
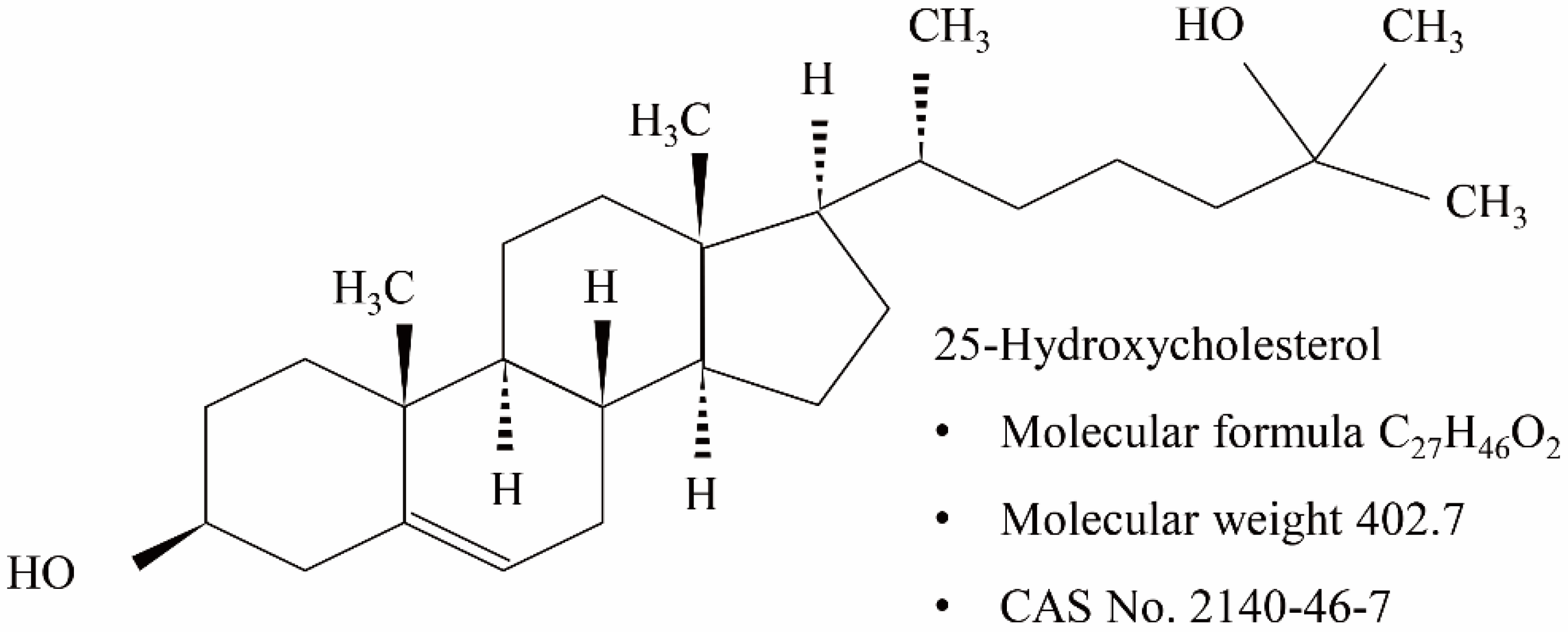
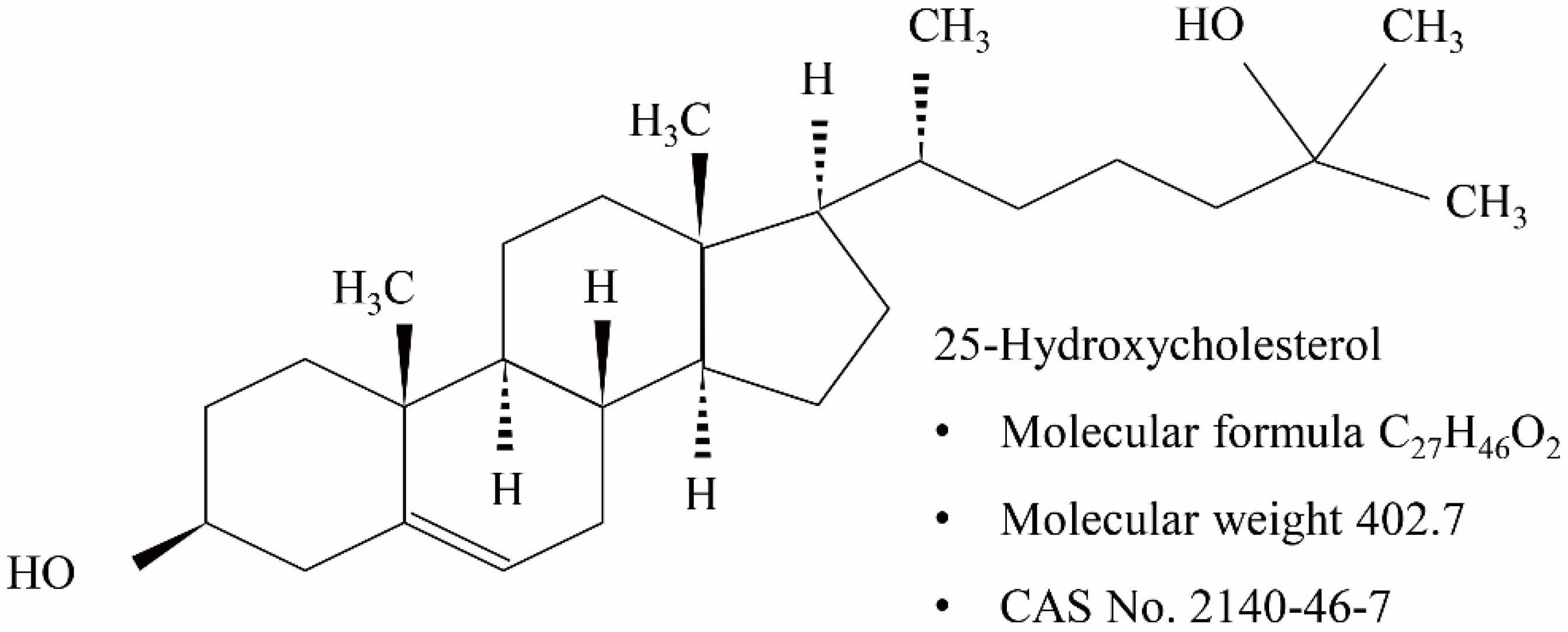
25-Hydroxycholesterol-Induced Oxiapoptophagy in L929 Mouse Fibroblast Cell Line

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
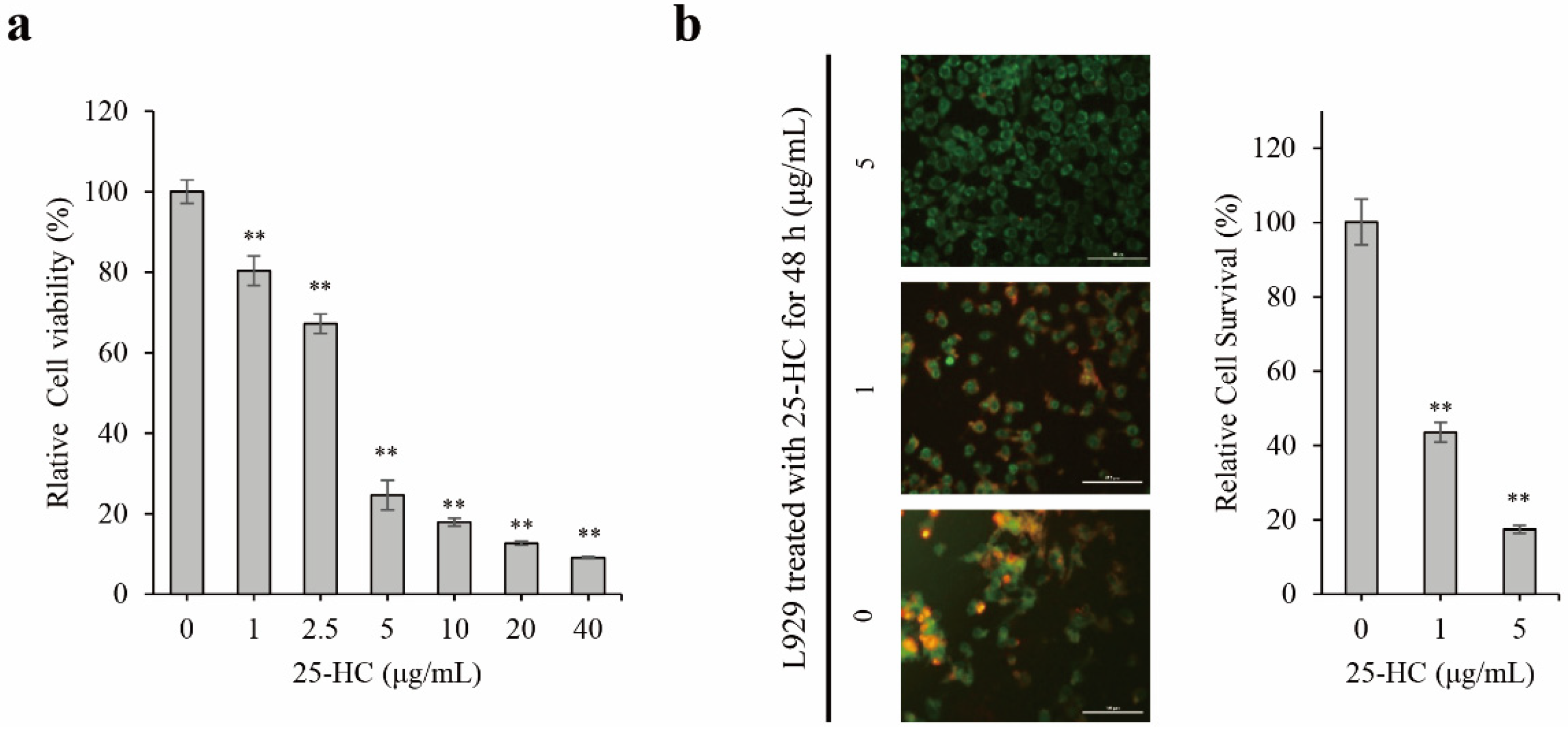
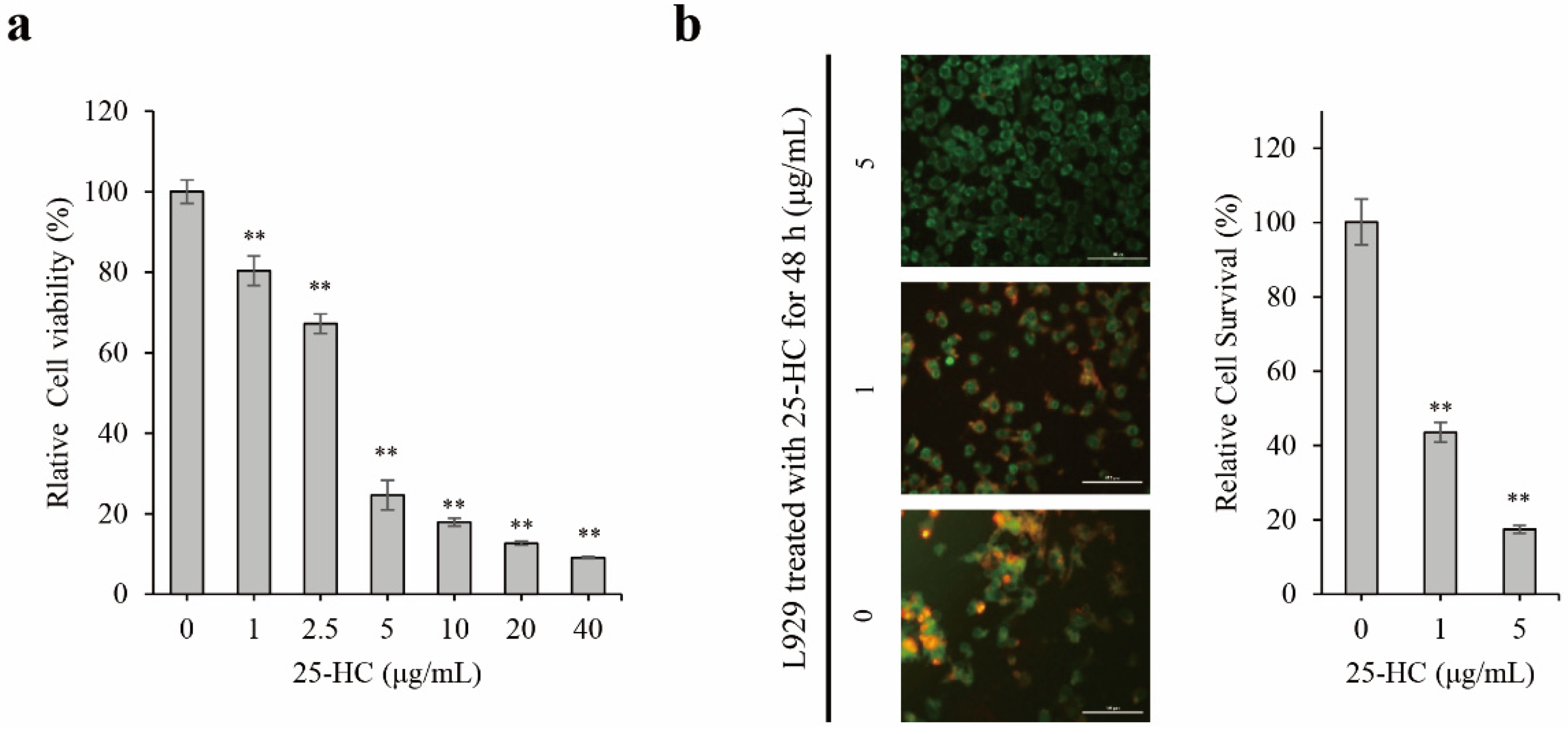
2.1. 25-HC Induced Cell Death through an Increase in Cytotoxicity and Decrease in Cell Survival in L929 Cells
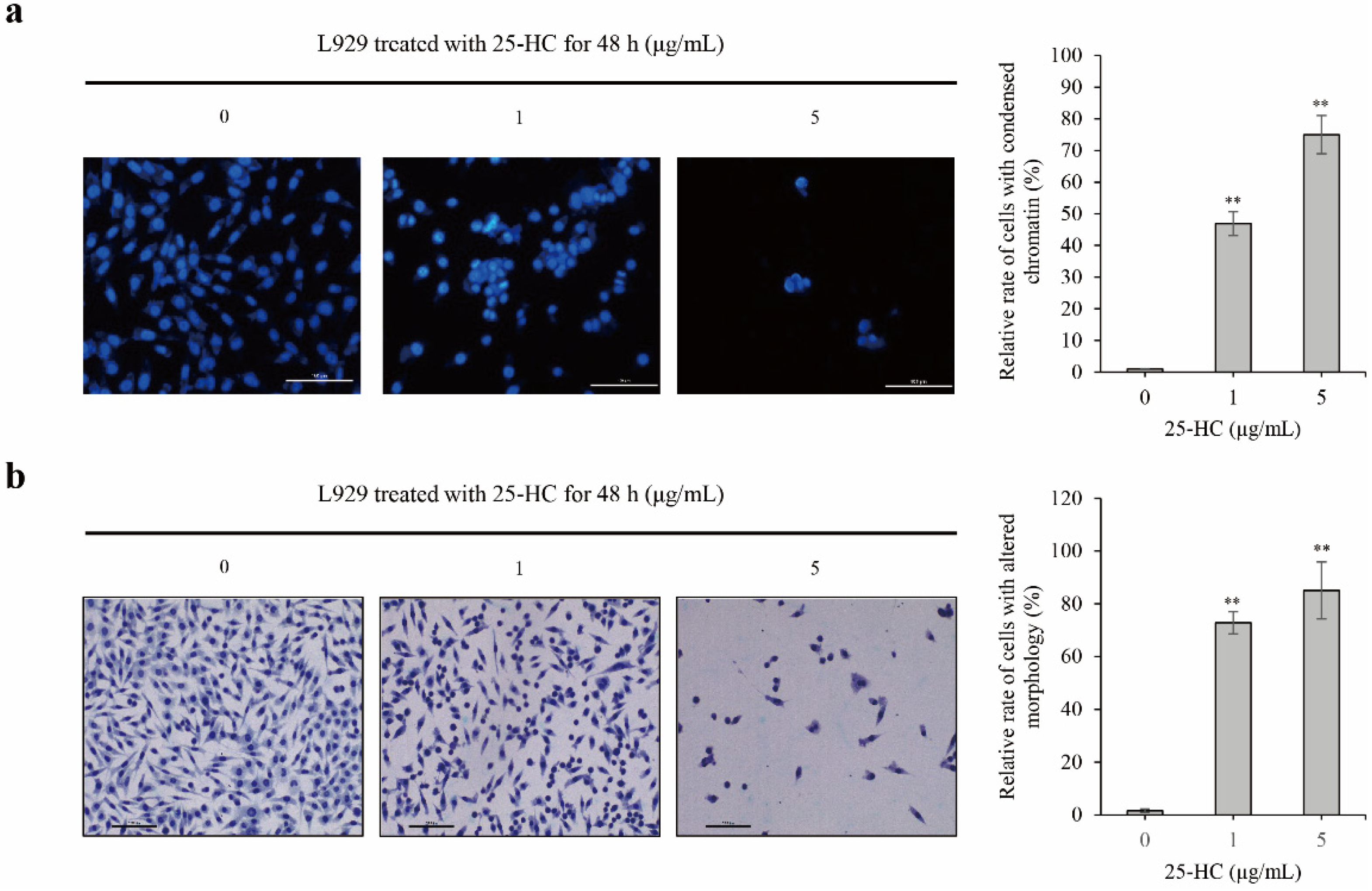
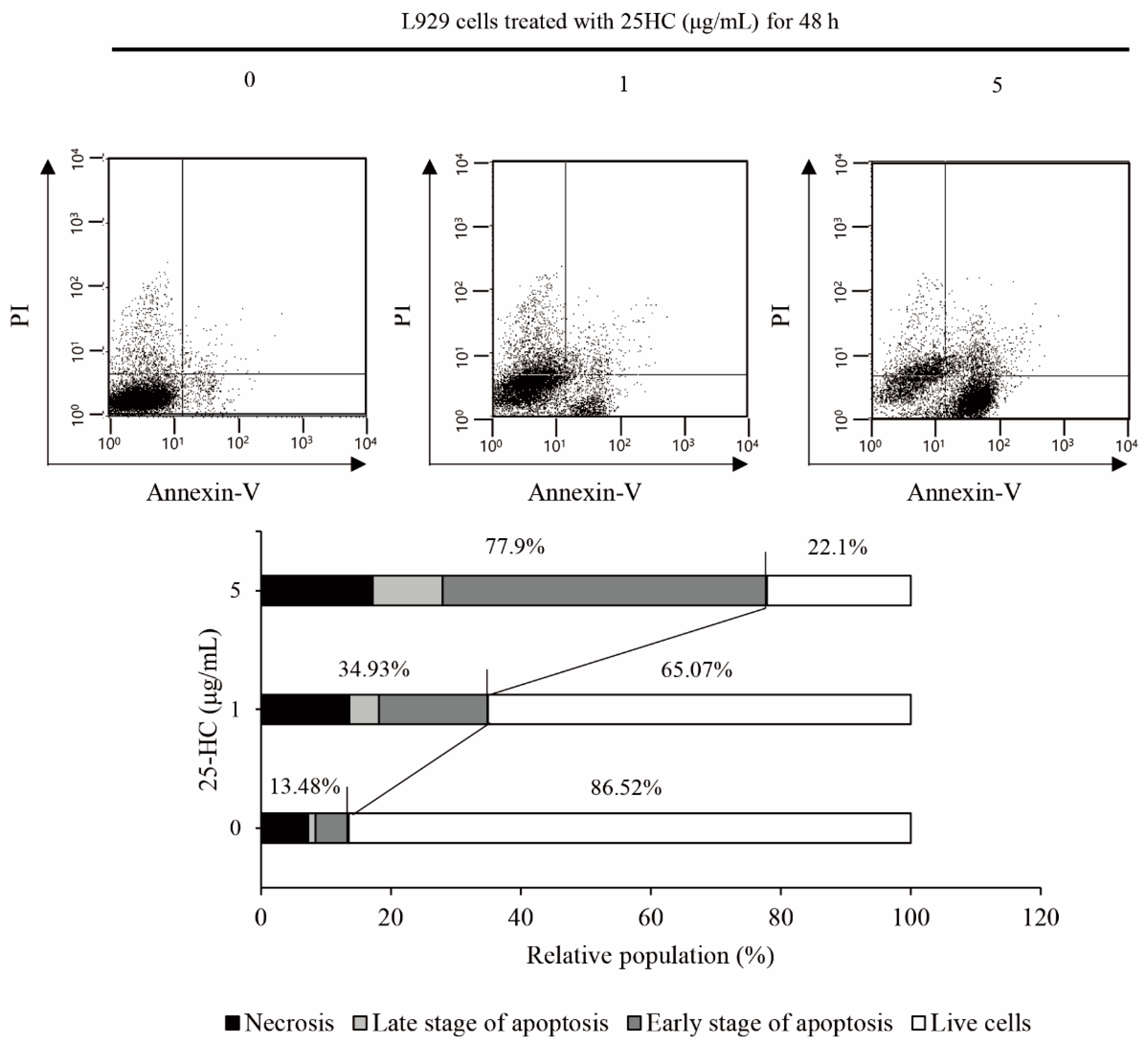
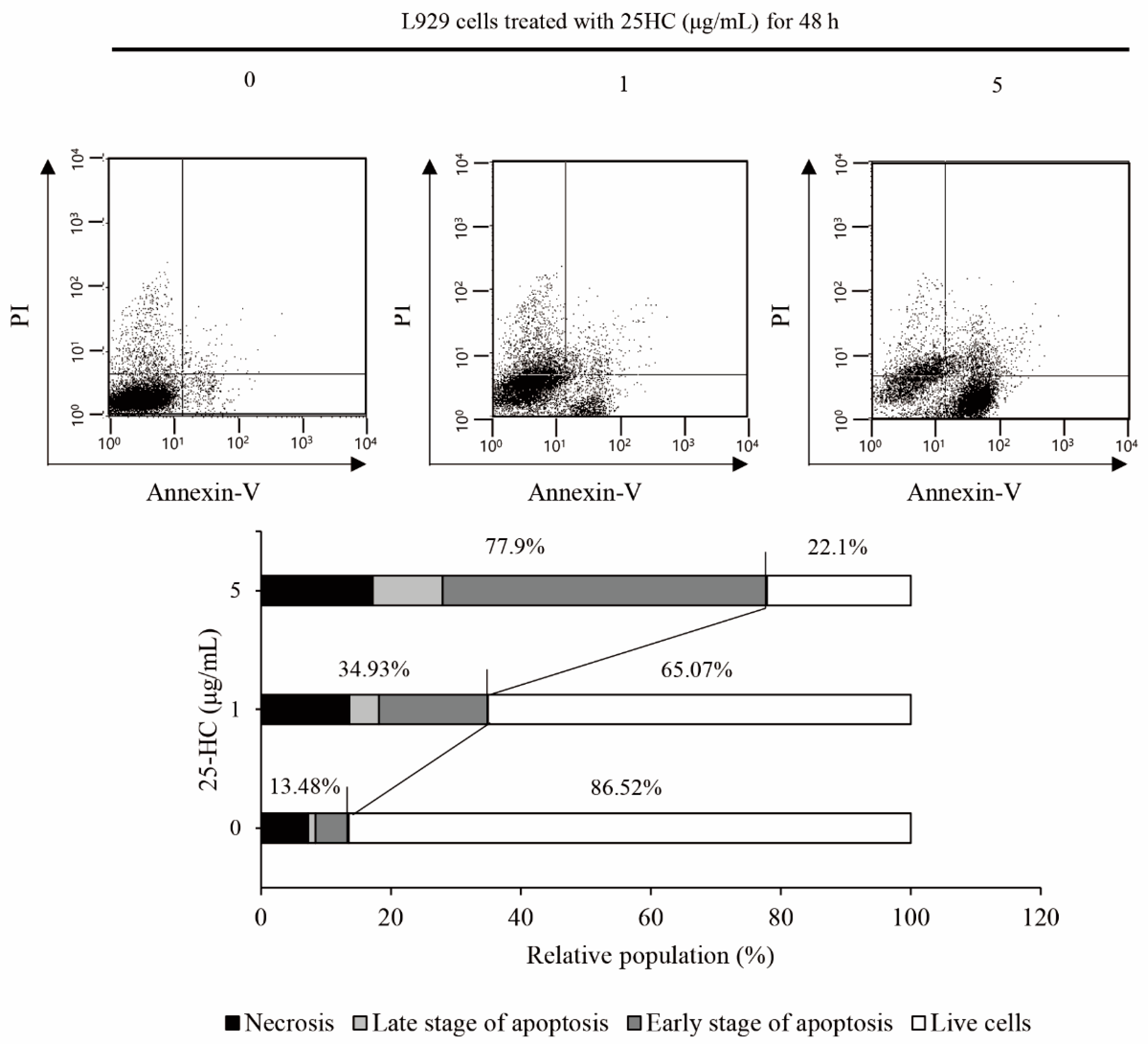
2.2. 25-HC-Induced Cell Death Involved in Apoptosis of L929 Cells
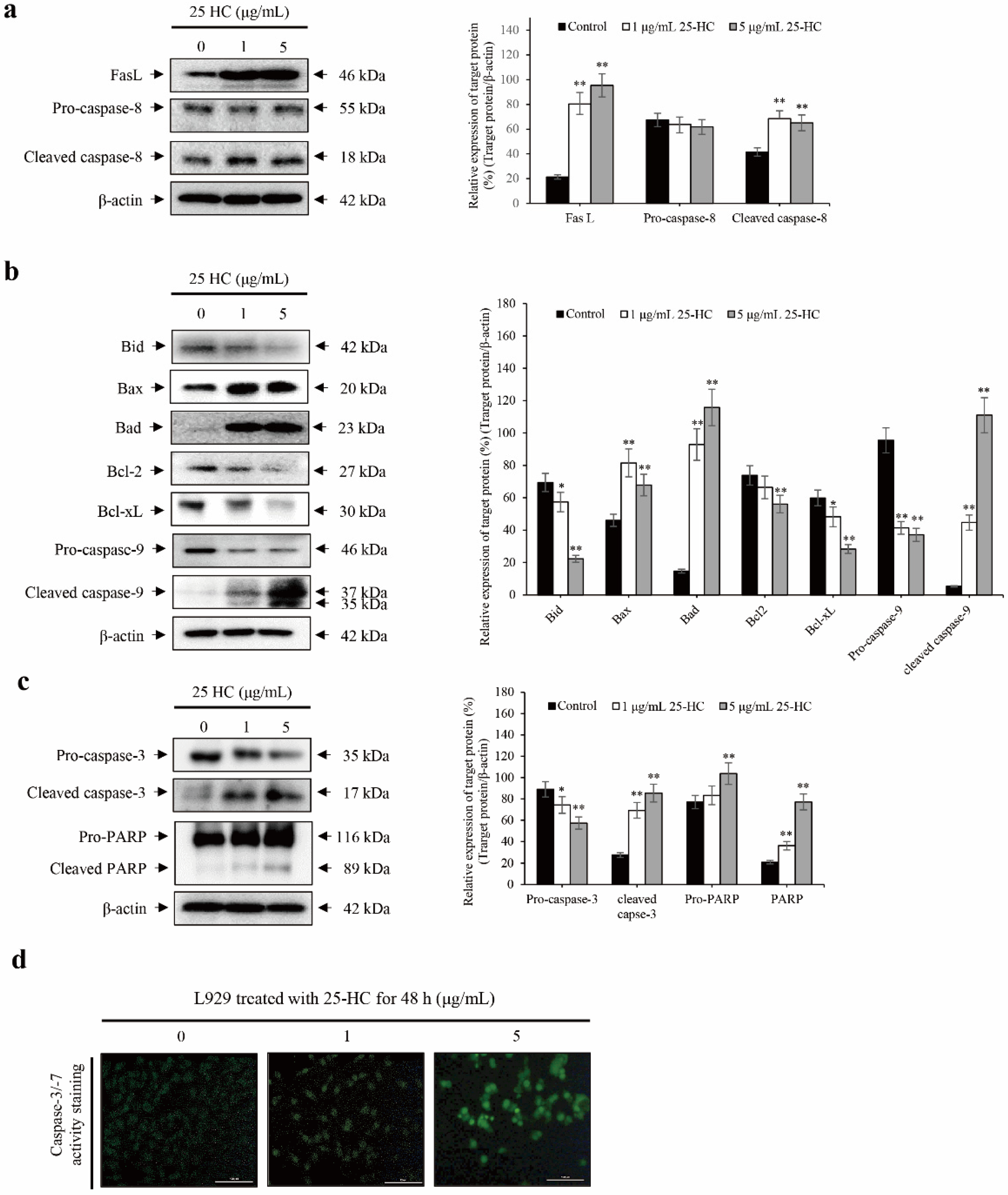
2.3. 25-HC-Induced Cell Death Is Mediated by FasL-Triggered Death Receptor-Dependent Extrinsic and Mitochondria-Dependent Intrinsic Apoptosis Pathways through the Cascade Activation of Caspases in L929 Cells
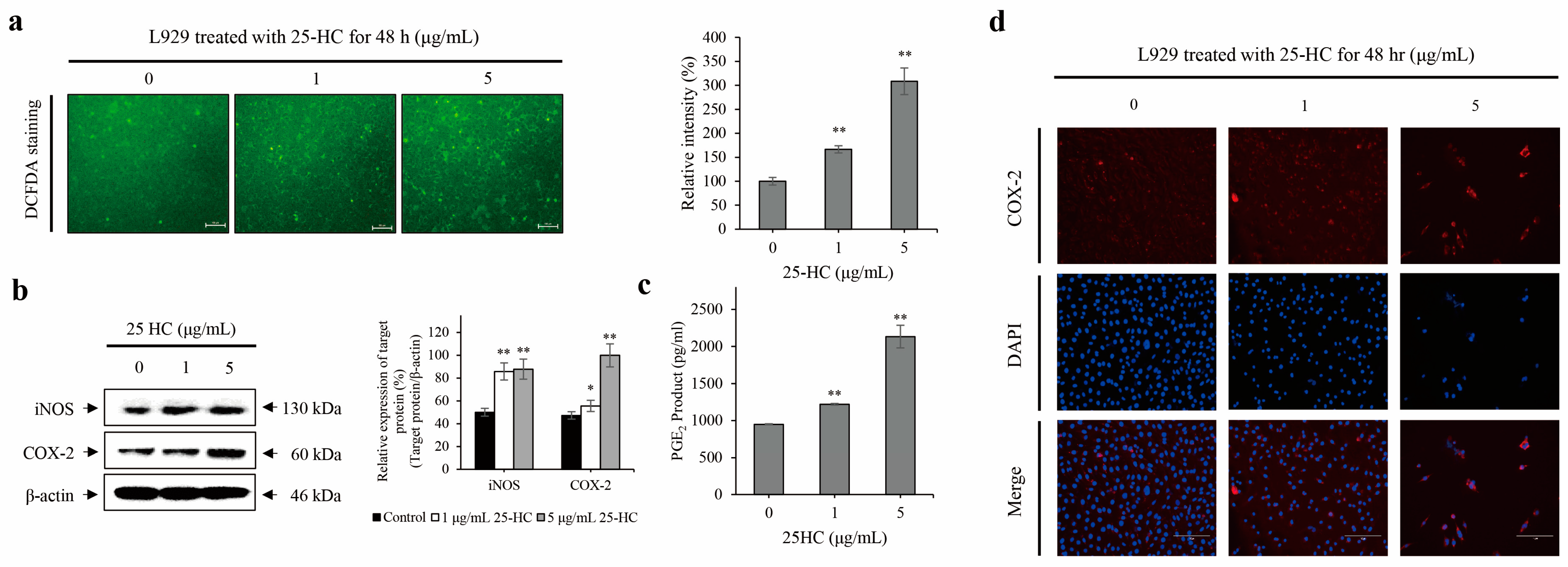
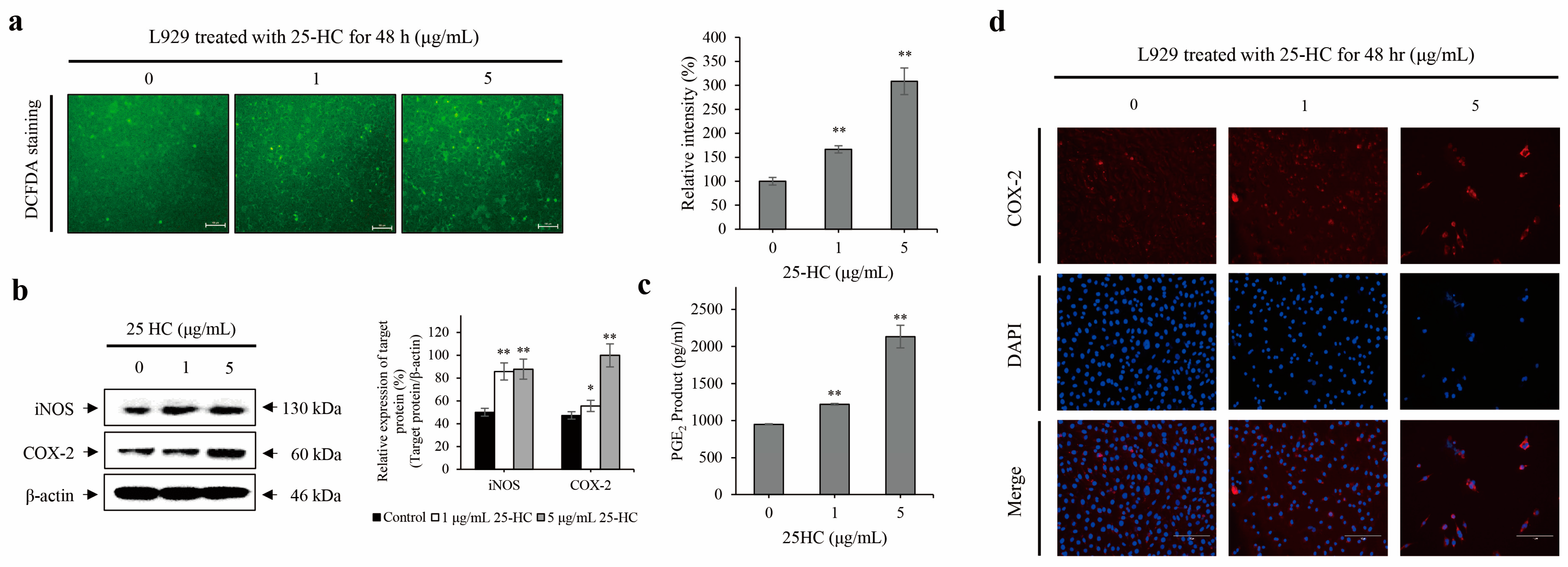
2.4. 25-HC Upregulated the Expression and Production of ROS and Inflammatory Mediators in L929 Cells
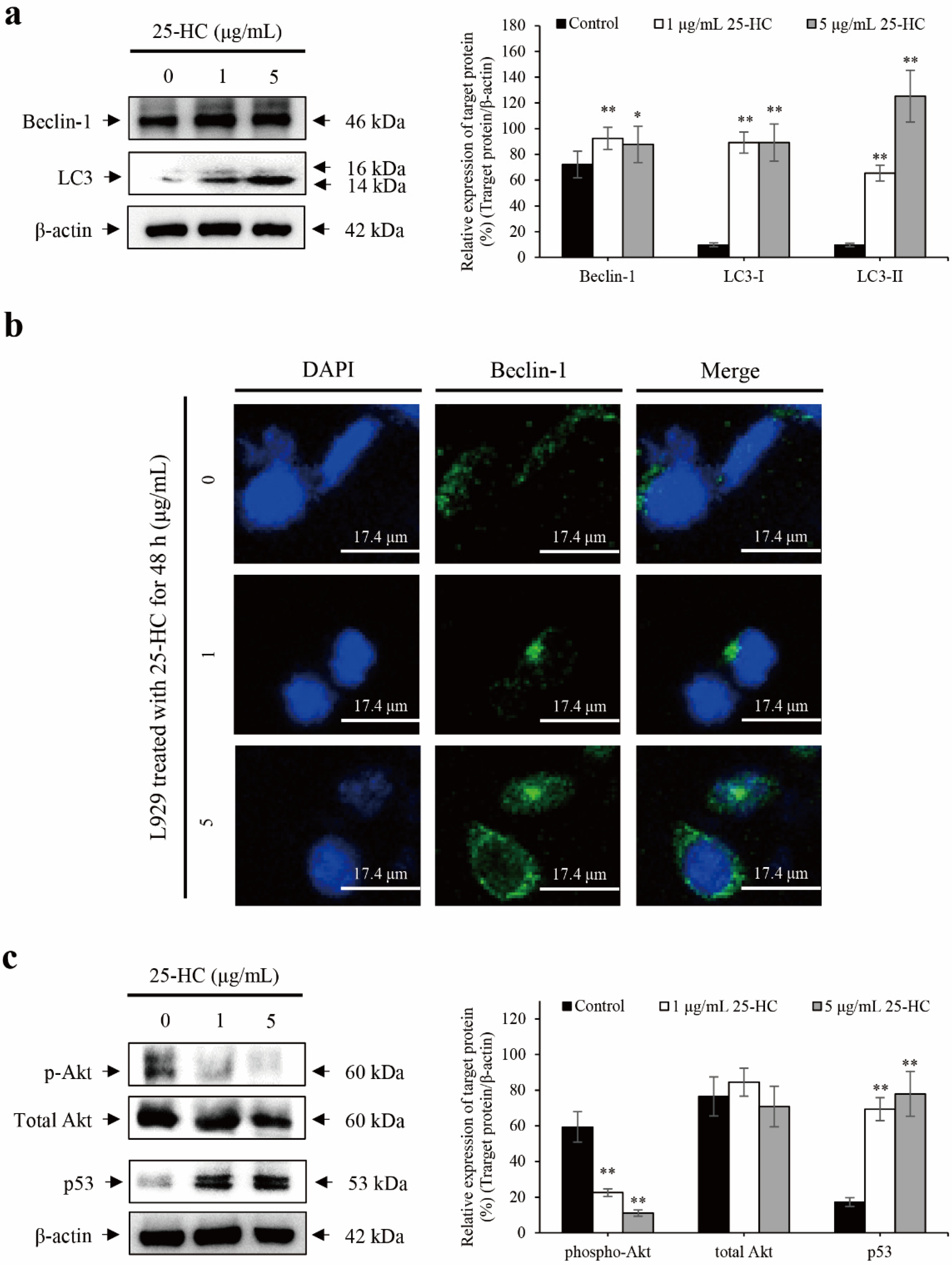
2.5. 25-HC-Induced Apoptosis Is Accompanied by p53-Dependent Autophagy through the Suppression of Akt Phosphorylation in L929 Cells
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Cell Viability Assay
4.3. Cell Survival Assay
4.4. Nuclear Staining
4.5. H&E Staining
4.6. FACS Analysis
4.7. Western Blot
4.8. NO Measurement
4.9. PGE2 Production Measurement
4.10. Detection of ROS
4.11. Caspase-3/-7 Activity Assay
4.12. Laser Confocal Scanning Microscopic Analysis
4.13. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Craig, M.; Yarrarapu, S.N.S.; Dimri, M. Biochemistry, Cholesterol; StatPearls: Treasure Island, FL, USA, 2021. [Google Scholar]
- Luo, J.; Yang, H.; Song, B.-L. Mechanisms and regulation of cholesterol homeostasis. Nat. Rev. Mol. Cell Biol. 2020, 21, 225–245. [Google Scholar] [CrossRef] [PubMed]
