
A New Oxadiazole-Based Topsentin Derivative Modulates Cyclin-Dependent Kinase 1 Expression and Exerts Cytotoxic Effects on Pancreatic Cancer Cells

 , ,
, ,  , ,
, ,  ,
,  and
and 
Abstract
:1. Introduction
2. Results and Discussion
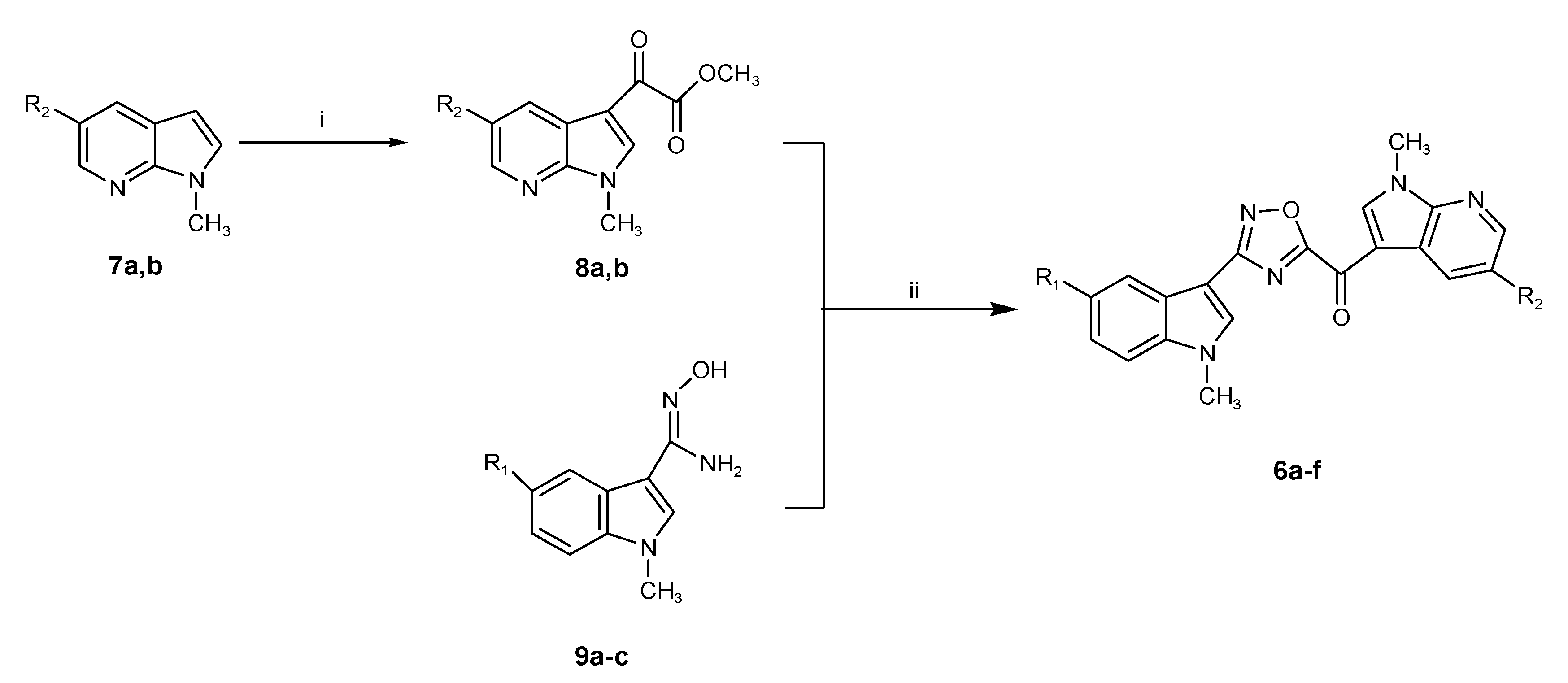
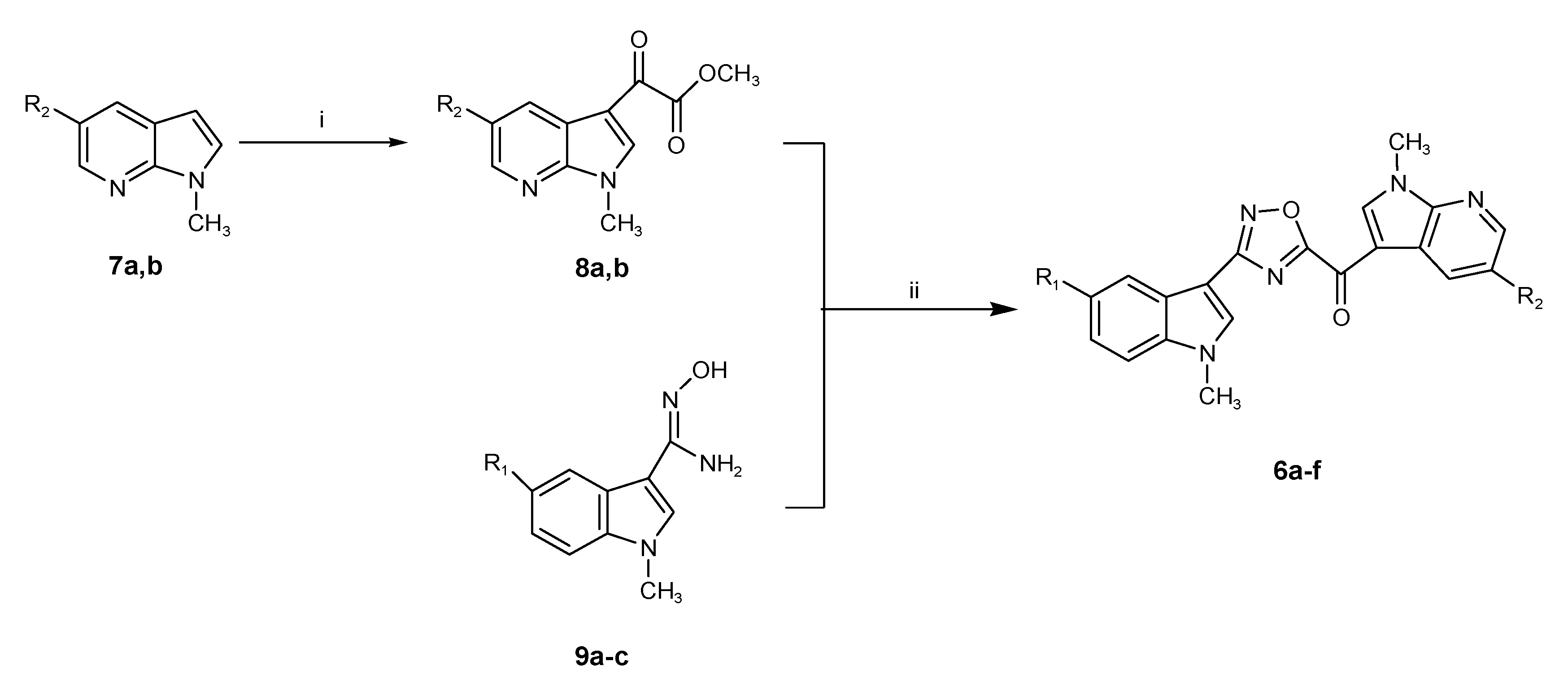
2.1. Chemistry
2.2. Biological Studies
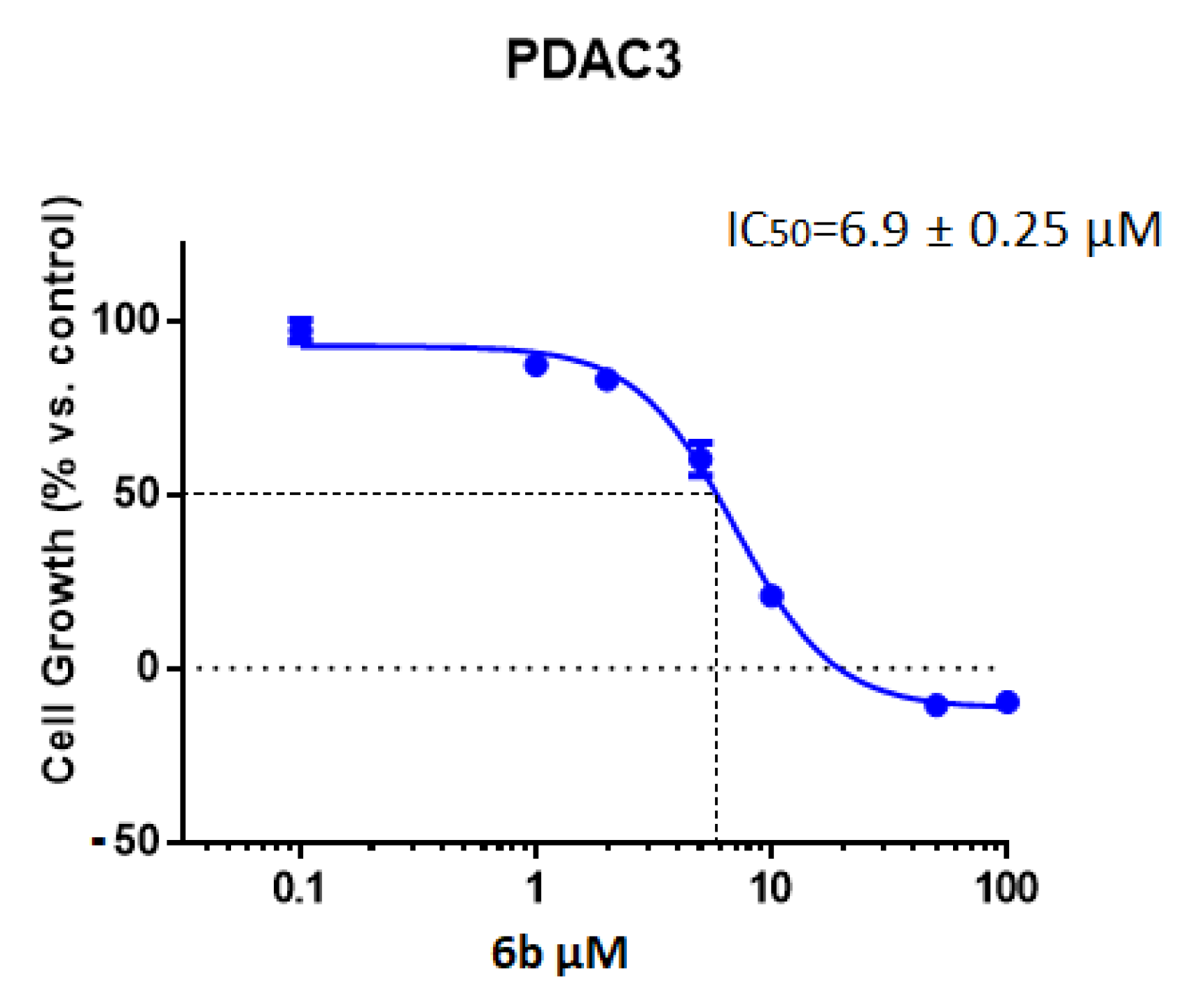
