
Imine Palladacycles: Synthesis, Structural Analysis and Application in Suzuki–Miyaura Cross Coupling in Semi-Aqueous Media

 , , and
, , and
Abstract
:1. Introduction
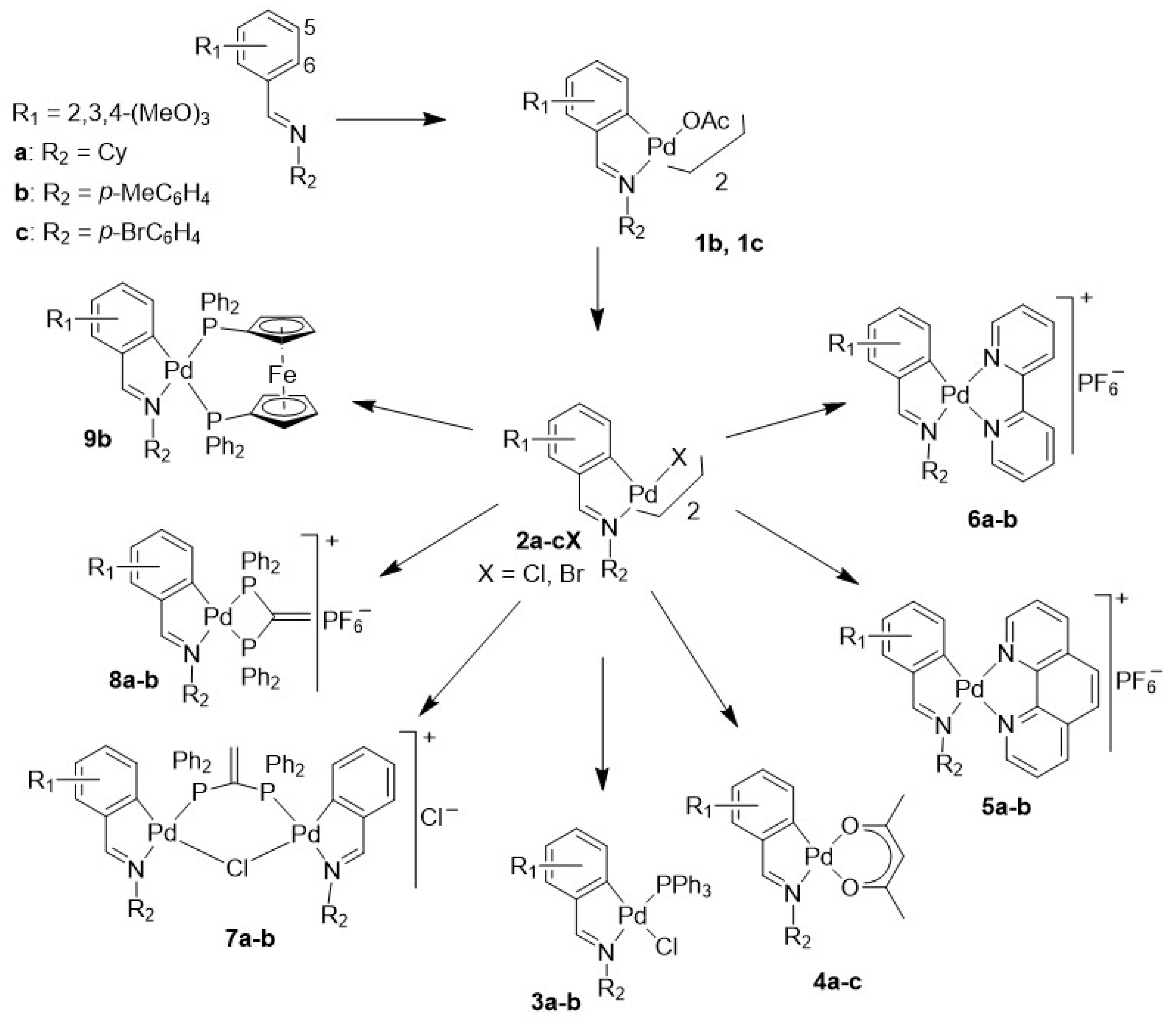
2. Results and Discussion
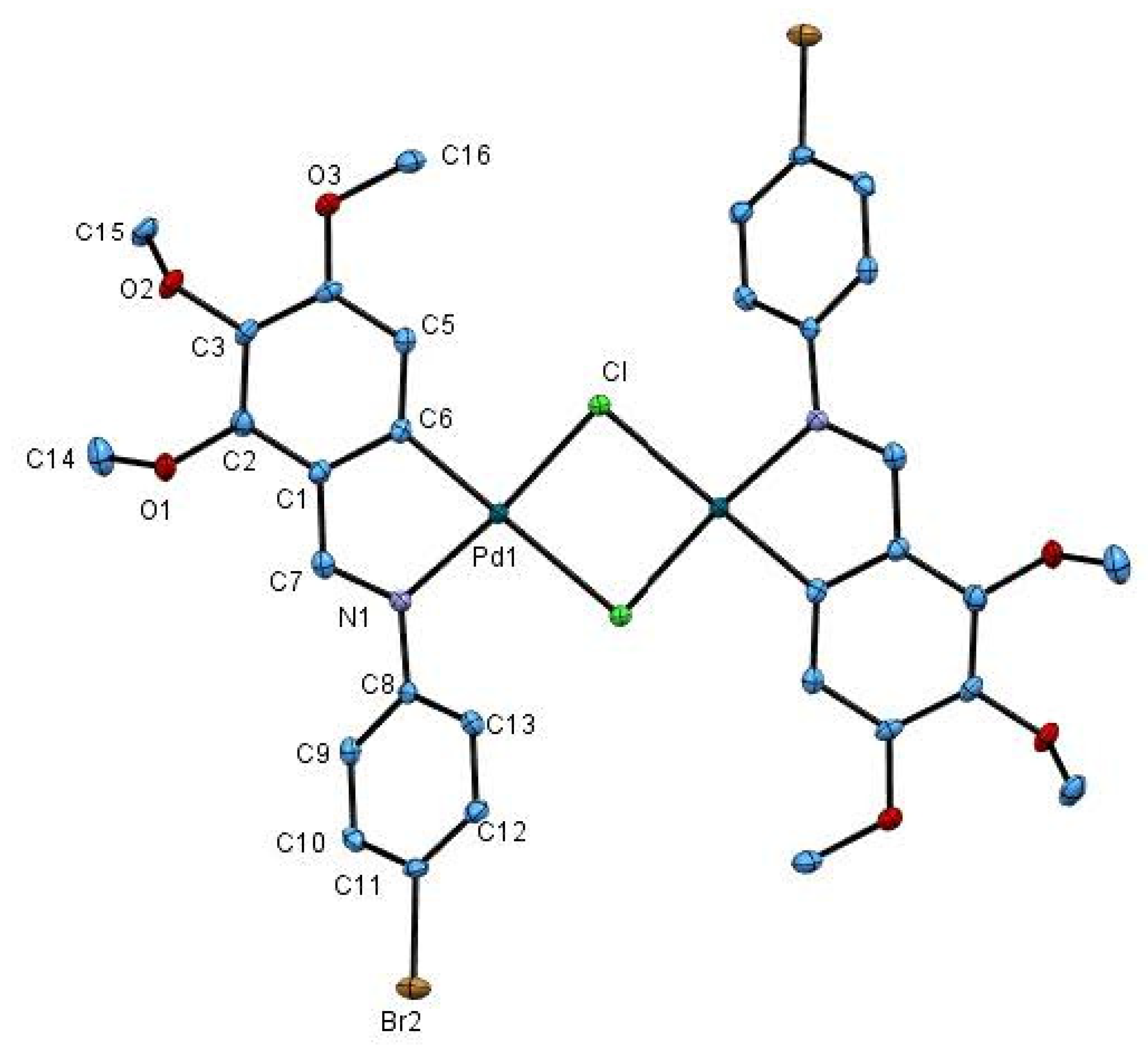
2.1. Crystal Structure of 1c
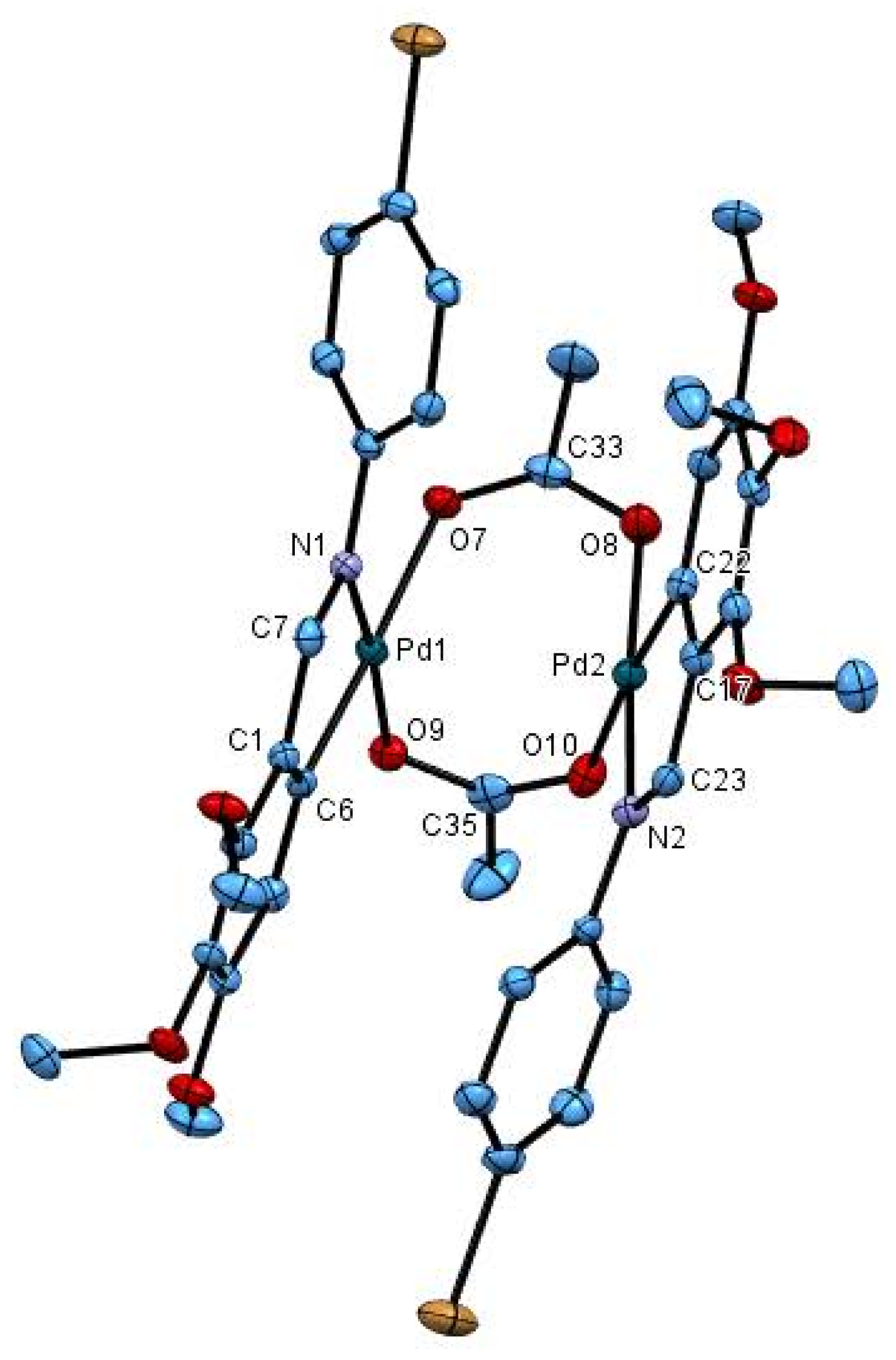
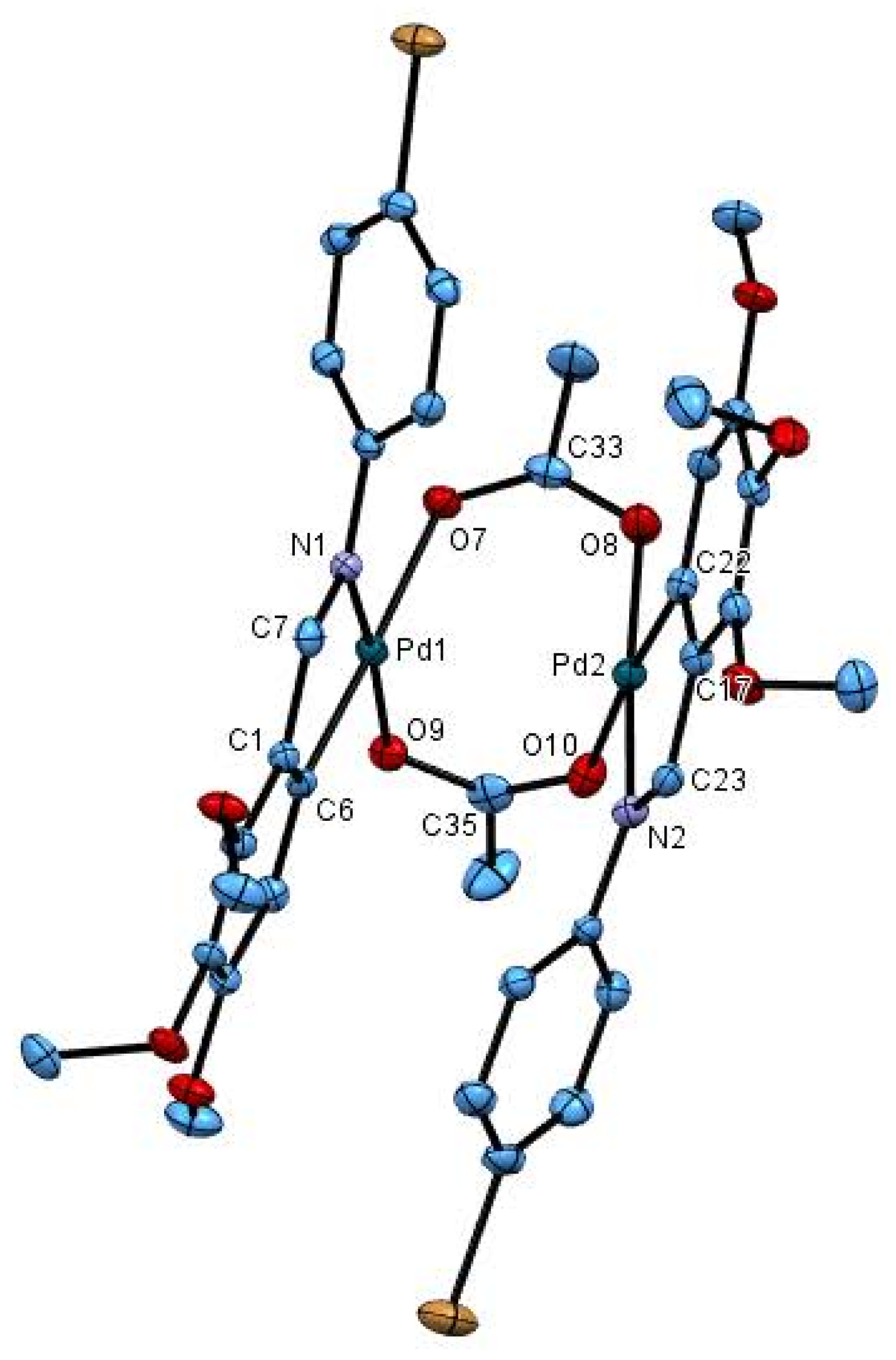
2.2. Crystal Structure of 2cCl
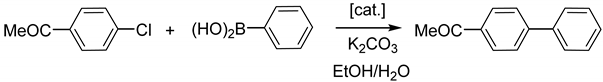
2.3. Catalytic Activity
3. Conclusions
4. Experimental Section
4.1. Preparation of the Ligands and Complexes
4.2. Crystal Structure Analysis and Details on Data Collection and Refinement
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cope, A.C.; Siekman, R.W. Formation of Covalent Bonds from Platinum or Palladium to Carbon by Direct Substitution. J. Am. Chem. Soc. 1965, 87, 3272. [Google Scholar] [CrossRef]
- Dupont, J.P. Palladacycles; Wiley-VCH: Weinheim, Germany, 2018. [Google Scholar]
- Sarma, K.; Devi, N.; Sutradhar, D.; Sarma, B.; Chandra, A.K.; Barman, P. Synthesis of a novel six membered CNS palladacycle; TD-DFT study and catalytic activity towards microwave-assisted selective oxidation of terminal olefin to aldehyde. J. Organomet. Chem. 2016, 822, 20. [Google Scholar] [CrossRef]
- Adams, M.; de Kock, C.; Smith, P.J.; Chibale, K.; Smith, G.S. Synthesis, characterization and antiplasmodial evaluation of cyclopalladated thiosemicarbazone complexes. J. Organomet. Chem. 2013, 736, 19. [Google Scholar] [CrossRef]
- Kozlov, V.A.; Aleksanyan, D.V.; Nelyubina, Y.V.; Lyssenko, K.A.; Vasilev, A.A.; Petrovskii, P.V.; Odinets, I.L. Cyclopalladation of meta-(Diphenylthiophosphoryloxy)benzaldimines: NCS and Unexpected NCO 5,6-Membered Pincer Palladium Complexes. Organometallics 2010, 29, 2054. [Google Scholar] [CrossRef]