- Huang, B.; Song, B.-L.; Xu, C. Cholesterol metabolism in cancer: Mechanisms and therapeutic opportunities. Nat. Metab. 2020, 2, 132–141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, T.-Y.; Chang, C.C.; Ohgami, N.; Yamauchi, Y. Cholesterol Sensing, Trafficking, and Esterification. Annu. Rev. Cell Dev. Biol. 2006, 22, 129–157. [Google Scholar] [CrossRef] [PubMed]
- Reiss, A.B.; Voloshyna, I. Regulation of cerebral cholesterol metabolism in Alzheimer disease. J. Investig. Med. 2012, 60, 576–582. [Google Scholar] [CrossRef] [PubMed]
- Mascitelli, L.; Pezzetta, F.; Goldstein, M.R.; Tuomilehto, J. Total cholesterol and the risk of parkinson disease. Neurology 2009, 72, 860–861. [Google Scholar] [CrossRef]
- Karasinska, J.M.; Hayden, M.R. Cholesterol metabolism in Huntington disease. Nat. Rev. Neurol. 2011, 7, 561–572. [Google Scholar] [CrossRef]
- Reiss, A.B.; Vernice, N.A.; Siegart, N.M.; De Leon, J.; Kasselman, L.J. Exosomes in Cholesterol Metabolism and Atherosclerosis. Cardiovasc. Hematol. Disord. Targets 2018, 17, 185–194. [Google Scholar] [CrossRef]
- van Schie, M.C.; Jainandunsing, S.; van Lennep, J.E.R. Monogenetic disorders of the cholesterol metabolism and premature cardiovascular disease. Eur. J. Pharmacol. 2017, 816, 146–153. [Google Scholar] [CrossRef]
- López-Bautista, F.; Posadas-Sánchez, R.; Vázquez-Vázquez, C.; Fragoso, J.; Rodríguez-Pérez, J.; Vargas-Alarcón, G. IL-37 Gene and Cholesterol Metabolism: Association of Polymorphisms with the Presence of Hypercholesterolemia and Cardiovascular Risk Factors. The GEA Mexican Study. Biomolecules 2020, 10, 1409. [Google Scholar] [CrossRef]
