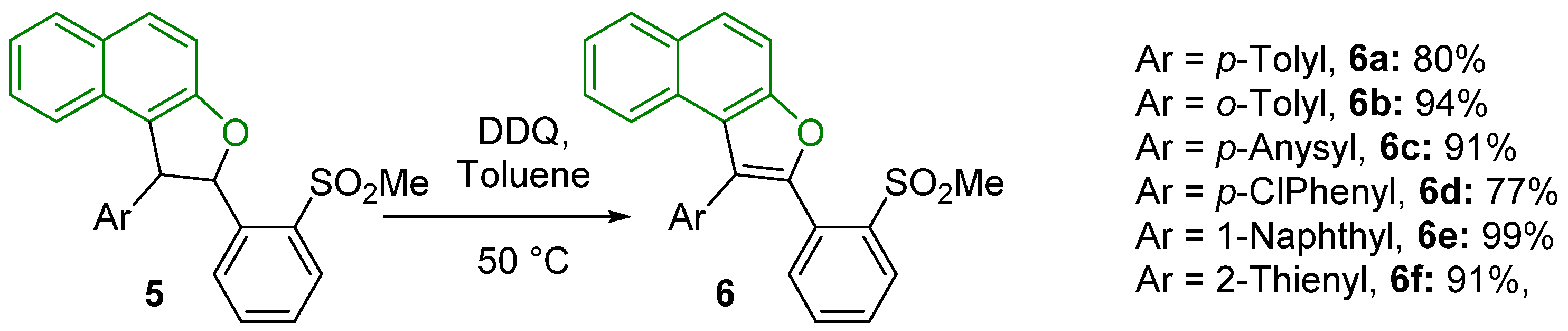
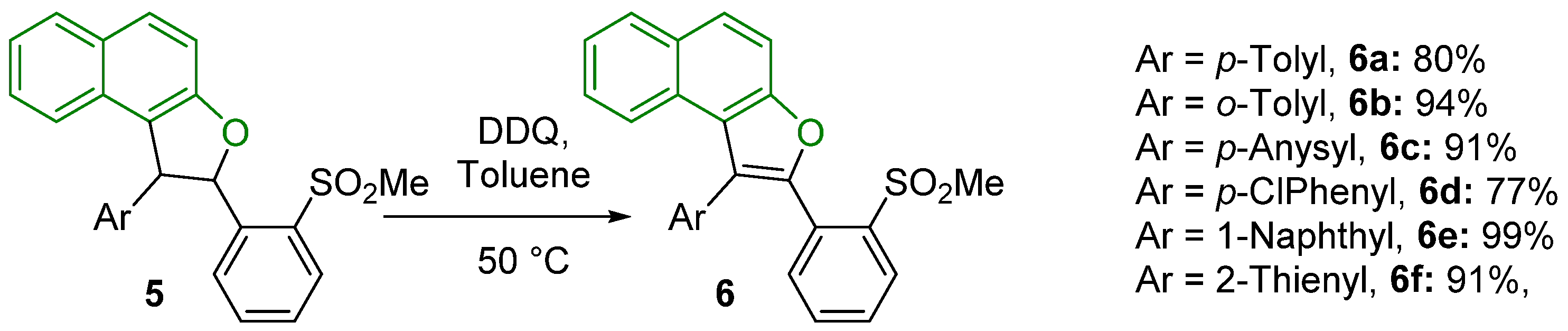
3.2. General Procedure for the Oxidative Aromatization Reaction of Dihydronaphthofurans 5a–f to Naphthofurans 6a–f
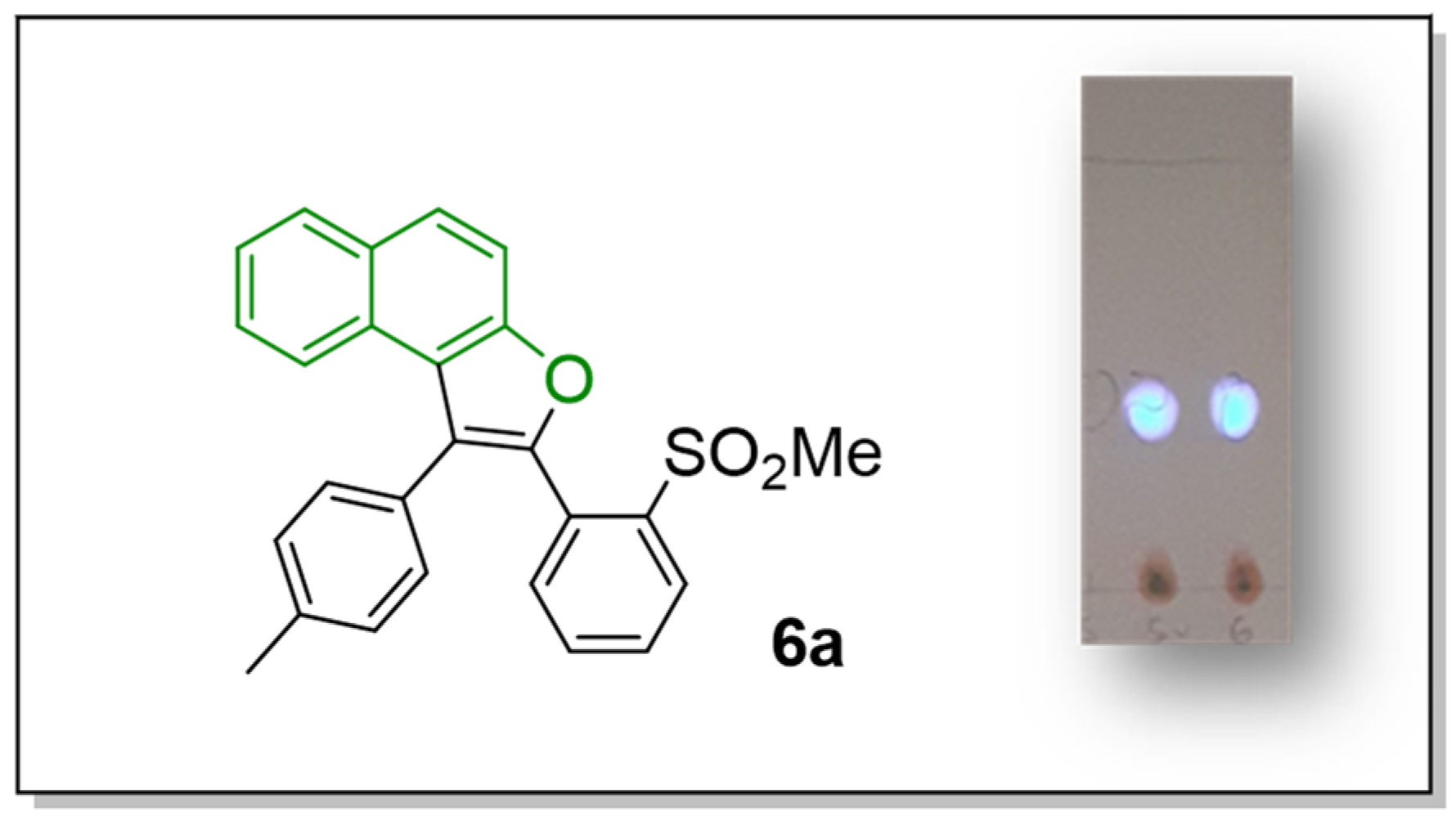
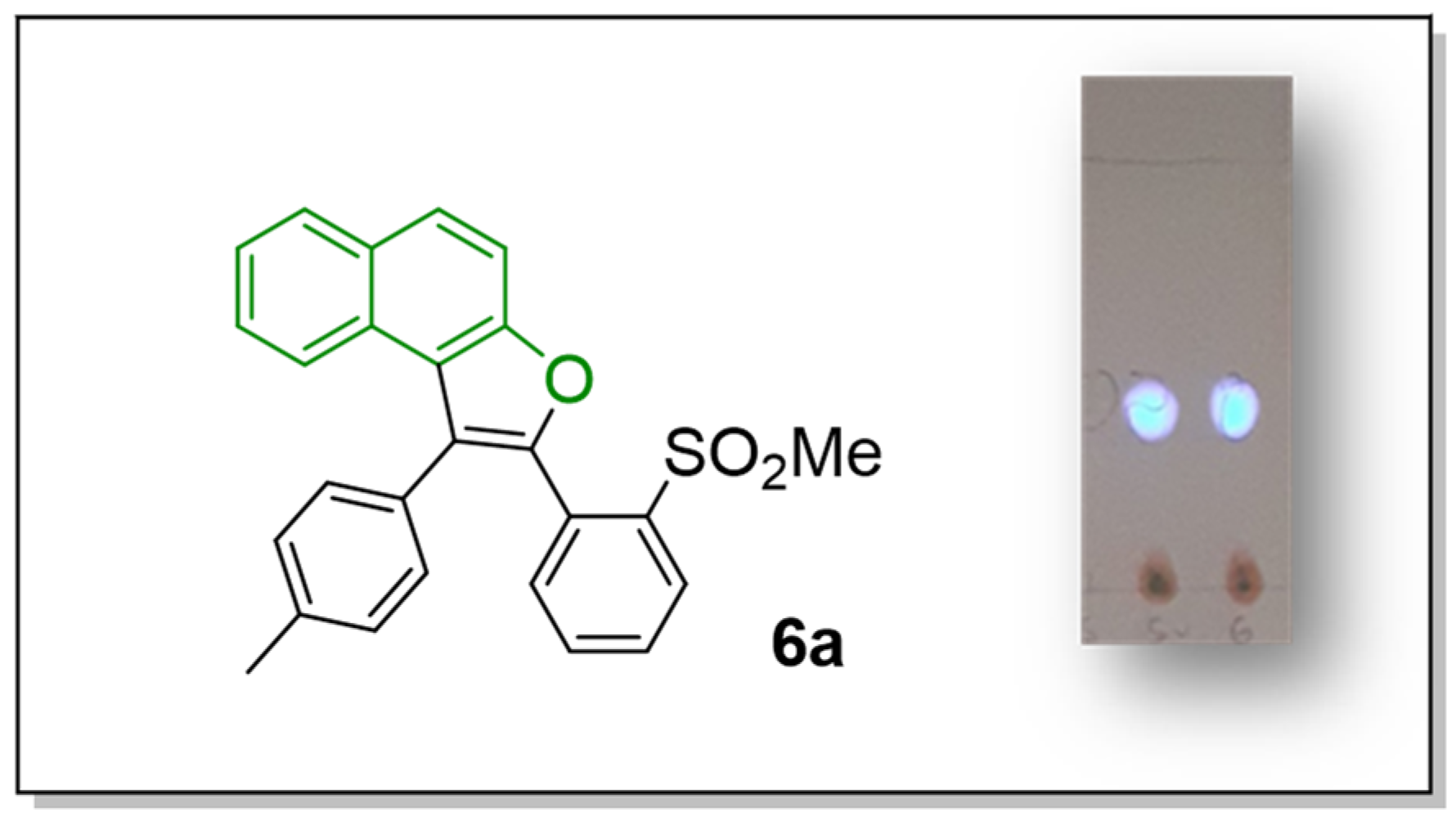
2-(2-(Methylsulfonyl)phenyl)-1-(p-tolyl)naphtho[2,1-b]furan (6a)
2-(2-(Methylsulfonyl)phenyl)-1-(o-tolyl)naphtho[2,1-b]furan (6b)
1-(4-Methoxyphenyl)-2-(2-(methylsulfonyl)phenyl)naphtho[2,1-b]furan (6c)
1-(4-Chlorophenyl)-2-(2-(methylsulfonyl)phenyl)naphtho[2,1-b]furan (6d)
2-(2-(Methylsulfonyl)phenyl)-1-(naphthalen-1-yl)naphtho[2,1-b]furan (6e)
2-(2-(Methylsulfonyl)phenyl)-1-(thiophen-2-yl)naphtho[2,1-b]furan (6f)
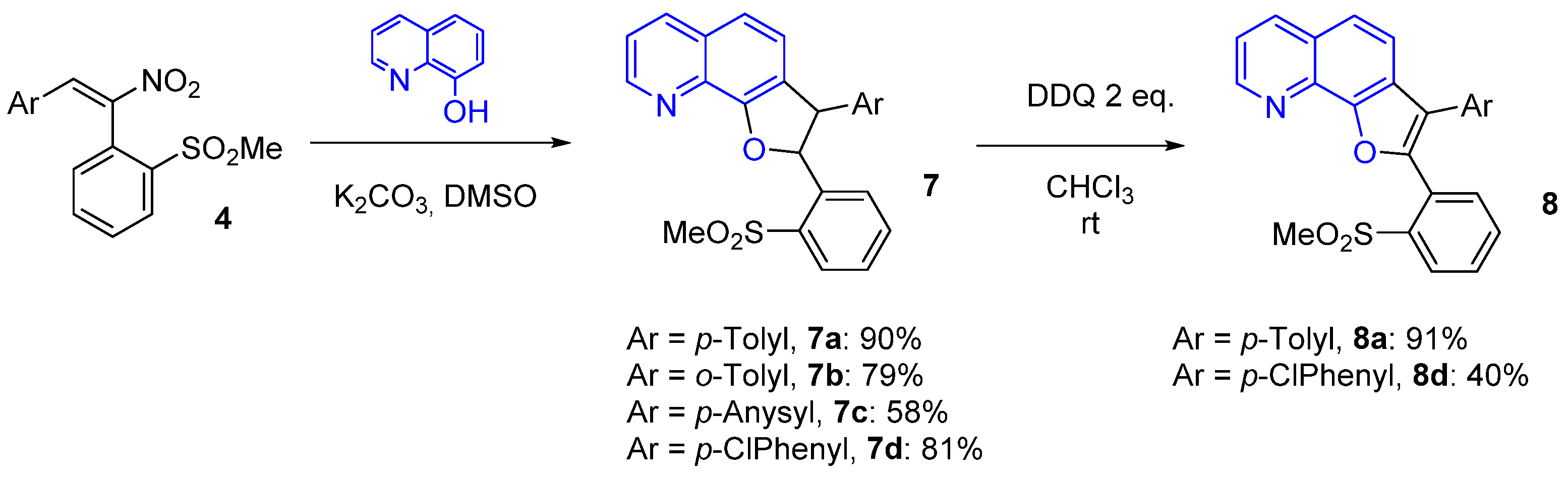
3.3. General Procedure for the Reaction of Substrates 4 with 8-Hydroxyquinoline
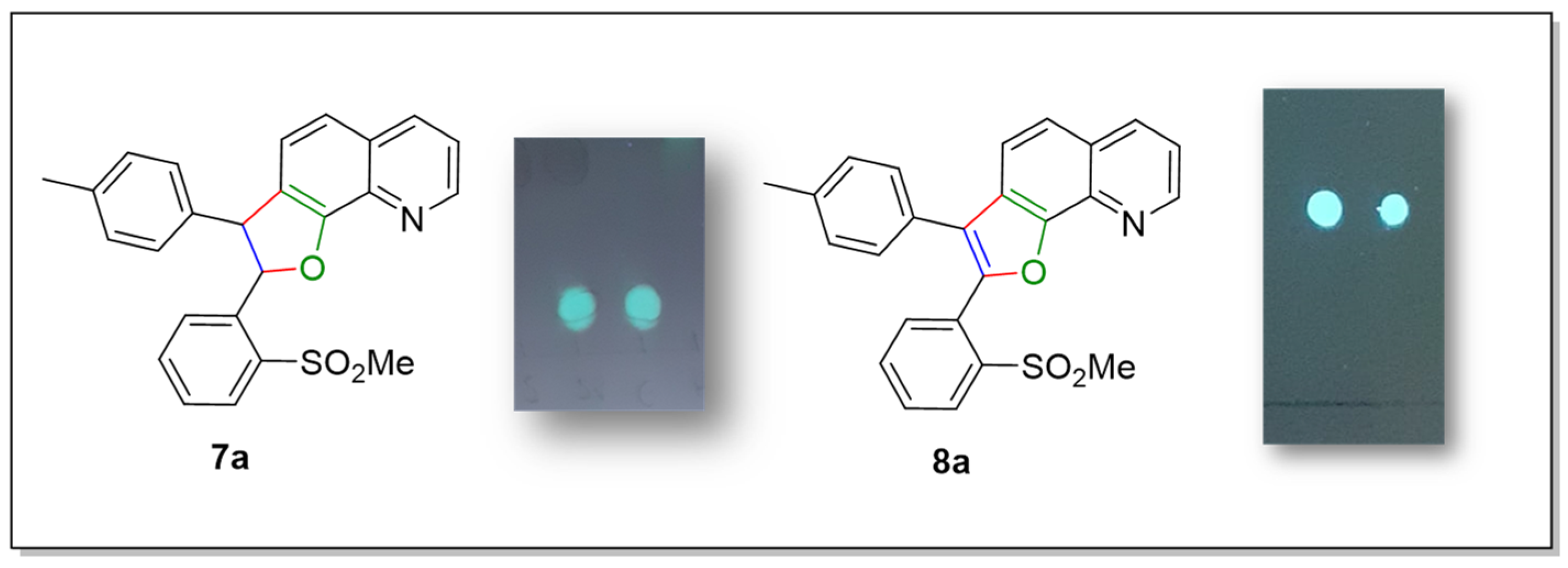
2-(2-(Methylsulfonyl)phenyl)-3-(p-tolyl)-2,3-dihydrofuro[3,2-h]quinoline (7a)
2-(2-(Methylsulfonyl)phenyl)-3-(o-tolyl)-2,3-dihydrofuro[3,2-h]quinoline (7b)
2-(2-(Methylsulfonyl)phenyl)-3-(p-anysyl)-2,3-dihydrofuro[3,2-h]quinoline (7c)
3-(4-Chlorophenyl)-2-(2-(methylsulfonyl)phenyl)-2,3-dihydrofuro[3,2-h]quinoline (7d)
3.4. General Procedure for the Oxidative Aromatization Reaction of Dihydrofuroquinolines 7a, d to Furoquinolines 8a, d
2-(2-(Methylsulfonyl)phenyl)-3-(p-tolyl)furo[3,2-h]quinoline (8a)
3-(4-Chlorophenyl)-2-(2-(methylsulfonyl)phenyl)furo[3,2-h]quinoline (8d)
3.5. Procedure for the Reaction of Substrate 4a with 4-Hydroxycoumarin
2-(2-(Methylsulfonyl)phenyl)-3-(p-tolyl)-2,3-dihydro-4H-furo[3,2-c]chromen-4-one (9a)
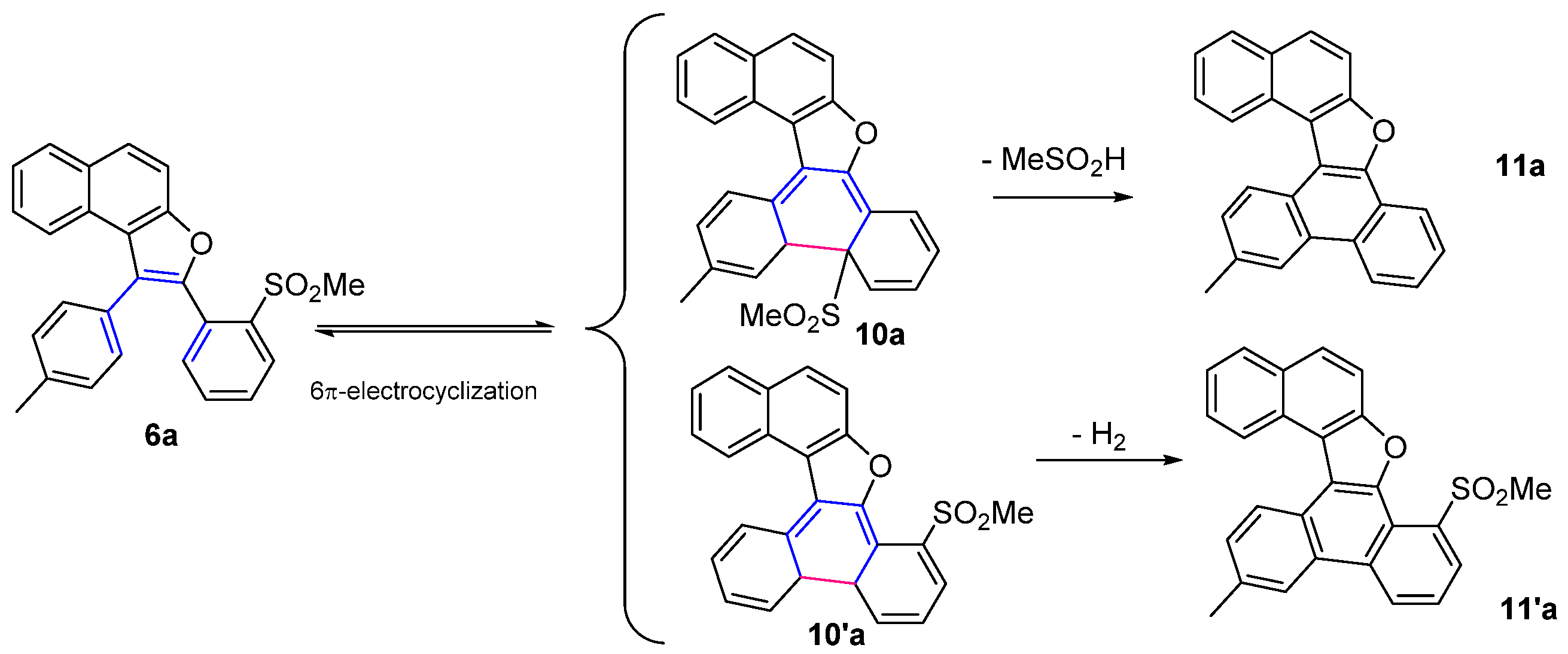
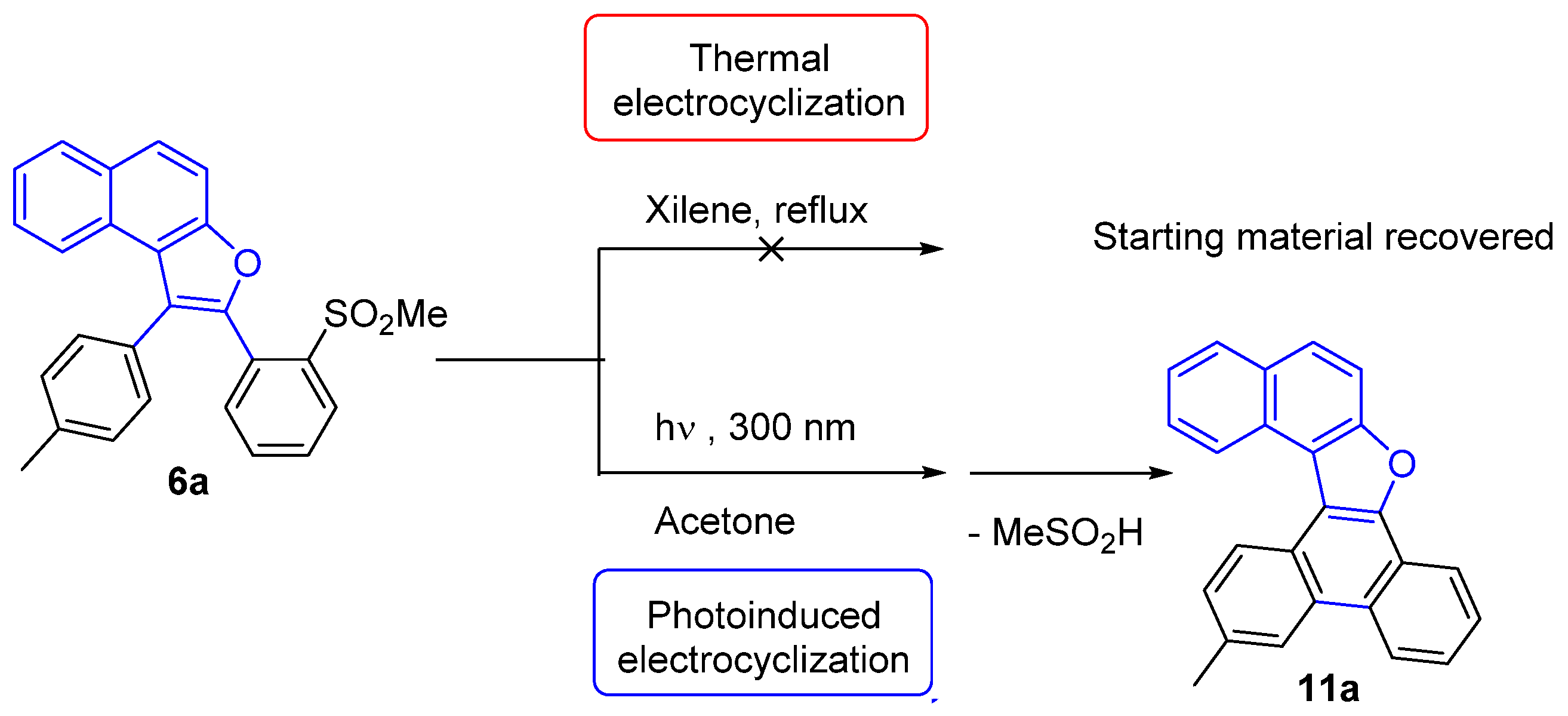
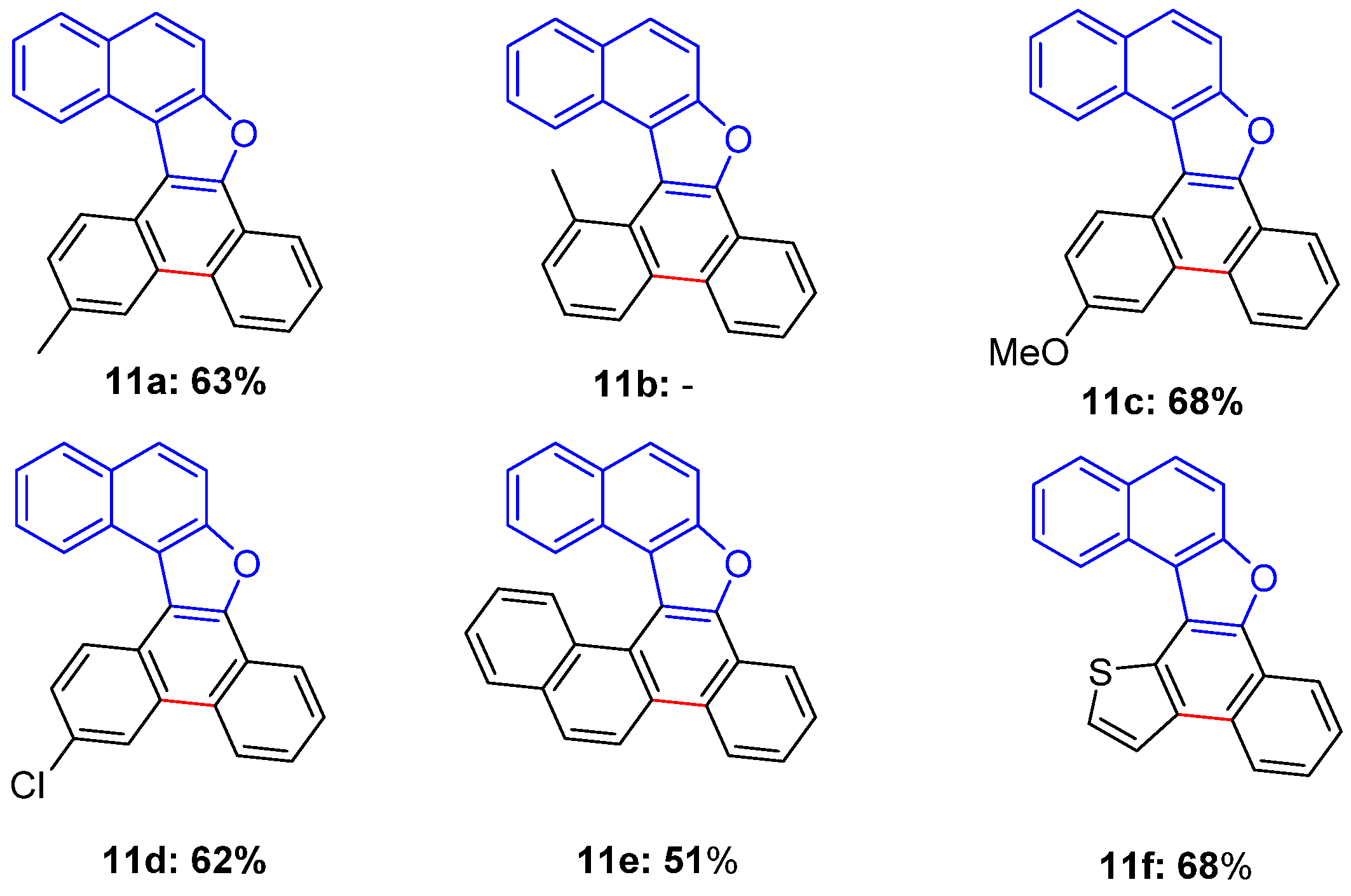
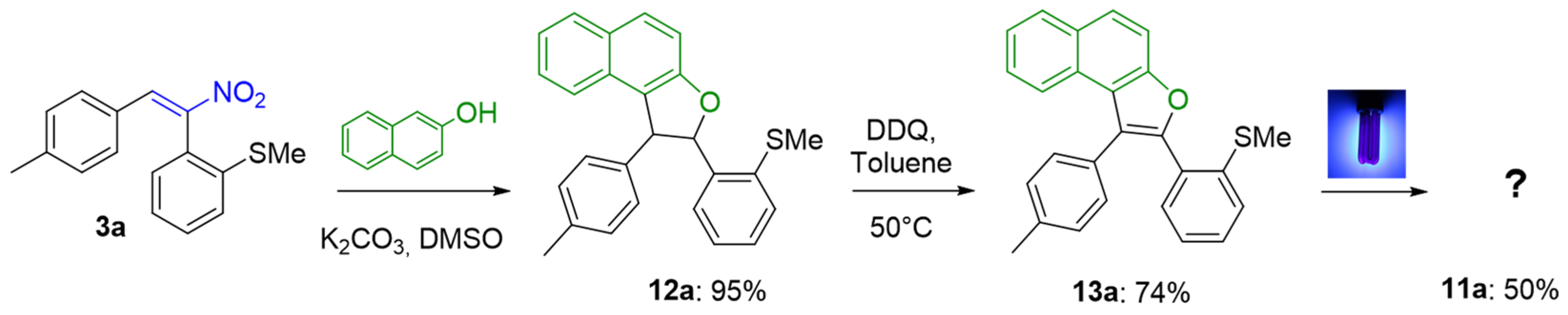
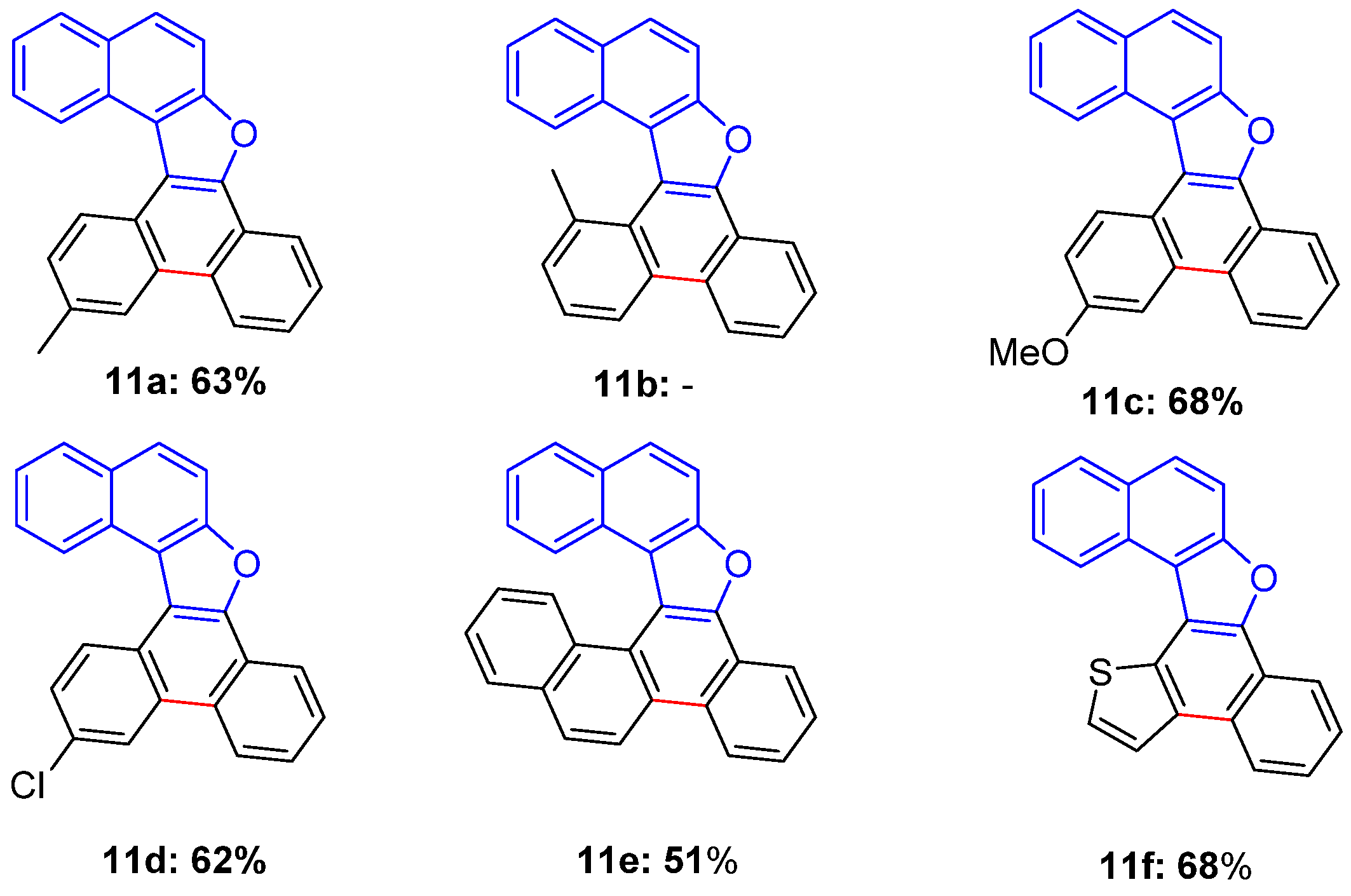
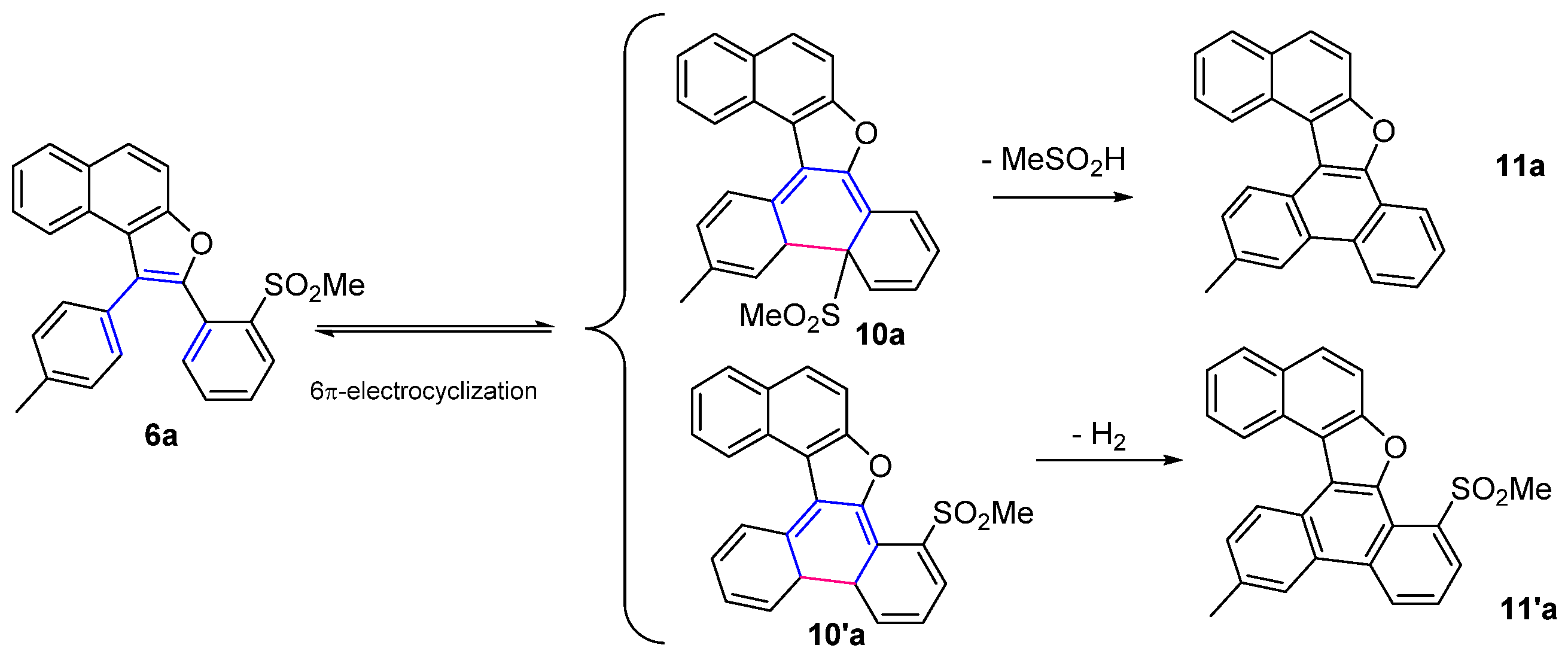
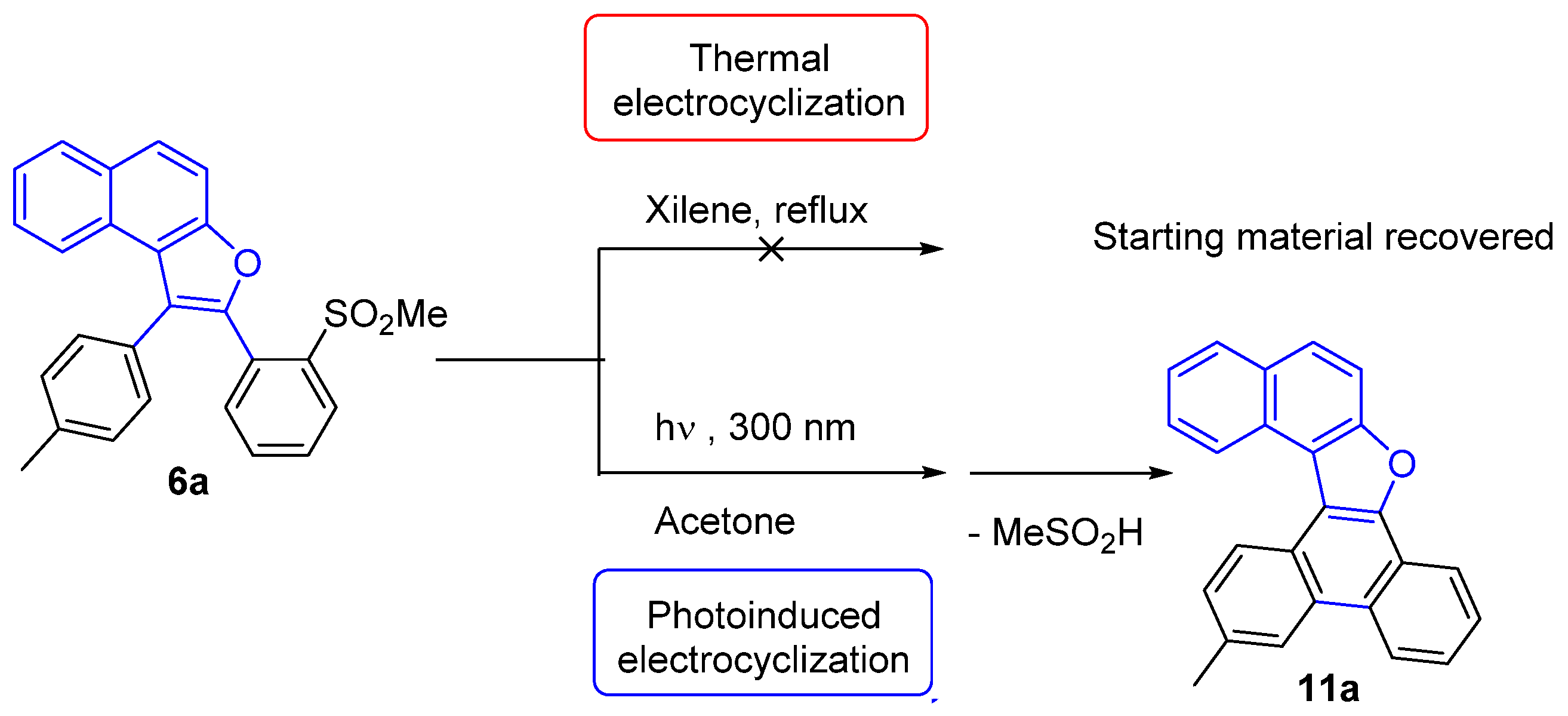
