
Perfluorination of Aromatic Compounds Reinforce Their van der Waals Interactions with Rare Gases: The Rotational Spectrum of Pentafluoropyridine-Ne





Abstract
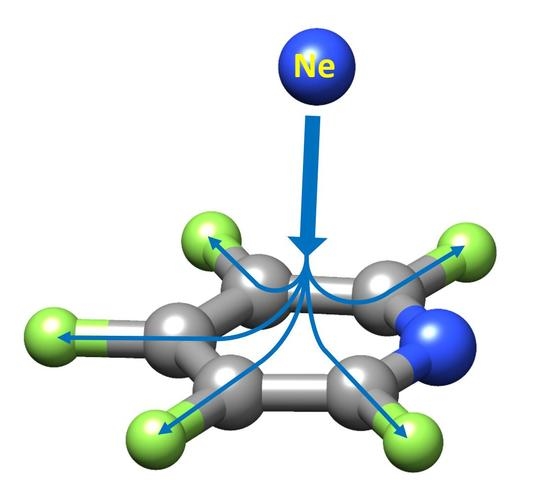
:
1. Introduction
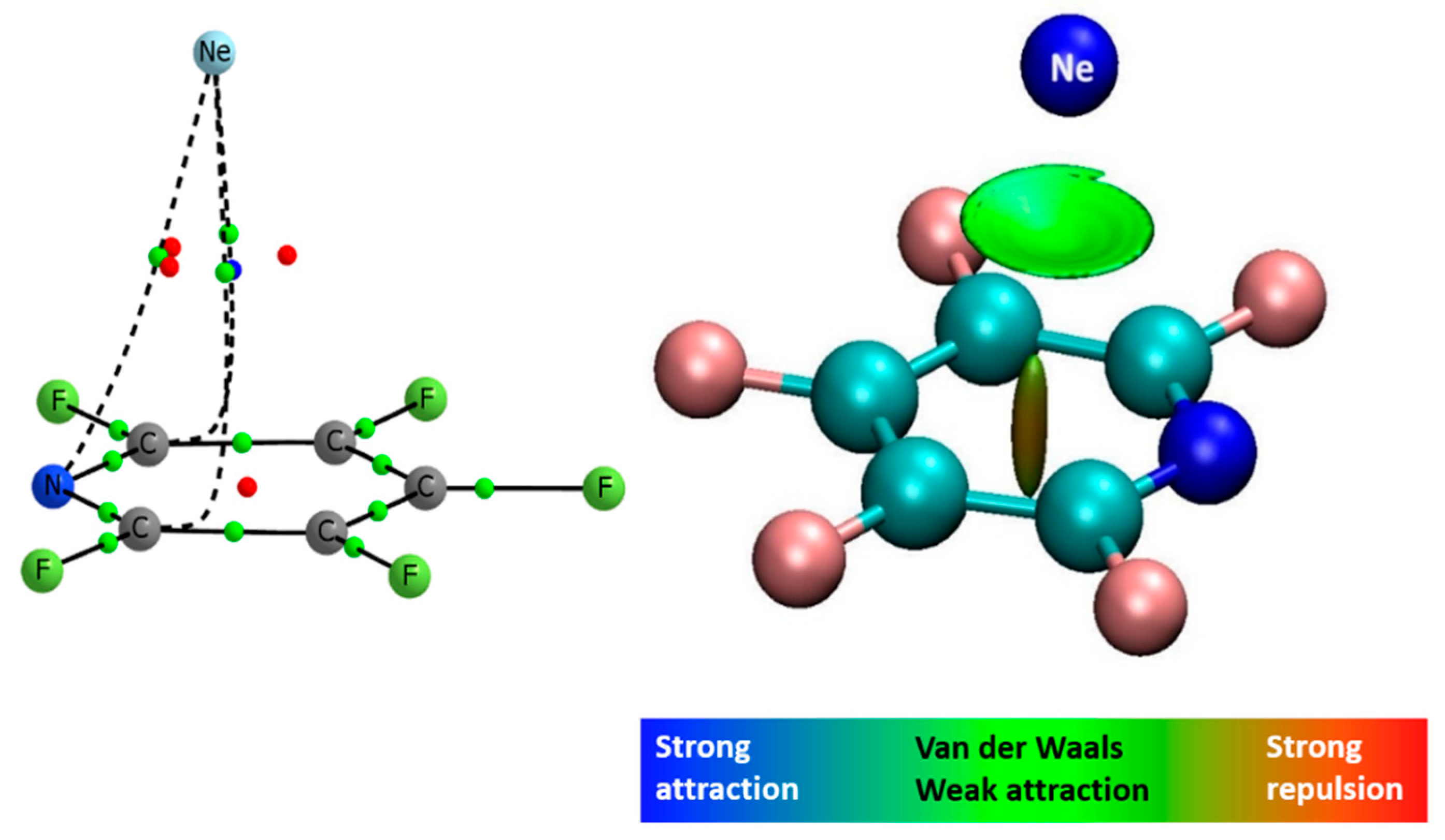
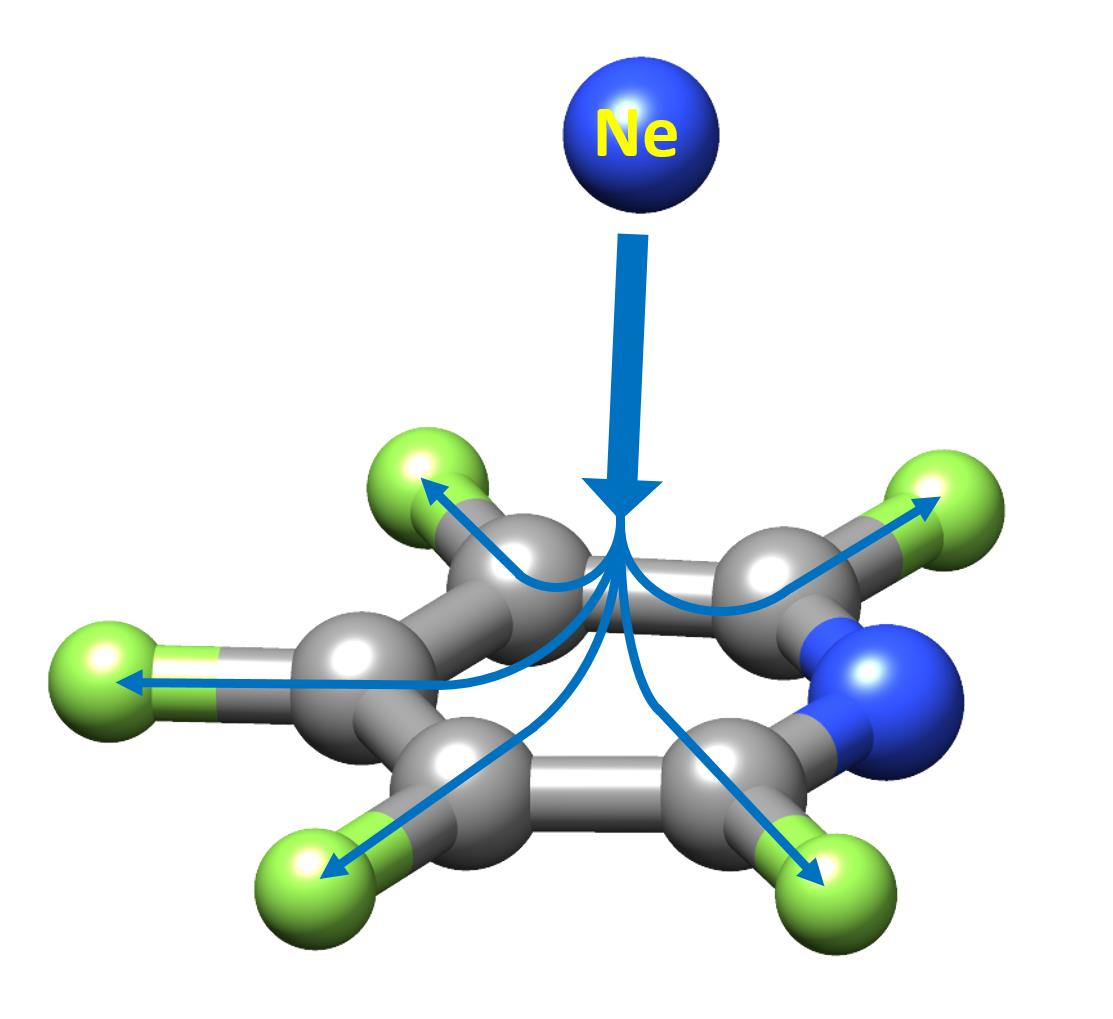
2. Results and Discussion
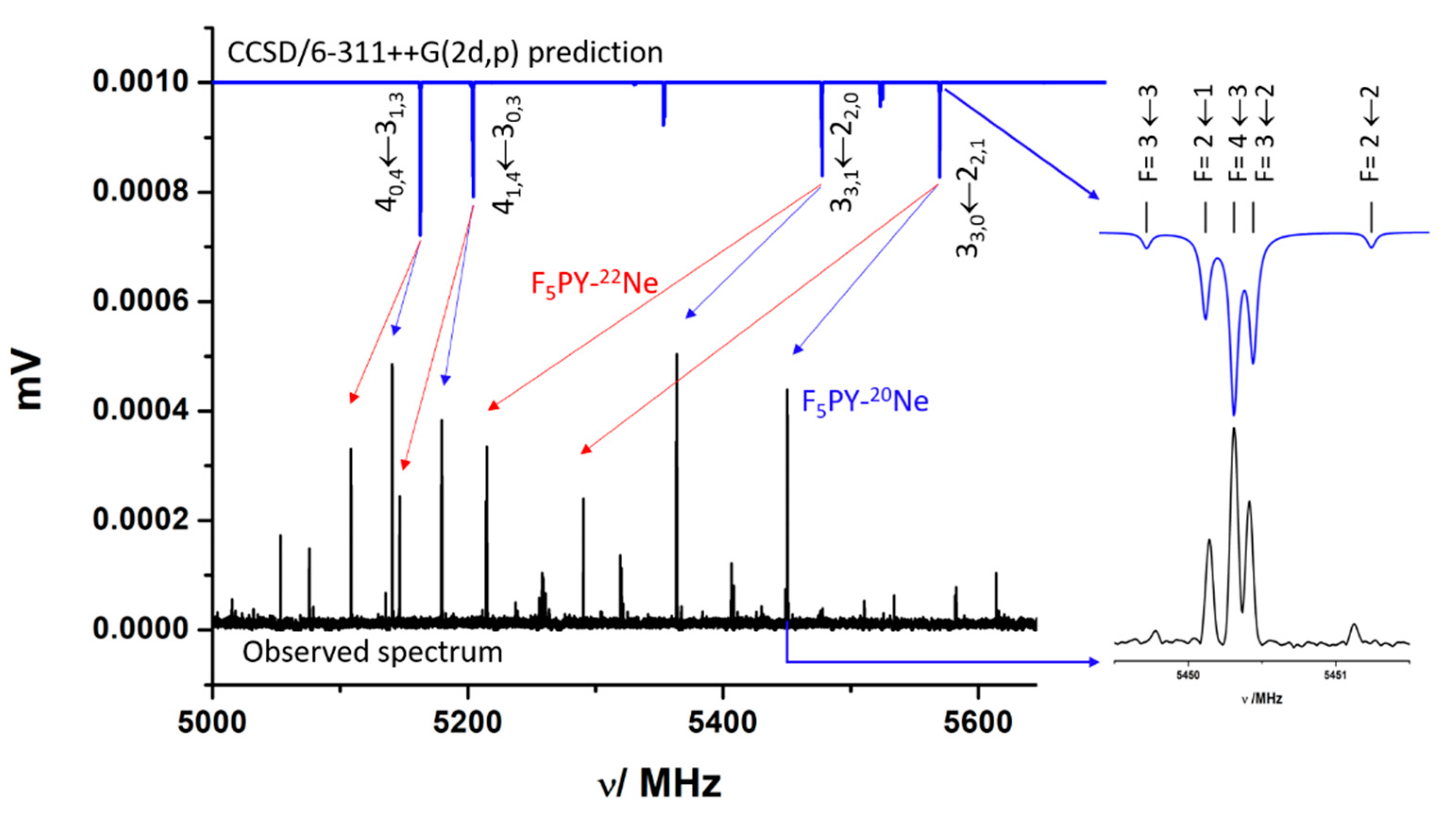
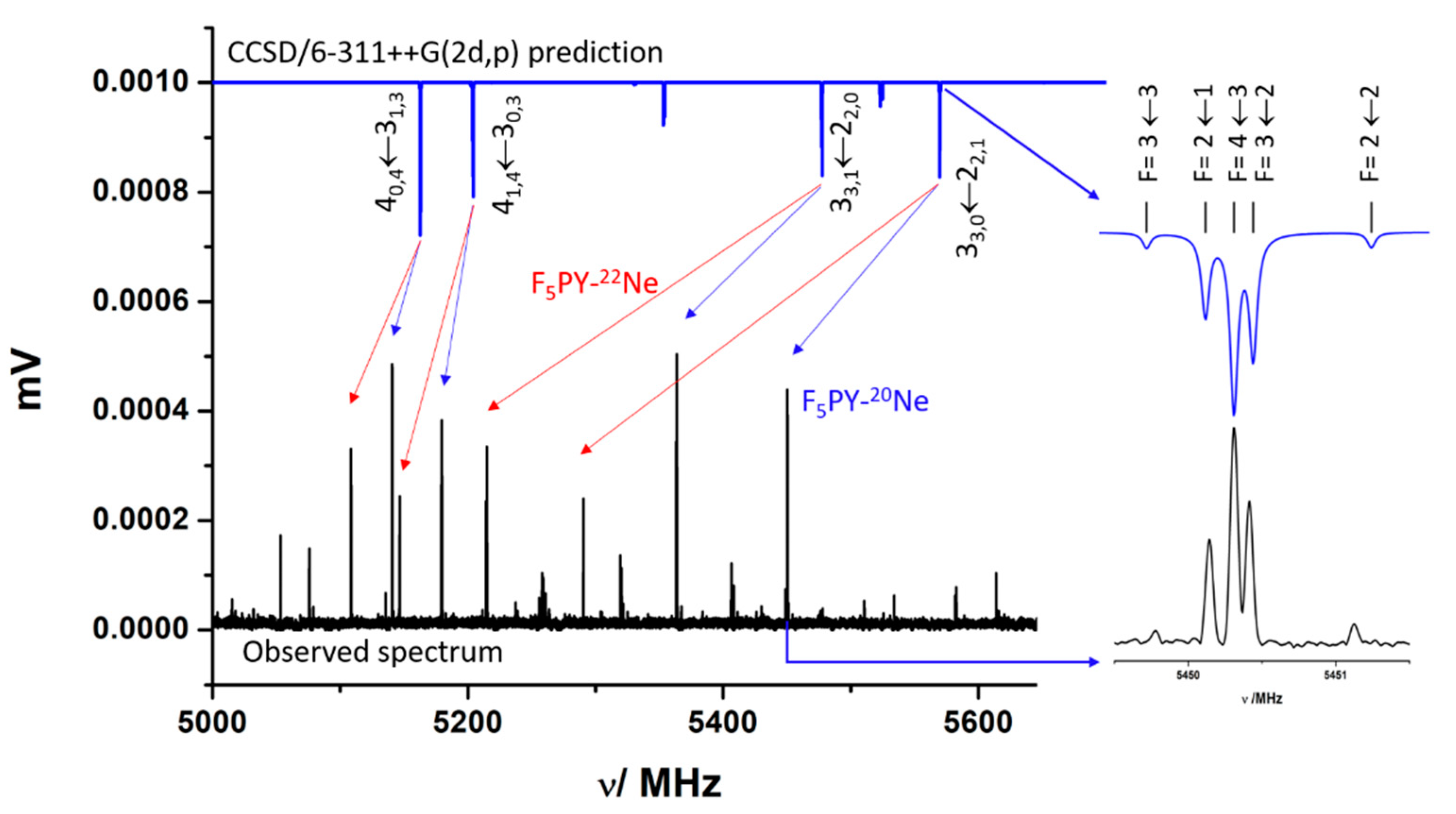
2.1. Rotational Spectrum
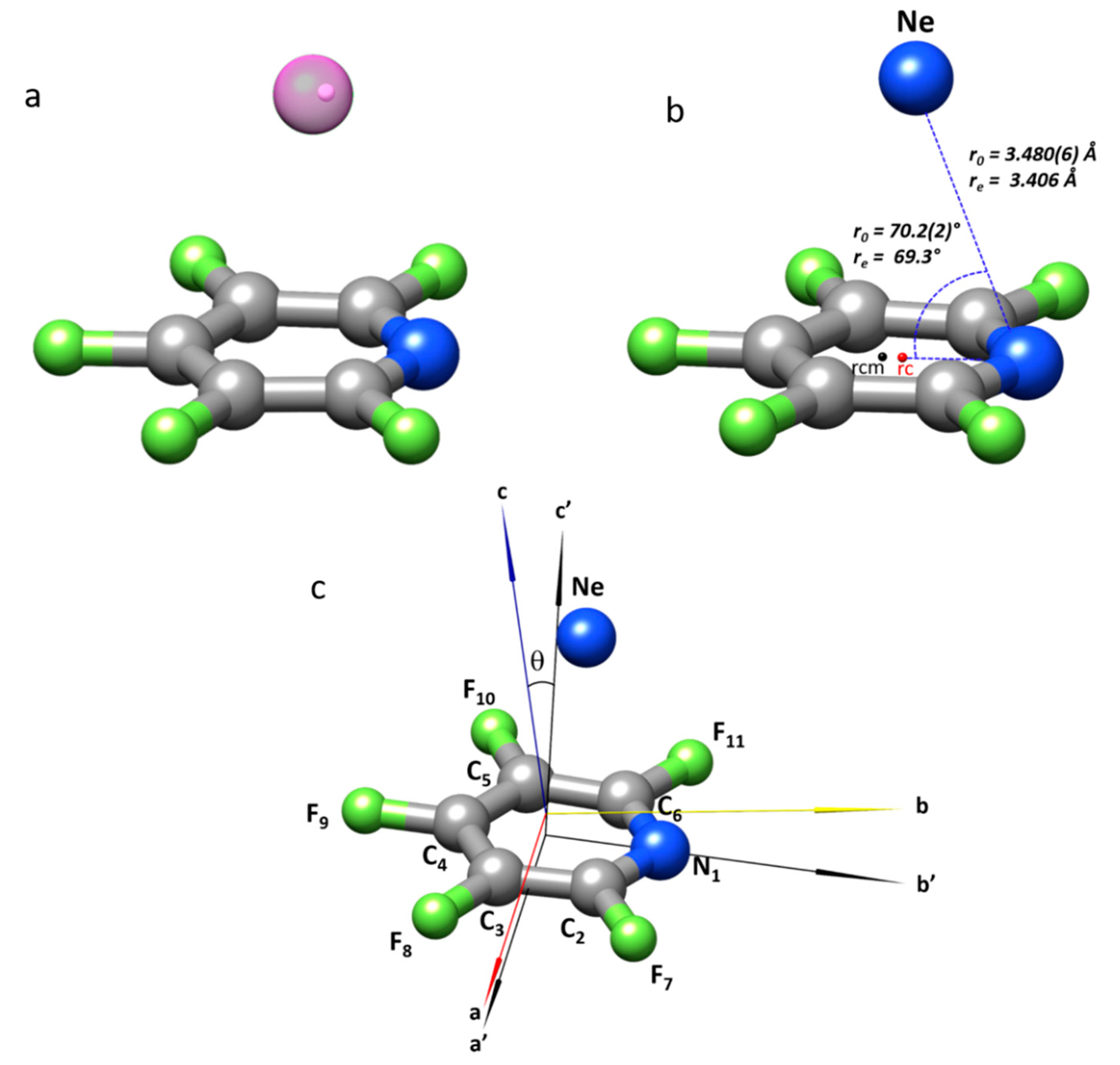
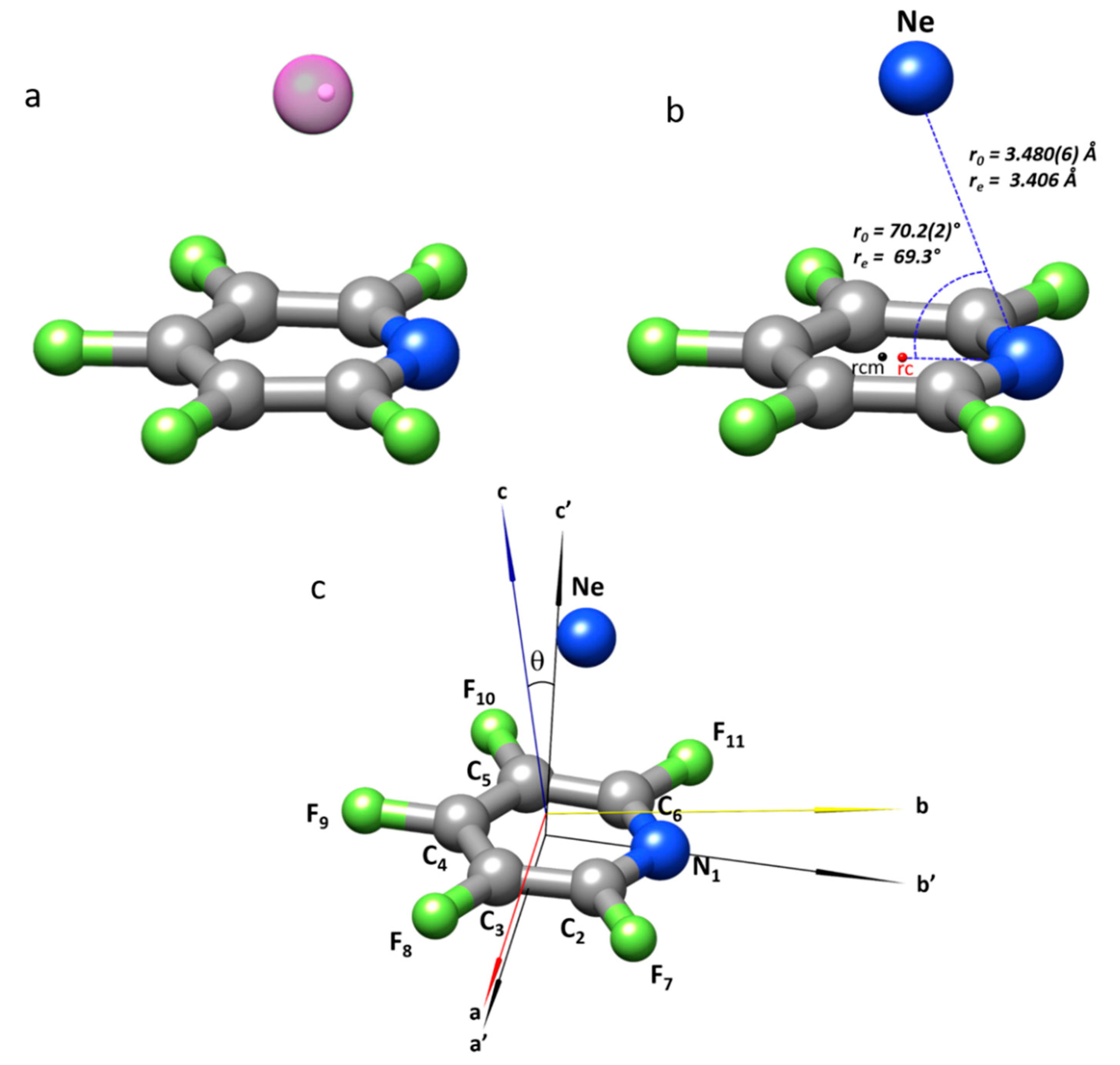
2.2. Structure of the Complex
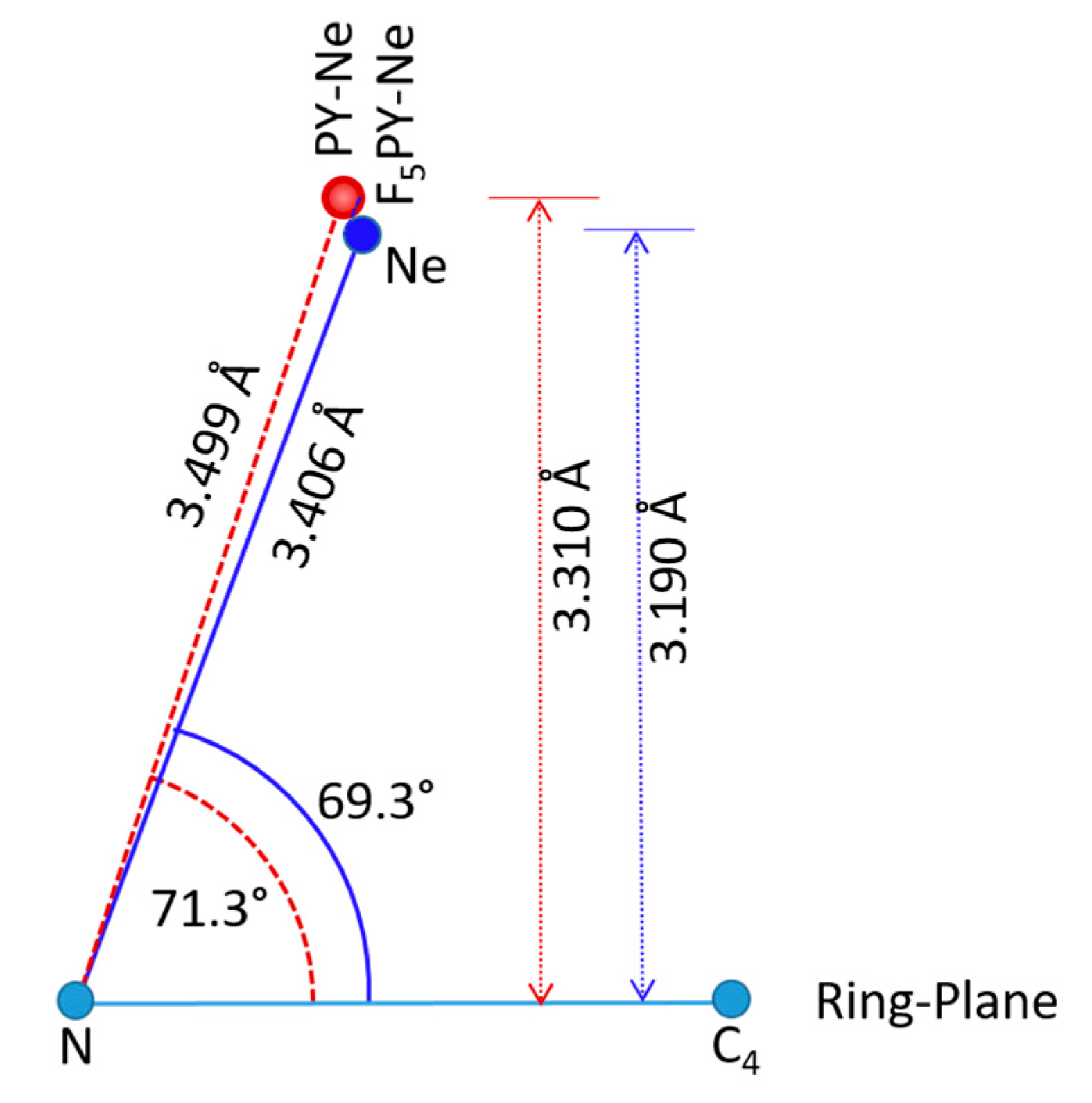
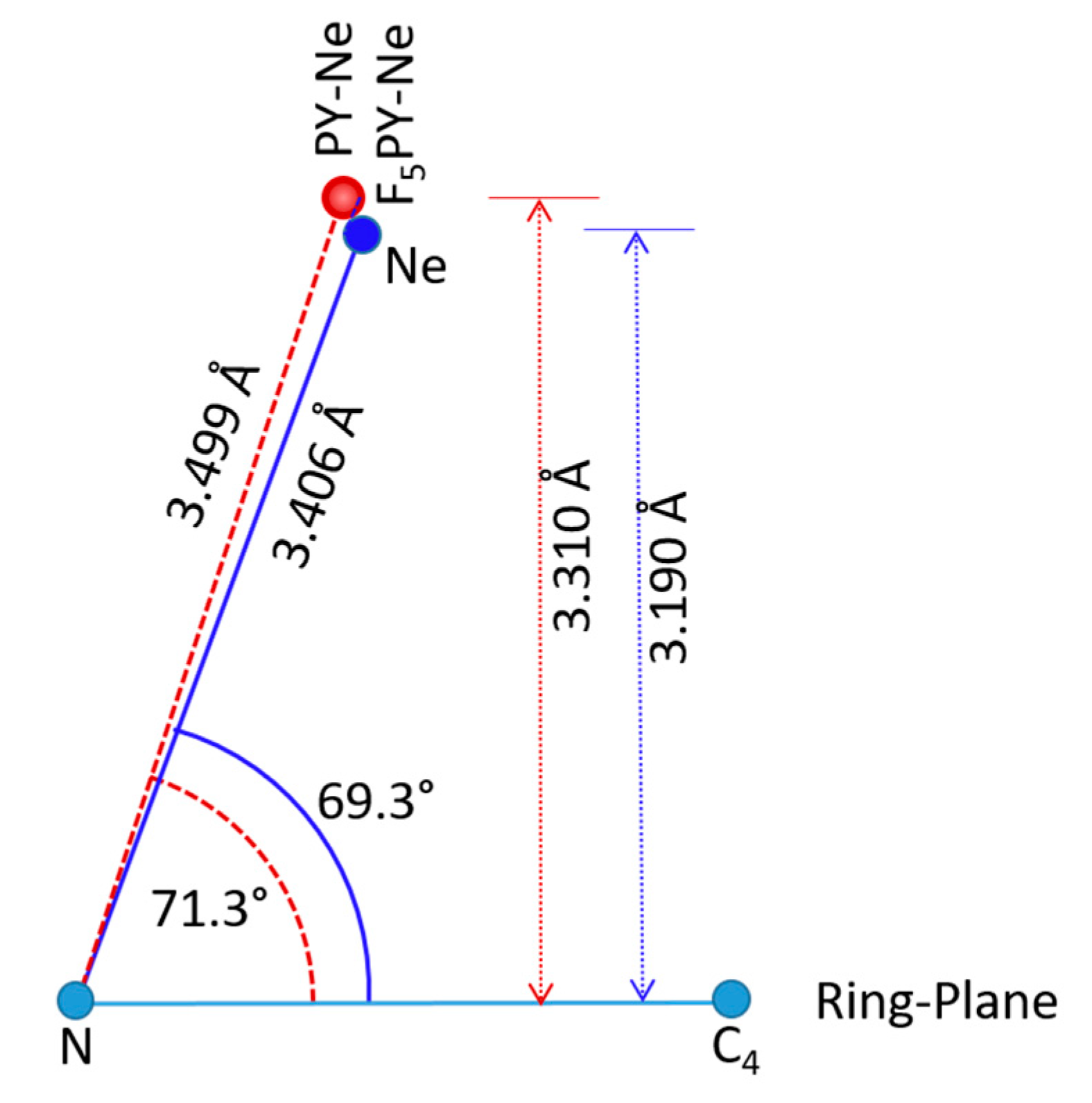
2.3. Comparison with Related Complexes
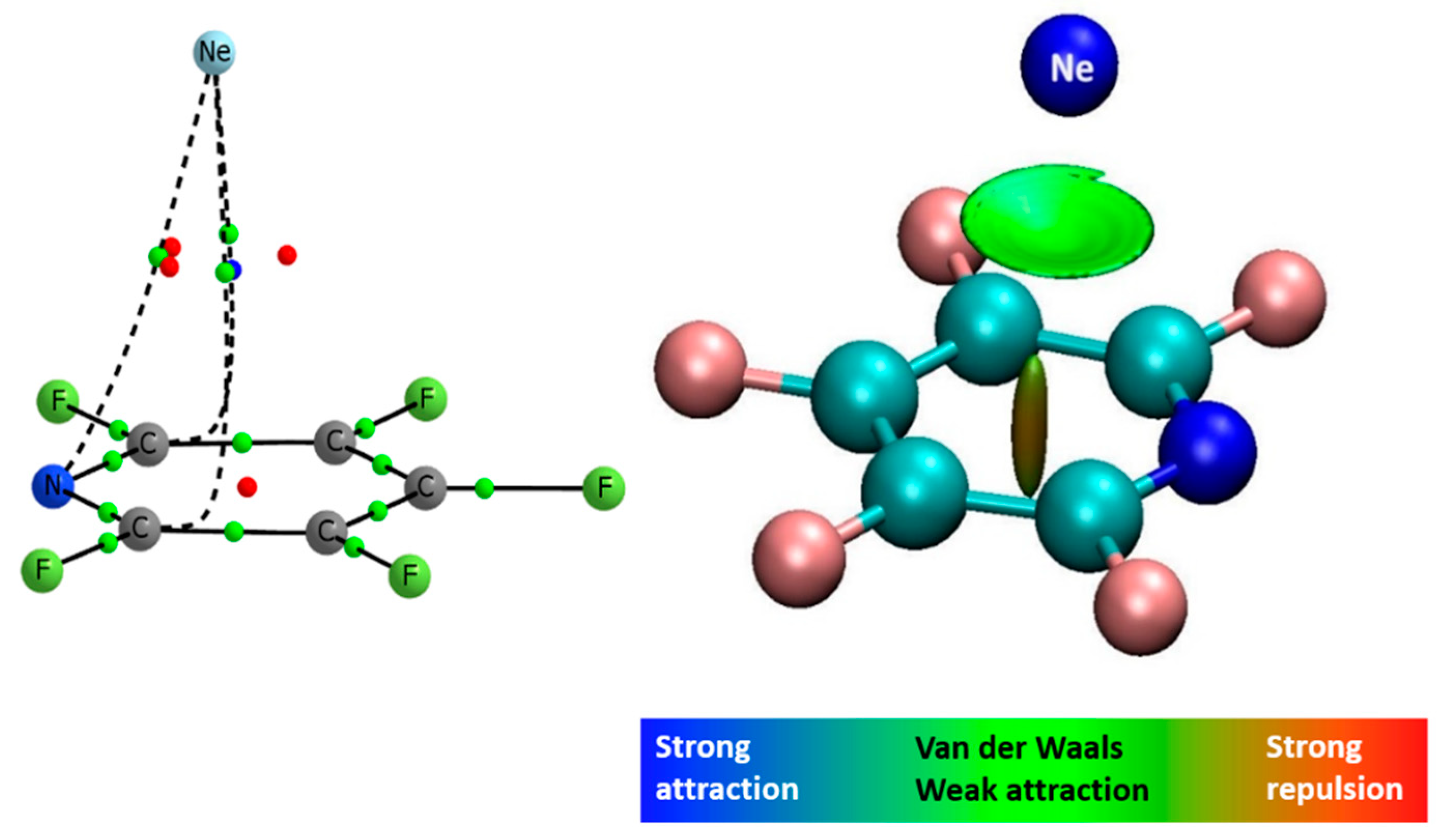
2.4. Binding Energies
3. Materials and Methods
3.1. Theoretical Methods
3.2. Experimental Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Grabow, J.-U.; Caminati, W. Microwave Spectroscopy: Experimental Techniques. Front. Mol. Spectrosc. 2009, 383–454. [Google Scholar] [CrossRef]
- Pate, B.H. Taking the Pulse of Molecular Rotational Spectroscopy. Science 2011, 333, 947–948. [Google Scholar] [CrossRef]
- Li, X.; Lu, T.; Obenchain, D.A.; Zhang, J.; Herbers, S.; Grabow, J.; Feng, G. The Characteristics of Disulfide-Centered Hydrogen Bonds. Angew. Chem. 2021, 133, 5902–5906. [Google Scholar] [CrossRef]
- Xu, Y.; Zhang, J.; Li, W.; Li, J.; Feng, G. High-Resolution Rotational Spectroscopy and Interstellar Search for Isopropyl Isothiocyanate. ACS Earth Space Chem. 2021, 5, 33–39. [Google Scholar] [CrossRef]
- Caminati, W.; Grabow, J.-U. Microwave Spectroscopy: Molecular Systems. In Frontiers of Molecular Spectroscopy; Laane, J., Ed.; Elsevier: Amsterdam, The Netherlands, 2009; pp. 455–552. [Google Scholar] [CrossRef]
- Blanco, S.; Macario, A.; García-Calvo, J.; Revilla-Cuesta, A.; Torroba, T.; López, J.C. Microwave Detection of Wet Triacetone Triperoxide (TATP): Non-Covalent Forces and Water Dynamics. Chem. Eur. J. 2021, 27, 1680–1687. [Google Scholar] [CrossRef] [PubMed]
- Puzzarini, C. Computational Approach to Rotational Spectroscopy. In Computational Strategies for Spectroscopy; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2011; pp. 261–307. [Google Scholar]
- Stone, A. The Theory of Intermolecular Forces; Oxford University Press: Oxford, UK, 2013. [Google Scholar]
