
Insights into Structural Modifications of Valproic Acid and Their Pharmacological Profile

 ,
,  ,
,
Abstract
:1. Introduction
2. Structure and Pharmacology of Valproic Acid
2.1. VPA Structural Elements
2.2. VPA Pharmacokinetics
2.3. VPA Pharmacodynamics
3. Structural Modification of Valproic Acid: Derivatives and Analogues
3.1. Amide Derivatives of VPA
3.1.1. Isovaleramide
3.1.2. Valpromide (VPD)
Valnoctamide (VCD, 2-Ethyl-3-methylpentanamide)
sec-Butyl-propylacetamide (SPD, 3-Methyl-2-propylpentanamide)
Propylisopropyl Acetamide (PID, Diisopropyl Acetamide)
3.2. Acid Analogues of VPA
3.2.1. 2-ene-VPA (2-Propyl-2-pentenoic Acid)
3.2.2. 4-ene-VPA (2-n-Propyl-4-pentenoic Acid)
3.3. Fluorinated Derivatives
3.4. Cyclic Analogues of VPA
3.4.1. Cyclooctylideneacetic Acid (2-Cycloctylideneacetic Acid)
3.4.2. Tetramethylcyclopropyl Analogues
2,2,3,3-Tetramethylcyclopropanecarboxylic Acid (TMCA)
2,2,3,3-Tetramethylcyclopropanecarboxamide (TMCD)
N-Methoxy-TMCD (MTMCD)
Alpha-Fluro-TMCD
3.5. Urea Derivatives of VPA
3.5.1. Valnoctylurea (VCU, 2-Ethyl-3-methylpentanoyl Urea)
3.5.2. Propyl Isopropylacetyl Urea (PIU)
3.5.3. Diisopropyl Acetyl Urea (DIU)
3.6. Conjugation Products of VPA
Valrocemide (TV1901, VGD)
3.7. Prodrugs (Sugar Esters of VPA)
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Perucca, E. Pharmacological and Therapeutic Properties of Valproate: A Summary after 35 Years of Clinical Experience. CNS Drugs 2002, 16, 695–714. Available online: https://pubmed.ncbi.nlm.nih.gov/12269862/ (accessed on 25 November 2021). [CrossRef]
- Ghodke-Puranik, Y.; Thorn, C.F.; Lamba, J.K.; Leeder, J.S.; Song, W.; Birnbaum, A.K.; Altman, R.B.; Klein, T.E. Valproic Acid Pathway: Pharmacokinetics and Pharmacodynamics. Pharm. Genom. 2013, 23, 236–241. Available online: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3696515/ (accessed on 29 September 2020). [CrossRef] [Green Version]
- López-Muñoz, F.; Baumeister, A.A.; Hawkins, M.F.; Álamo, C. The Role of Serendipity in the Discovery of the Clinical Effects of Psychotropic Drugs: Beyond of the Myth. Actas Esp. Psiquiatr. 2012, 40, 34–42. [Google Scholar]
- Meunier, H.; Carraz, G.; Neunier, Y.; Eymard, P.; Aimard, M. Pharmacodynamic Properties of N-dipropylacetic acid. Therapie 1963, 18, 435–438. Available online: http://europepmc.org/article/med/13935231 (accessed on 29 September 2020).
- Diederich, M.; Chateauvieux, S.; Morceau, F.; Dicato, M. Molecular and Therapeutic Potential and Toxicity of Valproic Acid. J. Biomed. Biotechnol. 2010. Available online: https://pubmed.ncbi.nlm.nih.gov/20798865/ (accessed on 29 September 2020).
- Emrich, H.M.; Von Zerssen, D.; Kissling, W.; Moeller, H.J. Therapeutic Effect of Valproate in Mania. Am. J. Psychiatry 1981, 138, 256. Available online: https://pubmed.ncbi.nlm.nih.gov/6779643/ (accessed on 29 September 2020).
- Calabresi, P.; Galletti, F.; Rossi, C.; Sarchielli, P.; Cupini, L.M. Antiepileptic drugs in Migraine: From Clinical Aspects to Cellular Mechanisms. Trends Pharmacol. Sci. 2007, 28, 188–195. Available online: https://linkinghub.elsevier.com/retrieve/pii/S0165614707000491 (accessed on 29 September 2020). [CrossRef]
- Tariot, P.N.; Loy, R.; Ryan, J.M.; Porsteinsson, A.; Ismail, S. Mood Stabilizers in Alzheimer’s Disease: Symptomatic and Neuroprotective Rationales. Adv. Drug Deliv. Rev. 2002, 54, 1567–1577. Available online: https://linkinghub.elsevier.com/retrieve/pii/S0169409×02001539 (accessed on 29 September 2020). [CrossRef]
- Loy, R.; Tariot, P.N. Neuroprotective Properties of Valproate: Potential Benefit for AD and Tauopathies. J. Mol. Neurosci. 2002, 19, 303–307. Available online: https://pubmed.ncbi.nlm.nih.gov/12540056/ (accessed on 25 November 2021). [CrossRef]
- Nielsen, N.M.; Svanström, H.; Stenager, E.; Magyari, M.; Koch-Henriksen, N.; Pasternak, B.; Hviid, A. The Use of Valproic Acid and Multiple Sclerosis. Pharmacoepidemiol. Drug Saf. 2015, 24, 262–268. Available online: https://pubmed.ncbi.nlm.nih.gov/25111895/ (accessed on 25 November 2021). [CrossRef] [PubMed]
- Blaheta, R.; Nau, H.; Michaelis, M.; Cinatl, J., Jr. Valproate and Valproate-Analogues: Potent Tools to Fight Against Cancer. Curr. Med. Chem. 2012, 9, 1417–1433. Available online: https://pubmed.ncbi.nlm.nih.gov/12173980/ (accessed on 29 September 2020). [CrossRef]
- Phiel, C.J.; Zhang, F.; Huang, E.Y.; Guenther, M.G.; Lazar, M.A.; Klein, P.S. Histone Deacetylase is a Direct Target of Valproic Acid, a Potent Anticonvulsant, Mood Stabilizer, and Teratogen. J. Biol. Chem. 2001, 276, 36734–36741. Available online: https://pubmed.ncbi.nlm.nih.gov/11473107/ (accessed on 10 September 2020). [CrossRef] [Green Version]
- Göttlicher, M.; Minucci, S.; Zhu, P.; Krämer, O.H.; Schimpf, A.; Giavara, S.; Sleeman, J.P.; Coco, F.L.; Nervi, C.; Pelicci, P.G.; et al. Valproic Acid Defines a Novel Class of HDAC Inhibitors inducing Differentiation of Transformed Cells. EMBO J. 2001, 20, 6969–6978. Available online: https://pubmed.ncbi.nlm.nih.gov/11742974/ (accessed on 22 November 2021). [CrossRef] [Green Version]
- Bradbury, C.A.; Khanim, F.L.; Hayden, R.; Bunce, C.M.; White, D.A.; Drayson, M.T.; Craddock, C.; Turner, B.M. Histone Deacetylases in Acute Myeloid Leukaemia Show a Distinctive Pattern of Expression that Changes Selectively in Response to Deacetylase Inhibitors. Leukemia 2005, 19, 1751–1759. Available online: https://pubmed.ncbi.nlm.nih.gov/16121216/ (accessed on 22 November 2021). [CrossRef] [PubMed]
- Jäger-Roman, E.; Deichl, A.; Jakob, S.; Hartmann, A.M.; Koch, S.; Rating, D.; Steldinger, R.; Nau, H.; Helge, H. Fetal Growth, Major Malformations, and Minor Anomalies in Infants Born to Women Receiving Valproic Acid. J. Pediatr. 1986, 108, 997–1004. Available online: https://pubmed.ncbi.nlm.nih.gov/3086531/ (accessed on 30 September 2020). [CrossRef]
- Terbach, N.; Shah, R.; Kelemen, R.; Klein, P.S.; Gordienko, D.; Brown, N.A.; Wilkinson, C.J.; Williams, R.S. Identifying an Uptake Mechanism for the Antiepileptic and Bipolar Disorder Treatment Valproic acid Using the Simple Biomedical Model Dictyostelium. J. Cell Sci. 2011, 124 Pt 13, 2267–2276. Available online: https://pubmed.ncbi.nlm.nih.gov/21652627/ (accessed on 22 November 2021). [CrossRef] [PubMed] [Green Version]