3.6. General Procedure for the 6π-Electrocyclization of Naphthofurans 6a–f
13-Methylnaphtho[2,1-b]phenanthro[9,10-d]furan (11a)
13-Methoxynaphtho[2,1-b]phenanthro[9,10-d]furan (11c)
13-Chloronaphtho[2,1-b]phenanthro[9,10-d]furan (11d)
Chryseno[6,5-b]naphtho[1,2-d]furan (11e)
Naphtho[2,1-b]thieno[2’,3’:3,4]naphtho[2,1-d]furan (11f)
2-(2-(Methylthio)phenyl)-1-(p-tolyl)-1,2-dihydronaphtho[2,1-b]furan (12a)
2-(2-(Methylthio)phenyl)-1-(p-tolyl)naphtho[2,1-b]furan (13a)
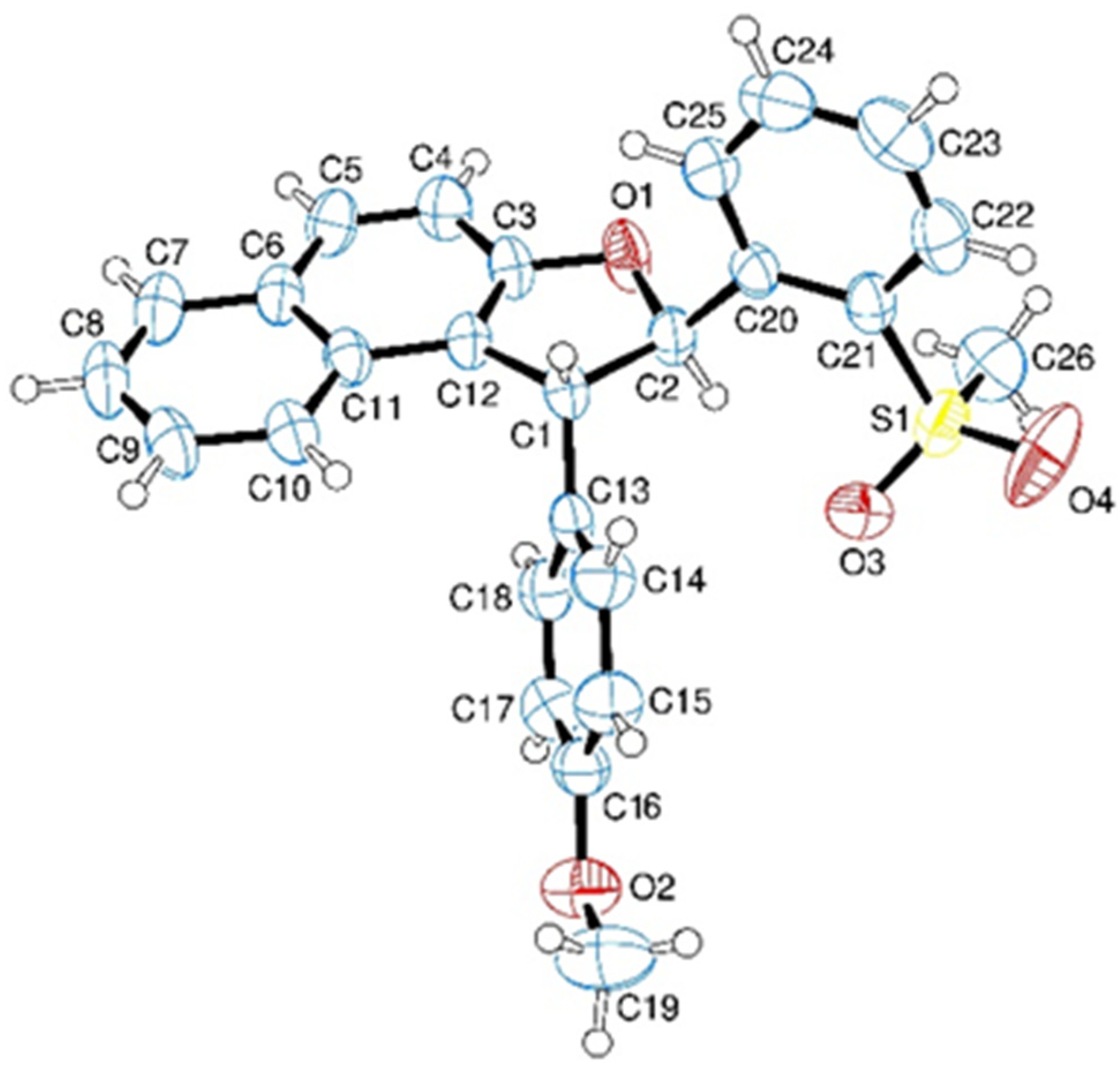
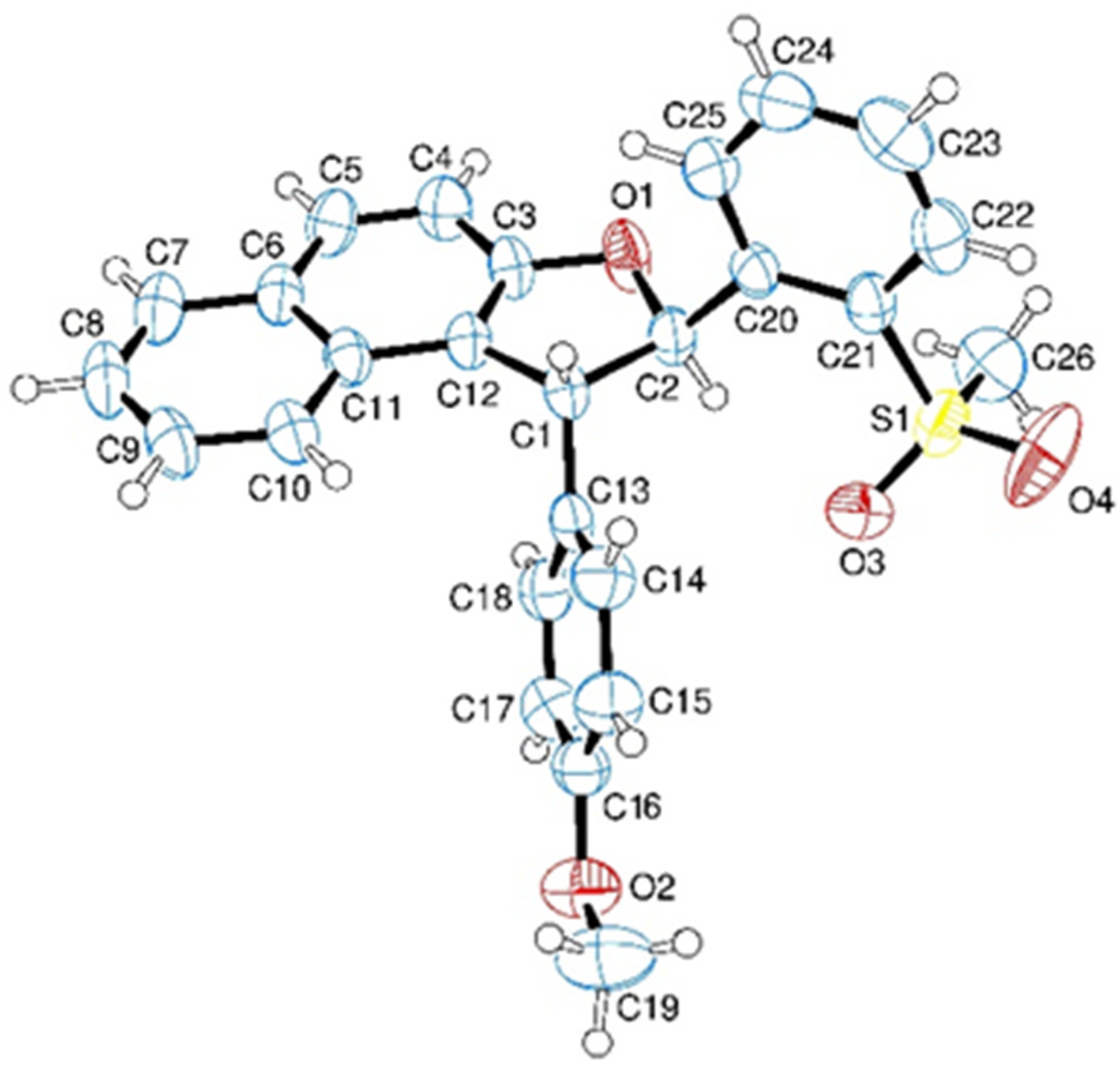
3.7. Crystal Structure of 5a
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Boto, A.; Alvarez, L. Furan and its derivatives. In Heterocycles in Natural Product Synthesis; Wiley: Weinheim, Germany, 2011; pp. 97–152. [Google Scholar]
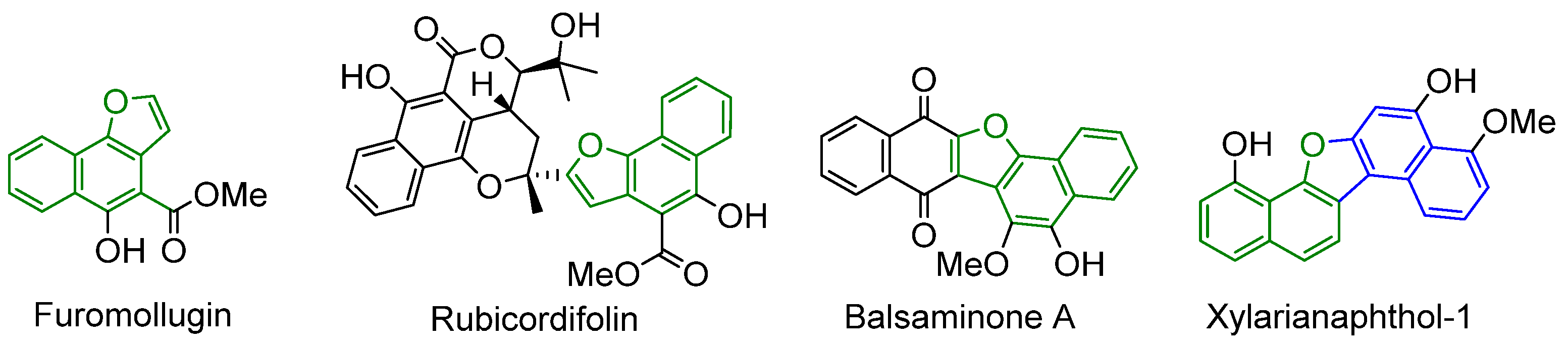
- Ho, L.-K.; Don, M.-J.; Chen, H.-C.; Yeh, S.-F.; Chen, J.-M. Inhibition of Hepatitis B Surface Antigen Secretion on Human Hepatoma Cells. Components from Rubia cordifolia. J. Nat. Prod. 1996, 59, 330–333. [Google Scholar] [CrossRef] [PubMed]
- Ishiguro, K.; Ohira, Y.; Oku, H. Antipruritic Dinaphthofuran-7,12-dione Derivatives from the Pericarp of Impatiens balsamina. J. Nat. Prod. 1998, 61, 1126–1129. [Google Scholar] [CrossRef] [PubMed]
- Kotoku, N.; Higashimoto, K.; Kurioka, M.; Arai, M.; Fukuda, A.; Sumii, Y.; Sowa, Y.; Sakai, T.; Kobayashi, M. Xylarianaphthol-1, a novel dinaphthofuran derivative, activates p21 promoter in a p53-independent manner. Bioorganic Med. Chem. Lett. 2014, 24, 3389–3391. [Google Scholar] [CrossRef] [PubMed]
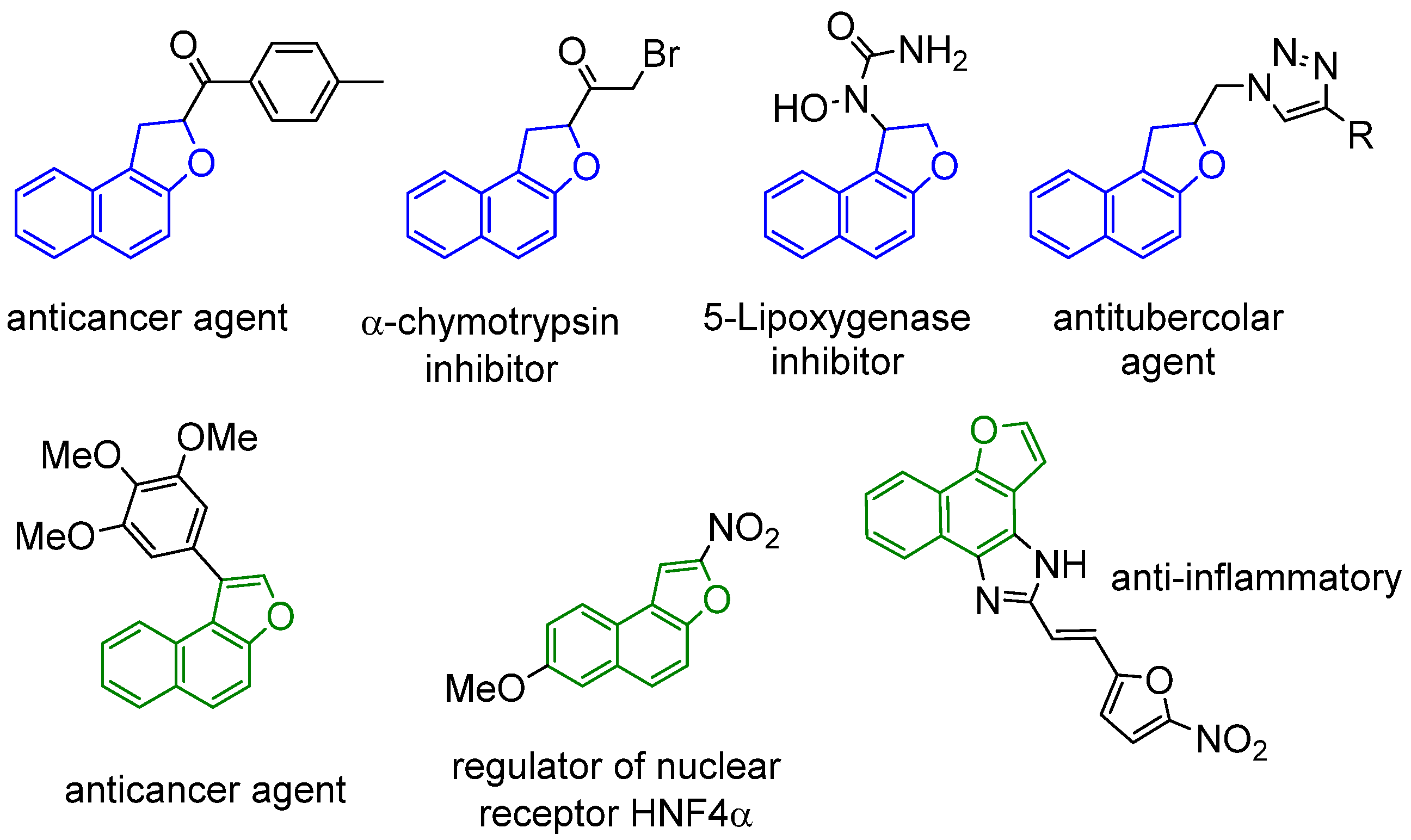
- Srivastava, V.; Negi, A.S.; Kumar, J.K.; Faridi, U.; Sisodia, B.S.; Darokar, M.P.; Luqman, S.; Khanuja, S.P.S. Synthesis of 1-(3′,4′,5′-trimethoxy) phenyl naphtho[2,1-b]furan as a novel anticancer agent. Bioorg. Med. Chem. Lett. 2006, 16, 911–914. [Google Scholar] [CrossRef]