- Wagner, J.P.; Schreiner, P.R. London Dispersion in Molecular Chemistry-Reconsidering Steric Effects. Angew. Chem. Int. Ed. 2015, 54, 12274–12296. [Google Scholar] [CrossRef] [PubMed]
- Minaev, B.F.; Panchenko, A.A. New Aspects of the Airglow Problem and Reactivity of the Dioxygen Quintet O2(5Πg) State in the MLT Region as Predicted by DFT Calculations. J. Phys. Chem. A 2020, 124, 9638–9655. [Google Scholar] [CrossRef]
- Hayashi, M.; Ohshima, Y. Quantum Tunneling of a He Atom above and below a Benzene Ring. J. Phys. Chem. Lett. 2020, 11, 9745–9750. [Google Scholar] [CrossRef] [PubMed]
- Arunan, E.; Emilsson, T.; Gutowsky, H.S. Rotational spectra and structures of Rg-C6H6-H 2O trimers and the Ne-C6H6 dimer (Rg = Ne, Ar, or Kr). J. Chem. Phys. 1994, 101, 861–868. [Google Scholar] [CrossRef]
- Brupbacher, T.; Makarewicz, J.; Bauder, A. Intermolecular dynamics of benzene-rare gas complexes as derived from microwave spectra. J. Chem. Phys. 1994, 101, 9736–9746. [Google Scholar] [CrossRef]
- Brupbacher, T.; Bauder, A. Rotational spectrum and dipole moment of the benzene-argon van der Waals complex. Chem. Phys. Lett. 1990, 173, 435–438. [Google Scholar] [CrossRef]
- Klots, T.D.; Emilsson, T.; Gutowsky, H.S. Rotational spectra, structure, Kr-83 nuclear quadrupole coupling constant, and the dipole moment of the Kr-benzene dimer. J. Chem. Phys. 1992, 97, 5335–5340. [Google Scholar] [CrossRef]
- Tanjaroon, C.; Jäger, W. High-resolution microwave spectrum of the weakly bound helium-pyridine complex. J. Chem. Phys. 2007, 127, 034302. [Google Scholar] [CrossRef]
- Maris, A.; Caminati, W.; Favero, P.G. Bond energy of complexes of neon with aromatic molecules: Rotational spectrum and dynamics of pyridine-neon. Chem. Commun. 1998, 2625–2626. [Google Scholar] [CrossRef]
- Evangelisti, L.; Favero, L.B.; Giuliano, B.M.; Tang, S.; Melandri, S.; Caminati, W. Microwave Spectrum of [1,1]-Pyridine−Ne2. J. Phys. Chem. A 2009, 113, 14227–14230. [Google Scholar] [CrossRef]
- Melandri, S.; Giuliano, B.M.; Maris, A.; Evangelisti, L.; Velino, B.; Caminati, W. Rotational Spectrum of the Mixed van der Waals Triad Pyridine-Ar-Ne. ChemPhysChem 2009, 10, 2503–2507. [Google Scholar] [CrossRef] [PubMed]
- Velino, B.; Caminati, W. Fourier transform microwave spectrum of pyridine–neon. J. Mol. Spectrosc. 2008, 251, 176–179. [Google Scholar] [CrossRef]