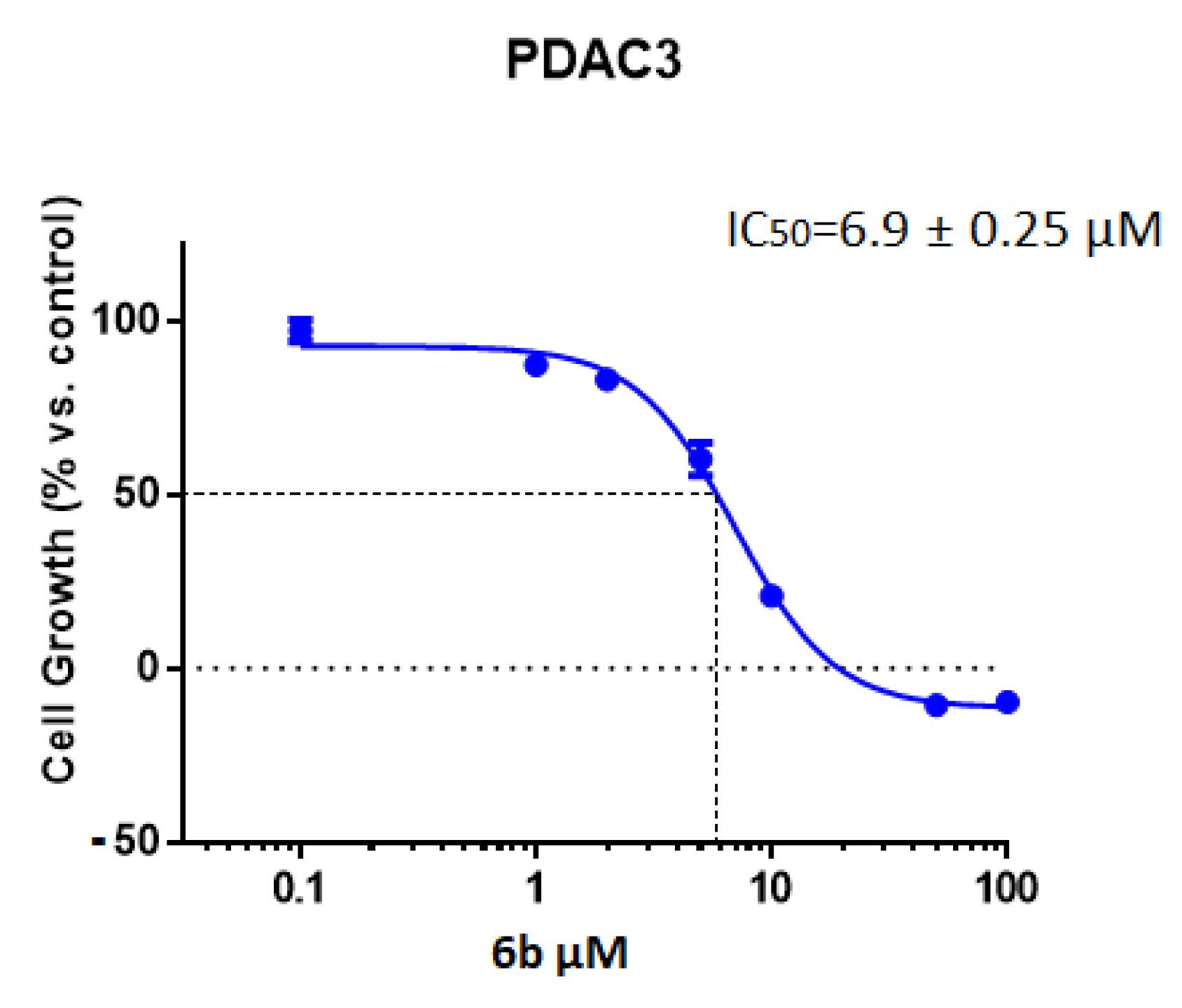
2.2.1. Antiproliferative Activity of the New 1,2,4-Oxadiazole Compounds 6a–f against PDAC3, PATU-T, Hs766T, and HPAF-II PDAC Cells
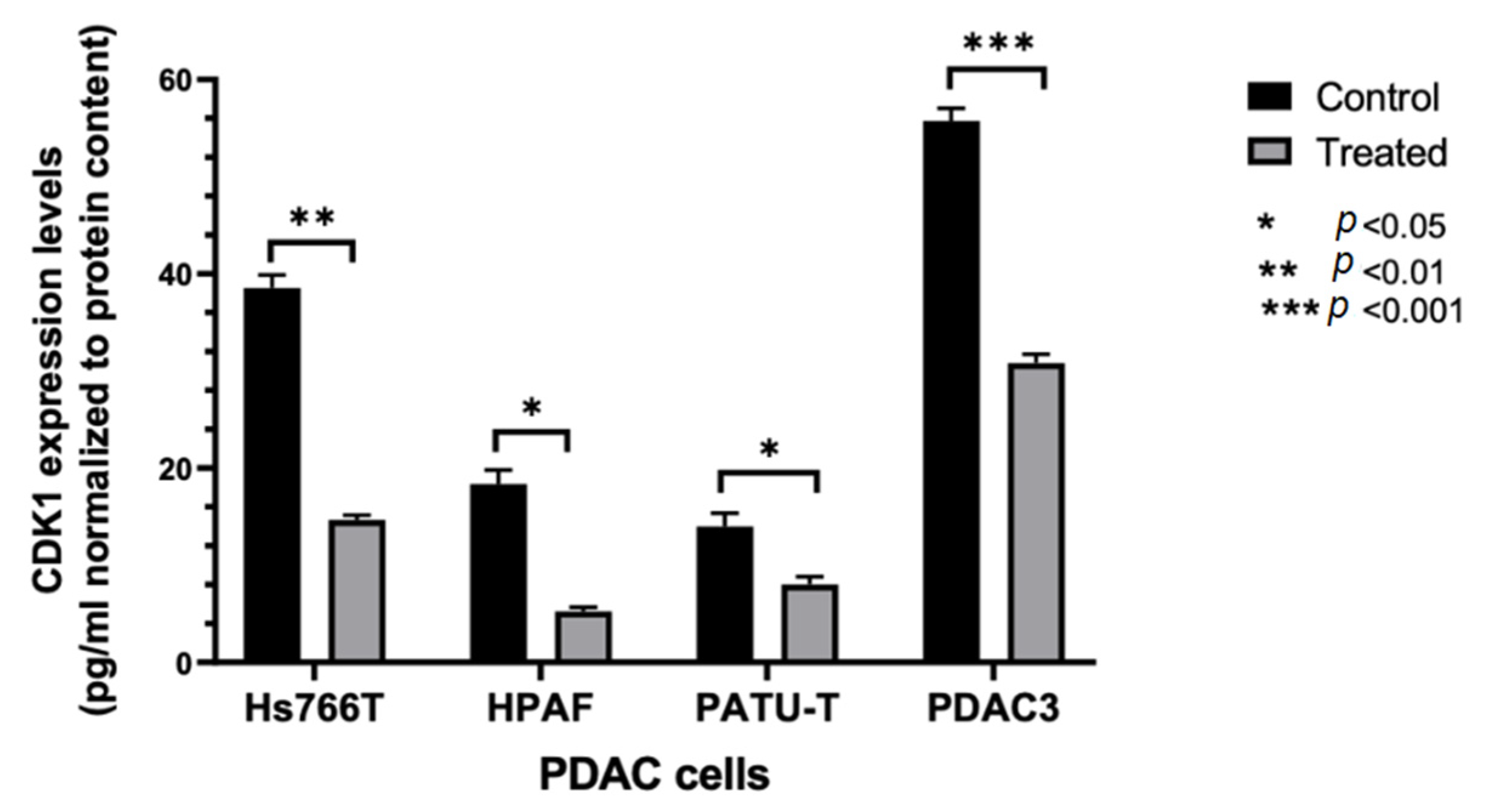
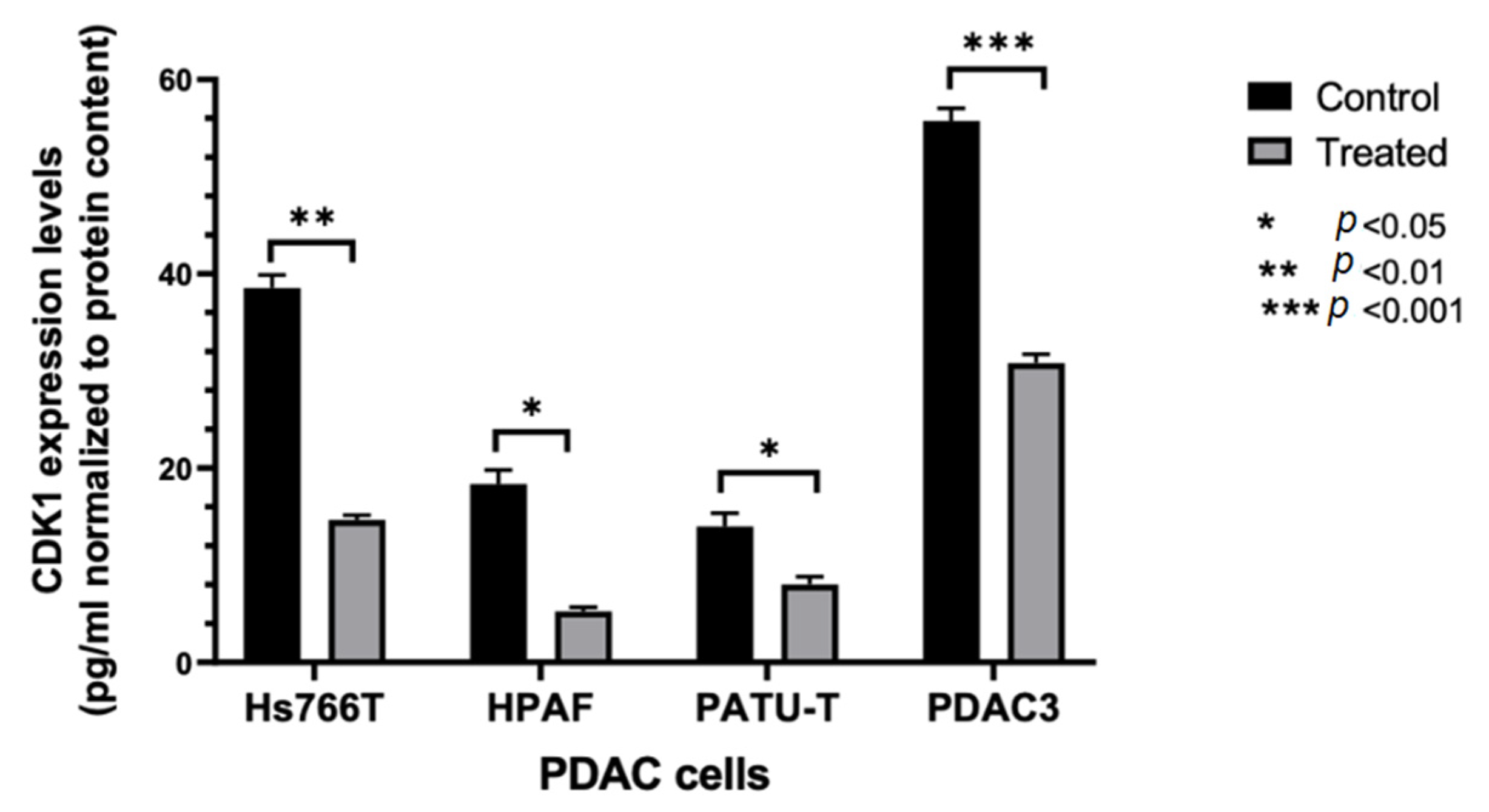
2.2.2. Modulation of CDK1 Expression by ELISA
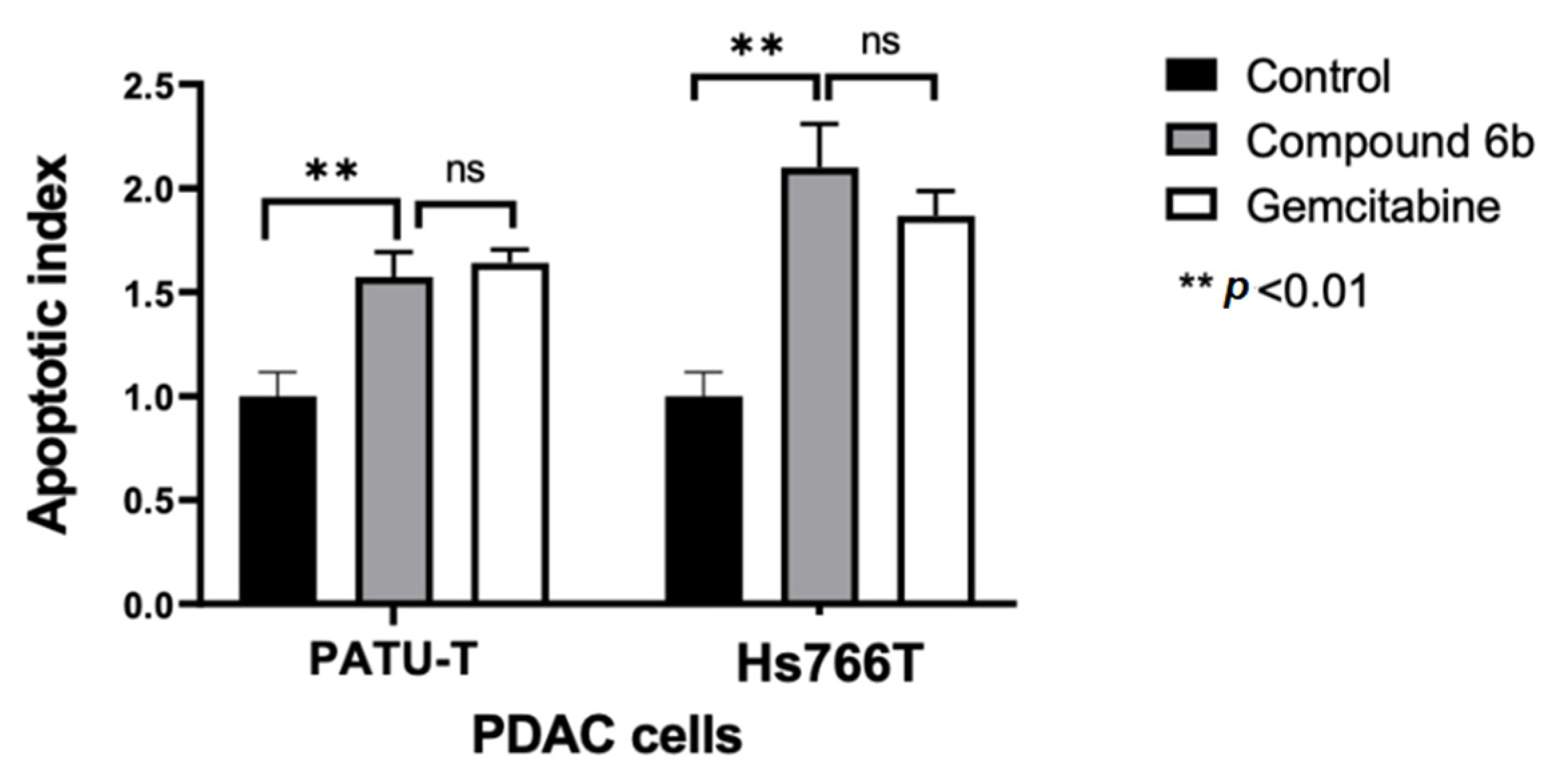
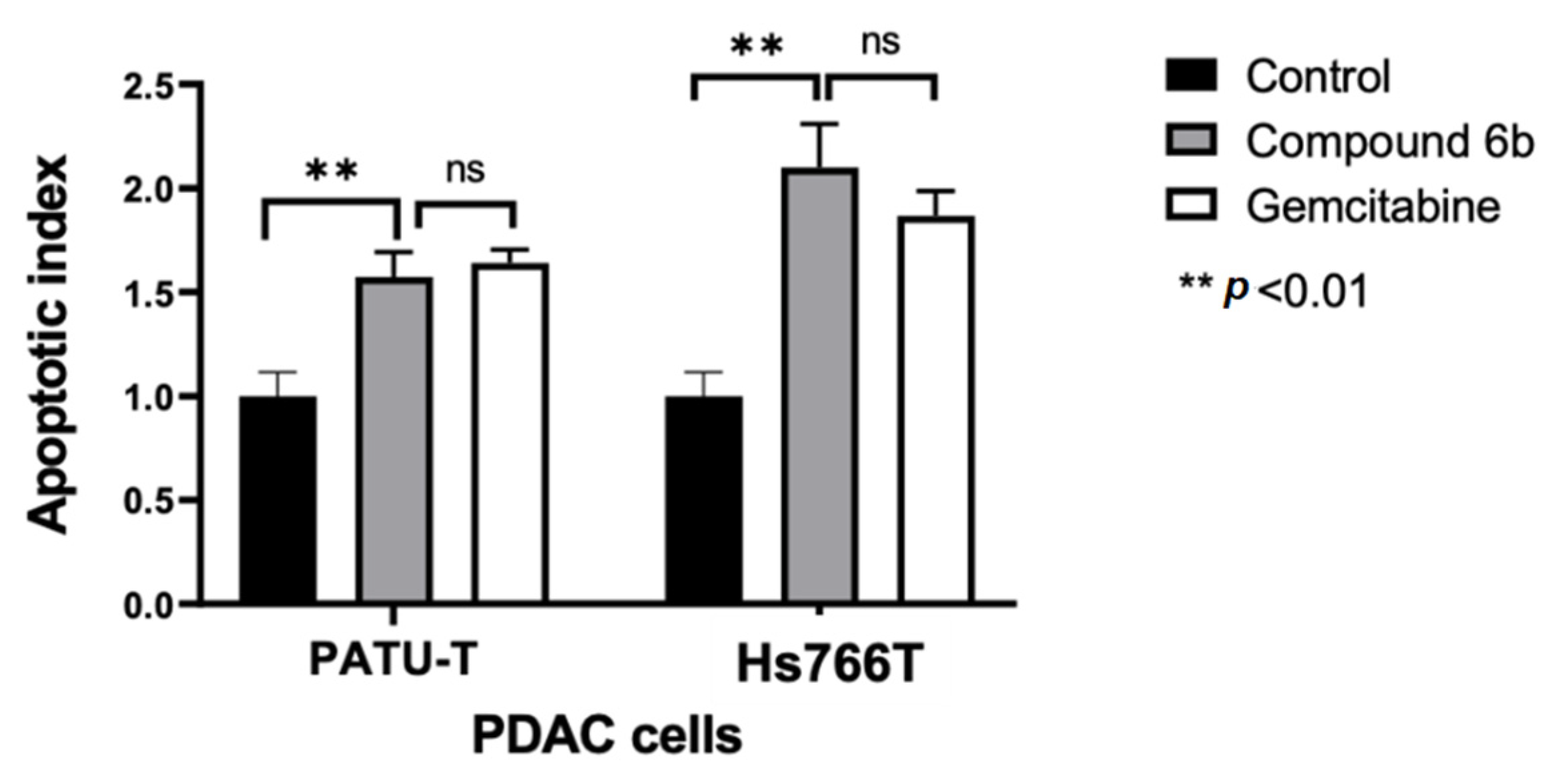
2.2.3. Induction of Apoptosis