- Notarnicola, M.; Messa, C.; Orlando, A.; D’Attoma, B.; Tutino, V.; Rivizzigno, R.; Caruso, M.G. Effect of genistein on cholesterol metabolism-related genes in a colon cancer cell line. Genes Nutr. 2008, 3, 35–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stadler, S.C.; Hacker, U.; Burkhardt, R. Cholesterol metabolism and breast cancer. Curr. Opin. Lipidol. 2016, 27, 200–201. [Google Scholar] [CrossRef] [PubMed]
- Vladimirov, S.; Gojkovic, T.; Zeljkovic, A.; Jelic-Ivanovic, Z.; Zeljkovic, D.; Antonic, T.; Trifunovic, B.; Spasojevic-Kalimanovska, V. Can non-cholesterol sterols indicate the presence of specific dysregulation of cholesterol metabolism in patients with colorectal cancer? Biochem. Pharmacol. 2021, 114595. [Google Scholar] [CrossRef] [PubMed]
- Brown, A.J.; Sharpe, L.J.; Rogers, M.J. Oxysterols: From physiological tuners to pharmacological opportunities. Br. J. Pharmacol. 2021, 178, 3089–3103. [Google Scholar] [CrossRef] [PubMed]
- Nury, T.; Zarrouk, A.; Yammine, A.; Mackrill, J.J.; Vejux, A.; Lizard, G. Oxiapoptophagy: A type of cell death induced by some oxysterols. Br. J. Pharmacol. 2021, 178, 3115–3123. [Google Scholar] [CrossRef] [PubMed]
- Cao, Q.; Liu, Z.; Xiong, Y.; Zhong, Z.; Ye, Q. Multiple Roles of 25-Hydroxycholesterol in Lipid Metabolism, Antivirus Process, Inflammatory Response, and Cell Survival. Oxidative Med. Cell. Longev. 2020, 2020, 8893305. [Google Scholar] [CrossRef]
- Choi, Y.K.; Kim, Y.S.; Choi, I.Y.; Kim, S.W.; Kim, W.K. 25-hydroxycholesterol induces mitochondria-dependent apoptosis via activation of glycogen synthase kinase-3beta in PC12 cells. Free Radic. Res 2008, 42, 544–553. [Google Scholar] [CrossRef]
- Seo, Y.-S.; Cho, I.-A.; Kim, T.-H.; You, J.-S.; Oh, J.-S.; Lee, G.-J.; Kim, D.K.; Kim, A.J.-S. Oxysterol 25-hydroxycholesterol as a metabolic pathophysiological factors of osteoarthritis induces apoptosis in primary rat chondrocytes. Korean J. Physiol. Pharmacol. 2020, 24, 249–257. [Google Scholar] [CrossRef] [PubMed]