- Klots, T.D.; Emilsson, T.; Ruoff, R.S.; Gutowsky, H.S. Microwave spectra of noble gas-pyridine dimers: Argon-pyridine and krypton-pyridine. J. Phys. Chem. 1989, 93, 1255–1261. [Google Scholar] [CrossRef]
- Melandri, S.; Maccaferri, G.; Maris, A.; Millemaggi, A.; Caminati, W.; Favero, P.G. Observation of the rotational spectra of van der Waals complexes by free jet absorption millimeter wave spectroscopy: Pyridine-argon. Chem. Phys. Lett. 1996, 261, 267–271. [Google Scholar] [CrossRef]
- Caminati, W.; Favero, P.G. Chemistry at Low Pressure and Low Temperature: Rotational Spectrum and Dynamics of Pyrimidine-Neon. Chem. Eur. J. 1999, 5, 811–814. [Google Scholar] [CrossRef]
- Caminati, W.; Favero, P.G.; Melandri, S.; Meyer, R. Free jet absorption millimeter wave spectrum of the pyrimidine-argon molecular complex. Chem. Phys. Lett. 1997, 268, 393–400. [Google Scholar] [CrossRef]
- Caminati, W.; Melandri, S.; Dell’Erba, A.; Favero, P.G. Bonding energies of rare gases with aromatic molecules: Rotational spectrum and dynamics of pyridazine⋯neon. PhysChemComm 2000, 3, 1–4. [Google Scholar] [CrossRef]
- Caminati, W.; Millemaggi, A.; Favero, P.G.; Makarewicz, J. Free jet absorption millimeter wave spectrum and van der Waals potential energy surface of the pyridazine-argon adduct. J. Phys. Chem. A 1997, 101, 9272–9275. [Google Scholar] [CrossRef]
- Stahl, W.; Grabow, J.U. The Rotational Spectrum of the Fluorobenzene-Argon Van der Waals Complex. Z. Naturforsch. Sect. A J. Phys. Sci. 1992, 47, 681–684. [Google Scholar] [CrossRef]
- Sussmann, R.; Neusser, H.J. The van der Waals rovibronic spectrum of p-difluorobenzene-Ar up to 125 cm−1 intermolecular energy: Assignment and character of van der Waals modes. J. Chem. Phys. 1995, 102, 3055–3063. [Google Scholar] [CrossRef]
- Jayasekharan, T.; Parmenter, C.S. Characteristics and relaxation dynamics of van der Waals complexes between p-difluorobenzene and Ne. J. Chem. Phys. 2004, 120, 11469–11478. [Google Scholar] [CrossRef]
- Sun, M.; Kamaee, M.; Van Wijngaarden, J. Rotational spectra and structures of the van der waals dimers of argon with 2-fluoropyridine and 3-fluoropyridine. J. Phys. Chem. A 2013, 117, 13429–13434. [Google Scholar] [CrossRef]
- Bettens, R.P.A.; Spycher, R.M.; Bauder, A. Intermolecular force field and approximate equilibrium structure of various complexes containing one or two rare gas atoms from microwave spectroscopic constants. Mol. Phys. 1995, 86, 487–511. [Google Scholar] [CrossRef]