- Islam, K.; Pal, K.; Debnath, U.; Basha, R.S.; Khan, A.T.; Jana, K.; Misra, A.K. Anti-cancer potential of (1,2-dihydronaphtho[2,1-b]furan-2-yl)methanone derivatives. Bioorg. Med. Chem. Lett. 2020, 30, 127476. [Google Scholar] [CrossRef]
- Tseng, C.-H.; Lin, C.-S.; Shih, P.-K.; Tsao, L.-T.; Wang, J.-P.; Cheng, C.-M.; Tzeng, C.-C.; Chen, Y.-L. Furo[3′,2′:3,4]naphtho[1,2-d]imidazole derivatives as potential inhibitors of inflammatory factors in sepsis. Bioorg. Med. Chem. Lett. 2009, 17, 6773–6779. [Google Scholar] [CrossRef]
- Tripathi, R.P.; Yadav, A.K.; Ajay, A.; Bisht, S.S.; Chaturvedi, V.; Sinha, S.K. Application of Huisgen (3 + 2) cycloaddition reaction: Synthesis of 1-(2,3-dihydrobenzofuran-2-yl-methyl [1,2,3]-triazoles and their antitubercular evaluations. Eur. J. Med. Chem. 2010, 45, 142–148. [Google Scholar] [CrossRef]
- Guével, R.L.; Oger, F.; Lecorgne, A.; Dudasova, Z.; Chevance, S.; Bondon, A.; Barath, P.; Simonneaux, G.; Salbert, G. Identification of small molecule regulators of the nuclear receptor HNF4α based on naphthofuran scaffolds. Bioorg. Med. Chem. Lett. 2009, 17, 7021–7030. [Google Scholar] [CrossRef]
- Pattabiramin, T.N.; Lawson, W.B. Stereochemistry of the active site of α-chymotrypsin: The effect of some tricyclic bromomethyl ketones on α-chymotrypsin. Biochim. Biophys. Acta 1972, 258, 548–553. [Google Scholar] [CrossRef]
- Adams, J.L.; Garigipati, R.S.; Sorenson, M.; Schmidt, S.J.; Brian, W.R.; Newton, F.J.; Tyrrell, K.A.; Garver, E.; Yodis, L.A.; Chabot-Fletcher, M.; et al. Bicyclic N-Hydroxyurea Inhibitors of 5-Lipoxygenase: Pharmacodynamic, Pharmacokinetic, and in Vitro Metabolic Studies Characterizing N-Hydroxy-N-(2,3-dihydro-6-(phenylmethoxy)-3-benzofuranyl)urea. J. Med. Chem. 1996, 39, 5035–5046. [Google Scholar] [CrossRef]
