
Eco-Friendly, Simple, Fast, and Sensitive UPLC-MS/MS Method for Determination of Pexidartinib in Plasma and Its Application to Metabolic Stability

 ,
,  ,
,
Abstract
:1. Introduction
2. Results and Discussion
2.1. Method Validation
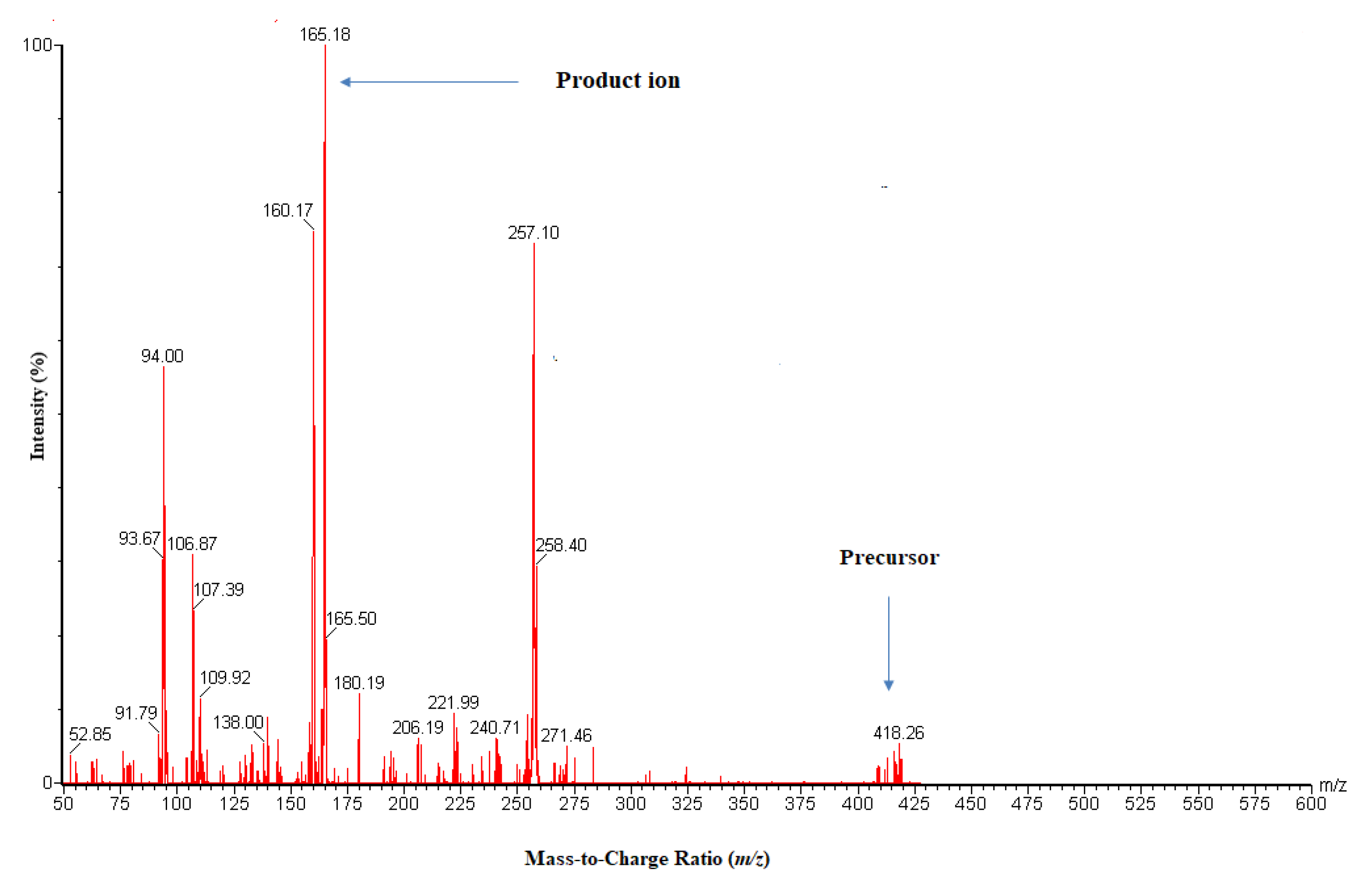
2.1.1. Optimization of Mass Spectrometry Conditions
2.1.2. Optimization of Extraction and Chromatographic Methods
2.1.3. Selectivity and Specificity
2.1.4. LLOQ and the Linearity of Plasma Calibration Curve
2.1.5. Accuracy and Precision
2.1.6. Recovery and Matrix Effects
2.1.7. Stability
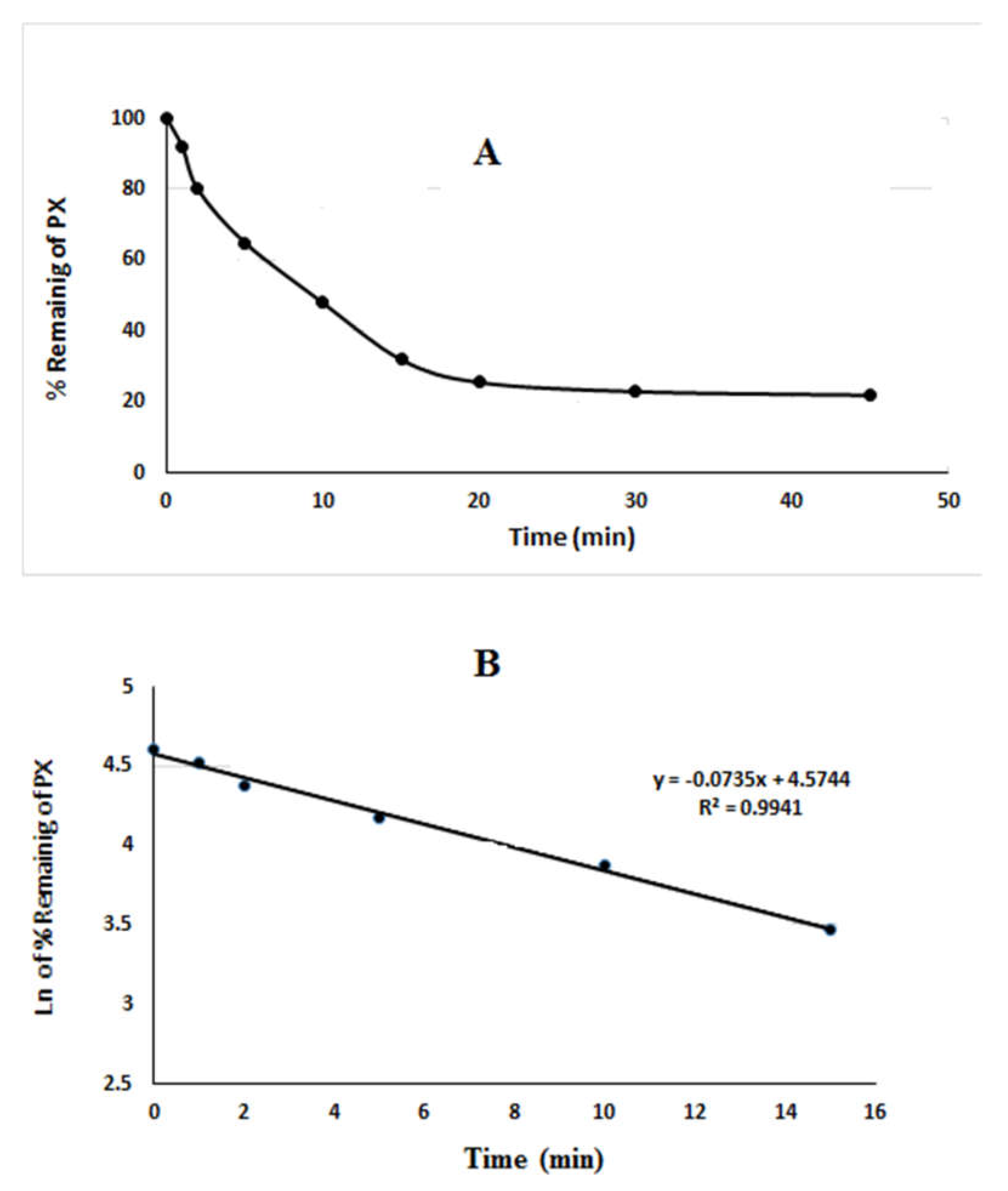
2.2. In Vitro Metabolic Stability Study
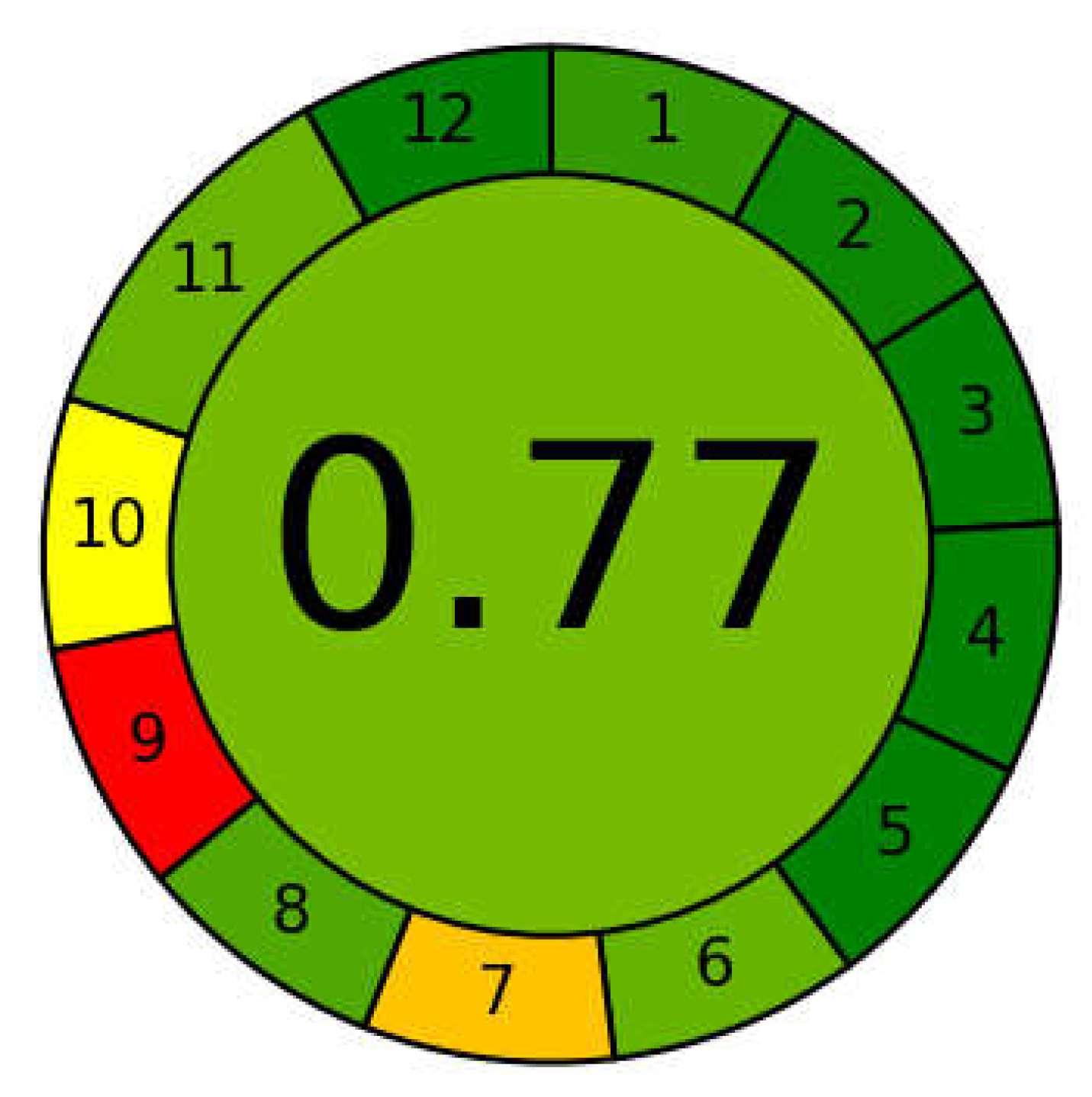