- You, J.-S.; Lim, H.; Kim, T.-H.; Oh, J.-S.; Lee, G.-J.; Seo, Y.-S.; Kim, D.K.; Yu, S.-K.; Kim, H.-J.; Kim, C.S.; et al. 25-Hydroxycholesterol Induces Death Receptor-mediated Extrinsic and Mitochondria-dependent Intrinsic Apoptosis in Head and Neck Squamous Cell Carcinoma Cells. Anticancer. Res. 2020, 40, 779–788. [Google Scholar] [CrossRef]
- Cao, Y.; Luo, Y.; Zou, J.; Ouyang, J.; Cai, Z.; Zeng, X.; Ling, H.; Zeng, T. Autophagy and its role in gastric cancer. Clin. Chim. Acta 2019, 489, 10–20. [Google Scholar] [CrossRef]
- Mrakovcic, M.; Fröhlich, L. p53-Mediated Molecular Control of Autophagy in Tumor Cells. Biomolecules 2018, 8, 14. [Google Scholar] [CrossRef] [Green Version]
- Hussain, G.; Wang, J.; Rasul, A.; Anwar, H.; Imran, A.; Qasim, M.; Zafar, S.; Kamran, S.K.S.; Razzaq, A.; Aziz, N.; et al. Role of cholesterol and sphingolipids in brain development and neurological diseases. Lipids Health Dis. 2019, 18, 26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Head, B.P.; Patel, H.H.; Insel, P.A. Interaction of membrane/lipid rafts with the cytoskeleton: Impact on signaling and function: Membrane/lipid rafts, mediators of cytoskeletal arrangement and cell signaling. Biochim. Biophys. Acta 2014, 1838, 532–545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lecerf, J.M.; de Lorgeril, M. Dietary cholesterol: From physiology to cardiovascular risk. Br. J. Nutr. 2011, 106, 6–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ansari, M.Y.; Ahmad, N.; Haqqi, T.M. Oxidative stress and inflammation in osteoarthritis pathogenesis: Role of polyphenols. Biomed. Pharmacother. 2020, 129, 110452. [Google Scholar] [CrossRef] [PubMed]
- Pitocco, D.; Zaccardi, F.; Di Stasio, E.; Romitelli, F.; Santini, S.A.; Zuppi, C.; Ghirlanda, G. Oxidative Stress, Nitric Oxide, and Diabetes. Rev. Diabet. Stud. 2010, 7, 15–25. [Google Scholar] [CrossRef] [Green Version]