- Matsunaga, N.; Kaku, T.; Ojida, A.; Tanaka, T.; Hara, T.; Yamaoka, M.; Kusaka, M.; Tasaka, A. C17,20-lyase inhibitors. Part 2: Design, synthesis and structure–activity relationships of (2-naphthylmethyl)-1H-imidazoles as novel C17,20-lyase inhibitors. Bioorg. Med. Chem. 2004, 12, 4313–4336. [Google Scholar] [CrossRef] [PubMed]
- Costa, D.; Sousa, C.M.; Coelho, P.J. Colour switching with photochromic vinylidene-naphthofurans. Tetrahedron 2018, 74, 7372–7379. [Google Scholar] [CrossRef]
- Tsuji, H.; Nakamura, E. Design and Functions of Semiconducting Fused Polycyclic Furans for Optoelectronic Applications. Acc. Chem. Res. 2017, 50, 396–406. [Google Scholar] [CrossRef] [PubMed]
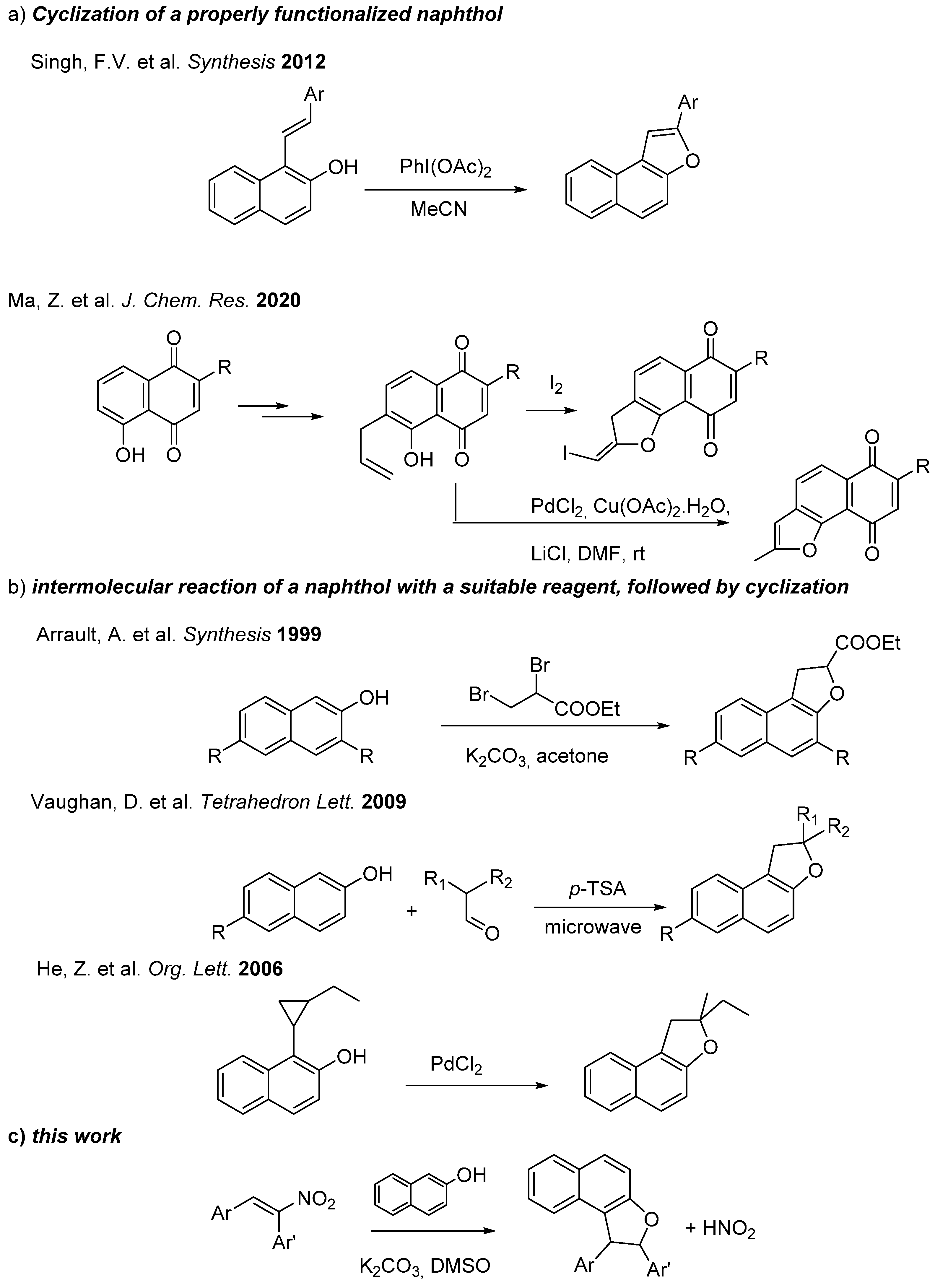
- Singh, F.V.; Wirth, T. Hypervalent iodine mediated oxidative cyclization of o-hydroxystilbenes into benzo-and naphthofurans. Synthesis 2012, 44, 1171–1177. [Google Scholar] [CrossRef]
- Mallavadhani, U.V.; Prasad, C.V.; Shrivastava, S.; Naidu, V.G.M. Synthesis and anticancer activity of some novel 5,6-fused hybrids of juglone based 1,4-naphthoquinones. Eur. J. Med. Chem. 2014, 83, 84–91. [Google Scholar] [CrossRef]
- Ma, Z.; Zhou, M.; Ma, L.; Zhang, M. Synthesis of benzofurans from the cyclodehydration of α-phenoxy ketones mediated by Eaton’s reagent. J. Chem. Res. 2020, 44, 426–436. [Google Scholar] [CrossRef]
- Arrault, A.; Touzeau, F.; Guillaumet, G.; Mérour, J.-Y. A Straightforward Synthesis of 1, 2-Dihydronaphtho [2, 1-b] furans from 2-Naphthols. Synthesis 1999, 1241–1245. [Google Scholar] [CrossRef]
- Vaughan, D.; Jha, A. Convenient synthesis of novel 2,2-dialkyl-1,2-dihydronaphtho[2,1-b]furans. Tetrahedron Lett. 2009, 50, 5709–5712. [Google Scholar] [CrossRef]
- Wang, B.; Zhang, Q.; Luo, J.; Gan, Z.; Jiang, W.; Tang, Q. One-Step Regioselective Synthesis of Benzofurans from Phenols and α-Haloketones. Molecules 2019, 24, 2187. [Google Scholar] [CrossRef] [Green Version]