2.2.4. Molecular Modeling
2.2.5. ADME Prediction
3. Materials and Methods
3.1. Chemistry
3.1.1. Synthesis of 1-methyl-1H-pyrrolo[2,3-b]pyridines (7a,b)
3.1.2. Synthesis of (1-methyl-1H-pyrrolo[2,3-b]pyridin-3-yl)-oxo-acetic Acid Methyl Esters (8a,b)
- (5-Bromo-1-methyl-1H-pyrrolo[2,3-b]pyridin-3-yl)-oxo-acetic acid methyl ester (8a)Yield: 62%; light yellow solid; mp: 140.5–141.5 °C; IR (cm−1): 1731 (CO), 1653 (CO); 1H-NMR (200 MHz, DMSO-d6) δ: 3.91 (3H, s, CH3), 3.92 (3H, s, OCH3), 8.54 (1H, d, J = 2.2 Hz, H-4), 8.57 (1H, d, J = 2.2 Hz, H-6), 8.80 (1H, s, H-2); 13C NMR (50 MHz, DMSO-d6) δ: 32.0 (q), 52.8 (q), 109.2 (s), 114.7 (s), 131.4 (d), 143.0 (d), 145.1 (d), 146.6 (s), 162.6 (s), 176.8 (s), 177.6 (s); Anal. calculated for C11H9BrN2O3 (MW: 297.10): C, 44.47; H, 3.05; N, 9.43%. Found: C, 44.58; H, 3.24; N, 9.65%.