- Keane, P.E.; Simiand, J.; Mendes, E.; Santucci, V.; Morre, M. The Effects of Analogues of Valproic Acid on Seizures Induced by Pentylenetetrazol and GABA Content in Brain of Mice. Neuropharmacology 1983, 22, 875–879. Available online: https://pubmed.ncbi.nlm.nih.gov/6413882/ (accessed on 22 November 2021). [CrossRef]
- Chapman, A.G.; Meldrum, B.S.; Mendes, E. Acute Anticonvulsant Activity of Structural Analogues of Valproic Acid and Changes in Brain GABA and Aspartate Content. Life Sci. 1983, 32, 2023–2031. Available online: https://pubmed.ncbi.nlm.nih.gov/6403794/ (accessed on 22 November 2021). [CrossRef]
- Chapman, A.G.; Croucher, M.J.; Meldrum, B.S. Anticonvulsant Activity of Intracerebroventricularly Administered Valproate and Valproate Analogues. A Dose-Dependent Correlation with Changes in Brain Aspartate and GABA Levels in DBA/2 Mice. Biochem. Pharmacol. 1984, 33, 1459–1463. Available online: https://pubmed.ncbi.nlm.nih.gov/6428419/ (accessed on 22 November 2021). [CrossRef]
- Comelli, N.C.; Duchowicz, P.R.; Lobayan, R.M.; Jubert, A.H.; Castro, E.A. QSPR Study of Valproic Acid and Its Functionalized Derivatives. Mol. Inform. 2012, 31, 181–188. Available online: https://pubmed.ncbi.nlm.nih.gov/27476963/ (accessed on 25 November 2021). [CrossRef] [PubMed]
- Nau, H.; Hauck, R.-S.; Ehlers, K. Valproic Acid-Induced Neural Tube Defects in Mouse and Human: Aspects of Chirality, Alternative Drug Development, Pharmacokinetics and Possible Mechanisms. Pharmacol. Toxicol. 1991, 69, 310–321. Available online: https://pubmed.ncbi.nlm.nih.gov/1803343/ (accessed on 22 November 2021). [CrossRef]
- Nau, H.; Löscher, W. Pharmacologic Evaluation of Various Metabolites and Analogs of Valproic Acid: Teratogenic Potencies in mice. Fundam. Appl. Toxicol. 1986, 6, 669–676. Available online: https://pubmed.ncbi.nlm.nih.gov/3086174/ (accessed on 22 November 2021). [CrossRef]
- Lloyd, K.A.; Sills, G. A Scientific Review: Mechanisms of Valproate-Mediated Teratogenesis. Biosci. Horiz. Int. J. Stud. Res. 2013, 6, 2013. Available online: https://academic.oup.com/biohorizons/article/doi/10.1093/biohorizons/hzt003/302011 (accessed on 25 November 2021). [CrossRef] [Green Version]
- Tang, W.; Palaty, J.; Abbott, F.S. Time Course of Alpha-Fluorinated Valproic Acid in Mouse Brain and Serum and Its Effect on Synaptosomal Gamma-Aminobutyric Acid Levels in Comparison to Valproic Acid. J. Pharmacol. Exp. Ther. 1997. Available online: https://pubmed.ncbi.nlm.nih.gov/9316822/ (accessed on 22 November 2021).
- Tang, W.; Borel, A.G.; Abbott, F.S.; Fujimiya, T. Fluorinated Analogues as Mechanistic Probes in Valproic Acid Hepatotoxicity: Hepatic Microvesicular Steatosis and Glutathione Status. Chem. Res. Toxicol. 1995, 8, 671–682. Available online: https://pubmed.ncbi.nlm.nih.gov/7548749/ (accessed on 22 November 2021). [CrossRef]
- Neuman, M.G.; Shear, N.H.; Jacobson-Brown, P.M.; Katz, G.G.; Neilson, H.K.; Malkiewicz, I.M.; Cameron, R.G.; Abbott, F. CYP2E1-Mediated Modulation of Valproic Acid-Induced Hepatocytotoxicity. Clin. Biochem. 2001, 34, 211–218. Available online: https://pubmed.ncbi.nlm.nih.gov/11408019/ (accessed on 22 November 2021). [CrossRef]
- Lee, M.S.; Lee, Y.J.; Kim, B.J.; Shin, K.J.; Chung, B.C.; Baek, D.J.; Jung, B.H. The Relationship between Glucuronide Conjugate Levels and Hepatotoxicity after Oral Administration of Valproic Acid. Arch Pharm. Res. 2009, 32, 1029–1035. Available online: https://pubmed.ncbi.nlm.nih.gov/19641884/ (accessed on 10 September 2020). [CrossRef]
- Zaccara, G.; Messori, A.; Moroni, F. Clinical Pharmacokinetics of Valproic Acid—1988. Clin. Pharmacokinet. 1988, 15, 367–389. Available online: https://pubmed.ncbi.nlm.nih.gov/3149565/ (accessed on 22 November 2021). [CrossRef]
- Romoli, M.; Mazzocchetti, P.; D’Alonzo, R.; Siliquini, S.; Rinaldi, V.E.; Verrotti, A.; Calabresi, P.; Costa, C. Valproic Acid and Epilepsy: From Molecular Mechanisms to Clinical Evidences. Curr. Neuropharmacol. 2019, 17, 926–946. Available online: https://pubmed.ncbi.nlm.nih.gov/30592252/ (accessed on 22 November 2021). [CrossRef]
- Levy, R.H.; Cenraud, B.; Loiseau, P.; Akbaraly, R.; Brachet-Liermain, A.; Guyot, M.; Gomeni, R.; Morselli, P.L. Meal-Dependent Absorption of Enteric-Coated Sodium Valproate. Epilepsia 1980, 21, 273–280. Available online: https://pubmed.ncbi.nlm.nih.gov/6769666/ (accessed on 22 November 2021). [CrossRef]
- Ito, M.; Ikeda, Y.; Arnez, J.G.; Finocchiaro, G.; Tanaka, K. The Enzymatic Basis for the Metabolism and Inhibitory Effects of Valproic Acid: Dehydrogenation of valproyl-CoA by 2-methyl-branched-chain acyl-CoA Dehydrogenase. Biochim. Biophys. Acta 1990, 1034, 213–218. Available online: https://pubmed.ncbi.nlm.nih.gov/2112956/ (accessed on 22 November 2021). [CrossRef]
- Argikar, U.A.; Remmel, R.P. Effect of Aging on Glucuronidation of Valproic acid in Human Liver Microsomes and the Role of UDP-glucuronosyltransferase UGT1A4, UGT1A8, and UGT1A10. Drug Metab. Dispos. 2009, 37, 229–236. Available online: https://pubmed.ncbi.nlm.nih.gov/18838507/ (accessed on 22 November 2021). [CrossRef] [Green Version]
- Tan, L.; Yu, J.T.; Sun, Y.P.; Ou, J.R.; Song, J.H.; Yu, Y. The influence of cytochrome oxidase CYP2A6, CYP2B6, and CYP2C9 Polymorphisms on the Plasma Concentrations of Valproic Acid in Epileptic Patients. Clin. Neurol. Neurosurg. 2010, 112, 320–323. Available online: https://pubmed.ncbi.nlm.nih.gov/20089352/ (accessed on 22 November 2021). [CrossRef]
- Rettie, A.E.; Rettenmeier, A.W.; Howald, W.N.; Baillie, T.A. Cytochrome P-450—Catalyzed Formation of Delta 4-VPA, a Toxic Metabolite of Valproic Acid. Science 1987, 235, 890–893. Available online: https://pubmed.ncbi.nlm.nih.gov/3101178/ (accessed on 22 November 2021). [CrossRef] [PubMed]
- Bialer, M.; Hussein, Z.; Raz, I.; Abramsky, O.; Herishanu, Y.; Pachys, F. Pharmacokinetics of Valproic Acid in Volunteers after a Single Dose Study. Biopharm. Drug Dispos. 1985, 6, 33–42. Available online: https://pubmed.ncbi.nlm.nih.gov/3921078/ (accessed on 22 November 2021). [CrossRef]
- Metabolite Pattern of Valproic Acid. Part I: Gaschromatographic Determination of the Valproic Acid Metabolite Artifacts, Heptanone-3, 4- and 5-Hydroxyvalproic Acid Lactone-PubMed. Available online: https://pubmed.ncbi.nlm.nih.gov/373770/ (accessed on 22 November 2021).