- He, Z.; Yudin, A.K. Palladium-Catalyzed Oxidative Activation of Arylcyclopropanes. Org. Lett. 2006, 8, 5829–5832. [Google Scholar] [CrossRef]
- Liu, G.; Lu, X. Palladium(II)-catalyzed intramolecular addition of arylboronic acids to ketone. Tetrahedron 2008, 64, 7324–7330. [Google Scholar] [CrossRef]
- van Otterlo, W.A.L.; Morgans, G.L.; Madeley, L.G.; Kuzvidza, S.; Moleele, S.S.; Thornton, N.; de Koning, C.B. An isomerization-ring-closing metathesis strategy for the synthesis of substituted benzofurans. Tetrahedron 2005, 61, 7746–7755. [Google Scholar] [CrossRef]
- Martin-Matute, C.B.; Nevado, C.; Cardenas, D.J.; Echavarren, A.M. Intramolecular Reactions of Alkynes with Furans and Electron Rich Arenes Catalyzed by PtCl2: The Role of Platinum Carbenes as Intermediates. J. Am. Chem. Soc. 2003, 125, 5757–7566. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Liu, L.; Shaq, Z.; Wu, Y.-C.; Wang, D.; Chen, Y.-J. InCl3-catalyzed propargylation of indoles and phenols with propargylic acetates: Application to the syntheses of benzofurans and naphthofurans. Synthesis 2007, 1961–1969. [Google Scholar] [CrossRef]
- Kundu, D.; Samim, M.; Majee, A.; Hajra, A. Indium triflate-catalyzed coupling between nitroalkenes and phenol/naphthols: A simple and direct synthesis of arenofurans by a cyclization reaction. Chem. Asian J. 2011, 6, 406–409. [Google Scholar] [CrossRef]
- Rao, V.K.; Shelke, G.M.; Tiwari, R.; Parang, K.; Kumar, A. A Simple and Efficient Synthesis of 2,3-Diarylnaphthofurans Using Sequential Hydroarylation/Heck Oxyarylation. Org. Lett. 2013, 15, 2190–2193. [Google Scholar] [CrossRef] [Green Version]
- Jana, R.; Paul, S.; Biswas, A.; Ray, J.K. Copper-catalyzed addition of water affording highly substituted furan and unusual formation of naphthofuran ring from 3-(1-alkenyl)-2-alkene-1-al. Tetrahedron Lett. 2010, 51, 273–276. [Google Scholar] [CrossRef]