2.3. Greenness of the Method
3. Materials and Methods
3.1. Chemicals and Reagents
3.2. Stock Solution, Calibration Standards, and Quality Control Sample Preparation
3.3. Sample Preparation
3.4. Chromatographic Conditions
3.5. Mass Spectrometry
3.6. Method Validation
3.7. In Vitro Metabolic Stability
3.8. Greenness of the Method
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Benner, B.; Good, L.; Quiroga, D.; Schultz, T.E.; Kassem, M.; Carson, W.E.; Cherian, M.A.; Sardesai, S.; Wesolowski, R. Pexidartinib, a Novel Small Molecule CSF-1R Inhibitor in Use for Tenosynovial Giant Cell Tumor: A Systematic Review of Pre-Clinical and Clinical Development. Drug Des. Dev. Ther. 2020, 14, 1693–1704. [Google Scholar] [CrossRef]
- Palmerini, E.; Longhi, A.; Donati, D.M.; Staals, E.L. Pexidartinib for the treatment of adult patients with symptomatic tenosynovial giant cell tumor: Safety and efficacy. Expert Rev. Anticancer Ther. 2020, 20, 441–445. [Google Scholar] [CrossRef]
- Lamb, Y.N. Pexidartinib: First Approval. Drugs 2019, 79, 1805–1812. [Google Scholar] [CrossRef]
- FDA Approves Pexidartinib for Tenosynovial Giant Cell Tumor. U.S. Food & Drug Administration Web Site; 2019. Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-pexidartinib-tenosynovial-giant-cell-tumor (accessed on 4 March 2021).