- Identification and Characterization of the Glutathione and N-Acetylcysteine Conjugates of (E)-2-propyl-2,4-pentadienoic Acid, a Toxic Metabolite of Valproic Acid, in Rats and Humans-PubMed. Available online: https://pubmed.ncbi.nlm.nih.gov/1676665/ (accessed on 22 November 2021).
- Kassahun, K.; Hu, P.; Grillo, M.P.; Davis, M.R.; Jin, L.; Baillie, T.A. Metabolic activation of unsaturated derivatives of valproic acid. Identification of novel glutathione adducts formed through coenzyme A-dependent and -independent processes. Chem. Biol. Interact. 1994, 90, 253–275. Available online: https://pubmed.ncbi.nlm.nih.gov/8168173/ (accessed on 23 November 2021). [CrossRef]
- Rettenmeier, A.W.; Prickett, K.S.; Gordon, W.P.; Bjorge, S.M.; Chang, S.L.; Levy, R.H.; Baillie, T.A. Studies on the Biotransformation in the Perfused Rat Liver of 2-n-Propyl-4-pentenoic Acid, a Metabolite of the Antiepileptic Drug Valproic Acid. Evidence for the Formation of Chemically Reactive Intermediates-PubMed. Available online: https://pubmed.ncbi.nlm.nih.gov/2858383/ (accessed on 22 November 2021).
- Baillie, T.A. Metabolic Activation of Valproic Acid and Drug-Mediated Hepatotoxicity. Role of the Terminal Olefin, 2-n-propyl-4-pentenoic Acid. Chem. Res. Toxicol. 1988, 1, 195–199. Available online: https://pubmed.ncbi.nlm.nih.gov/2979731/ (accessed on 22 November 2021). [CrossRef] [PubMed]
- Peterson, G.M.; Naunton, M. Valproate: A Simple Chemical with so Much to Offer. J. Clin. Pharm. Ther. 2005, 30, 417–421. Available online: https://pubmed.ncbi.nlm.nih.gov/16164485/ (accessed on 22 November 2021). [CrossRef]
- Riva, R.; Albani, F.; Contin, M.; Baruzzi, A.; Altomare, M.; Merlini, G.P.; Perucca, E. Mechanism of Altered Drug Binding to Serum Proteins in Pregnant Women: Studies with Valproic Acid. Ther. Drug Monit. 1984, 6, 25–30. Available online: https://pubmed.ncbi.nlm.nih.gov/6424276/ (accessed on 22 November 2021). [CrossRef]
- Svensson, C.K.; Woodruff, M.N.; Baxter, J.G.; Lalka, D. Free Drug Concentration Monitoring in Clinical Practice. Rationale and Current Status. Clin. Pharmacokinet. 1986, 11, 450–469. Available online: https://pubmed.ncbi.nlm.nih.gov/3542337/ (accessed on 22 November 2021). [CrossRef] [PubMed]
- Kiang, T.K.L.; Ho, P.C.; Anari, M.R.; Tong, V.; Abbott, F.S.; Chang, T.K.H. Contribution of CYP2C9, CYP2A6, and CYP2B6 to Valproic Acid Metabolism in Hepatic Microsomes from Individuals with the CYP2C9*1/*1 Genotype. Toxicol. Sci. 2006, 94, 261–271. Available online: https://pubmed.ncbi.nlm.nih.gov/16945988/ (accessed on 10 September 2020). [CrossRef] [PubMed] [Green Version]
- VanDongen, A.M.J.; VanErp, M.G.; Voskuyl, R.A. Valproate Reduces Excitability by Blockage of Sodium and Potassium Conductance. Epilepsia 1986, 27, 177–182. Available online: https://pubmed.ncbi.nlm.nih.gov/3084227/ (accessed on 22 November 2021). [CrossRef] [PubMed]
- Johannessen, C.U.; Johannessen, S.I. Valproate: Past, Present, and Future. CNS Drug Rev. 2003, 9, 199–216. Available online: https://pubmed.ncbi.nlm.nih.gov/12847559/ (accessed on 22 November 2021). [CrossRef]
- Van den Berg, R.J.; Kok, P.; Voskuyl, R.A. Valproate and Sodium Currents in Cultured Hippocampal Neurons. Exp. Brain Res. 1993, 93, 279–287. Available online: https://pubmed.ncbi.nlm.nih.gov/8387930/ (accessed on 22 November 2021). [CrossRef]
- Tan, N.N.; Tang, H.L.; Lin, G.W.; Chen, Y.H.; Lu, P.; Li, H.J.; Gao, M.M.; Zhao, Q.H.; Yi, Y.H.; Liao, W.P.; et al. Epigenetic Downregulation of Scn3a Expression by Valproate: A Possible Role in Its Anticonvulsant Activity. Mol. Neurobiol. 2017, 154, 2831–2842. Available online: https://pubmed.ncbi.nlm.nih.gov/27013471/ (accessed on 25 November 2021). [CrossRef]
- Bialer, M.; Johannessen, S.I.; Kupferberg, H.J.; Levy, R.H.; Perucca, E.; Tomson, T. Progress Report on New Antiepileptic Drugs: A Summary of the Seventh Eilat Conference (EILAT VII). Epilepsy Res. 2004, 61, 1–48. Available online: https://pubmed.ncbi.nlm.nih.gov/15570674/ (accessed on 22 November 2021). [CrossRef] [PubMed]
- Blotnik, S.; Bergman, F.; Bialer, M. Disposition of Valpromide, Valproic Acid, and Valnoctamide in the Brain, Liver, Plasma, and Urine of Rats-PubMed. Available online: https://pubmed.ncbi.nlm.nih.gov/8723737/ (accessed on 22 November 2021).