- Lowry, M.S.; Bernhard, S. Synthetically Tailored Excited States: Phosphorescent, Cyclometalated Iridium(III) Complexes and Their Applications. Chem. A Eur. J. 2006, 12, 7970. [Google Scholar] [CrossRef]
- Navarro-Ranninger, C.; López-Solera, I.; González, V.M.; Pérez, J.M.; Álvarez-Vales, A.; Martín, A.; Raithby, P.; Masaguer, J.R.; Alonso, C. Cyclometalated Complexes of Platinum and Palladium with N-(4-Chlorophenyl)-α-benzoylbenzylideneamine. In Vitro Cytostatic Activity, DNA Modification, and Interstrand Cross-Link Studies. Inorg. Chem. 1996, 35, 5181. [Google Scholar] [CrossRef]
- Cutillas, N.; Yellol, G.S.; de Haro, C.; Vicente, C.; Rodríguez, V.; Ruiz, J. Anticancer cyclometalated complexes of platinum group metals and gold. Chem. Rev. 2013, 257, 2784. [Google Scholar] [CrossRef]
- Takahashi, H.; Tsuji, J. Organic syntheses by means of noble metal compounds: XXXIII. Carbonylation of azobenzene-palladium chloride complexes. J. Organomet. Chem. 1967, 10, 511. [Google Scholar] [CrossRef]
- Hiraky, K.; Fuchita, Y.; Takakura, S. Syntheses and characterization of cyclopalladated complexes of 2-phenylthiazole. J. Organomet. Chem. 1981, 210, 273. [Google Scholar] [CrossRef]
- Vicente, J.; Saura-Llamas, I.; Turpín, J.; Bautista, D.; Ramirez de Arellano, C.; Jones, P.G. Insertion of One, Two, and Three Molecules of Alkyne into the Pd− C Bond of Ortho-palladated Primary and Secondary Arylalkylamines. Organometallics 2009, 28, 4175. [Google Scholar] [CrossRef]
- Beller, M.; Fischer, H.; Herrmann, W.A.; Öfele, K.; Brossmer, C. Palladacycles as efficient catalysts for Aryl Coupling Reactions. Angew. Chemie Int. Ed. Engl. 1995, 34, 1848. [Google Scholar] [CrossRef]
- Herrmann, W.A.; Brossmer, C.; Öfele, K.; Reisinger, C.-P.; Priermeier, T.; Beller, M.; Fischer, H. Palladacycles as Structurally Defined Catalysts for the Heck Olefination of Chloro- and Bromoarenes. Angew. Chemie Int. Ed. Engl. 1995, 34, 1844. [Google Scholar] [CrossRef]