- Evangelisti, L.; Brendel, K.; Mäder, H.; Caminati, W.; Melandri, S. Rotational Spectroscopy Probes Water Flipping by Full Fluorination of Benzene. Angew. Chem. Int. Ed. 2017, 56, 13699–13703. [Google Scholar] [CrossRef]
- Calabrese, C.; Gou, Q.; Maris, A.; Caminati, W.; Melandri, S. Probing the Lone Pair·π-Hole Interaction in Perfluorinated Heteroaromatic Rings: The Rotational Spectrum of Pentafluoropyridine·Water. J. Phys. Chem. Lett. 2016, 7, 1513–1517. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Usabiaga, I.; Calabrese, C.; Evangelisti, L.; Maris, A.; Favero, L.B.; Melandri, S. Characterizing the lone pair⋯π–hole interaction in complexes of ammonia with perfluorinated arenes. Phys. Chem. Chem. Phys. 2021, 23, 9121–9129. [Google Scholar] [CrossRef] [PubMed]
- López, J.C.; Macario, A.; Maris, A.; Alkorta, I.; Blanco, S. How Aromatic Fluorination Exchanges the Interaction Role of Pyridine with Carbonyl Compounds: The Formaldehyde Adduct. Chem. Eur. J. 2021, 27, 13870–13878. [Google Scholar] [CrossRef]
- Bauzá, A.; Mooibroek, T.J.; Frontera, A. The Bright Future of Unconventional σ/π-Hole Interactions. ChemPhysChem 2015, 16, 2496–2517. [Google Scholar] [CrossRef]
- Alkorta, I.; Rozas, I.; Elguero, J. An Attractive Interaction between the π-Cloud of C6F6 and Electron-Donor Atoms. J. Org. Chem. 1997, 62, 4687–4691. [Google Scholar] [CrossRef]
- Gallivan, J.P.; Dougherty, D.A. Can Lone Pairs Bind to a π System? The Water···Hexafluorobenzene Interaction. Org. Lett. 1999, 1, 103–106. [Google Scholar] [CrossRef] [PubMed]
- Alkorta, I.; Rozas, I.; Elguero, J. Interaction of Anions with Perfluoro Aromatic Compounds. J. Am. Chem. Soc. 2002, 124, 8593–8598. [Google Scholar] [CrossRef]
- Quiñonero, D.; Garau, C.; Rotger, C.; Frontera, A.; Ballester, P.; Costa, A.; Deyà, P.M. Anion–π Interactions: Do They Exist? Angew. Chem. Int. Ed. 2002, 41, 3389–3392. [Google Scholar] [CrossRef]
- Frontera, A.; Gamez, P.; Mascal, M.; Mooibroek, T.J.; Reedijk, J. Putting Anion-π Interactions Into Perspective. Angew. Chem. Int. Ed. 2011, 50, 9564–9583. [Google Scholar] [CrossRef] [PubMed]
- Mascal, M.; Armstrong, A.; Bartberger, M.D. Anion-aromatic bonding: A case for anion recognition by π-acidic rings. J. Am. Chem. Soc. 2002, 124, 6274–6276. [Google Scholar] [CrossRef]