- White, H.S.; Alex, A.B.; Pollock, A.; Hen, N.; Shekh-Ahmad, T.; Wilcox, K.S.; McDonough, J.H.; Stables, J.P.; Kaufmann, D.; Yagen, B.; et al. A New Derivative of Valproic Acid Amide Possesses a Broad-Spectrum Antiseizure Profile and Unique Activity against Status Epilepticus and Organophosphate Neuronal Damage. Epilepsia 2012, 53, 134–146. Available online: https://pubmed.ncbi.nlm.nih.gov/22150444/ (accessed on 10 September 2020). [CrossRef] [Green Version]
- Bialer, M.; Johannessen, S.I.; Kupferberg, H.J.; Levy, R.H.; Perucca, E.; Tomson, T. Progress Report on New Antiepileptic Drugs: A Summary of the Eigth Eilat Conference (EILAT VIII). Epilepsy Res. 2007, 73, 1–52. Available online: http://www.ncbi.nlm.nih.gov/pubmed/17158031 (accessed on 22 November 2021). [CrossRef] [PubMed]
- Düsing, R.H. Single-Dose Tolerance and Pharmacokinetics of 2-n-propyl-2(E)-pentenoate (delta 2(E)-valproate) in Healthy Male Volunteers. Pharm. Weekbl. Sci. 1992, 14, 152–158. Available online: https://pubmed.ncbi.nlm.nih.gov/1502017/ (accessed on 22 November 2021). [CrossRef]
- Hauck, R.S.; Nau, H. Asymmetric Synthesis and Enantioselective Teratogenicity of 2-n-propyl-4-pentenoic Acid (4-en-VPA), an Active Metabolite of the Anticonvulsant Drug, Valproic Acid. Toxicol. Lett. 1989, 49, 41–48. Available online: https://pubmed.ncbi.nlm.nih.gov/2510370/ (accessed on 10 September 2020). [CrossRef]
- Palaty, J.; Abbott, F.S. Structure-Activity Relationships of Unsaturated Analogues of Valproic Acid. J. Med. Chem. 1995, 38, 3398–3406. Available online: https://pubmed.ncbi.nlm.nih.gov/7650693/ (accessed on 18 November 2021). [CrossRef]
- Shimshoni, J.A.; Bialer, M.; Wlodarczyk, B.; Finnell, R.H.; Yagen, B. Potent Anticonvulsant Urea Derivatives of Constitutional Isomers of Valproic Acid. J. Med. Chem. 2007, 50, 6419–6427. Available online: https://pubmed.ncbi.nlm.nih.gov/17994680/ (accessed on 10 September 2020). [CrossRef] [PubMed]
- Isoherranen, N.; Woodhead, J.H.; White, H.S.; Bialer, M. Anticonvulsant Profile of Valrocemide (TV1901): A New Antiepileptic Drug. Epilepsia 2001, 42, 831–836. [Google Scholar] [CrossRef] [Green Version]
- O’Shea, S.; Johnson, K.; Clark, R.; Sliwkowski, M.X.; Erickson, S.L. Effects of In Vivo Heregulin Beta1 Treatment in Wild-Type and ErbB Gene-Targeted Mice Depend on Receptor Levels and Pregnancy. Am. J. Pathol. 2001, 158, 1871–1880. Available online: http://www.ncbi.nlm.nih.gov/pubmed/11337386 (accessed on 10 September 2020). [CrossRef]
- Armand, V.; Louvel, J.; Pumain, R.; Heinemann, U. Effects of New Valproate Derivatives on Epileptiform Discharges Induced by Pentylenetetrazole or Low Mg2+ in Rat Entorhinal Cortex-Hippocampus Slices. Epilepsy Res. 1998, 32, 345–355. Available online: https://pubmed.ncbi.nlm.nih.gov/9839774/ (accessed on 22 November 2021). [CrossRef]
- Armand, V.; Louvel, J.; Pumain, R.; Ronco, G.; Villa, P. Effects of Various Valproic Acid Derivatives on Low-Calcium Spontaneous Epileptiform Activity in Hippocampal Slices. Epilepsy Res. 1995, 22, 185–192. Available online: https://pubmed.ncbi.nlm.nih.gov/8991785/ (accessed on 22 November 2021). [CrossRef]
- Gravemann, U.; Volland, J.; Nau, H. Hydroxamic acid and Fluorinated Derivatives of Valproic Acid: Anticonvulsant Activity, Neurotoxicity and Teratogenicity. Neurotoxicol. Teratol. 2008, 30, 390–394. Available online: https://pubmed.ncbi.nlm.nih.gov/18455366/ (accessed on 10 November 2021). [CrossRef]
- Winkler, I.; Sobol, E.; Yagen, B.; Steinman, A.; Devor, M.; Bialer, M. Efficacy of antiepileptic tetramethylcyclopropyl analogues of valproic acid amides in a rat model of neuropathic pain. Neuropharmacology 2005, 49, 1110–1120. Available online: https://pubmed.ncbi.nlm.nih.gov/16055160/ (accessed on 18 November 2021). [CrossRef]
- Blotnik, S.; Bergman, F.; Bialer, M. Disposition of two tetramethylcyclopropane analogues of valpromide in the brain, liver, plasma and urine of rats. Eur. J. Pharm. Sci. 1998, 6, 93–98. Available online: https://pubmed.ncbi.nlm.nih.gov/9795021/ (accessed on 18 November 2021). [CrossRef]
- Okada, A.; Onishi, Y.; Yagen, B.; Shimshoni, J.A.; Kaufmann, D.; Bialer, M.; Fujiwara, M. Tetramethylcyclopropyl Analogue of the Leading Antiepileptic Drug, Valproic Acid: Evaluation of the Teratogenic Effects of Its Amide Derivatives in NMRI Mice. Birth Defects Res. Part A Clin. Mol. Teratol. 2008, 82, 610–621. Available online: https://pubmed.ncbi.nlm.nih.gov/18671279/ (accessed on 18 November 2021). [CrossRef]
- Pessah, N.; Bialer, M.; Wlodarczyk, B.; Finnell, R.H.; Yagen, B. Alpha-fluoro-2,2,3,3-tetramethylcyclopropanecarboxamide, a Novel Potent Anticonvulsant Derivative of a Cyclic Analogue of Valproic Acid. J. Med. Chem. 2009, 52, 2233–2242. Available online: https://pubmed.ncbi.nlm.nih.gov/19296679/ (accessed on 18 November 2021). [CrossRef]
- Shekh-Ahmad, T.; Mawasi, H.; McDonough, J.H.; Yagen, B.; Bialer, M. The Potential of Sec-Butylpropylacetamide (SPD) and Valnoctamide and their Individual Stereoisomers in Status Epilepticus. Epilepsy Behav. 2015, 49, 298–302. Available online: https://pubmed.ncbi.nlm.nih.gov/25979572/ (accessed on 22 November 2021). [CrossRef]
- Safety and Efficacy of NPS 1776 in the Acute Treatment of Migraine Headaches-Full Text View-ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT00172094?id=NCT00172094&draw=2&rank=1&load=cart (accessed on 28 September 2020).
