1. Introduction
The liver-expressed antimicrobial peptide 2 (LEAP2) was originally isolated from human hemofiltrates [
1]. Its encoding gene is composed of three exons and two introns located at chromosome 5q31 in humans and chromosome 11 in mice. There are two different transcripts of LEAP2 in humans; the one mainly found in the liver, small intestine, kidney, colon, and gastric antrum is the 350bp transcript, whereas, the lung, heart, and trachea express the 550bp transcript [
1,
2]. The murine
Leap2 RNA encodes for a 76 amino acid protein, although the mature LEAP2 peptide is composed of 40 amino acid residues [
1]. The peptide is positively charged and shows similar characteristics to other cationic peptides with antimicrobial activity in vitro. It is composed of an N-terminal (1–14 amino acids) and a C-terminal (15–40 amino acids) domain and two disulfide bonds. LEAP2 shares structural characteristics with antimicrobial peptides such as defensins and LEAP1/hepcidin [
1]. Several Gram-positive bacteria, such as
Bacillus megaterium and
Bacillus subtilis, are sensitive to treatment with a synthetic LEAP2 peptide, which did not affect the growth of Gram-negative
Escherichia coli and
Pseudomonas [
1]. The antimicrobial activity of LEAP2 was suggested to be dependent on the amino-terminal domain and not on the induction of membrane destabilization and/or pore formation under physiological conditions [
3], indicating that the peptide may bind to an intracellular target to exert this activity.
The strong sequence conservation of LEAP2 in vertebrates might indicate a physiological role unrelated to antimicrobial activity [
3,
4]. Whilst it was shown that the peptide is not mitogenic for epithelial cells and does not function directly to link the innate and adaptive immune systems [
2,
5], regulation of the action of ghrelin, a key hormone produced from the stomach and acting in the brain to regulate food intake, reward, and other fundamental central nervous system (CNS) functions, was recently suggested as an additional function of LEAP2. Ge et al. [
6] used a mouse model of vertical sleeve gastrectomy (VSG) as a tool to identify secreted proteins and peptides that might act as metabolic regulators. They analyzed various genes that encode for secreted proteins and peptides in the stomach and intestines and found that one set of genes exhibited inverse regulation between the stomach and duodenum. Among these genes,
Leap2 expression increased by 52-fold in the stomach and decreased by 94% in the duodenum following VSG [
6]. Subsequently, it was found that LEAP2 acts as an endogenous antagonist of the ghrelin receptor by specifically inhibiting ghrelin binding to its receptor, the GHSR, in a non-competitive manner, thus blocking ghrelin-mediated GH release, food intake, and glucose mobilization [
6]. This discovery led to the proposal of LEAP2 as a new potential therapeutic target for uncontrolled ghrelin signaling-related diseases, such as obesity and diabetes, cachexia, anorexia, alcohol abuse, and Prader-Willi Syndrome.
Two other reciprocally interacting players in energy metabolism and its pathological disturbances are: (1) the endocannabinoidome (eCBome), which includes: (a) the endocannabinoids anandamide (AEA), 2-arachidonoylglycerol (2-AG), and noladin ether; (b) the cannabinoid CB1 and CB2 receptors; (c) endocannabinoid-related mediators, such as the
N-acylethanolamines (NAE), like AEA, and other long-chain fatty acid amides and esters in general, such as the 2-monoacyl-glycerols (2-MAGs), like 2-AG, and the
N-acyl amino acids, the
N-acyl-glycines, as well as their receptors (which encompass peroxisome proliferator-activated receptors [PPARs], transient receptor potential [TRP] channels and orphan G-protein-coupled receptors [GPRs]) and anabolic/catabolic enzymes [
7]; and (2) the gut microbiome (mBIome), which encompasses thousands of commensal intestinal microorganism species with their armamentarium of genes, proteins and metabolites signaling to the host [
8]. Both these systems have been related to ghrelin function, for example through the following mechanisms: (1) ghrelin has been shown to enhance food intake partly via endocannabinoid biosynthesis and CB1 receptor activation, e.g., in the hypothalamus [
9,
10], while, vice versa, some central effects of CB1 receptors have been suggested to be mediated by activation of the ghrelin receptor, GHSR [
11]; additionally, the anorexigenic NAE,
N-oleoyl-ethanolamine (OEA), which is inactive at cannabinoid receptors and activates instead PPARα, TRPV1 and GPR119, or CB1 activation by endocannabinoids, were suggested to inhibit or stimulate, respectively, ghrelin release from the stomach [
12,
13,
14]; interestingly, in human plasma, similar or opposing alterations in the levels of ghrelin and 2-AG or non-endocannabinoid NAEs, respectively, occur following exposure to palatable food of lean volunteers [
15], whereas in obese individuals exposed to chocolate, the plasma levels of both orexigenic and anorexigenic eCBome mediators (i.e., AEA and OEA, respectively) were directly correlated to ghrelin levels [
16]; and (2) through some of its specific metabolites, such as the short chain fatty acids (SCFAs), the gut microbiome impacts on ghrelin action at GHSR, whereas concomitant changes in circulating ghrelin levels and specific gut microbiota taxa are also known to occur under different experimental conditions; however, the mechanisms by which the gut microbiota interacts with ghrelin secretion and signaling are still largely unknown [
17]. These, and many other previously published data, strongly suggest that both the eCBome and the gut mBIome, through their multiple signaling mechanisms, are likely to regulate energy metabolism also via interactions with ghrelin.