- Sosna, J.; Philipp, S.; Albay, R.; Reyes-Ruiz, J.M.; Baglietto-Vargas, D.; LaFerla, F.M.; Glabe, C.G. Early long-term administration of the CSF1R inhibitor PLX3397 ablates microglia and reduces accumulation of intraneuronal amyloid, neuritic plaque deposition and pre-fibrillar oligomers in 5XFAD mouse model of Alzheimer’s disease. Mol. Neurodegener. 2018, 13, 11. [Google Scholar] [CrossRef]
- Lee, J.H.; Chen, T.W.-W.; Hsu, C.-H.; Yen, Y.-H.; Yang, J.C.-H.; Cheng, A.-L.; Sasaki, S.I.; Chiu, L.L.; Sugihara, M.; Ishizuka, T.A. Phase I study of pexidartinib, a colony-stimulating factor 1 receptor inhibitor, in Asian patients with advanced solid tumors. Investig. New Drugs 2020, 38, 99–110. [Google Scholar] [CrossRef] [Green Version]
- Gelderblom, H.; de Sande, M.V. Pexidartinib: First approved systemic therapy for patients with tenosynovial giant cell tumor. Future Oncol. 2020, 16, 2345–2356. [Google Scholar] [CrossRef]
- Lewis, J.H.; Gelderblom, H.; Van de Sande, M.; Stacchiotti, S.; Healey, J.H.; Tap, W.D.; Wagner, A.J.; Pousa, A.L.; Druta, M.; Lin, C.C. Pexidartinib long-term hepatic safety profile in TGCT patients. Oncologist 2020, 26, e863–e873. [Google Scholar] [CrossRef]
- Tap, W. ENLIVEN study: Pexidartinib for tenosynovial giant cell tumor (TGCT). Future Oncol. 2020, 16, 1875–1878. [Google Scholar] [CrossRef]
- Tap, W.D.; Gelderblom, H.; Palmerini, E.; Desai, J.; Bauer, S.; Blay, J.-Y.; Alcindor, T.; Ganjoo, K.; Martín-Broto, J.; Ryan, C.W. Pexidartinib versus placebo for advanced tenosynovial giant cell tumour (ENLIVEN): A randomised phase 3 trial. Lancet 2019, 394, 478–487. [Google Scholar] [CrossRef]
- Zahir, H.; Kobayash, I.F.; Zamora, C.; Gajee, R.; Gordon, M.S.; Babikr, H.M.; Wang, Q.; Greenberg, J.; Wagner, A.J. Evaluation of Potential Drug-Drug Interaction Risk of Pexidartinib with Substrates of Cytochrome P450 and P-Glycoprotein. J. Clin. Pharmacol. 2021, 61, 298–306. [Google Scholar] [CrossRef]
- Shi, L.; Jiang, Z.; Zhang, B.; Tang, C.; Xu, R.A. Development of UPLC-MS/MS method for studying the pharmacokinetic interactions of pexidartinib with antifungal drugs in rats. J. Pharm. Biomed. Anal. 2020, 188, 113386. [Google Scholar] [CrossRef]
- Manji, G.A.; Van Tine, B.A.; Lee, S.M.; Raufi, A.G.; Pellicciotta, I.; Hirbe, A.C.; Pradhan, J.; Chen, A.; Rabadan, R.; Schwartz, G.K. A Phase I Study of the Combination of Pexidartinib and Sirolimus to Target Tumor-Associated Macrophages in Unresectable Sarcoma and Malignant Peripheral Nerve Sheath Tumors. Clin. Cancer Res. 2021, 27, 5519–5527. [Google Scholar] [CrossRef]