- Löscher, W.; Nau, H. Pharmacological Evaluation of Various Metabolites and Analogues of Valproic Acid. Anticonvulsant and Toxic Potencies in Mice. Neuropharmacology 1985, 24, 427–435. Available online: https://pubmed.ncbi.nlm.nih.gov/3927183/ (accessed on 22 November 2021). [CrossRef]
- Okada, A.; Aoki, Y.; Kushima, K.; Kurihara, H.; Bialer, M.; Fujiwara, M. Polycomb Homologs are Involved in Teratogenicity of Valproic Acid in Mice. Birth Defects Res. Part A Clin. Mol. Teratol. 2004, 70, 870–879. Available online: https://pubmed.ncbi.nlm.nih.gov/15523661/ (accessed on 10 September 2020). [CrossRef]
- Paradis, F.H.; Hales, B.F. Exposure to Valproic Acid Inhibits Chondrogenesis and Osteogenesis in Mid-Organogenesis Mouse Limbs. Toxicol. Sci. 2013, 131, 234–241. Available online: https://pubmed.ncbi.nlm.nih.gov/23042728/ (accessed on 10 September 2020). [CrossRef] [Green Version]
- Bialer, M.; Rubinstein, A. Pharmacokinetics of Valpromide in Dogs after Various Modes of Administration. Biopharm. Drug Dispos. 1984, 5, 177–183. Available online: https://pubmed.ncbi.nlm.nih.gov/6430363/ (accessed on 25 November 2021). [CrossRef]
- Bialer, M.; Rubinstein, A.; Raz, I.; Abramsky, O. Pharmacokinetics of Valpromide after Oral Administration of a Solution and a Tablet to Healthy Volunteers. Eur. J. Clin. Pharmacol. 1984, 27, 501–503. Available online: https://pubmed.ncbi.nlm.nih.gov/6440792/ (accessed on 25 November 2021). [CrossRef]
- Lindekens, H.; Smolders, I.; Khan, G.M.; Bialer, M.; Ebinger, G.; Michotte, Y. In Vivo Study of the Effect of Valpromide and Valnoctamide in the Pilocarpine Rat Model of Focal Epilepsy. Pharm. Res. 2000, 17, 1408–1413. Available online: https://pubmed.ncbi.nlm.nih.gov/11205735/ (accessed on 10 September 2020). [CrossRef]
- Kaufmann, D.; Yagen, B.; Minert, A.; Wlodarczyk, B.; Finnell, R.H.; Schurig, V.; Devor, M.; Bialer, M. Evaluation of the Antiallodynic, Teratogenic and Pharmacokinetic Profile of Stereoisomers of Valnoctamide, an Amide Derivative of a Chiral Isomer of Valproic Acid. Neuropharmacology 2010, 58, 1228–1236. Available online: https://pubmed.ncbi.nlm.nih.gov/20230843/ (accessed on 10 September 2020). [CrossRef]
- Okada, A.; Kushima, K.; Aoki, Y.; Bialer, M.; Fujiwara, M. Identification of Early-Responsive Genes Correlated to Valproic Acid-Induced Neural Tube Defects in Mice. Birth Defects Res. Part A Clin. Mol. Teratol. 2005, 73, 229–238. Available online: https://pubmed.ncbi.nlm.nih.gov/15799026/ (accessed on 10 September 2020). [CrossRef]
- Bialer, M.; White, H.S. Key Factors in the Discovery and Development of New Antiepileptic Drugs. Nat. Rev. Drug Discov. 2010, 9, 68–82. Available online: https://pubmed.ncbi.nlm.nih.gov/20043029/ (accessed on 25 November 2021). [CrossRef]
- Bialer, M.; Yagen, B. Valproic Acid: Second Generation. Neurotherapeutics 2007, 4, 130–137. Available online: https://pubmed.ncbi.nlm.nih.gov/17199028/ (accessed on 25 November 2021). [CrossRef]
- Kaufmann, D.; West, P.J.; Smith, M.D.; Yagen, B.; Bialer, M.; Devor, M.; White, H.S.; Brennan, K.C. sec-Butylpropylacetamide (SPD), a New Amide Derivative of Valproic Acid for the Treatment of Neuropathic and Inflammatory Pain. Pharmacol. Res. 2017, 117, 129–139. Available online: https://pubmed.ncbi.nlm.nih.gov/27890817/ (accessed on 25 November 2021). [CrossRef] [Green Version]
- Hen, N.; Shekh-Ahmad, T.; Yagen, B.; McDonough, J.H.; Finnell, R.H.; Wlodarczyk, B.; Bialer, M. Stereoselective Pharmacodynamic and Pharmacokinetic Analysis of sec-Butylpropylacetamide (SPD), a New CNS-Active Derivative of Valproic Acid with Unique Activity against Status Epilepticus. J. Med. Chem. 2013, 2256, 6467–6477. Available online: https://pubmed.ncbi.nlm.nih.gov/23879329/ (accessed on 22 November 2021). [CrossRef]
- Haj-Yehia, A.; Bialer, M. Structure–Pharmacokinetic Relationships in a Series of Valpromide Derivatives with Antiepileptic Activity. Pharm. Sci. 1989, 6, 683–689. Available online: https://pubmed.ncbi.nlm.nih.gov/2510141/ (accessed on 10 September 2020).
- Isoherranen, N.; Yagen, B.; Woodhead, J.H.; Spiegelstein, O.; Blotnik, S.; Wilcox, K.S.; Finnell, R.H.; Bennett, G.D.; White, H.S.; Bialer, M. Characterization of the Anticonvulsant Profile and Enantioselective Pharmacokinetics of the Chiral Valproylamide Propylisopropyl Acetamide in Rodents. Br. J. Pharmacol. 2003, 138, 602–613. Available online: https://pubmed.ncbi.nlm.nih.gov/12598414/ (accessed on 10 September 2020). [CrossRef] [Green Version]
- Mareš, P.; Kubová, H.; Hen, N.; Yagen, B.; Bialer, M. Derivatives of Valproic Acid are Active against Pentetrazol-Induced Seizures in Immature Rats. Epilepsy Res. 2013, 106, 64–73. Available online: https://pubmed.ncbi.nlm.nih.gov/23815889/ (accessed on 10 September 2020). [CrossRef]
- Spiegelstein, O.; Yagen, B.; Levy, R.H.; Finnell, R.H.; Bennett, G.D.; Roeder, M.; Schurig, V.; Bialer, M. Stereoselective Pharmacokinetics and Pharmacodynamics of Propylisopropyl Acetamide, a CNS-Active Chiral Amide Analog of Valproic Acid. Pharm. Res. 1999, 16, 1582–1588. Available online: https://pubmed.ncbi.nlm.nih.gov/10554101/ (accessed on 10 September 2020). [CrossRef]
- Spiegelstein, O.; Bialer, M.; Radatz, M.; Nau, H.; Yagen, B. Enantioselective Synthesis and Teratogenicity of Propylisopropyl Acetamide, a CNS-Active Chiral Amide Analogue of Valproic Acid. Chirality 1999, 11, 645–650. Available online: https://pubmed.ncbi.nlm.nih.gov/10467316/ (accessed on 10 September 2020). [CrossRef]
- Bialer, M.; Johannessen, S.I.; Levy, R.H.; Perucca, E.; Tomson, T.; White, H.S. Progress Report on New Antiepileptic Drugs: A Summary of the Tenth Eilat Conference (EILAT X). Epilepsy Res. 2010, 92, 89–124. Available online: http://www.ncbi.nlm.nih.gov/pubmed/20970964. (accessed on 22 November 2021). [CrossRef]
- Loescher, W. Anticonvulsant Activity of Metabolites of Valproic Acid-PubMed. Available online: https://pubmed.ncbi.nlm.nih.gov/6784685/ (accessed on 22 November 2021).
- Loscher, W.; Nau, H. Distribution of Valproic Acid and Its Metabolites in Various Brain Areas of Dogs and Rats after Acute and Prolonged Treatment-PubMed. Available online: https://pubmed.ncbi.nlm.nih.gov/6411902/ (accessed on 22 November 2021).