- (1-Methyl-1H-pyrrolo[2,3-b]pyridin-3-yl)-oxo-acetic acid methyl ester (8b)Yield: 73%; white solid; mp: 97.8–98.8 °C; IR (cm−1): 1729 (CO), 1650 (CO); 1H-NMR (200 MHz, DMSO-d6) δ: 3.91 (3H, s, CH3), 3.93 (3H, s, OCH3), 7.38 (1H, dd, J = 7.8, 4.8 Hz, H-4), 8.44–8.51 (2H, m, H-5 and H-6), 8.74 (1H, s, H-2); 13C NMR (50 MHz, DMSO-d6) δ: 31.8 (q), 52.7 (q), 109.7 (s), 118.3 (s), 119.3 (d), 129.8 (d), 141.8 (d), 144.9 (d) 148.1 (s), 163.1 (s), 176.8 (s), 178.0 (s); Anal. calculated for C11H10N2O3 (MW: 218.21): C, 60.55; H, 4.62; N, 12.84%. Found: C, 60.38; H, 4.78; N, 12.72%.
3.1.3. Synthesis of (1-methyl-1H-pyrrolo[2,3-b]pyridine-3-yl)-[3-(1-methyl-1H-indol-3-yl)-[1,2,4]oxadiazol-5-yl]-methanones (6a–f)
- (5-Bromo-1-methyl-1H-pyrrolo[2,3-b]pyridin-3-yl)-[3-(1-methyl-1H-indol-3-yl)-[1,2,4]oxadiazol-5-yl]-methanone (6a)Yield: 60%; yellow solid; mp: 258 °C (dec); IR (cm−1): 1630 (CO); 1H-NMR (200 MHz, DMSO-d6) δ: 3.90 (3H, s, CH3), 3.98 (3H, s, CH3), 7.21–7.33 (2H, m, H-5′ and H-6′), 7.56–7.60 (1H, m, H-7′), 7.99–8.06 (1H, m, H-4′), 8.37 (1H, s, H-2′), 8.56 (1H, d, J = 2.2 Hz, H-4), 8.68 (1H, d, J = 2.2 Hz, H-6), 9.24 (1H, s, H-2); Anal. calculated for C20H14BrN5O2 (MW: 436.26): C, 55.06; H, 3.23; N, 16.05%. Found: C, 55.18; H, 3.04; N, 15.89%.
- (5-Bromo-1-methyl-1H-pyrrolo[2,3-b]pyridin-3-yl)-[3-(5-fluoro-1-methyl-1H-indol-3-yl)-[1,2,4]oxadiazol-5-yl]-methanone (6b)Yield: 67%; yellow solid; mp: 246 °C (dec); IR (cm−1): 1635 (CO); 1H-NMR (200 MHz, DMSO-d6) δ: 3.97 (3H, s, CH3), 4.04 (3H, s, CH3), 7.17–7.28 (1H, m, H-6′), 7.65–7.78 (2H, m, H-7′ and H-4′), 8.50 (1H, s, H-2′), 8.61 (1H, d, J = 1.8 Hz, H-4), 8.73 (1H, d, J = 1.8 Hz, H-6), 9.30 (1H, s, H-2); Anal. calculated for C20H13BrFN5O2 (MW: 454.25): C, 52.88; H, 2.88; N, 15.42%. Found: C, 55.80; H, 3.00; N, 15.55%.
- (5-Bromo-1-methyl-1H-pyrrolo[2,3-b]pyridin-3-yl)-[3-(5-methoxy-1-methyl-1H-indol-3-yl)-[1,2,4]oxadiazol-5-yl]-methanone (6c)Yield: 85%; yellow solid; mp: 288 °C (dec); IR (cm−1): 1637 (CO); 1H-NMR (200 MHz, DMSO-d6) δ: 3.86 (3H, s, CH3), 3.92 (3H, s, CH3), 4.02 (3H, s, OCH3), 6.95–7.08 (1H, m, H-6′), 7.54–7.55 (2H, m, H-7′ and H-4′), 8.35 (1H, s, H-2′), 8.60 (1H, s, H-4), 8.72 (1H, s, H-6), 9.29 (1H, s, H-2); Anal. calculated for C21H16BrN5O3 (MW: 466.29): C, 54.09; H, 3.46; N, 15.02%. Found: C, 54.28; H, 3.30; N, 15.15%.
- (1-Methyl-1H-pyrrolo[2,3-b]pyridin-3-yl)-[3-(1-methyl-1H-indol-3-yl)-[1,2,4]oxadiazol-5-yl]-methanone (6d)Yield: 77%; yellow solid; mp: 230 °C (dec); IR (cm−1): 1620 (CO); 1H-NMR (200 MHz, DMSO-d6) δ: 3.97 (3H, s, CH3), 4.05 (3H, s, CH3), 7.28–7.40 (2H, m, H-5′ and H-6′), 7.46 (1H, dd, J = 9.9, 4.8 Hz, H-4), 7.65 (1H, dd, J = 6.5, 2.1 Hz, H-7′), 8.11 (1H, dd, J = 6.2, 2.4 Hz, H-4′), 8.44 (1H, s, H-2′), 8.51 (1H, dd, J = 4.7, 1.3 Hz, H-6), 8.64 (1H, dd, J = 7.9, 1.3 Hz, H-5), 9.27 (1H, s, H-2); Anal. calculated for C20H15N5O2 (MW: 357.37): C, 67.22; H, 4.23; N, 19.60%. Found: C, 67.08; H, 4.04; N, 19.78%.
- (1-Methyl-1H-pyrrolo[2,3-b]pyridin-3-yl)-[3-(5-fluoro-1-methyl-1H-indol-3-yl)-[1,2,4]oxadiazol-5-yl]-methanone (6e)Yield: 68%; yellow solid; mp: 257 °C (dec); IR (cm−1): 1638 (CO); 1H-NMR (200 MHz, DMSO-d6) δ: 3.98 (3H, s, CH3), 4.05 (3H, s, CH3), 7.18–7.28 (1H, m, H-6′), 7.42–7.50 (1H, m, H-4), 7.66–7.79 (2H, m, H-7′ and H-4′), 8.50 (1H, s, H-2′), 8.52–8.67 (2H, m, H-5 and H-6), 9.26 (1H, s, H-2); Anal. calculated for C20H14FN5O2 (MW: 375.36): C, 64.00; H, 3.76; N, 18.66%. Found: C, 64.20; H, 3.64; N, 18.50%.
- (1-Methyl-1H-pyrrolo[2,3-b]pyridin-3-yl)-[3-(5-methoxy-1-methyl-1H-indol-3-yl)-[1,2,4]oxadiazol-5-yl]-methanone (6f)Yield: 80%; yellow solid; mp: 231 °C (dec); IR (cm−1): 1623 (CO); 1H-NMR (200 MHz, DMSO-d6) δ: 3.87 (3H, s, CH3), 3.92 (3H, s, CH3), 4.04 (3H, s, OCH3), 6.95–7.08 (1H, m, H-6′), 7.40–7.63 (3H, m, H-7′, H-4′ and H-4), 8.31–8.67 (3H, m, H-2′, H-5 and H-6), 9.29 (1H, s, H-2); Anal. calculated for C21H17N5O3 (MW: 387.39): C, 65.11; H, 4.42; N, 18.08%. Found: C, 65.28; H, 4.60; N, 18.25%.