Based on this background, we hypothesized that some of the effects of the eCBome and the gut mBIome on ghrelin action may occur via changes in Leap2 expression in the liver and, particularly, the intestine, and investigated this hypothesis either in vivo or in vitro. In particular, we first aimed at identifying a direct effect of the gut mBIome by studying Leap2 expression in germ-free (GF) mice before and after the reinstatement of functional gut microbiota by fecal microbiota transfer (FMT) from conventionally raised (CR) mice. Next, we investigated if the effect of the gut mBIome on Leap2 expression in the gut was due to its previous effect on the gut eCBome, by looking at how several eCBome mediators, or eCBome receptor activating and inactivating pharmacological tools, affected Leap2 expression in differentiated intestinal epithelial CaCo-2/15 cells or organoids prepared from the mouse small intestine, in the presence or absence of a gut microbiota-derived pro-inflammatory signal, i.e., lipopolysaccharide (LPS). Finally, we investigated whether, and how, intestinal Leap2 expression is altered in mouse models of obesity/type 2 diabetes and how such alterations relate to those of the eCBome in the same models, where profound gut microbiota perturbations, sometimes referred to as dysbiosis, also exist.
3. Discussion
LEAP2 is a recently discovered, a potential endogenous regulator of food intake and energy metabolism through antagonism of ghrelin action [
6]. In agreement with these properties of this peptide, originally isolated from the liver as an antimicrobial agent, are the following previous findings: (1) selectively in female mice subjected to a high-fat diet,
Leap2 deletion raises body weight, food intake, lean mass, and hepatic fat, and reduces O
2 consumption, heat production, and locomotor activity during the first part of the dark period [
27]; (2) in both female and male lean mice,
Leap2 deletion renders the animals more sensitive to the hyperphagic actions of ghrelin [
27]; LEAP2 administration also reduces blood glucose levels in lean mice [