- Herrmann, W.A.; Böhm, P.W.; Reisinger, C.-P. Application of palladacycles in Heck type reactions. J. Organomet. Chem. 1999, 576, 23. [Google Scholar] [CrossRef]
- Miyaura, N.; Suzuki, A. Stereoselective synthesis of arylated (E)-alkenes by the reaction of alk-1-enylboranes with aryl halides in the presence of palladium catalyst. Chem. Commun. 1979, 866–867. [Google Scholar] [CrossRef]
- Suzuki, A. Carbon–carbon bonding made easy. Chem. Commun. 2005, 4759–4763. [Google Scholar] [CrossRef]
- Cívicos, J.F.; Alonso, D.A.; Nájera, C. Oxime–Palladacycle-Catalyzed Suzuki–Miyaura Arylation and Alkenylation of Aryl Imidazolesulfonates under Aqueous and Phosphane-Free Conditions. Eur. J. Org. Chem. 2012, 2012, 3670–3676. [Google Scholar] [CrossRef]
- Lucio-Martínez, F.; Adrio, L.A.; Polo-Ces, P.; Ortigueira, J.M.; Fernández, J.J.; Adams, H.; Pereira, M.T.; Vila, J.M. Palladacycle catalysis: An innovation to the Suzuki–Miyaura cross-coupling reaction. Dalt. Trans. 2016, 45, 17598. [Google Scholar] [CrossRef]
- Firinci, R.; Günay, M.E.; Gökçe, A.G. Synthesis, characterization and catalytic activity in Suzuki-Miyaura coupling of palladacycle complexes with n -butyl-substituted N-heterocyclic carbene ligands. Appl. Organomet. Chem. 2018, 32, e4109. [Google Scholar] [CrossRef]
- Alonso, D.A.; Nájera, C.; Pacheco, M.C. Oxime-Derived Palladium Complexes as Very Efficient Catalysts for the Heck–Mizoroki Reaction. Adv. Synth. Catal. 2002, 344, 172. [Google Scholar] [CrossRef]
- Yang, Y.; Oldenhuis, N.J.; Buchwald, S.L. Mild and general conditions for negishi cross-coupling enabled by the use of palladacycle precatalysts. Angew. Chemie Int. Ed. Engl. 2013, 52, 615. [Google Scholar] [CrossRef] [Green Version]