- Pickett, H.M. The fitting and prediction of vibration-rotation spectra with spin interactions. J. Mol. Spectrosc. 1991, 148, 371–377. [Google Scholar] [CrossRef]
- Watson, J.K.G. Aspects of Quartic and Sextic Centrifugal Effects on Rotational Energy Levels. In Vibrational Spectra and Structure a Series of Advances; Durig, J.R., Ed.; Elsevier: New York, NY, USA, 1977; Volume 6, pp. 1–89. [Google Scholar]
- Gordy, W.; Cook, R.L. Microwave Molecular Spectra; Wiley-Interscience: New York, NY, USA, 1984; Volume 11, ISBN 0471086819. [Google Scholar]
- Doraiswamy, S.; Sharma, S.D. Microwave spectrum, centrifugal distortion constants, dipole moment and quadrupole coupling constants of pentafluoropyridine. Chem. Phys. 1974, 6, 76–86. [Google Scholar] [CrossRef]
- Kraitchman, J. Determination of Molecular Structure from Microwave Spectroscopic Data. Am. J. Phys. 1953, 21, 17–24. [Google Scholar] [CrossRef]
- van Eijck, B.P. Influence of molecular vibrations on substitution coordinates. J. Mol. Spectrosc. 1982, 91, 348–362. [Google Scholar] [CrossRef]
- Kisiel, Z. Least-squares mass-dependence molecular structures for selected weakly bound intermolecular clusters. J. Mol. Spectrosc. 2003, 218, 58–67. [Google Scholar] [CrossRef]
- Watson, J.K.; Roytburg, A.; Ulrich, W. Least-Squares Mass-Dependence Molecular Structures. J. Mol. Spectrosc. 1999, 196, 102–119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stephens, P.J.; Devlin, F.J.; Chabalowski, C.F.; Frisch, M.J. Ab Initio calculation of vibrational absorption and circular dichroism spectra using density functional force fields. J. Phys. Chem. 1994, 98, 11623–11627. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef]
- Møller, C.; Plesset, M.S. Note on an approximation treatment for many-electron systems. Phys. Rev. 1934, 46, 618–622. [Google Scholar] [CrossRef] [Green Version]
- Frisch, M.J.; Pople, J.A.; Binkley, J.S. Self-consistent molecular orbital methods 25. Supplementary functions for Gaussian basis sets. J. Chem. Phys. 1984, 80, 3265–3269. [Google Scholar] [CrossRef]
- Blanco, S.; Pinacho, P.; López, J.C. Hydrogen-Bond Cooperativity in Formamide 2-Water: A Model for Water-Mediated Interactions. Angew. Chem. Int. Ed. 2016, 55, 9331–9335. [Google Scholar] [CrossRef] [Green Version]