6]; (3) in humans and mice, circulating LEAP2 levels display an inverse pattern compared to ghrelin, by increasing with food intake and obesity, and decreasing upon fasting and weight loss [
28,
29]; refeeding decreases circulating levels of ghrelin, while LEAP2 goes up to baseline levels; and (4) like other anorectic signals, LEAP2 is also produced by the small intestine [
2]. Others have reported that all regions of the mouse small intestine express
Leap2, with the highest expression having been found in the jejunum (whereas we detected slightly higher levels in the duodenum), with next to no detectable levels expressed in the stomach [
6]. Regardless, after bariatric surgery (vertical sleeve gastrectomy), the expression in the jejunum decreased significantly, while expression in the stomach increased over 50-fold. This shows that
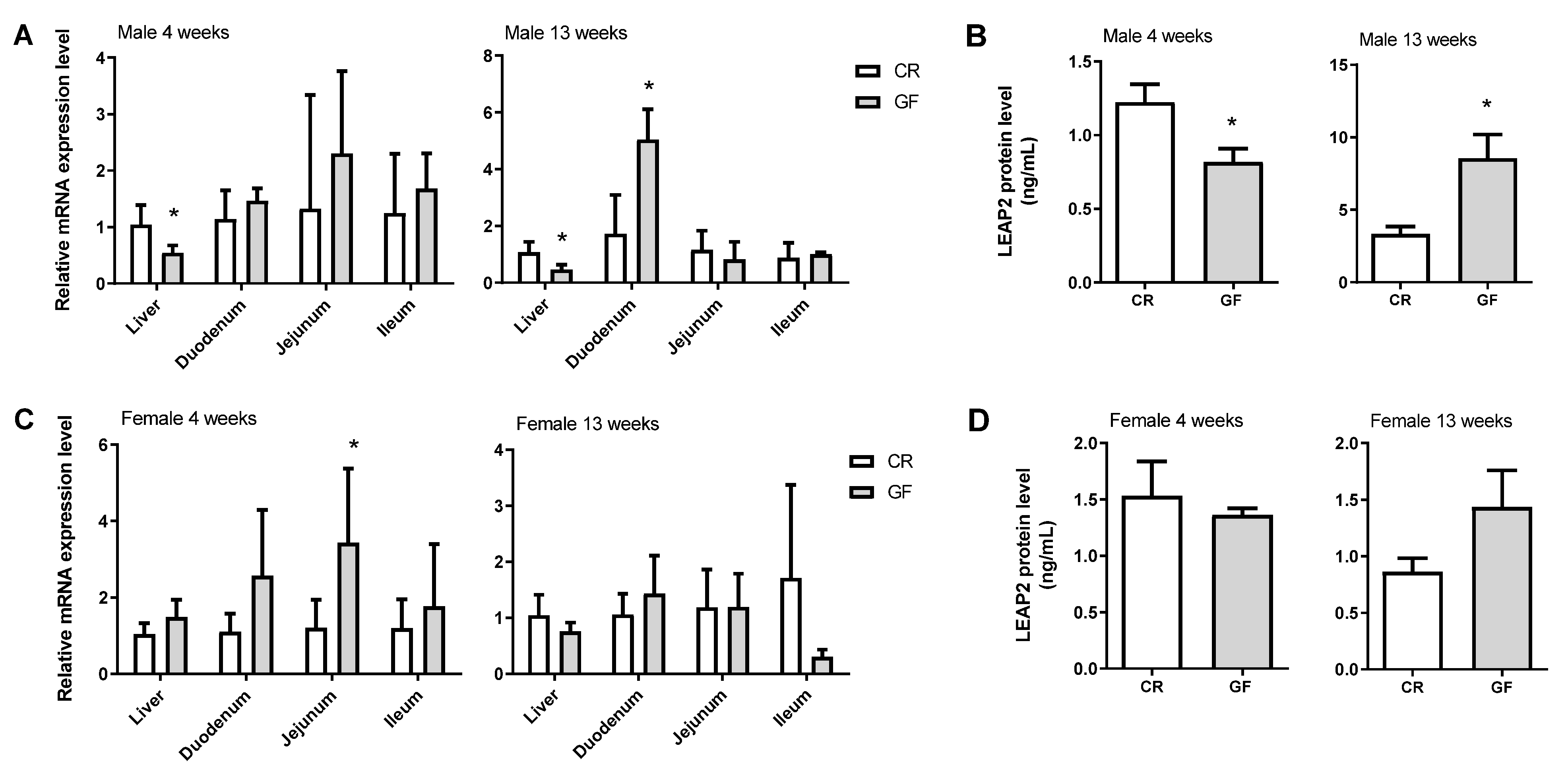
Leap2 expression can respond to various experimental paradigms in an intestinal-region-specific manner. However, LEAP2 regulation in this tissue has not yet been investigated, nor have its potential interactions therein with two major and inter-related players in obesity, i.e., the eCBome and the gut mBIome. Here we have reported evidence suggesting that: (1) the presence or absence of the gut microbiota strongly and directly affects
Leap2 mRNA expression in the liver and duodenum, and LEAP2 circulating levels, in 13 week-old male, but not female, mice (gender differences are known to influence gene expression [
30] and their targeting by transcription [
31] and translation regulators that ultimately lead to different mRNAs or protein products [
32]); (2) despite the fact that the gut microbiota also strongly and directly affects small intestinal eCBome signaling, generalized modulation of eCBome receptors does not result in significant changes of
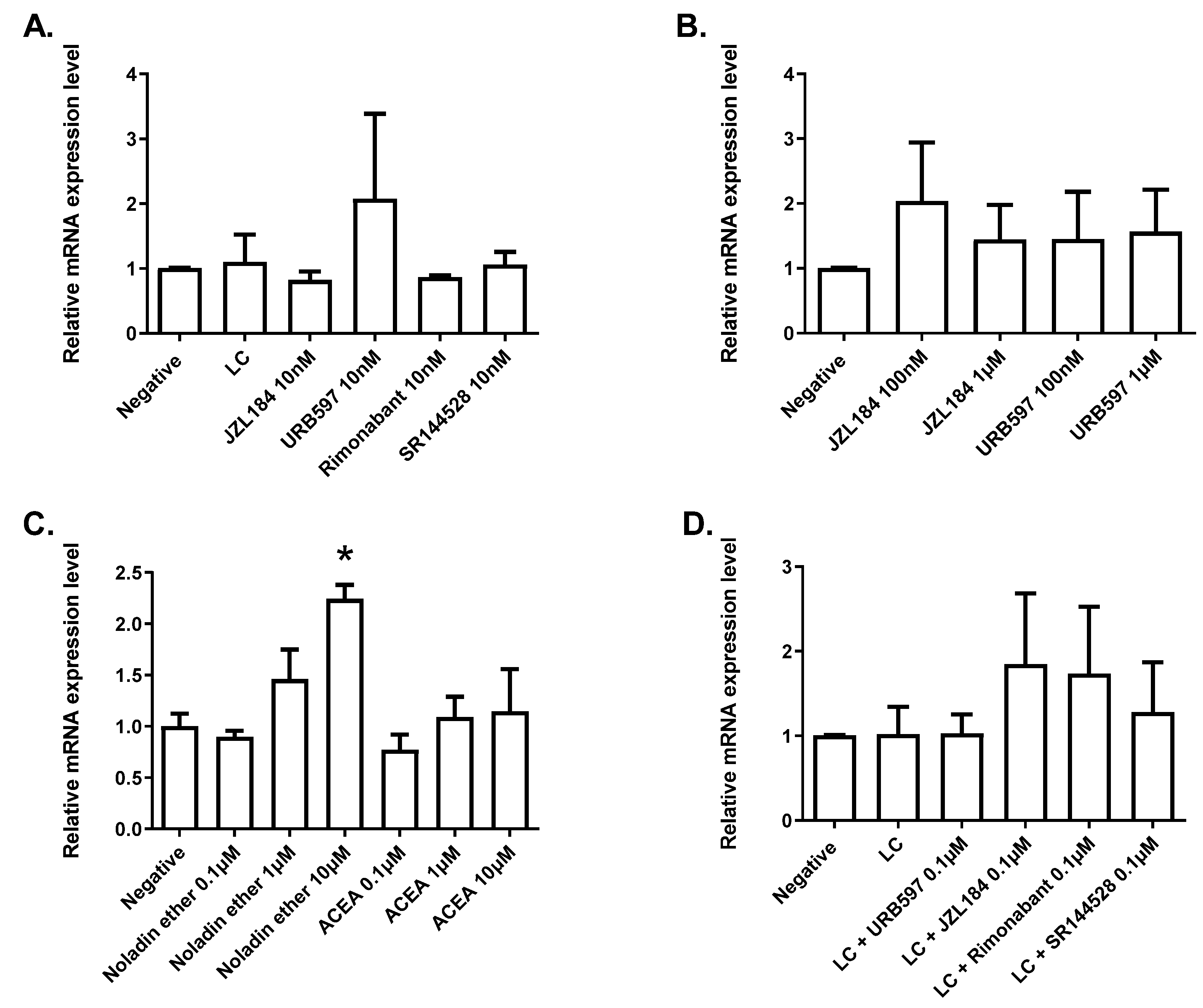
Leap2 expression in a model of human enterocytes, except for the activation by the synthetic (i.e., rosiglitazone) agonist of PPARγ, which caused a strong upregulation; additionally, noladin ether, another endocannabinoid discovered by Raphael Mechoulam and his group in 2001 [