- Yang, F.; Cui, X.; Li, Y.-N.; Zhang, J.; Ren, G.-R.; Wu, Y. Cyclopalladated ferrocenylimines: Efficient catalysts for homocoupling and Sonogashira reaction of terminal alkynes. Tetrahedron 2007, 63, 1963. [Google Scholar] [CrossRef]
- Schiff, H. Mittheilungen aus dem Universitätslaboratorium in Pisa: Eine neue Reihe organischer Basen. Ann. Chem. Pharm. 1864, 131, 118. [Google Scholar] [CrossRef] [Green Version]
- Vázquez-García, D.; Fernández, A.; López-Torres, M.; Rodríguez, A.; Gómez-Blanco, N.; Viader, C.; Vila, J.M.; Fernández, J.J. Versatile Behavior of the Schiff Base Ligand 2,5-Me2C6H3C(H)═N(2,4,6-Me3C6H2) toward Cyclometalation Reactions: C(sp2,phenyl)−H vs C(sp3,methyl)−H Activation. Organometallics 2010, 29, 3303. [Google Scholar] [CrossRef]
- Teijido, B.; Fernández, A.; López-Torresa, M.; Castro-Juiz, S.; Suárez, A.; Ortigueira, J.M.; Vila, J.M.; Fernández, J.J. Influence of phenyl ring substituents in the cyclometallation of Schiff base ligands: Crystal and molecular structures of [Pd-{3,4-(OCH2O)C6H2C(H) N(Cy)-C2,N}(μ-O2CMe)]2 and [Pd-{3,4-(OCH2CH2O)C6H2C(H) N(Cy)-C6,N}(μ-O2CMe)]2. J. Organomet. Chem. 2000, 598, 71. [Google Scholar] [CrossRef]
- Vila, J.M.; Gayoso, M.; Pereira, M.T.; López, M.; Alonso, G.; Fernández, J.J. Cyclometallated complexes of PdII and MnI with N,N-terephthalylidenebis(cyclohexylamine). J. Organomet. Chem. 1993, 445, 287. [Google Scholar] [CrossRef]
- Wang, H.; Yang, J. Synthesis and characterizations of arsine– and stibine–ligated Schiff base palladacycles and their applications in Suzuki–Miyaura cross-coupling reactions. Appl. Organomet. Chem. 2016, 30, 262. [Google Scholar] [CrossRef]
- Serrano, J.L.; García, L.; Pérez, J.; Pérez, E.; García, J.; Sánchez, G.; Sehnal, P.; De Ornellas, S.; Williams, T.J.; Fairlamb, I.J.S. Synthesis and Characterization of Imine-Palladacycles Containing Imidate “Pseudohalide” Ligands: Efficient Suzuki–Miyaura Cross-Coupling Precatalysts and Their Activation To Give Pd0Ln Species (L = Phosphine). Organometallics 2011, 30, 5095–5109. [Google Scholar] [CrossRef]