- Kendall, R.A.; Dunning, T.H.; Harrison, R.J. Electron affinities of the first-row atoms revisited. Systematic basis sets and wave functions. J. Chem. Phys. 1992, 96, 6796–6806. [Google Scholar] [CrossRef] [Green Version]
- Scuseria, G.E.; Janssen, C.L.; Schaefer, H.F. An efficient reformulation of the closed-shell coupled cluster single and double excitation (CCSD) equations. J. Chem. Phys. 1988, 89, 7382–7387. [Google Scholar] [CrossRef]
- Frisch, M.J.; Frisch, M.J.; Trucks, M.J.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; et al. Gaussian 16, Revision, A.03; Gaussian Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Jansen, G.; Hesselmann, A.; Williams, H.L.; Chabalowski, C.F. Comment on “using Kohn-Sham orbitals in symmetry-adapted perturbation theory to investigate intermolecular interactions”. J. Phys. Chem. A 2001, 105, 11156–11157. [Google Scholar] [CrossRef]
- Hesselmann, A. DFT-SAPT Intermolecular Interaction Energies Employing Exact-Exchange Kohn-Sham Response Methods. J. Chem. Theory Comput. 2018, 14, 1943–1959. [Google Scholar] [CrossRef]
- Werner, H.J.; Knowles, P.J.; Manby, F.R.; Black, J.A.; Doll, K.; Heßelmann, A.; Kats, D.; Köhn, A.; Korona, T.; Kreplin, D.A.; et al. The Molpro quantum chemistry package. J. Chem. Phys. 2020, 152, 144107. [Google Scholar] [CrossRef] [Green Version]
- Adamo, C.; Barone, V. Toward reliable density functional methods without adjustable parameters: The PBE0 model. J. Chem. Phys. 1999, 110, 6158–6170. [Google Scholar] [CrossRef]
- Bader, R.F.W. A quantum theory of molecular structure and its applications. Chem. Rev. 1991, 91, 893–928. [Google Scholar] [CrossRef]
- Contreras-García, J.; Johnson, E.R.; Keinan, S.; Chaudret, R.; Piquemal, J.P.; Beratan, D.N.; Yang, W. NCIPLOT: A program for plotting noncovalent interaction regions. J. Chem. Theory Comput. 2011, 7, 625–632. [Google Scholar] [CrossRef]
- Brown, G.G.; Dian, B.C.; Douglass, K.O.; Geyer, S.M.; Shipman, S.T.; Pate, B.H. A broadband Fourier transform microwave spectrometer based on chirped pulse excitation. Rev. Sci. Instrum. 2008, 79, 053103. [Google Scholar] [CrossRef] [PubMed]
- Pinacho, P.; Blanco, S.; López, J.C. The complete conformational panorama of formanilide–water complexes: The role of water as a conformational switch. Phys. Chem. Chem. Phys. 2019, 21, 2177–2185. [Google Scholar] [CrossRef] [PubMed]