- Varghese, J.F.; Patel, R.; Yadav, U.C. Novel Insights in the Metabolic Syndrome-induced Oxidative Stress and Inflammation-mediated Atherosclerosis. Curr. Cardiol. Rev. 2018, 14, 4–14. [Google Scholar] [CrossRef]
- Griffiths, W.J.; Wang, Y. Oxysterol research: A brief review. Biochem. Soc. Trans. 2019, 47, 517–526. [Google Scholar] [CrossRef] [Green Version]
- Zarrouk, A.; Hammouda, S.; Ghzaiel, I.; Hammami, S.; Khamlaoui, W.; Ahmed, S.H.; Lizard, G.; Hammami, M. Association Between Oxidative Stress and Altered Cholesterol Metabolism in Alzheimer’s Disease Patients. Curr. Alzheimer Res. 2021, 17, 823–834. [Google Scholar] [CrossRef]
- Zarrouk, A.; Vejux, A.; Mackrill, J.; O’Callaghan, Y.; Hammami, M.; O’Brien, N.; Lizard, G. Involvement of oxysterols in age-related diseases and ageing processes. Ageing Res. Rev. 2014, 18, 148–162. [Google Scholar] [CrossRef]
- Russo, L.; Muir, L.; Geletka, L.; Delproposto, J.; Baker, N.; Flesher, C.; O’Rourke, R.; Lumeng, C.N. Cholesterol 25-hydroxylase (CH25H) as a promoter of adipose tissue inflammation in obesity and diabetes. Mol. Metab. 2020, 39, 100983. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Chen, J.; Li, M.; Chen, M.; Sun, C. Multifaceted Functions of CH25H and 25HC to Modulate the Lipid Metabolism, Immune Responses, and Broadly Antiviral Activities. Viruses 2020, 12, 727. [Google Scholar] [CrossRef]
- Karuna, R.; Christen, I.; Sailer, A.W.; Bitsch, F.; Zhang, J. Detection of dihydroxycholesterols in human plasma using HPLC–ESI-MS/MS. Steroids 2015, 99, 131–138. [Google Scholar] [CrossRef]
- Ichikawa, T.; Sugiura, H.; Koarai, A.; Kikuchi, T.; Hiramatsu, M.; Kawabata, H.; Akamatsu, K.; Hirano, T.; Nakanishi, M.; Matsunaga, K.; et al. 25-hydroxycholesterol promotes fibroblast-mediated tissue remodeling through NF-κB dependent pathway. Exp. Cell Res. 2013, 319, 1176–1186. [Google Scholar] [CrossRef]