- Löscher, W.; Nau, H.; Marescaux, C.; Vergnes, M. Comparative Evaluation of Anticonvulsant and Toxic Potencies of Valproic Acid and 2-en-valproic Acid in Different Animal Models of Epilepsy. Eur. J. Pharmacol. 1984, 99, 211–218. Available online: https://pubmed.ncbi.nlm.nih.gov/6428923/ (accessed on 10 September 2020). [CrossRef]
- Strichartz, G.R.; Wang, G.K. Rapid Voltage-Dependent Dissociation of Scorpion Alpha-Toxins Coupled to Na Channel Inactivation in Amphibian Myelinated Nerves. J. Gen. Physiol. 1986, 88, 413–435. Available online: https://pubmed.ncbi.nlm.nih.gov/2428923/ (accessed on 22 November 2021). [CrossRef] [Green Version]
- Isoherranen, N.; Yagen, B.; Bialer, M. New CNS-Active Drugs Which are Second-Generation Valproic Acid: Can They Lead to the Development of a Magic Bullet? Curr. Opin. Neurol. 2003, 16, 203–211. Available online: https://pubmed.ncbi.nlm.nih.gov/12644750/ (accessed on 10 September 2020). [CrossRef]
- Tang, W.; Abbott, F.S. A Comparative Investigation of 2-Propyl-4-Pentenoic Acid (4-ene VPA) and its α-Fluorinated Analogue: Phase II Metabolism and Pharmacokinetics. Drug Metab. Dispos. 1997, 25, 219–227. [Google Scholar]
- Kesterson, J.W.; Granneman, G.R.; Machinist, J.M. The Hepatotoxicity of Valproic acid and Its Metabolites in Rats. I. Toxicologic, Biochemical and Histopathologic Studies. Hepatology 1984, 4, 1143–1152. [Google Scholar] [CrossRef]
- Sobol, E.; Bialer, M.; Yagen, B. Tetramethylcyclopropyl Analogue of a Leading Antiepileptic Drug, Valproic acid. Synthesis and Evaluation of Anticonvulsant Activity of Its Amide Derivatives. J. Med. Chem. 2004, 47, 4316–4326. Available online: https://pubmed.ncbi.nlm.nih.gov/15294003/ (accessed on 18 November 2021). [CrossRef]
- Bialer, M.; Hadad, S.; Kadry, B.; Abdul-Hai, A.; Haj-Yehia, A.; Sterling, J.; Herzig, Y.; Yagen, B. Pharmacokinetic Analysis and Antiepileptic Activity of Tetra-Methylcyclopropane Analogues of Valpromide. Pharm. Res. 1996, 13, 284–289. Available online: https://pubmed.ncbi.nlm.nih.gov/8932450/ (accessed on 18 November 2021). [CrossRef]
- Bialer, M.; Johannessen, S.I.; Levy, R.H.; Perucca, E.; Tomson, T.; White, H.S. Progress Report on New Antiepileptic Drugs: A Summary of the Ninth Eilat Conference (EILAT IX). Epilepsy Res. 2009, 83, 1–43. Available online: https://pubmed.ncbi.nlm.nih.gov/19008076/ (accessed on 25 November 2021). [CrossRef]
- Blotnik, S.; Bergman, F.; Bialer, M. The Disposition of Valproyl Glycinamide and Valproyl Glycine in Rats. Pharm. Res. 1997, 14, 873–878. [Google Scholar] [CrossRef]
- Bialer, M.; Johannessen, S.I.; Kupferberg, H.J.; Levy, R.H.; Loiseau, P.; Perucca, E. Progress Report on New Antiepileptic Drugs: A Summary of the Fifth Eilat Conference (EILAT V). Epilepsy Res. 2001, 43, 11–58. Available online: https://pubmed.ncbi.nlm.nih.gov/11137386/ (accessed on 22 November 2021). [CrossRef]