- Cozzi, P.G. Metal–Salen Schiff base complexes in catalysis: Practical aspects. Chem. Soc. Rev. 2004, 33, 410. [Google Scholar] [CrossRef]
- Gupta, K.C.; Sutar, A.K. Catalytic activities of Schiff base transition metal complexes. Coord. Chem. Rev. 2008, 252, 1420. [Google Scholar] [CrossRef]
- Gupta, K.C.; Sutar, A.K.; Lin, C.-C. Polymer-supported Schiff base complexes in oxidation reactions. Coord. Chem. Rev. 2009, 253, 1926. [Google Scholar] [CrossRef]
- Rosner, T.; Pfaltz, A.; Blackmond, D.G. Observation of Unusual Kinetics in Heck Reactions of Aryl Halides: The Role of Non-Steady-State Catalyst Concentration. J. Am. Chem. Soc. 2001, 123, 4621. [Google Scholar] [CrossRef] [PubMed]
- Rosner, T.; Le Bars, J.; Pfaltz, A.; Blackmond, D.G. Kinetic Studies of Heck Coupling Reactions Using Palladacycle Catalysts: Experimental and Kinetic Modeling of the Role of Dimer Species. J. Am. Chem. Soc. 2001, 123, 1848. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, K.; Shiraishi, R. Clean and efficient condensation reactions of aldehydes and amines in a water suspension medium. Green Chem. 2000, 2, 272. [Google Scholar] [CrossRef]
- Onoue, H.; Moritani, I. ortho-Metalation reactions of N-phenylbenzaldimine and its related compounds by palladium(II) acetate. J. Organomet. Chem. 1972, 43, 431. [Google Scholar] [CrossRef]
- Ustinyuk, Y.A.; Chertov, V.A.; Barinov, I.V. The interaction of nickelocene with benzal anilines. J. Organomet. Chem. 1971, 29, C53. [Google Scholar] [CrossRef]
- Nakamoto, K. Infrared Raman Spectra Inorg. Coord. Compd., 6th ed.; Wiley: New York, NY, USA, 2009. [Google Scholar]