33], caused a significant elevation of
Leap2 expression in organoids from the mouse duodenum, through yet to be investigated mechanisms; and (3)
Leap2 expression in the ileum of HFHS- and genetically (leptin signaling deficiency)-induced obesity is increased concomitantly with glucose intolerance rather than obesity, and in a manner variedly and strongly correlated with the ileal levels of either an eCBome mediator (2-PG, positive) or mRNAs of proteins that participate in either NAE or 2-MAG biosynthesis (
Gde1, positive;
Plcb1, negative) or actions (
Ppara, positive;
Trpv2, negative), or in NAE and
N-acyl-glycine inactivation (
Faah, positive).
The gut microbiota is known to affect host physiology, and metabolism in particular, through a plethora of microorganism-derived molecules, of which eCB-like mediators different from those produced by the host are also part [
34]. Therefore, it was not surprising to find previously that the gut microbiota is a strong determinant of eCBome signaling in the mouse intestine [
18], although this action of intestinal microorganisms could also be mediated by other, non-eCBome-related molecules, such as, for example, short-chain fatty acids, tryptophan metabolites and secondary bile acids [
35,
36]. Likewise, the tonic inhibitory effect of the gut microbiota on duodenal
Leap2 expression, described here for the first time in adult male mice, might be both eCBome- and non-eCBome-mediated. It is noteworthy that the intestinal mBIome is markedly varied along the length of the gastrointestinal tract, with there being relatively few bacteria in the duodenum, and the number and diversity of bacteria increasing distally [
37,
38]. This of course does not discount the ability of the duodenal microbiome, by being significantly modified with obesity [
38,
39], to impact both the host physiology and small intestinal bacterial overgrowth [
40]. Indeed, we have reported that fecal microbiota transfer into germ-free mice reconstitutes the small intestinal bacterial communities and significantly modulates the eCBome of the small intestine, including the duodenum [
18].