- Wesolowski, R.; Sharma, N.; Reebel, L.; Rodal, M.B.; Peck, A.; West, B.L.; Marimuthu, A.; Severson, P.; Karlin, D.A.; Dowlati, A.; et al. Phase Ib study of the combination of pexidartinib (PLX3397), a CSF-1R inhibitor, and paclitaxel in patients with advanced solid tumors. Ther. Adv. Med. Oncol. 2019, 11. [Google Scholar] [CrossRef]
- Boal, L.H.; Glod, J.; Spencer, M.; Kasai, M.; Derdak, J.; Dombi, E.; Ahlman, M.; Beury, D.W.; Merchant, M.S.; Persenaire, C.; et al. Pediatric PK/PD Phase I Trial of Pexidartinib in Relapsed and Refractory Leukemias and Solid Tumors Including Neurofibromatosis Type I-Related Plexiform Neurofibromas. Clin. Cancer Res. 2020, 26, 6112–6121. [Google Scholar] [CrossRef]
- Panchal, P.; Mochi, N.; Patel, H.U.; Patel, O. Stability Indicating Analytical Method Development and Validation for The Estimation of Pexidartinib in Pharmaceutical Dosage Form. World J. Pharm. Pharm. Sci. 2020, 9, 1161–1173. [Google Scholar]
- Yin, O.; Wagner, A.J.; Kang, J.; Knebel, W.; Zahir, H.; van de Sande, M.; Tap, W.D.; Gelderblom, H.; Healey, J.H.; Shuster, D.; et al. Population Pharmacokinetic Analysis of Pexidartinib in Healthy Subjects and Patients with Tenosynovial Giant Cell Tumor or Other Solid Tumors. J. Clin. Pharmacol. 2021, 61, 480–492. [Google Scholar] [CrossRef]
- Shankarappa, P.S.; Peer, C.J.; Odabas, A.; McCully, C.L.; Garcia, R.C.; Figg, W.D.; Warren, K.E. Cerebrospinal fluid penetration of the colony-stimulating factor-1 receptor (CSF-1R) inhibitor, pexidartinib. Cancer Chemother. Pharmacol. 2020, 85, 1003–1007. [Google Scholar] [CrossRef]
- Liang, Y.; Guan, T.; Zhou, Y.; Liu, Y.; Xing, L.; Zheng, X.; Dai, C.; Du, P.; Rao, T.; Zhou, L.; et al. Effect of mobile phase additives on qualitative and quantitative analysis of ginsenosides by liquid chromatography hybrid quadrupole-time of flight mass spectrometry. J. Chromatogr. A 2013, 1297, 29–36. [Google Scholar] [CrossRef]
- Lu, X.; Aurand, C.; Bell, D.S. Impact of Mobile Phase Additives on LC-MS Sensitivity, Demonstrated using Spice Cannabinoids. Report. US 2012, 30, 8–10. Available online: https://www.sigmaaldrich.com/SA/en/technical-documents/technical-article/analytical-chemistry/small-molecule-hplc/lcms-spice-cannabinoids (accessed on 3 February 2021).