- Wlodarczyk, B.J.; Ogle, K.; Lin, L.Y.; Bialer, M.; Finnell, R.H. Comparative Teratogenicity Analysis of Valnoctamide, Risperidone, and Olanzapine in Mice. Bipolar Disord. 2015, 17, 615–625. Available online: https://pubmed.ncbi.nlm.nih.gov/26292082/ (accessed on 26 November 2021). [CrossRef] [Green Version]
- Bialer, M.; Johannessen, S.I.; Levy, R.H.; Perucca, E.; Tomson, T.; White, H.S. Progress Report on New Antiepileptic Drugs: A Summary of the Eleventh Eilat Conference (EILAT XI). Epilepsy Res. 2013, 103, 2–30. Available online: https://pubmed.ncbi.nlm.nih.gov/23219031/ (accessed on 26 November 2021). [CrossRef] [Green Version]
- Mawasi, H.; Shekh-Ahmad, T.; Finnell, R.H.; Wlodarczyk, B.J.; Bialer, M. Pharmacodynamic and Pharmacokinetic Analysis of CNS-Active Constitutional Isomers of Valnoctamide and sec-Butylpropylacetamide—Amide Derivatives of Valproic Acid. Epilepsy Behav. 2015, 46, 72–78. Available online: https://pubmed.ncbi.nlm.nih.gov/25863940/ (accessed on 26 November 2021). [CrossRef]
- Pouliot, W.; Bialer, M.; Hen, N.; Shekh-Ahmad, T.; Kaufmann, D.; Yagen, B.; Ricks, K.; Roach, B.; Nelson, C.; Dudek, F.E. A Comparative Electrographic Analysis of the Effect of sec-butyl-propylacetamide on Pharmacoresistant Status Epilepticus. Neuroscience 2013, 231, 145–156. Available online: https://pubmed.ncbi.nlm.nih.gov/23159312/ (accessed on 26 November 2021). [CrossRef]
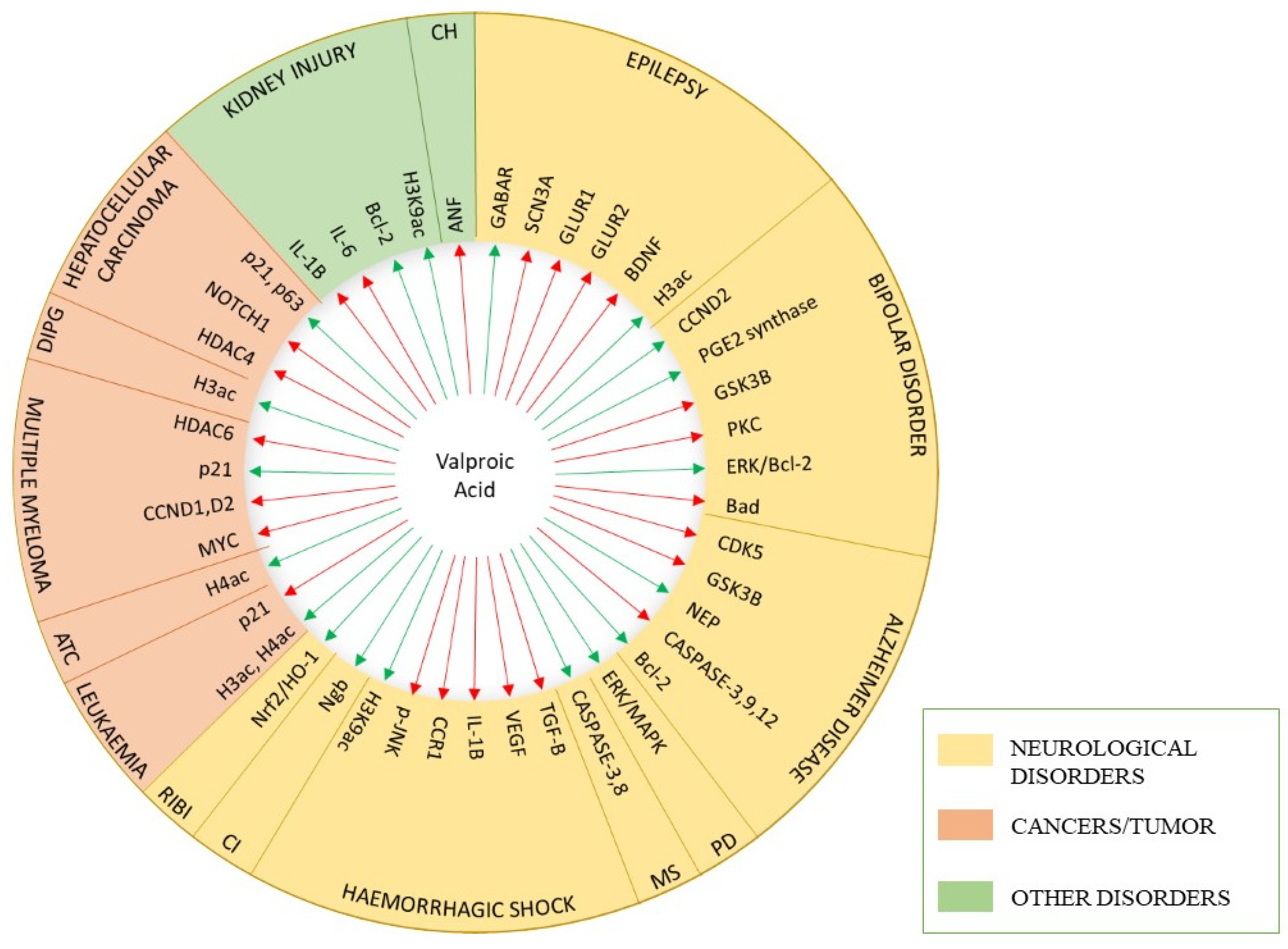
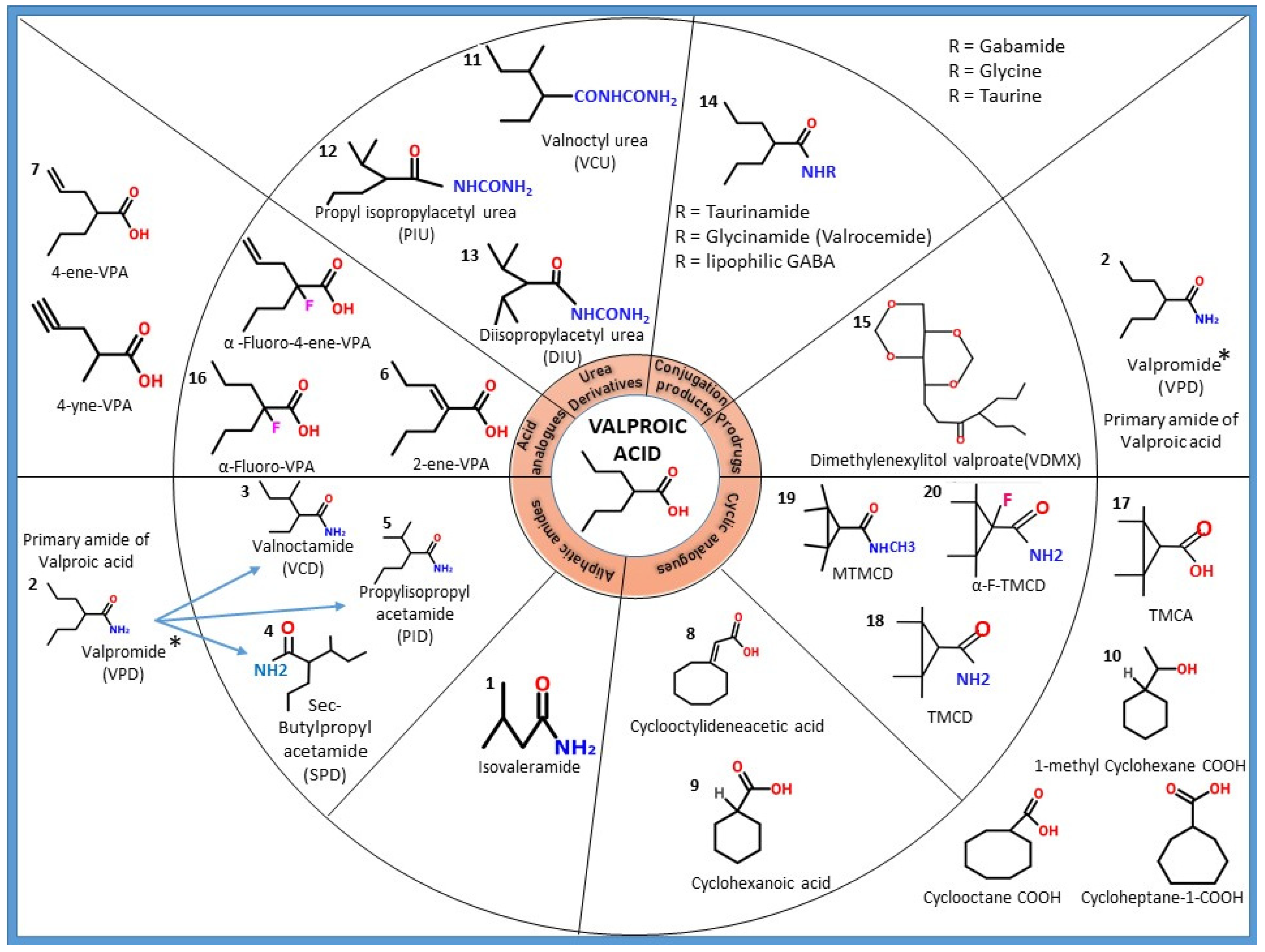

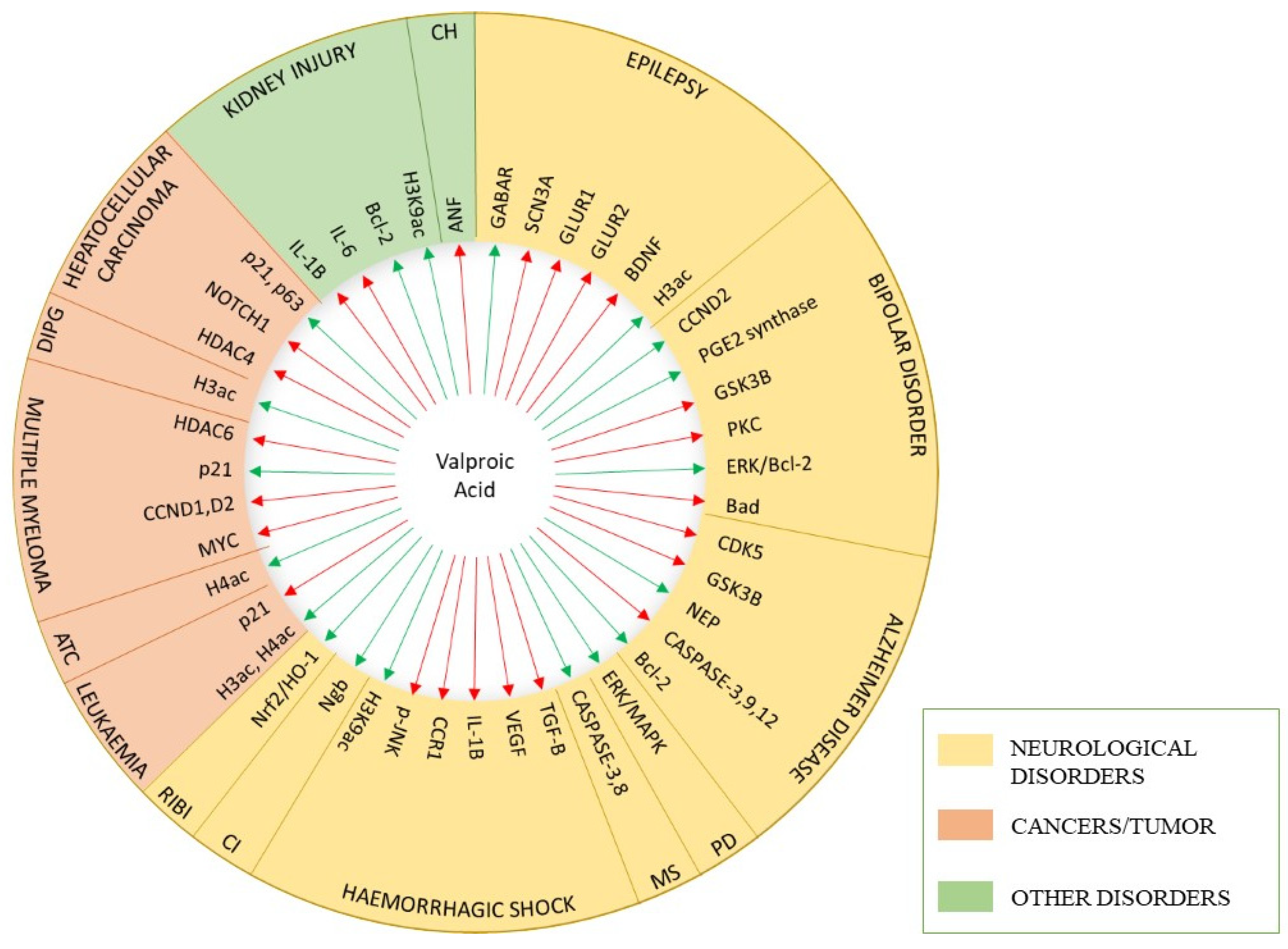{kind=link}
{kind=link}
| S. No | Candidate Compound | Parent Compound | Classification | Chemical Formula | Molecular Weight (g/mol) | Anticonvulsant Property | Clinical Trial Phase | Teratogenicity | Hepatotoxicity | Reference |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | Isovaleramide | Isovaleric acid | Aliphatic amide | C5H11NO | 101.149 | Reduced | Phase I, a Phase II | Reduced | N.D. | [49] |
| 2 | Valpromide | Valproic acid | Aliphatic amide | C8H17NO | 143.23 | Increased | NA | * No change in humans | * No change in humans | [50] |
| 3 | Valnoctamide | Valpromide | Aliphatic amide | C8H17NO | 143.23 | Increased | b Phase III completed | Reduced | Reduced | [50] |
| 4 | sec-Butyl-propyl acetamide | Valpromide | Aliphatic amide | C9H19NO | 157.25 | Increased | Phase II | Reduced | Reduced | [51] |
| 5 | Propyl isopropyl acetamide | Valpromide | Aliphatic amide | C8H17NO | 143.23 | Increased | NA | Reduced | N.D. | [52] |
| 6 | 2-ene-VPA | Valproic acid | Acid analogue | C8H14O2 | 142.2 | Similar | c Phase I | Reduced | Reduced | [53] |
| 7 | 4-ene-VPA | Valproic acid | Acid analogue | C8H14O2 | 142.2 | Similar | NA | Increased | Increased | [27,54] |