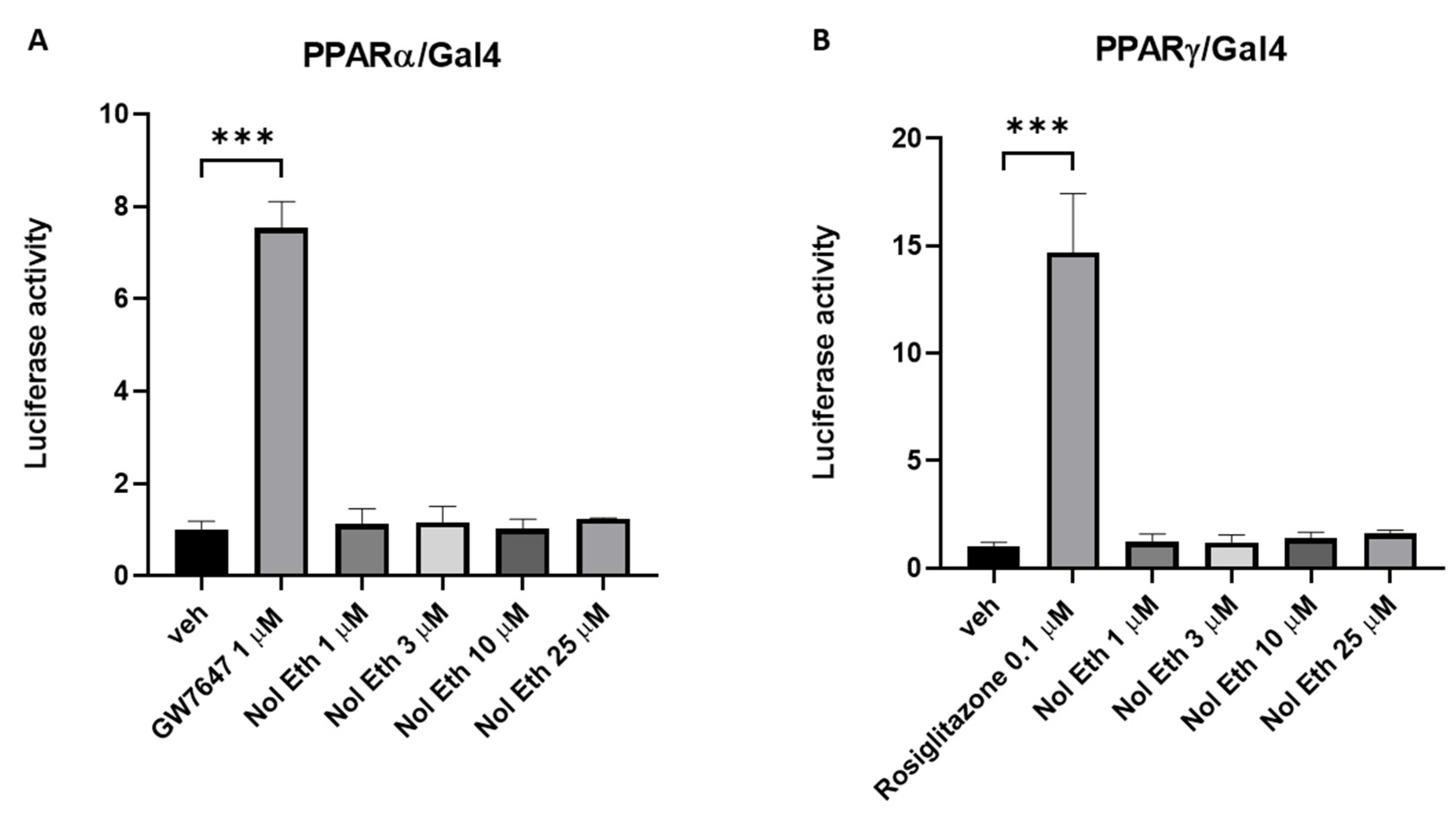
We found that at least one eCBome mediator, noladin ether, can stimulate
Leap2 expression in duodenal organoids, which, together with the previous observation that the gut microbiota tonically reduces duodenal PPARα expression in the small intestine of adult male mice [
18], may suggest that this receptor and its endogenous ligands might mediate in part gut microbiota tonic inhibition of duodenal
Leap2 expression. However, we could not confirm the proposed mechanism of action of noladin ether in organoids, as this compound did not activate recombinant PPARγ or PPARα in a functional assay, nor could we show that it stimulates
Leap2 expression in a model of human enterocytes. Additionally: (1) PPARγ (an alternative AEA target), although involved in stimulating
Leap2 expression in vitro, particularly when rosiglitazone was used as an agonist, is not significantly altered by the lack of presence of the gut microbiota in terms of mRNA expression in the duodenum [
18]; (2) GPR55 (another proposed target for AEA and noladin ether) and GPR18 (a proposed target for NAGly) are tonically stimulated, rather than inhibited, in the duodenum by the gut microbiota [
18], and hence cannot partake in microbiota tonic inhibition of
Leap2 expression; and (3) also the duodenal expression of
Cnr1 or
Cnr2, the two preferential targets for eCBs, is not significantly altered by the gut microbiota [
18]. In sum, present and previous data suggest that the effects of the gut microbiota on
Leap2 expression and eCBome signaling might be two unrelated, or only very partially related, phenomena. Nevertheless, the increase in
Leap2 expression observed in GF mice might still account, at least in part, for their more favorable metabolic and glycemic profile under conditions of DIO [
41].
Indeed, we observed here, again for the first time, that increased RNA levels encoding for this peptide are produced in the ileum 3 days following an HFHS diet, concomitantly with the appearance of hyperglycemia, and in both
ob/ob and
db/db mice that, apart from being obese, are also glucose intolerant. This finding may suggest that LEAP2 is overproduced by the ileum to counteract glucose intolerance induced by an HFHS diet or hyperphagia. Since the ileum of HFHS and of
ob/ob and
db/db mice presents with inter-related alterations in gut eCBome signaling (present results and [
23]) and microbiota composition [
23,
42,
43], we hypothesized a role of either system in the modulation of
Leap2 expression. However, also, in this case, the only potential eCBome signaling pathway that changed in a manner similar to
Leap2 expression following HFHS and in
ob/ob and
db/db mice was that mediated by PPARα and/or its ligands. Conversely, as mentioned above, the only eCBome signaling pathway that was clearly implicated in
Leap2 expression stimulation in human enterocytes was that mediated by PPARγ, whose expression, however, peaks negatively in HFHS mice when
Leap2 expression peaks positively. This may suggest that also in the case of obesity and hyperglycemia the regulation of
Leap2 expression and eCBome signaling might be two unrelated phenomena, and not controlled in a coordinated manner by gut dysbiosis.