- Pitt, J.J. Principles and applications of liquid chromatography-mass spectrometry in clinical biochemistry. Clin. Biochem. Rev. 2009, 30, 19–34. [Google Scholar]
- Ezzeldin, E.; Iqbal, M.; Herqash, R.N.; ElNahhas, T. Simultaneous quantitative determination of seven novel tyrosine kinase inhibitors in plasma by a validated UPLC-MS/MS method and its application to human microsomal metabolic stability study. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2020, 1136, 121851. [Google Scholar] [CrossRef]
- Scott, W.J.; Hentemann, M.F.; Rowley, R.B.; Bull, C.O.; Jenkins, S.; Bullion, A.M.; Johnson, J.; Redman, A.; Robbins, A.H.; Esler, W.; et al. Discovery and SAR of Novel 2,3-Dihydroimidazo[1, 2-c]quinazoline PI3K Inhibitors: Identification of Copanlisib (BAY 80-6946). ChemMedChem 2016, 11, 1517–1530. [Google Scholar] [CrossRef] [Green Version]
- Attwa, M.W.; Kadi, A.A.; Darwish, H.W.; Abdelhameed, A.S. Investigation of the metabolic stability of olmutinib by validated LC-MS/MS: Quantification in human plasma. RSC Adv. 2018, 8, 40387–40394. [Google Scholar] [CrossRef] [Green Version]
- Van Aken, K.; Strekowski, L.; Patiny, L. EcoScale, a semi-quantitative tool to select an organic preparation based on economical and ecological parameters. Beilstein J. Org. Chem. 2006, 2, 3. [Google Scholar] [CrossRef] [Green Version]
- Emergency Planning and Community Right-to-Know Act. United States Environmental Protection Agency Web Site. Available online: https://www.epa.gov/epcra#:~:text=The%20Emergency%20Planning%20and%20Community%2C%20state%2C%20and%20local%20governments (accessed on 25 February 2021).
- Sargent, M. (Ed.) Guide to Achieving Reliable Quantitative LC-MS Measurements, RSC Analytical Methods Committee, First Edition. Royal Society of Chemistry Web Site. 2013. Available online: https://www.rsc.org/images/AMC%20LCMS%20Guide_tcm18-240030.pdf (accessed on 25 February 2021).
- Center for Drug Evaluation and Research (CDER). Guidance for Industry on Bioanalytical Method Validation. U.S. Food & Drug Administration Web Site; 2018. Available online: https://www.fda.gov/downloads/drugs/guidances/ucm070107.pdf (accessed on 25 February 2021).
- Baranczewski, P.; Stanczak, A.; Sundberg, K.; Svensson, R.; Wallin, A.; Jansson, J.; Garberg, P.; Postlind, H. Introduction to in vitro estimation of metabolic stability and drug interactions of new chemical entities in drug discovery and development. Pharmacol. Rep. 2006, 58, 453–472. [Google Scholar]
- Hill, J.R. In vitro drug metabolism using liver microsomes. Curr. Protoc. Pharmacol. 2004, 7, 7.8.1–7.8.11. [Google Scholar] [CrossRef]
- Asha, S.; Vidyavathi, M. Role of human liver microsomes in in vitro metabolism of drugs-a review. Appl. Biochem. Biotechnol. 2010, 160, 1699–1722. [Google Scholar] [CrossRef]
- Manzo, A.; Montanino, A.; Costanzo, R. EGFR mutations: Best results from second- and third-generation tyrosine kinase inhibitors. In Oncogenomics; Dammacco, F., Silvestris, F., Eds.; Academic Press: Cambridge, MA, USA, 2019; pp. 477–486. [Google Scholar]
- Pena-Pereira, F.; Wojnowski, W.; Tobiszewski, M. AGREE-Analytical GREEnness Metric Approach and Software. Anal. Chem. 2020, 92, 10076–10082. [Google Scholar] [CrossRef]
- Tobiszewski, M.; Marć, M.; Gałuszka, A.; Namieśnik, J. Green Chemistry Metrics with Special Reference to Green Analytical Chemistry. Molecules 2015, 20, 10928–10946. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameters | PX | Gifitinib (IS) |