- Olyaei, A.; Sadeghpourb, M. Dihydronaphthofurans: Synthetic strategies and applications. RSC Adv. 2020, 10, 5794–5826. [Google Scholar] [CrossRef]
- Omelchuk, O.A.; Tikhomirov, A.S.; Shchekotikhin, A.E. Annelation of furan rings to arenes. Russ. Chem. Rev. 2016, 85, 817–835. [Google Scholar] [CrossRef]
- Mane, V.; Pandey, J.; Ayyagari, N.; Dey, C.; Kale, R.; Namboothiri, I.N.N. Synthesis of hydrazinoheterocycles from Morita–Baylis–Hillman adducts of nitroalkenes with azodicarboxylates. Org. Biomol. Chem. 2016, 14, 2427–2438. [Google Scholar] [CrossRef]
- Dell’Erba, C.; Spinelli, D.; Leandri, G. Ring-opening reaction in the thiophen series: Reaction between 3,4-dinitrothiophen and secondary amines. J. Chem. Soc. Chem. Commun. 1969, 10, 549. [Google Scholar] [CrossRef]
- Bianchi, L.; Maccagno, M.; Petrillo, G.; Rizzato, E.; Sancassan, F.; Severi, E.; Spinelli, D.; Tavani, C.; Viale, M. (Eds.) Versatile nitrobutadienic building-blocks from the ring opening of 2- and 3-nitrothiophenes. In Targets in Heterocyclic System-Chemistry and Properties; Italian Society of Chemistry: Rome, Italy, 2007; Volume 11, pp. 1–20. [Google Scholar]
- Bianchi, L.; Maccagno, M.; Petrillo, G.; Sancassan, F.; Spinelli, D. (Eds.) Tavani,2,3-Dinitro-1,3-butadienes: Versatile building-blocks from the ring opening of 3,4-dinitrothiophene. In Targets in Heterocyclic Systems-Chemistry and Properties; Italian Society of Chemistry: Rome, Italy, 2006; Volume 10, pp. 1–23. [Google Scholar]
- Petrillo, G.; Benzi, A.; Bianchi, L.; Maccagno, M.; Pagano, A.; Tavani, C.; Spinelli, D. Recent advances in the use of conjugated nitro or dinitro-1,3-butadienes as building-blocks for the synthesis of heterocycles. Tetrahedron Lett. 2020, 61, 152297–152309. [Google Scholar] [CrossRef]