- Yap, J.S.L.; Li, B.B.; Wong, J.; Li, Y.; Pullarkat, S.A.; Leung, P.-H. Development of a novel chiral palladacycle and its application in asymmetric hydrophosphination reaction. Dalt. Trans. 2014, 43, 5777. [Google Scholar] [CrossRef] [PubMed]
- Fernández, A.; Pereira, E.; Fernández, J.J.; López-Torres, M.; Suárez, A.A.; Mosteiro, R.; Pereira, M.T.; Vila, J.J. Sterically controlled reactivity of palladium(ii) tetranuclear cyclometallated complexes. Crystal and molecular structure of the novel tetranuclear compound [Pd2{1,3-[C(H)=NCH2C4H7O]2C6H2}(μ-Cl)(Cl)(PPh3)]2. New J. Chem. 2002, 26, 895. [Google Scholar] [CrossRef]
- Pregosin, P.S.; Kunz, R.W. 31P 13C NMR of Transition Met. Phosphine Complexes, NMR 16; Diehl, P., Fluck, E., Kosfeld, R., Eds.; Springer: Berlin, Germany, 1979. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
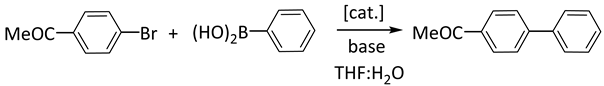 | |||||||||
| Entry | [cat.] | T (h) | T (°C) | Yield c | Entry | [cat.] | T (h) | T (°C) | Yield c |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 1b | 5 | 80 | 100 | 20 | 4b | 5 | rt | 47 |
| 2 | 1c | 5 | 80 | 98 | 21 | 4c | 5 | rt | 52 |
| 3 | 2aCl | 5 | 80 | 100 | 22 | 5a | 24 | 40 | 18 |
| 4 | 2aBr | 0.5 | 80 | 100 | 23 | 5a | 24 | 80 | 100 |
| 5 | 2aBr | 2 | rt | 100 | 24 | 5a | 24 | rt | 0 |
| 6 | 2bCl | 5 | 80 | 100 | 25 | 5b | 24 | 80 | 66 |
| 7 | 2bCl | 1 | rt | 80 | 26 | 5b | 24 | rt | 0 |
| 8 | 2bBr | 1 | rt | 83 | 27 | 6a | 24 | 80 | 33 |
| 9 | 2cCl | 2 | rt | 88 | 28 | 6a | 24 | rt | 0 |
| 10 | 3a | 1 | 80 | 100 | 29 | 6b | 24 | 80 | 20 |
| 11 | 3a | 1 | rt | 35 | 30 | 6b | 24 | rt | 0 |
| 12 | 3a | 3 | rt | 100 | 31 | 7a | 5 | 80 | 100 |
| 13 | 3b | 1 | rt | 37 | 32 | 7a | 24 | rt | 99 |
| 14 | 3b | 3 | rt | 90 | 33 | 7b | 5 | 80 | 100 |
| 15 | 3b | 1 | rt | 37 | 34 | 7b | 24 | rt | 95 |
| 16 | 3b | 3 | rt | 94 | 35 | 8a | 5 | 80 | 95 |
| 17 | 4a | 2 | 80 | 83 | 36 | 8b | 5 | 80 | 90 |
| 18 | 4a | 24 | rt | 100 | 37 | 8b | 24 | rt | 3 |