A limitation of this study is that the selected in vitro systems for the small intestine do not necessarily reflect what is going on in vivo in the ileum. The organoids were prepared from the duodenum and not from the ileum, and therefore results obtained in this system may only be relevant to those in GF mice, where the strongest reduction in Leap2 expression was indeed found in this small intestinal section. Yet, the geometry of organoids is such that their basolateral side, rather than the apical one, is the one that is exposed to treatments, which may have altered the effects of the latter. Differentiated Caco-2 cells, instead, model only one cell type (enterocytes) among the several ones that are found in the small intestinal mucosa. Therefore, the findings in these two in vitro systems may not necessarily be relevant to the actual in vivo regulation of Leap2 expression by the gut microbiota in the dysbiosis typical of GF and hyperglycemic/obese mice, thus leaving still open the possibility that the eCBome, per se or following its modulation by the gut microbiome, might be an important determinant of LEAP2 production. In this sense, it is noteworthy that the mRNA expression of this potentially metabolically beneficial endogenous antagonist of ghrelin action is stimulated in vitro by, and correlates in vivo with, non-CB1-mediated (and hence non-metabolically “noxious”) eCBome signaling, i.e., respectively: (1) activation of PPARγ, which is known to be an intermediate in anti-diabetic and anti-glucose intolerance drugs, and (2) expression of PPARα and the levels of one of its endogenous agonists, i.e., 2-PG, with potential anorectic and anti-dyslipidemic actions.
In summary, we have provided here unprecedented evidence for the existence of gut microbiota tonic in vivo control over intestinal Leap2 expression, which persists into adulthood, at least in the duodenum, of male mice. We also provided preliminary in vitro data suggesting that stimulation of the levels of the mRNA encoding for this metabolically beneficially peptide may be exerted by non-CB1-mediated eCBome signaling. It remains to be clarified whether the gut microbiota-eCBome axis is involved in the control of intestinal LEAP2 levels, especially during obesity and hyperglycemia, where, as shown previously and confirmed here, all the members of this triangle undergo adaptive regulation.
4. Materials and Methods
4.1. Animals and Housing
Conventionally Raised (CR) and Germ-free (GF) C57BL/6NTac mice were purchased from Taconic (Taconic Bioscience, NY, USA) and maintained in the animal facility of the Institut Universitaire de Cardiologie et Pneumologie de Québec (IUCPQ, QC, Canada). All animals have grouped 3–4 mice per cage under a 12 h:12 h light-dark cycle with ad libitum access to NIH-31 Open Formula Autoclavable Diet (Zeigler, PA, USA) and water. GF mice were housed in axenic status and fecal samples were weekly tested for microbes and parasites by the facility’s staff to ensure that the GF unit was indeed sterile. Both GF and CR mice were acclimatized for at least one week prior to starting the procedures.
4.2. Animal Experiments and Fecal Microbial Transplant (FMT)
Twelve (6 male and 6 female) CR and GF mice at 4 and 13 weeks of age, were intraperitoneally anesthetized with a cocktail of ketamine/xylazine/acepromazine at a dose of 50/10/1.7 mg/kg body weight and euthanized by cervical dislocation, following an intra-cardiac puncture. Whole blood was collected in K3-EDTA tubes. The abdominal cavity was opened and the whole digestive tract was carefully aligned from the stomach up to the colon. Once the stomach was removed, the small (duodenum, jejunum, ileum) and large (cecum and colon) intestine were carefully excised and separated and the intestinal contents were harvested by flushing with 1ml of sterile PBS without Ca/Mg (Thermo Fisher Scientific, MA, USA) and snap-frozen. The liver was also isolated from the abdominal cavity. Sections of the liver as well as small and large intestine were stored either in RNALater (Thermo Fisher Scientific, MA, USA) for RNA stabilization or immediately snap-frozen and stored at −80 °C for further analysis. This common procedure was concluded, for each mouse, within a maximum of 15 min, to ensure the preservation of mRNA and lipid for further analysis.
For fecal microbiota transplant (FMT) experiments only 12 weeks old male mice were utilized. GF mice were randomly divided into two groups at the age of 12 weeks: those gavaged with sterile PBS (SHAM; 5 mice) and those gavaged with fecal material (FMT; 6 mice). Material gavaged for FMT consisted of a cocktail of the intestinal contents and stools of a single and 4 CR donor mice, respectively. Briefly, the intestinal contents of the duodenum, jejunum, ileum, colon, and cecum were collected from one 12-week-old CR donor mouse and mixed with stool pellets from all the CR mice to be used as controls. The mixture was well homogenized, weighed, suspended at 1:10 in sterile PBS, and centrifuged at 805×
g for 10 min at room temperature. The supernatant was used to gavage the mice (200 µL of homogenate per mouse) [
18]. The FMT mice were then housed (3 per cage, like for CR mice) for one week in conventional conditions in cages contaminated with used litter coming from donor mouse cages. SHAM mice were submitted to a similar gavage with saline solution, but then kept in the germ-free facility for a week in the same conditions as GF mice. CR mice were euthanized the day of the gavages, while SHAM and FMT mice were sacrificed one week after the gavage; the tissues were collected from all animals as previously described.