- Tavani, C.; Bianchi, L.; Giorgi, G.; Maccagno, M.; Petrillo, G. Densely Functionalized 2-Methylideneazetidines from Nitrodienic Building Blocks. Eur. J. Org. Chem. 2018, 126–136. [Google Scholar] [CrossRef]
- Bianchi, L.; Giorgi, G.; Maccagno, M.; Petrillo, G.; Scapolla, C.; Tavani, C. On the behavior of bis(sulfonyl)nitrobutadienes towards primary amines: A convenient access to 1-alkyl-2-aryl-4-(phenylsulfonyl)pyrroles. Tetrahedron 2016, 72, 7050–7058. [Google Scholar] [CrossRef]
- Bianchi, L.; Carloni-Garaventa, A.; Maccagno, M.; Pani, M.; Petrillo, G.; Scapolla, C.; Tavani, C. Synthesis of poly-functionalized pyrazoles and pyridazines from nitrobutadienes: An interesting dichotomy of practical relevance. Tetrahedron 2015, 71, 7550–7561. [Google Scholar] [CrossRef]
- Bianchi, L.; Maccagno, M.; Pani, M.; Petrillo, G.; Scapolla, C.; Tavani, C. A straight access to functionalized carbazoles by tandem reaction between indole and nitrobutadienes. Tetrahedron 2015, 71, 7421–7435. [Google Scholar] [CrossRef]
- Benzi, A.; Bianchi, L.; Giorgi, G.; Maccagno, M.; Petrillo, G.; Tavani, C. 2-Aryl-3-Vinyl Substituted Imidazo[1,2-a]pyridines and Fluorescent Electrocyclization Derivatives therefrom. ChemistrySelect 2020, 5, 4552–4558. [Google Scholar] [CrossRef]
- Benzi, A.; Bianchi, L.; Maccagno, M.; Pagano, A.; Petrillo, G.; Tavani, C. Sequential Annulations to Interesting Novel Pyrrolo[3,2-c]carbazoles. Molecules 2019, 24, 3802. [Google Scholar] [CrossRef] [Green Version]
- Dell’Erba, C.; Gabellini, A.; Novi, M.; Petrillo, G.; Tavani, C.; Cosimelli, B.; Spinelli, D. Ring opening of 2-substituted 4-nitrothiophenes with pyrrolidine. Access to new functionalized nitro-unsaturated building blocks. Tetrahedron 2001, 57, 8159–8165. [Google Scholar] [CrossRef]
- Armstrong, K.J.; Martin-Smith, M.; Brown, N.M.D.; Brophy, G.C.; Sternhell, S. Benzo[b]thiophen derivatives. Part IX. Nitration of benzo[b]thiophen and the isomeric nitrobenzo[b]thiophens. J. Chem. Soc. C Org. 1969, 1766–1775. [Google Scholar] [CrossRef]