3.2. Biology
3.2.1. Drugs and Chemicals
3.2.2. Cell Cultures
3.2.3. Inhibition of Cell Growth
3.2.4. Enzyme-Linked Immunosorbent Assay (ELISA) for CDK1 Expression
3.2.5. Apoptosis
3.2.6. Statistical Analysis
3.3. In Silico Studies
3.3.1. Molecular Modelling and Docking
3.3.2. ADME Studies
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Conroy, T.; Desseigne, F.; Ychou, M.; Bouché, O.; Guimbaud, R.; Bécouarn, Y.; Adenis, A.; Raoul, J.L.; Gourgou-Bourgade, S.; de la Fouchardière, C.; et al. FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N. Engl. J. Med. 2011, 364, 1817–1825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caparello, C.; Meijer, L.L.; Garajova, I.; Falcone, A.; Le Large, T.Y.; Funel, N.; Kazemier, G.; Peters, G.J.; Vasile, E.; Giovannetti, E. FOLFIRINOX and translational studies: Towards personalized therapy in pancreatic cancer. World J. Gastroenterol. 2016, 22, 6987–7005. [Google Scholar] [CrossRef]
- Von Hoff, D.D.; Ervin, T.; Arena, F.P.; Chiorean, E.G.; Infante, J.; Moore, M.; Seay, T.; Tjulandin, S.A.; Ma, W.W.; Saleh, M.N.; et al. Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine. N. Engl. J. Med. 2013, 369, 1691–1703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le Large, T.Y.S.; Bijlsma, M.F.; El Hassouni, B.; Mantini, G.; Lagerweij, T.; Henneman, A.A.; Funel, N.; Kok, B.; Pham, T.V.; de Haas, R.; et al. Focal adhesion kinase inhibition synergizes with nab-paclitaxel to target pancreatic ductal adenocarcinoma. J. Exp. Clin. Cancer Res. 2021, 40, 91. [Google Scholar] [CrossRef]
- Roskoski, R., Jr. Properties of FDA-approved small molecule protein kinase inhibitors: A 2020 update. Pharmacol. Res. 2019, 152, 104609. [Google Scholar] [CrossRef] [PubMed]
- Roskoski, R., Jr. Properties of FDA-approved small molecule protein kinase inhibitors. Pharmacol. Res. 2019, 144, 19–50. [Google Scholar] [CrossRef]
- Wu, P.; Nielsen, T.E.; Clausen, M.H. Small-molecule kinase inhibitors: An analysis of FDA-approved drugs. Drug Discov. Today 2016, 21, 5–10. [Google Scholar] [CrossRef]
- Wu, P.; Nielsen, T.E.; Clausen, M.H. FDA-approved small-molecule kinase inhibitors. Trends Pharmacol. Sci. 2015, 36, 422–439. [Google Scholar] [CrossRef] [Green Version]
- Lim, S.; Kaldis, P. Cdks, cyclins and CKIs: Roles beyond cell cycle regulation. Development 2013, 140, 3079–3093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wijnen, R.; Pecoraro, C.; Carbone, D.; Fiuji, H.; Avan, A.; Peters, G.J.; Giovannetti, E.; Diana, P. Cyclin Dependent Kinase-1 (CDK-1) Inhibition as a Novel Therapeutic Strategy against Pancreatic Ductal Adenocarcinoma (PDAC). Cancers 2021, 13, 4389. [Google Scholar] [CrossRef] [PubMed]
- Pelosi, E.; Castelli, G.; Testa, U. Pancreatic Cancer: Molecular Characterization, Clonal Evolution and Cancer Stem Cells. Biomedicines 2017, 5, 65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakemi, S.; Sun, H.H. Nortopsentins A, B, and C. Cytotoxic and antifungal imidazolediylbis[indoles] from the sponge Spongosorites ruetzleri. J. Org. Chem. 1991, 56, 4304–4307. [Google Scholar] [CrossRef]
- Cascioferro, S.; Attanzio, A.; Di Sarno, V.; Musella, S.; Tesoriere, L.; Cirrincione, G.; Diana, P.; Parrino, B. New 1,2,4-Oxadiazole Nortopsentin Derivatives with Cytotoxic Activity. Mar. Drugs 2019, 17, 35. [Google Scholar] [CrossRef] [Green Version]
- Pecoraro, C.; Carbone, D.; Aiello, D.; Carbone, A. Synthesis and cytotoxic activity of 3-[2-(1H-Indol-3-yl)-1,3-thiazol-4-yl]-1H-pyrrolo[3,2-c]pyridine hydrobromides, analogues of marine alkaloid nortopsentin. Arkivoc 2021. [Google Scholar] [CrossRef]
- Di Franco, S.; Parrino, B.; Gaggianesi, M.; Pantina, V.D.; Bianca, P.; Nicotra, A.; Mangiapane, L.R.; Lo Iacono, M.; Ganduscio, G.; Veschi, V.; et al. CHK1 inhibitor sensitizes resistant colorectal cancer stem cells to nortopsentin. iScience 2021, 24, 102664. [Google Scholar] [CrossRef]
- Li Petri, G.; Cascioferro, S.; El Hassouni, B.; Carbone, D.; Parrino, B.; Cirrincione, G.; Peters, G.J.; Diana, P.; Giovannetti, E. Biological Evaluation of the Antiproliferative and Anti-migratory Activity of a Series of 3-(6-Phenylimidazo[2,1-b][1,3,4]thiadiazol-2-yl)-1H-indole Derivatives against Pancreatic Cancer Cells. Anticancer Res. 2019, 39, 3615–3620. [Google Scholar] [CrossRef]