4.3. RNA Isolation, Reverse Transcription, and qPCR
RNA was extracted from tissues and cells with the RNeasy Plus Mini Kit (Qiagen, Hilden, Germany) following the manufacturer’s instruction and eluted in 50 μL of UltraPure Distilled Water (Invitrogen, CA, USA). The concentration and purity of RNA were determined by measuring the absorbance of the RNA in a Biodrop at 260 nm and 280 nm, and RNA integrity was assessed by an Agilent 2100 Bioanalyzer, using the Agilent RNA 6000 Nano Kit (Agilent Technologies, CA, USA). 1 µg of total RNA was reverse transcribed using the iScript cDNA synthesis kit (Bio-Rad, CA, USA) in a reaction volume of 20 μL. Leap2 mRNA expression levels were determined using primer pairs (Mm.PT.58.6158455.g, IDT, IA, USA) on a CFX384 touch qPCR System (BioRad) using PowerUp SYBR Green qPCR master mix (Thermo Fisher Scientific, CA, USA) in duplicate reactions. Hprt1 (Mm.PT.39a.22214828, IDT, IA, USA) was used as a reference gene. Because the Ct of the reference gene was the same in all the 13 weeks old control samples (first and FMT experiments), we merged the two control sets into 13 weeks control group. Gene expression levels in mice and cells (see below) were evaluated by the 2-ΔΔCt method and represented as fold increase with respect to the baseline of the relevant control. Data were analyzed by ANOVA followed by Dunnett’s multiple comparisons.
4.4. Organoid Preparation and Treatment
Crypt-derived organoids were mechanically separated from sacrificed black male C57BL/6 mouse duodenum. Murine small intestines were opened longitudinally, scratched, and washed with cold phosphate-buffered saline (PBS) (Gibco Life Technologies, Carlsbad, CA, USA). Organoids were incorporated in a solid matrix (Corning® Matrigel®) (Corning, Corning, NY, USA) and were incubated with advanced DMEM media (Dulbecco’s Modified Eagle’s Medium) (Gibco Life Technologies, Carlsbad, CA, USA) supplemented with HEPES buffer (Thermoscientific, Waltham, MA, USA), Glutamine (GlutaMAX) (Gibco Thermoscientific, Waltham, Ma, USA), antibiotic Pen-Strep (Millipore Sigma, Oakville, ON, Canada), Noggin (Millipore Sigma, Oakville, ON, Canada), mRSPO (Peprotech, Cranbury, NJ, USA), B27 supplement (Gibco Life Technologies, Carlsbad, CA, USA), EGF (Cedarlane, Burlington, ON, Canada, and N-acetyl-cystein (Millipore Sigma, Oakville, ON, Canada) and Ly-27. Organoids were split 1 into 3 every week, in 24 wells plates, and media was changed every 2 days. on Mature organoid cultures, 4 days following plating, were exposed 24 h to treatment or vehicle.
4.5. Caco-2/15Cell Differentiation and Treatment
Human Caco-2/15 intestinal cells (kindly provided by Dr. Jean-François Beaulieu (Université de Sherbrooke, Sherbrooke, Canada) were cultured in high glucose Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 10% Fetal Bovine Serum (FBS, Neuromics), 10 mM HEPES, 1X GlutaMAX, and 100 U/mL Penicillin/Streptomycin. Cell lines were grown until confluence, split, and plated 2 × 105 cell/well for differentiation for 21 days to induce the enterocyte phenotype. On the day of the experiment (after 21 days of differentiation), cells were treated for 24 or 48 h with media containing compounds at the indicated concentrations. DMSO controls were at 0.1%. For the LPS induction, 24 h prior to the experiment, cells were treated with 10ug/mL of LPS (Sigma) before being incubated for 24 h with the indicated compounds at the indicated concentrations. After incubation, media was removed, cells were washed with 1X PBS and frozen for RNA extraction as above.
4.6. PPAR-Luciferase Assays
Human embryonic kidney 293 cells (ATCC CRL-1573) were propagated in a growth medium (GM) composed of Dulbecco’s modified Eagle’s medium (DMEM cat. n. 41966029; Thermo Fisher, Monza, Italy) supplemented with 10% fetal bovine serum (cat. n. 16000044; Thermo Fisher, Monza, Italy) and 1% Pen/Strep (cat. n. 15140122 Thermo Fisher, Monza, Italy) under standard conditions. After plating (in 24 well plate density; 5 × 104 cells/well), the cells were transfected on the next day with the following plasmids: (a) pM1-hPPARα-Gal4 or pM1-hPPARγ-Gal4; (b) TK-MH100 × 4-Luc containing the UAS enhancer elements and; (c) Renilla luciferase (pRL, Cat. E2231; Promega, Milan, Italy) using lipofectamine 2000 (cat. n. 11668027; Life Technologies; Milan, Italy). The next day, the growth media was replaced with fresh media containing vehicle (Dimethyl sulfoxide, DMSO ≤ 0.03%) or compounds of interest (noladin ether Cat. No. 1411, GW7647 Cat. No. 1677/10 and Rosiglitazone Cat. No. 5325/10, purchased from TOCRIS, Abingdon, UK). On day 3, the cells were harvested and processed for analysis of luciferase activity using a GloMax Luminometer instrument (Promega, Milan, Italy) and the Dual-Luciferase Reporter Assay kit (cat. n. E1910 Promega, Milan, Italy) following published procedures [
26].