- Bianchi, L.; Dell’Erba, C.; Maccagno, M.; Morganti, S.; Novi, M.; Petrillo, G.; Rizzato, E.; Spinelli, D.; Tavani, C. Easy access to 4-nitrothiochroman S,S-dioxides via ring-enlargement from 3-nitrobenzo[b]thiophene. Tetrahedron 2004, 60, 4967–4973. [Google Scholar] [CrossRef]
- Sheldrick, G.M. A short history of SHELX. Acta Cryst. Sect. A 2008, 64, 112–122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Cryst. Sect. C Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
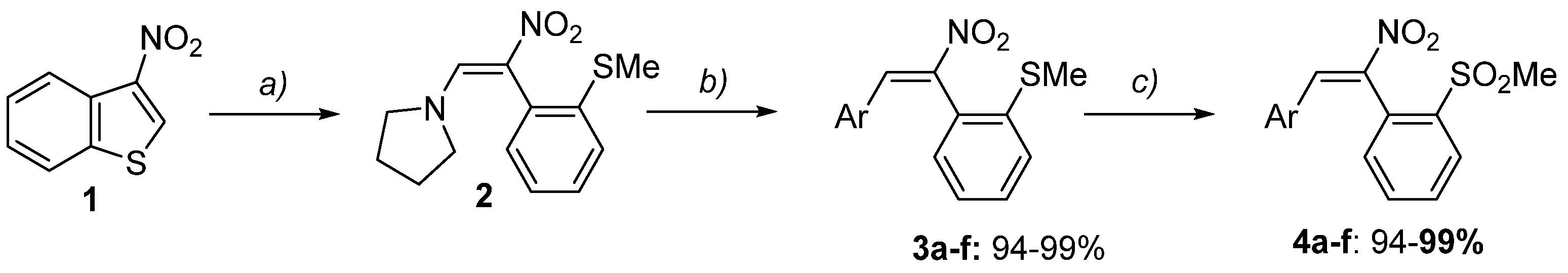{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 4 | Ar in 4 | 5 (Yields %) a |
|---|---|---|
| 4a | p-MeC6H4 | 5a, 92 |
| 4b | o-MeC6H4 | 5b, 89 |
| 4c | p-MeOC6H4 | 5c, 94 |
| 4d | p-ClC6H4 | 5d, 93 |
| 4e | 1-Naphthyl | 5e, 87 |
| 4f | 2-Thienyl | 5f, 96 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Benzi, A.; Bianchi, L.; Giorgi, G.; Maccagno, M.; Petrillo, G.; Spinelli, D.; Tavani, C. An Easy Access to Furan-Fused Polyheterocyclic Systems. Molecules 2022, 27, 3147. https://doi.org/10.3390/molecules27103147
Benzi A, Bianchi L, Giorgi G, Maccagno M, Petrillo G, Spinelli D, Tavani C. An Easy Access to Furan-Fused Polyheterocyclic Systems. Molecules. 2022; 27(10):3147. https://doi.org/10.3390/molecules27103147
Chicago/Turabian StyleBenzi, Alice, Lara Bianchi, Gianluca Giorgi, Massimo Maccagno, Giovanni Petrillo, Domenico Spinelli, and Cinzia Tavani. 2022. "An Easy Access to Furan-Fused Polyheterocyclic Systems" Molecules 27, no. 10: 3147. https://doi.org/10.3390/molecules27103147
APA StyleBenzi, A., Bianchi, L., Giorgi, G., Maccagno, M., Petrillo, G., Spinelli, D., & Tavani, C. (2022). An Easy Access to Furan-Fused Polyheterocyclic Systems. Molecules, 27(10), 3147. https://doi.org/10.3390/molecules27103147