| 19 | 4b | 5 | 80 | 90 | 38 | 9b | 24 | 80 | 15 |
| Entry | Base | Solvent b | T (°C) | Yield c |
|---|---|---|---|---|
| 1 | K2CO3 | Toluene | 80 | 65 |
| 2 | K2CO3 | THF:H2O d | 80 | 100 |
| 3 | K2CO3 | H2O | 80 | 91 |
| 4 | K2CO3 | EtOH:H2O e | 80 | 100 |
| 5 | K2CO3 | THF:H2O c | rt | 41 |
| 6 | K3PO4 | THF:H2O c | rt | 41 |
| 7 | K2CO3 | EtOH:H2O d | rt | 100 |
| 8 | K3PO4 | EtOH:H2O d | rt | 100 |
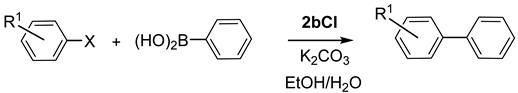 | ||||||
| Entry | Aryl Halide | Product | No. | T (h) | T (°C) | Yield b |
|---|---|---|---|---|---|---|
| 1 | 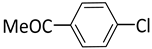 | 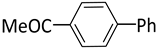 | 1 | 24 | 100 | 98 |
| 2 | 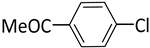 | 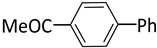 | 1 | 24 | rt | 57 |
| 3 | 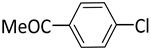 | 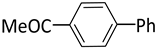 | 1 | 48 | rt | 64 |
| 4 | 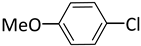 | 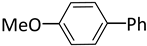 | 2 | 24 | 100 | 13 |
| 5 | 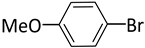 | 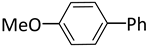 | 2 | 24 | rt | 100 |
| 6 | 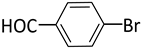 | 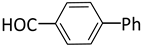 | 3 | 1 | 100 | 100 |
| 7 | 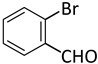 | 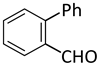 | 4 | 1 | 100 | 100 |
| 8 | 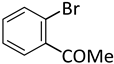 | 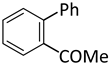 | 5 | 1 | 100 | 100 |
| 9 | 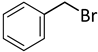 | 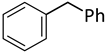 | 6 | 4 | 100 | 98 |
| 10 c | 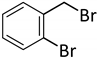 | 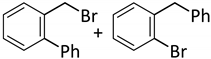 | 7 | 4 | 100 | 60/33 (7/8) |
| 11 c | 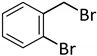 | 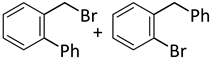 | 7 | 24 | 100 | 60/33 (7/8) |