- Cascioferro, S.; Li Petri, G.; Parrino, B.; El Hassouni, B.; Carbone, D.; Arizza, V.; Perricone, U.; Padova, A.; Funel, N.; Peters, G.J.; et al. 3-(6-Phenylimidazo [2,1-b][1,3,4]thiadiazol-2-yl)-1H-Indole Derivatives as New Anticancer Agents in the Treatment of Pancreatic Ductal Adenocarcinoma. Molecules 2020, 25, 329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vitaku, E.; Smith, D.T.; Njardarson, J.T. Analysis of the Structural Diversity, Substitution Patterns, and Frequency of Nitrogen Heterocycles among U.S. FDA Approved Pharmaceuticals. J. Med. Chem. 2014, 57, 10257–10274. [Google Scholar] [CrossRef]
- Carbone, D.; Parrino, B.; Cascioferro, S.; Pecoraro, C.; Giovannetti, E.; Di Sarno, V.; Musella, S.; Auriemma, G.; Cirrincione, G.; Diana, P. 1,2,4-Oxadiazole Topsentin Analogs with Antiproliferative Activity against Pancreatic Cancer Cells, Targeting GSK3β Kinase. ChemMedChem 2021, 16, 537–554. [Google Scholar] [CrossRef]
- Parrino, B.; Carbone, D.; Cascioferro, S.; Pecoraro, C.; Giovannetti, E.; Deng, D.; Di Sarno, V.; Musella, S.; Auriemma, G.; Cusimano, M.G.; et al. 1,2,4-Oxadiazole topsentin analogs as staphylococcal biofilm inhibitors targeting the bacterial transpeptidase sortase A. Eur. J. Med. Chem. 2021, 209, 112892. [Google Scholar] [CrossRef]
- Avan, A.; Caretti, V.; Funel, N.; Galvani, E.; Maftouh, M.; Honeywell, R.J.; Lagerweij, T.; Van Tellingen, O.; Campani, D.; Fuchs, D.; et al. Crizotinib inhibits metabolic inactivation of gemcitabine in c-Met-driven pancreatic carcinoma. Cancer Res. 2013, 73, 6745–6756. [Google Scholar] [CrossRef] [Green Version]
- Maftouh, M.; Belo, A.I.; Avan, A.; Funel, N.; Peters, G.J.; Giovannetti, E.; Van Die, I. Galectin-4 expression is associated with reduced lymph node metastasis and modulation of Wnt/β-catenin signalling in pancreatic adenocarcinoma. Oncotarget 2014, 5, 5335–5349. [Google Scholar] [CrossRef] [Green Version]
- Le Large, T.Y.; Mantini, G.; Meijer, L.L.; Pham, T.V.; Funel, N.; van Grieken, N.C.; Kok, B.; Knol, J.; van Laarhoven, H.W.; Piersma, S.R.; et al. Microdissected pancreatic cancer proteomes reveal tumor heterogeneity and therapeutic targets. JCI Insight 2021, 5, e138290. [Google Scholar] [CrossRef] [PubMed]
- Parry, D.; Guzi, T.; Shanahan, F.; Davis, N.; Prabhavalkar, D.; Wiswell, D.; Seghezzi, W.; Paruch, K.; Dwyer, M.P.; Doll, R.; et al. Dinaciclib (SCH 727965), a Novel and Potent Cyclin-Dependent Kinase Inhibitor. Mol. Cancer Ther. 2010, 9, 2344–2353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shendge, A.K.; Chaudhuri, D.; Mandal, N. The natural flavones, acacetin and apigenin, induce Cdk-Cyclin mediated G2/M phase arrest and trigger ROS-mediated apoptosis in glioblastoma cells. Mol. Biol. Rep. 2021, 48, 539–549. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Chen, P.; Liu, K.; Liu, J.; Zhou, B.; Wu, R.; Peng, Q.; Liu, Z.-X.; Li, C.; Kroemer, G.; et al. CDK1/2/5 inhibition overcomes IFNG-mediated adaptive immune resistance in pancreatic cancer. Gut 2020, 70, 890–899. [Google Scholar] [CrossRef]
- Navarro-Retamal, C.; Caballero, J. Flavonoids as CDK1 Inhibitors: Insights in Their Binding Orientations and Structure-Activity Relationship. PLoS ONE 2016, 11, e0161111. [Google Scholar] [CrossRef] [PubMed]
- SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [CrossRef] [Green Version]
- Rovithi, M.; Avan, A.; Funel, N.; Leon, L.G.; Gomez, V.E.; Wurdinger, T.; Griffioen, A.W.; Verheul, H.M.; Giovannetti, E. Development of bioluminescent chick chorioallantoic membrane (CAM) models for primary pancreatic cancer cells: A platform for drug testing. Sci. Rep. 2017, 7, 44686. [Google Scholar] [CrossRef]
- Sciarrillo, R.; Wojtuszkiewicz, A.; Kooi, I.E.; Gómez, V.E.; Boggi, U.; Jansen, G.; Kaspers, G.J.; Cloos, J.; Giovannetti, E. Using RNA-sequencing to Detect Novel Splice Variants Related to Drug Resistance in In Vitro Cancer Models. J. Vis. Exp. 2016, 118, 54714. [Google Scholar] [CrossRef]