- Taylor, F.R.; Kandutsch, A.A. Metabolism of 25-hydroxycholesterol in mammalian cell cultures. Side-chain scission to pregnenolone in mouse L929 fibroblasts. J. Lipid Res. 1989, 30, 899–905. [Google Scholar] [CrossRef]
- Torres, S.A.-; Moller, P.C.; Johnson, B.H.; Thompson, E. Characteristics of 25-Hydroxycholesterol-Induced Apoptosis in the Human Leukemic Cell Line CEM. Exp. Cell Res. 1997, 235, 35–47. [Google Scholar] [CrossRef] [PubMed]
- Trousson, A.; Bernard, S.; Petit, P.X.; Liere, P.; Pianos, A.; El Hadri, K.; Lobaccaro, J.-M.A.; Ghandour, M.S.; Raymondjean, M.; Schumacher, M.; et al. 25-hydroxycholesterol provokes oligodendrocyte cell line apoptosis and stimulates the secreted phospholipase A2 type IIA via LXR beta and PXR. J. Neurochem. 2009, 109, 945–958. [Google Scholar] [CrossRef]
- Ares, M.P.; Porn-Ares, M.I.; Thyberg, J.; Juntti-Berggren, L.; Berggren, P.O.; Diczfalusy, U.; Kallin, B.; Bjorkhem, I.; Orrenius, S.; Nilsson, J. Ca2+ channel blockers verapamil and nifedipine inhibit apoptosis induced by 25-hydroxycholesterol in human aortic smooth muscle cells. J. Lipid. Res. 1997, 38, 2049–2061. [Google Scholar] [CrossRef]
- Travert, C.; Carreau, S.; Le Goff, D. Induction of apoptosis by 25-hydroxycholesterol in adult rat Leydig cells: Protective effect of 17beta-estradiol. Reprod. Toxicol. 2006, 22, 564–570. [Google Scholar] [CrossRef] [PubMed]
- Olivier, E.; Dutot, M.; Regazzetti, A.; Laprévote, O.; Rat, P. 25-Hydroxycholesterol induces both P2X7-dependent pyroptosis and caspase-dependent apoptosis in human skin model: New insights into degenerative pathways. Chem. Phys. Lipids 2017, 207, 171–178. [Google Scholar] [CrossRef] [PubMed]
- Jaouadi, O.; Limam, I.; Abdelkarim, M.; Berred, E.; Chahbi, A.; Caillot, M.; Sola, B.; Ben Aissa-Fennira, F. 5,6-Epoxycholesterol Isomers Induce Oxiapoptophagy in Myeloma Cells. Cancers 2021, 13, 3747. [Google Scholar] [CrossRef] [PubMed]