|---|---|---|
| I. Compound Parameters | ||
| Precursor | 418.06 | 447.09 |
| Product ion | 165.0 | 128.0 |
| Dwell time (s) | 0.005 | 0.05 |
| Cone voltage (V) | 50 | 40 |
| Collision energy eV | 34 | 24 |
| II. Instrument Parameters | ||
| Collision gas flow rate (mL/min) | 0.1 | 0.1 |
| Nitrogen flow rate | 600 L/h | 600 L/h |
| Source temperature (°C) | 150 | 150 |
| Desolvation temperature (°C) | 350 | 350 |
| Conc. (ng/mL) | Interday | Intraday | ||||
|---|---|---|---|---|---|---|
| Mean ± SD | Precision (CV %) | Accuracy (%) | Mean ± SD | Precision (CV %) | Accuracy (%) | |
| 0.5 | 0.41 ± 0.04 | 9.75 | 82.57 | 0.42 ± 0.033 | 7.85 | 82.80 |
| 1.5 | 1.39 ± 0.12 | 8.63 | 92.63 | 1.36± 0.09 | 7.14 | 90.66 |
| 150 | 127.85 ± 11.48 | 9.26 | 85.23 | 130.02 ± 3.36 | 2.58 | 86.68 |
| 750 | 664.91 ± 37.10 | 5.58 | 88.65 | 644.16 ± 84.57 | 13.12 | 85.88 |
| Compound | Nominal Conc. (ng/mL) | Extraction Recovery | Matrix Effects | ||
|---|---|---|---|---|---|
| Mean | RSD * | Mean | RSD * | ||
| (%) | (%) | (%) | (%) | ||
| PX | 1.5 | 85.90 | 5.4 | 88.50 | 12.68 |
| 150 | 85.40 | 2.10 | 87.54 | 5.55 | |
| 750 | 81.4 | 13.3 | 89.46 | 10.01 | |
| IS | 100 | 76.5 | 6.5 | 89.20 | 8.90 |
| Stability Type | Nominal Con. (ng/mL) | Measured Con. (ng/mL) | Precision (%) | Accuracy (%) |
|---|---|---|---|---|
| Short-term | 150 | 131.5 ± 9.2 | 7.07 | 87.66 |
| 750 | 652.21 ± 32.44 | 4.97 | 86.96 | |
| Long-term | 150 | 129.9 ± 9.9 | 7.62 | 86.6 |
| 750 | 665.39 ± 63.04 | 9.47 | 88.71 | |
| Thaw and freeze | 150 | 129.8 ± 10.3 | 7.94 | 86.53 |
| 750 | 647.52 ± 73.69 | 11.38 | 86.33 | |
| Auto-sampler (24) h | 150 | 132.5 ± 12.9 | 9.74 | 86.33 |
| 750 | 638.60 ± 41.00 | 6.42 | 85.15 |
| Parameter | Value | Penalty Points |
|---|---|---|
| Dimethyl sulfoxide | <10 mL (g) | 1 |
| Acetonitrile | <10 m L (g) | 4 |
| Methanol | <10 mL (g) | 6 |
| Ammonium acetate | <10 mL (g) | 1 |
| Mmethyl tert-butyl ether | <10 mL (g) | 3 |
| Waste | 1.0 mL /run (g) | 3 |
| Instrument energy | More than 1.5 kw/h | 2 |
| Total penalty points | 20 | |
| Eco-scale score | 80 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ezzeldin, E.; Iqbal, M.; Asiri, Y.A.; Mostafa, G.A.E.; Sayed, A.Y.A. Eco-Friendly, Simple, Fast, and Sensitive UPLC-MS/MS Method for Determination of Pexidartinib in Plasma and Its Application to Metabolic Stability. Molecules 2022, 27, 297. https://doi.org/10.3390/molecules27010297
Ezzeldin E, Iqbal M, Asiri YA, Mostafa GAE, Sayed AYA. Eco-Friendly, Simple, Fast, and Sensitive UPLC-MS/MS Method for Determination of Pexidartinib in Plasma and Its Application to Metabolic Stability. Molecules. 2022; 27(1):297. https://doi.org/10.3390/molecules27010297
Chicago/Turabian StyleEzzeldin, Essam, Muzaffar Iqbal, Yousif A. Asiri, Gamal A. E. Mostafa, and Ahmed Y. A. Sayed. 2022. "Eco-Friendly, Simple, Fast, and Sensitive UPLC-MS/MS Method for Determination of Pexidartinib in Plasma and Its Application to Metabolic Stability" Molecules 27, no. 1: 297. https://doi.org/10.3390/molecules27010297
APA StyleEzzeldin, E., Iqbal, M., Asiri, Y. A., Mostafa, G. A. E., & Sayed, A. Y. A. (2022). Eco-Friendly, Simple, Fast, and Sensitive UPLC-MS/MS Method for Determination of Pexidartinib in Plasma and Its Application to Metabolic Stability. Molecules, 27(1), 297. https://doi.org/10.3390/molecules27010297