- Massihnia, D.; Avan, A.; Funel, N.; Maftouh, M.; van Krieken, A.; Granchi, C.; Raktoe, R.; Boggi, U.; Aicher, B.; Minutolo, F.; et al. Phospho-Akt overexpression is prognostic and can be used to tailor the synergistic interaction of Akt inhibitors with gemcitabine in pancreatic cancer. J. Hematol. Oncol. 2017, 10, 9. [Google Scholar] [CrossRef]
- Bianco, C.; Giovannetti, E.; Ciardiello, F.; Mey, V.; Nannizzi, S.; Tortora, G.; Troiani, T.; Pasqualetti, F.; Eckhardt, G.; de Liguoro, M.; et al. Synergistic antitumor activity of ZD6474, an inhibitor of vascular endothelial growth factor receptor and epidermal growth factor receptor signaling, with gemcitabine and ionizing radiation against pancreatic cancer. Clin. Cancer Res. 2006, 12, 7099–7107. [Google Scholar] [CrossRef] [Green Version]
- Brown, N.R.; Korolchuk, S.; Martin, M.P.; Stanley, W.A.; Moukhametzianov, R.; Noble, M.; Endicott, J.A. CDK1 structures reveal conserved and unique features of the essential cell cycle CDK. Nat. Commun. 2015, 6, 6769. [Google Scholar] [CrossRef] [PubMed]
- American Cancer Society. Key Statistics for Pancreatic Cancer. Available online: https://www.cancer.org/cancer/pancreatic-cancer/about/key-statistics.html (accessed on 2 November 2021).
- Le Large, T.Y.S.; Bijlsma, M.F.; Kazemier, G.; van Laarhoven, H.W.M.; Giovannetti, E.; Jimenez, C.R. Key biological processes driving metastatic spread of pancreatic cancer as identified by multi-omics studies. Semin. Cancer Biol. 2017, 44, 153–169. [Google Scholar] [CrossRef] [PubMed]
- Dong, S.; Huang, F.; Zhang, H.; Chen, Q. Overexpression of BUB1B, CCNA2, CDC20, and CDK1 in tumor tissues predicts poor survival in pancreatic ductal adenocarcinoma. Biosci. Rep. 2019, 39, BSR20182306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elnaggar, M.; Giovannetti, E.; Peters, G.J. Molecular targets of gemcitabine action: Rationale for development of novel drugs and drug combinations. Curr. Pharm. Des. 2012, 18, 2811–2829. [Google Scholar] [CrossRef] [PubMed]
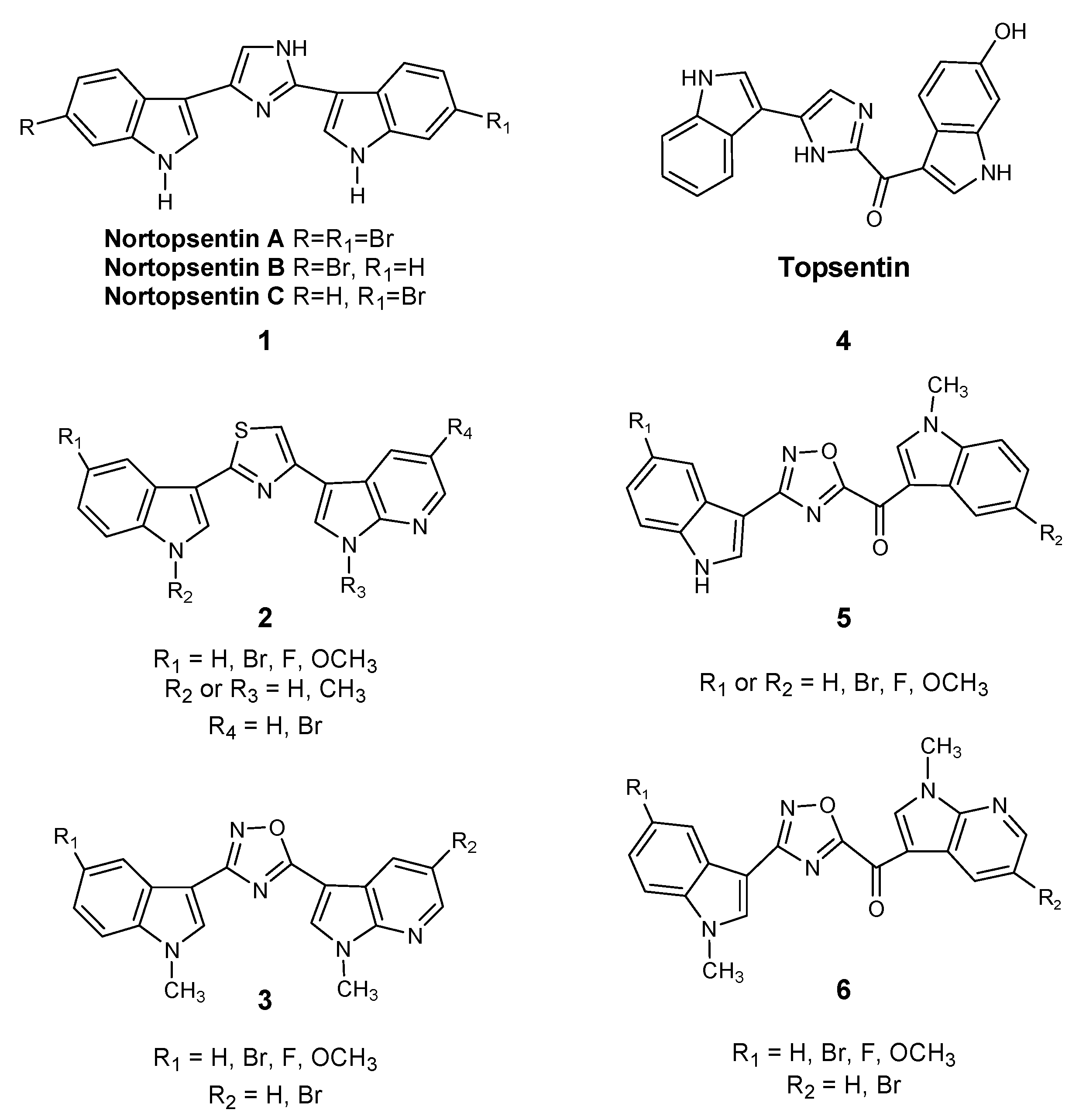


{kind=link}
{kind=link}
{kind=link}
{kind=link}
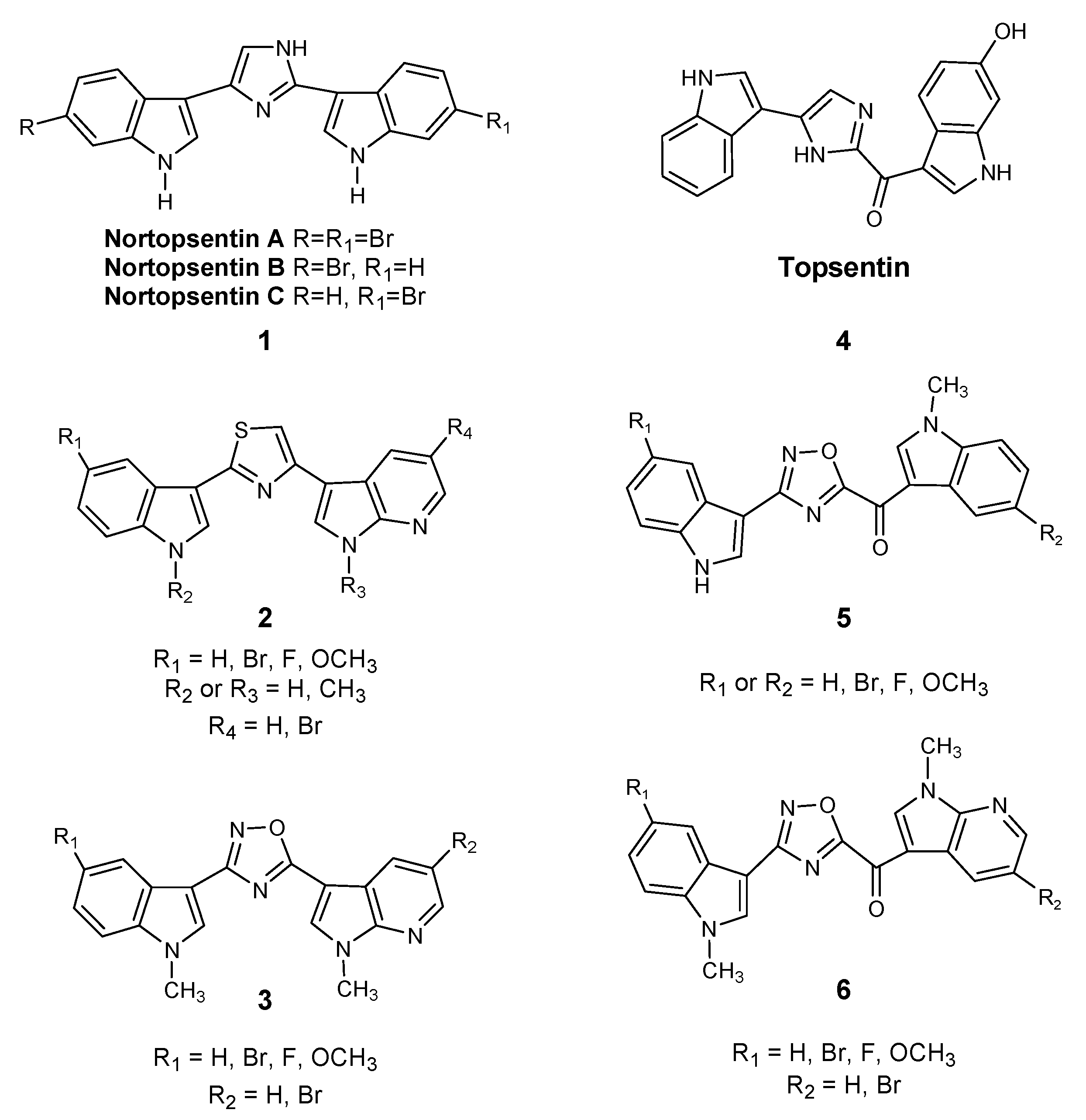{kind=link}
{kind=link}
| Compound | R1 | R2 | Yield (%) |
|---|---|---|---|
| 6a | H | Br | 60% |
| 6b | F | Br | 67% |
| 6c | OCH3 | Br | 85% |
| 6d | H | H | 77% |
| 6e | F | H | 68% |
| 6f | OCH3 | H | 80% |
| IC50 a (µM) ± SEM | ||
|---|---|---|
| Compound | Cell Line | IC50 ± SEM |
| 6b | Hs766T | 5.7 ± 0.60 |
| PDAC3 | 6.9 ± 0.25 | |
| HPAF-II | 9.8 ± 0.70 | |
| PATU-T | 10.7 ± 0.16 | |
| Parameters | Score |
|---|---|
| n. H-bond acceptor | 6 |
| n. H-bond donor | 0 |
| n. rotable bonds | 3 |
| LogPo/w(iLOGP) | 3.67 |
| Lipinski’s rule violation | No |
| Bioavailability score | 0.55 |
| GI absorption | High |
| BBB permeation | no |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pecoraro, C.; Parrino, B.; Cascioferro, S.; Puerta, A.; Avan, A.; Peters, G.J.; Diana, P.; Giovannetti, E.; Carbone, D. A New Oxadiazole-Based Topsentin Derivative Modulates Cyclin-Dependent Kinase 1 Expression and Exerts Cytotoxic Effects on Pancreatic Cancer Cells. Molecules 2022, 27, 19. https://doi.org/10.3390/molecules27010019
Pecoraro C, Parrino B, Cascioferro S, Puerta A, Avan A, Peters GJ, Diana P, Giovannetti E, Carbone D. A New Oxadiazole-Based Topsentin Derivative Modulates Cyclin-Dependent Kinase 1 Expression and Exerts Cytotoxic Effects on Pancreatic Cancer Cells. Molecules. 2022; 27(1):19. https://doi.org/10.3390/molecules27010019
Chicago/Turabian StylePecoraro, Camilla, Barbara Parrino, Stella Cascioferro, Adrian Puerta, Amir Avan, Godefridus J. Peters, Patrizia Diana, Elisa Giovannetti, and Daniela Carbone. 2022. "A New Oxadiazole-Based Topsentin Derivative Modulates Cyclin-Dependent Kinase 1 Expression and Exerts Cytotoxic Effects on Pancreatic Cancer Cells" Molecules 27, no. 1: 19. https://doi.org/10.3390/molecules27010019
APA StylePecoraro, C., Parrino, B., Cascioferro, S., Puerta, A., Avan, A., Peters, G. J., Diana, P., Giovannetti, E., & Carbone, D. (2022). A New Oxadiazole-Based Topsentin Derivative Modulates Cyclin-Dependent Kinase 1 Expression and Exerts Cytotoxic Effects on Pancreatic Cancer Cells. Molecules, 27(1), 19. https://doi.org/10.3390/molecules27010019