4.7. Lipid Extraction and HPLC-MS/MS for the Analysis of eCBome Mediators
Lipids were extracted from ileum samples as previously described [
18,
23,
26]. Briefly, about 10 mg of ileum was sampled and homogenized in 1 mL of a 1:1 Tris-HCl 50 mM pH 7: methanol solution containing 0.1 M acetic acid and 5 ng of deuterated standards. One ml of chloroform was then added to each sample, which was then vortexed for 30 s and centrifuged at 3000×
g for 5 min. The organic phase was collected and another 1 mL of chloroform was added to the inorganic one. This was repeated twice to ensure the maximum collection of the organic phase. The organic phases were pooled and evaporated under a stream of nitrogen and then suspended in 50 µL of mobile phase containing 50% of solvent A (water + 1 mM ammonium acetate +0.05% acetic acid) and 50% of solvent B (acetonitrile/water 95/5 + 1 mM ammonium acetate +0.05% acetic acid). Forty μL of each sample were finally injected onto an HPLC column (Kinetex C8, 150 × 2.1 mm, 2.6 μm, Phenomenex) and eluted at a flow rate of 400 μL/min using a discontinuous gradient of solvent A and solvent B [
18,
23,
26]. Quantification of eCBome-related mediator, was carried out by HPLC interfaced with the electrospray source of a Shimadzu 8050 triple quadrupole mass spectrometer and using multiple reaction monitoring in positive ion mode for the compounds and their deuterated homologs [
18,
23,
26].
4.8. Experiments in Mice Undergoing a High Fat High Sucrose Diet
As previously described [
20], sixty 6-week-old C57BL/6J male mice were fed ad libitum with a low-fat, low-sucrose purified diet (10% fat and 7% sucrose [LFLS]; Research Diet, NJ, USA) for a 10-day acclimatization period in the animal facility of the Institute of Nutrition and Functional Foods. Mice were then randomly assigned to 6 groups (n = 12) fed a high-fat, high-sucrose purified diet (45% fat and 17% sucrose [HFHS]; Research Diet, NJ, USA) for up to 56 days. These dye-free diets harbor comparable fiber contents, and while the HFHS diet had, by design, a higher fatty acid content, the omega-3/omega-6 ratios were comparable. Six-hour-fasted mice were sacrificed by cardiac puncture to retrieve plasma (1780×
g, 10 min) at either 0 (baseline), 3, 10, 21, or 56 days following HFHS diet initiation. Duodenum was collected 2 cm of the pylorus, while jejunum and ileum were collected 10 cm and 2 cm, respectively, from the ileocecal junction. All samples were stored at −80 °C until batch analysis.
4.9. Experiments in ob/ob and db/db Mice
As previously described [
36], male homozygous
ob/
ob mice (B6.V-Lepob/ob/JRj) were used as a leptin-deficient obese model, and their lean littermates served as controls (CT ob); (n = 9–10 per group). Male homozygous
db/
db mice (BKS-Lepr/db/db/JOrlRj) functionally deficient for the long-form leptin receptor were used as a hyperleptinemic obese type 2 diabetic model, and their lean littermates served as controls (CT db); (n = 9–10 per group). Mice were purchased at the same time and from the same supplier (Janvier Laboratories, Le Genest-Saint-Isle, France) at the age of 6 weeks. Mice were housed in a specific pathogen and opportunistic free (SOPF) controlled environment (room temperature of 22 ± 2 °C, humidity 55 ± 10%, 12 h daylight cycle, lights off at 6 p.m.) in groups of two mice per cage, with free access to sterile food and sterile water. Upon delivery, mice underwent an acclimation period of one week, during which they were fed a standard diet containing 10% calories from fat (D12450Ji; Research Diet; New Brunswick, NJ, USA) and were then kept ad libitum on the same diet for 7 weeks. Milli-Q water filtered by a Millipak
® Express 40 with a 0.22 µm membrane filter (Merck Millipore, Burlington, MA, USA) was autoclaved and provided ad libitum. All mouse experiments were approved by and performed in accordance with the guideline of the local ethics committee (Ethics committee of the Université Catholique de Louvain for Animal Experiments specifically approved this study that received the agreement number 2017/UCL/MD/005). Housing conditions were specified by the Belgian Law of 29 May 2013, regarding the protection of laboratory animals (agreement number LA1230314).


 ,
, 


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}