- Balle, T.J.; Flygare, W.H. Fabry-Perot cavity pulsed Fourier transform microwave spectrometer with a pulsed nozzle particle source. Rev. Sci. Instrum. 1981, 52, 33–45. [Google Scholar] [CrossRef]
- Alonso, J.; Lorenzo, F.J.; López, J.C.; Lesarri, A.; Mata, S.; Dreizler, H. Construction of a molecular beam Fourier transform microwave spectrometer used to study the 2,5-dihydrofuran-argon van der Waals complex. Chem. Phys. 1997, 218, 267–275. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| F5PY-20Ne | F5PY-22Ne | ||
|---|---|---|---|
| Param. a | exp | CCSD | exp |
| A/MHz | 940.26871(21) b | 962.2 | 911.57618(20) |
| B/MHz | 769.84484(19) | 780.8 | 752.84325(17) |
| C/MHz | 617.16139(13) | 618.2 | 615.66886(11) |
| κ | −0.05 | −0.05 | −0.07 |
| Paa/uÅ2 | 468.93082(23) | 469.7 | 468.87713(21) |
| Pbb/uÅ2 | 349.94576(23) | 347.7 | 351.98461(21) |
| Pcc/uÅ2 | 187.53784(23) | 177.5 | 202.41668(21) |
| ΔJ/kHz | 0.4055(25) | 0.4890(23) | |
| ΔJK/kHz | 6.329(10) | 6.1360(91) | |
| ΔK/kHz | −5.5700(68) | −5.4288(59) | |
| δJ/kHz | 0.1045(12) | 0.1066(11) | |
| δK/kHz | −4.2430(84) | −3.9140(73) | |
| 3/2(χaa)/MHz | 2.9536(29) | 3.11 | 2.9595(26) |
| 1/4(χbb-χcc)/MHz | −1.37697(75) | −1.51 | −1.33170(67) |
| χaa/MHz | 1.9687(19) | 2.07 | 1.9730(17) |
| χbb/MHz | −3.7383(25) | −4.05 | −3.6499(22) |
| χcc/MHz | 1.7696(25) | 1.98 | 1.6769(22) |
| n | 139 | 118 | |
| σ/kHz | 3.2 | 2.6 | |
| Parameters | F5PY a | F5PY-20Ne | F5PY-22Ne |
|---|---|---|---|
| A/MHz | 1481.58184(19) b | 940.26871(21) | 911.57618(20) |
| B/MHz | 1075.37335(17) | 769.84484(19) | 752.84325(17) |
| C/MHz | 623.11194(16) | 617.16139(13) | 615.66886(11) |
| Ia/uÅ2 | 341.107724(44) | 537.80275(12) | 554.73049(12) |
| Ib/uÅ2 | 469.956791(74) | 656.85847(16) | 671.69241(15) |
| Ic/uÅ2 | 811.05653(21) | 819.36283(17) | 821.34916(15) |
| Paa/uÅ2 | 469.95280(16) | 468.93082(23) | 468.87713(21) |
| Pbb/uÅ2 | 341.10373(16) | 349.94576(23) | 351.98461(21) |
| Pcc/uÅ2 | 0.00399(16) | 187.53784(23) | 202.41668(21) |
| χaa/MHz | 1.9664(53) | 1.9687(19) | 1.9730(17) |
| χbb/MHz | −3.9534(72) | −3.7383(25) | −3.6499(22) |
| χcc/MHz | 1.9870(72) | 1.7696(25) | 1.6769(22) |
| rs Coordinates | a | b | c |
|---|---|---|---|
| [0.0000] a | 0.9672(15) b | 2.75959(54) | |
| 0.0000 | 0.911(23) | 2.8134(64) | |
| 0.0000 | 0.814 | 2.757022 | |
| Parameters | r0 | rs | |
| r(N···Ne)/Å | 3.480(6) | 3.359 | |