- Caillot, M.; Dakik, H.; Mazurier, F.; Sola, B. Targeting Reactive Oxygen Species Metabolism to Induce Myeloma Cell Death. Cancers 2021, 13, 2411. [Google Scholar] [CrossRef]
- Marioli-Sapsakou, G.-K.; Kourti, M. Targeting Production of Reactive Oxygen Species as an Anticancer Strategy. Anticancer. Res. 2021, 41, 5881–5902. [Google Scholar] [CrossRef]
- Michallet, A.-S.; Mondiere, P.; Taillardet, M.; Leverrier, Y.; Genestier, L.; Defrance, T. Compromising the Unfolded Protein Response Induces Autophagy-Mediated Cell Death in Multiple Myeloma Cells. PLoS ONE 2011, 6, e25820. [Google Scholar] [CrossRef] [Green Version]
- Nury, T.; Zarrouk, A.; Vejux, A.; Doria, M.; Riedinger, J.M.; Delage-Mourroux, R.; Lizard, G. Induction of oxiapoptophagy, a mixed mode of cell death associated with oxidative stress, apoptosis and autophagy, on 7-ketocholesterol-treated 158N murine oligodendrocytes: Impairment by α-tocopherol. Biochem. Biophys. Res. Commun. 2014, 446, 714–719. [Google Scholar] [CrossRef]
- Debbabi, M.; Zarrouk, A.; Bezine, M.; Meddeb, W.; Nury, T.; Badreddine, A.; Karym, E.M.; Sghaier, R.; Bretillon, L.; Guyot, S.; et al. Comparison of the effects of major fatty acids present in the Mediterranean diet (oleic acid, docosahexaenoic acid) and in hydrogenated oils (elaidic acid) on 7-ketocholesterol-induced oxiapoptophagy in microglial BV-2 cells. Chem. Phys. Lipids 2017, 207, 151–170. [Google Scholar] [CrossRef]
- D’Arcy, M.S. Cell death: A review of the major forms of apoptosis, necrosis and autophagy. Cell Biol. Int. 2019, 43, 582–592. [Google Scholar] [CrossRef]
- Yu, L.; Chen, Y.; Tooze, S.A. Autophagy pathway: Cellular and molecular mechanisms. Autophagy 2018, 14, 207–215. [Google Scholar] [CrossRef] [Green Version]
- Codogno, P.; Meijer, A.J. Autophagy and signaling: Their role in cell survival and cell death. Cell Death Differ. 2005, 12, 1509–1518. [Google Scholar] [CrossRef]