| 8 | Cyclooctylideneacetic acid | Valproic acid | Cyclic analogue | C10H16O2 | 168.23 | Increased | NA | N.D. | N.D. | [55] |
| 9 | Cyclohexane carboxylic acid | Valproic acid | Cyclic analogue | C7H12O2 | 128.17 | Similar | NA | Reduced | N.D. | [55] |
| 10 | 1-methyl cyclohexane carboxylic acid | Cyclohexane carboxylic acid | Cyclic analogue | C8H14O2 | 142.2 | Increased | NA | N.D. | N.D. | [55] |
| 11 | Valnoctyl urea | Valnoctic acid | Urea Derivative | C9H18N2O2 | 186.25 | Increased | NA | Reduced | N.D. | [56] |
| 12 | Propyl isopropylacetyl urea | Diisopropyl acetamide | Urea Derivative | C9H18N2O2 | 186.25 | Increased | NA | Reduced | N.D. | [56] |
| 13 | Diisopropyl acetyl urea | Valproic acid | Urea Derivative | C9H18N2O2 | 186.25 | Increased | NA | N.D. | N.D. | [56] |
| 14 | Valrocemide | Valproic acid | Conjugation product | C10H20N2O2 | 200.282 | Increased | d Phase II | Absent | N.D. | [57,58] |
| 15 | Dimethylenexylitol valproate | Valproic acid | Sugar ester | C15H26O6 | 302.36 | Increased | NA | N.D. | N.D. | [59,60] |
| 16 | α-Floro-VPA | Valproic acid | Acid analogue | C8H15O2F | 162.20 | Reduced | NA | Reduced | Reduced | [61] |
| 17 | TMCA | Valproic acid | Cyclic analogue | C8H14O2 | 142.20 | Reduced | NA | N.D. | N.D. | [62] |
| 18 | TMCD | TMCA | Cyclic analogue | C8H15NO | 141 | Increased | NA | Reduced | N.D. | [62,63] |
| 19 | MTMCD | TMCA | Cyclic analogue | C9H17NO | 154 | Increased | NA | Reduced | N.D. | [63,64] |
| 20 | α-Floro-TMCD | TMCA | Cyclic analogue | C8H14FNO | 159.20 | Increased | NA | Reduced | N.D. | [24,65] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mishra, M.K.; Kukal, S.; Paul, P.R.; Bora, S.; Singh, A.; Kukreti, S.; Saso, L.; Muthusamy, K.; Hasija, Y.; Kukreti, R. Insights into Structural Modifications of Valproic Acid and Their Pharmacological Profile. Molecules 2022, 27, 104. https://doi.org/10.3390/molecules27010104
Mishra MK, Kukal S, Paul PR, Bora S, Singh A, Kukreti S, Saso L, Muthusamy K, Hasija Y, Kukreti R. Insights into Structural Modifications of Valproic Acid and Their Pharmacological Profile. Molecules. 2022; 27(1):104. https://doi.org/10.3390/molecules27010104
Chicago/Turabian StyleMishra, Manish Kumar, Samiksha Kukal, Priyanka Rani Paul, Shivangi Bora, Anju Singh, Shrikant Kukreti, Luciano Saso, Karthikeyan Muthusamy, Yasha Hasija, and Ritushree Kukreti. 2022. "Insights into Structural Modifications of Valproic Acid and Their Pharmacological Profile" Molecules 27, no. 1: 104. https://doi.org/10.3390/molecules27010104
APA StyleMishra, M. K., Kukal, S., Paul, P. R., Bora, S., Singh, A., Kukreti, S., Saso, L., Muthusamy, K., Hasija, Y., & Kukreti, R. (2022). Insights into Structural Modifications of Valproic Acid and Their Pharmacological Profile. Molecules, 27(1), 104. https://doi.org/10.3390/molecules27010104