| ∠(CNNe)/° | 70.2(2) | 69.2 | |
| r(rc···Ne) c/Å | 3.278(8) | 3.192 | |
| R = r(rcm···Ne) d/Å | 3.307(9) | 3.215 | 3.260 |
| ∠(Ne···rc···N)/° | 86.8(5) | 86.8 | |
| ∠(Ne···rcm···N)/° | 82.8(5) | 82.2 | |
| φ e/° | 7.2(5) | 7.2 | 8.1 |
| θf/° | 9.8 | 8.6 | |
| Fit | experimental | Residuals g | |
| 20Ne A/MHz | 940.26871(21) | −1.56 | |
| 20Ne B/MHz | 769.84484(19) | 0.41 | |
| 20Ne C/MHz | 617.16139(13) | 0.60 | |
| 22Ne A/MHz | 911.57618(20) | −0.84 | |
| 22Ne B/MHz | 752.84325(17) | 1.13 | |
| 22Ne C/MHz | 615.66886(11) | 0.47 |
| Param. | Method | F5PY-Ne | PY-Ne | F6BZ-Ne | BZ-Ne |
|---|---|---|---|---|---|
| R/Å | CCSD | 3.215 | 3.325 | 3.199 | 3.313 |
| (rs/e, r0) | (3.260 a, 3.302) | (3.316 b, 3.400 c) | (3.2989 d, 3.462 e) | ||
| φ/° | CCSD | 7.8 | 4.6 | 0.0 | 0.0 |
| (rs/e , r0) | (7.2 a, 8.1) | (4.2 b, 6.2 c) | (0.0 d, 0.0 e) |
| BE[CCSD(T)] | Elect. | Exchange | Induction | Dispersion | δHF | |
|---|---|---|---|---|---|---|
| PY-Ne | −2.9 | −0.66 | 2.21 | 0.02 | −3.05 | −0.10 |
| BZ-Ne | −2.8 | −0.81 | 2.67 | 0.01 | −3.27 | −0.14 |
| F5PY-Ne | −4.9 | −0.87 | 2.91 | −0.04 | −3.74 | −0.13 |
| F6BZ-Ne | −4.9 | −1.03 | 3.33 | 0.01 | −3.87 | −0.16 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Macario, A.; Blanco, S.; Alkorta, I.; López, J.C. Perfluorination of Aromatic Compounds Reinforce Their van der Waals Interactions with Rare Gases: The Rotational Spectrum of Pentafluoropyridine-Ne. Molecules 2022, 27, 17. https://doi.org/10.3390/molecules27010017
Macario A, Blanco S, Alkorta I, López JC. Perfluorination of Aromatic Compounds Reinforce Their van der Waals Interactions with Rare Gases: The Rotational Spectrum of Pentafluoropyridine-Ne. Molecules. 2022; 27(1):17. https://doi.org/10.3390/molecules27010017
Chicago/Turabian StyleMacario, Alberto, Susana Blanco, Ibon Alkorta, and Juan Carlos López. 2022. "Perfluorination of Aromatic Compounds Reinforce Their van der Waals Interactions with Rare Gases: The Rotational Spectrum of Pentafluoropyridine-Ne" Molecules 27, no. 1: 17. https://doi.org/10.3390/molecules27010017
APA StyleMacario, A., Blanco, S., Alkorta, I., & López, J. C. (2022). Perfluorination of Aromatic Compounds Reinforce Their van der Waals Interactions with Rare Gases: The Rotational Spectrum of Pentafluoropyridine-Ne. Molecules, 27(1), 17. https://doi.org/10.3390/molecules27010017