- Takahashi, M.; Kakudo, Y.; Takahashi, S.; Sakamoto, Y.; Kato, S.; Ishioka, C. Overexpression of DRAM enhances p53-dependent apoptosis. Cancer Med. 2013, 2, 1–10. [Google Scholar] [CrossRef]
- Tanida, I.; Ueno, T.; Kominami, E. LC3 and Autophagy. Methods Mol. Biol. 2008, 445, 77–88. [Google Scholar] [PubMed]
- Kim, S.-M.; Noh, M.-Y.; Kim, H.; Cheon, S.-Y.; Lee, K.M.; Lee, J.; Cha, E.; Park, K.S.; Lee, K.-W.; Sung, J.-J.; et al. 25-Hydroxycholesterol is involved in the pathogenesis of amyotrophic lateral sclerosis. Oncotarget 2017, 8, 11855–11867. [Google Scholar] [CrossRef] [PubMed] [Green Version]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
You, J.-S.; Lim, H.; Seo, J.-Y.; Kang, K.-R.; Kim, D.K.; Oh, J.-S.; Seo, Y.-S.; Lee, G.-J.; Kim, J.-S.; Kim, H.-J.; et al. 25-Hydroxycholesterol-Induced Oxiapoptophagy in L929 Mouse Fibroblast Cell Line. Molecules 2022, 27, 199. https://doi.org/10.3390/molecules27010199
You J-S, Lim H, Seo J-Y, Kang K-R, Kim DK, Oh J-S, Seo Y-S, Lee G-J, Kim J-S, Kim H-J, et al. 25-Hydroxycholesterol-Induced Oxiapoptophagy in L929 Mouse Fibroblast Cell Line. Molecules. 2022; 27(1):199. https://doi.org/10.3390/molecules27010199
Chicago/Turabian StyleYou, Jae-Seek, HyangI Lim, Jeong-Yeon Seo, Kyeong-Rok Kang, Do Kyung Kim, Ji-Su Oh, Yo-Seob Seo, Gyeong-Je Lee, Jin-Soo Kim, Heung-Joong Kim, and et al. 2022. "25-Hydroxycholesterol-Induced Oxiapoptophagy in L929 Mouse Fibroblast Cell Line" Molecules 27, no. 1: 199. https://doi.org/10.3390/molecules27010199
APA StyleYou, J.-S., Lim, H., Seo, J.-Y., Kang, K.-R., Kim, D. K., Oh, J.-S., Seo, Y.-S., Lee, G.-J., Kim, J.-S., Kim, H.-J., Yu, S.-K., & Kim, J.-S. (2022). 25-Hydroxycholesterol-Induced Oxiapoptophagy in L929 Mouse Fibroblast Cell Line. Molecules, 27(1), 199. https://doi.org/10.3390/molecules27010199