| 12 d | 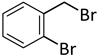 | 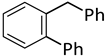 | 8 | 4 | 100 | 33/62 (8/7) |
| 13 d | 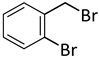 | 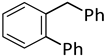 | 8 | 24 | 100 | 53/38 (8/7) |
| 14 d | 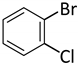 | 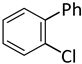 | 9 | 2 | 100 | 88 |
 | |||||
| Entry | [cat.] | [cat.] mol% | T (h) | T (°C) | Yield c |
|---|---|---|---|---|---|
| 1 | 2bCl | 2% | 24 | 100 | 98 |
| 2 | 2bCl | 2% | 48 | rt | 64 |
| 3 | Pd(OAc)2 | 4% | 24 | 100 | 64 |
| 4 b | Pd(OAc)2 + b | 4% | 24 | 100 | 80 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bermúdez-Puente, B.; Adrio, L.A.; Lucio-Martínez, F.; Reigosa, F.; Ortigueira, J.M.; Vila, J.M. Imine Palladacycles: Synthesis, Structural Analysis and Application in Suzuki–Miyaura Cross Coupling in Semi-Aqueous Media. Molecules 2022, 27, 3146. https://doi.org/10.3390/molecules27103146
Bermúdez-Puente B, Adrio LA, Lucio-Martínez F, Reigosa F, Ortigueira JM, Vila JM. Imine Palladacycles: Synthesis, Structural Analysis and Application in Suzuki–Miyaura Cross Coupling in Semi-Aqueous Media. Molecules. 2022; 27(10):3146. https://doi.org/10.3390/molecules27103146
Chicago/Turabian StyleBermúdez-Puente, Brais, Luis A. Adrio, Fátima Lucio-Martínez, Francisco Reigosa, Juan M. Ortigueira, and José M. Vila. 2022. "Imine Palladacycles: Synthesis, Structural Analysis and Application in Suzuki–Miyaura Cross Coupling in Semi-Aqueous Media" Molecules 27, no. 10: 3146. https://doi.org/10.3390/molecules27103146
APA StyleBermúdez-Puente, B., Adrio, L. A., Lucio-Martínez, F., Reigosa, F., Ortigueira, J. M., & Vila, J. M. (2022). Imine Palladacycles: Synthesis, Structural Analysis and Application in Suzuki–Miyaura Cross Coupling in Semi-Aqueous Media. Molecules, 27(10), 3146. https://doi.org/10.3390/molecules27103146