
Investigation of Antifungal Properties of Synthetic Dimethyl-4-Bromo-1-(Substituted Benzoyl) Pyrrolo[1,2-a] Quinoline-2,3-Dicarboxylates Analogues: Molecular Docking Studies and Conceptual DFT-Based Chemical Reactivity Descriptors and Pharmacokinetics Evaluation

 ,
,  ,
,  ,
,  , ,
, ,  , ,
, ,  , , ,
, , ,  add
Show full author list
add
Show full author list
Abstract
1. Introduction
2. Results and Discussion
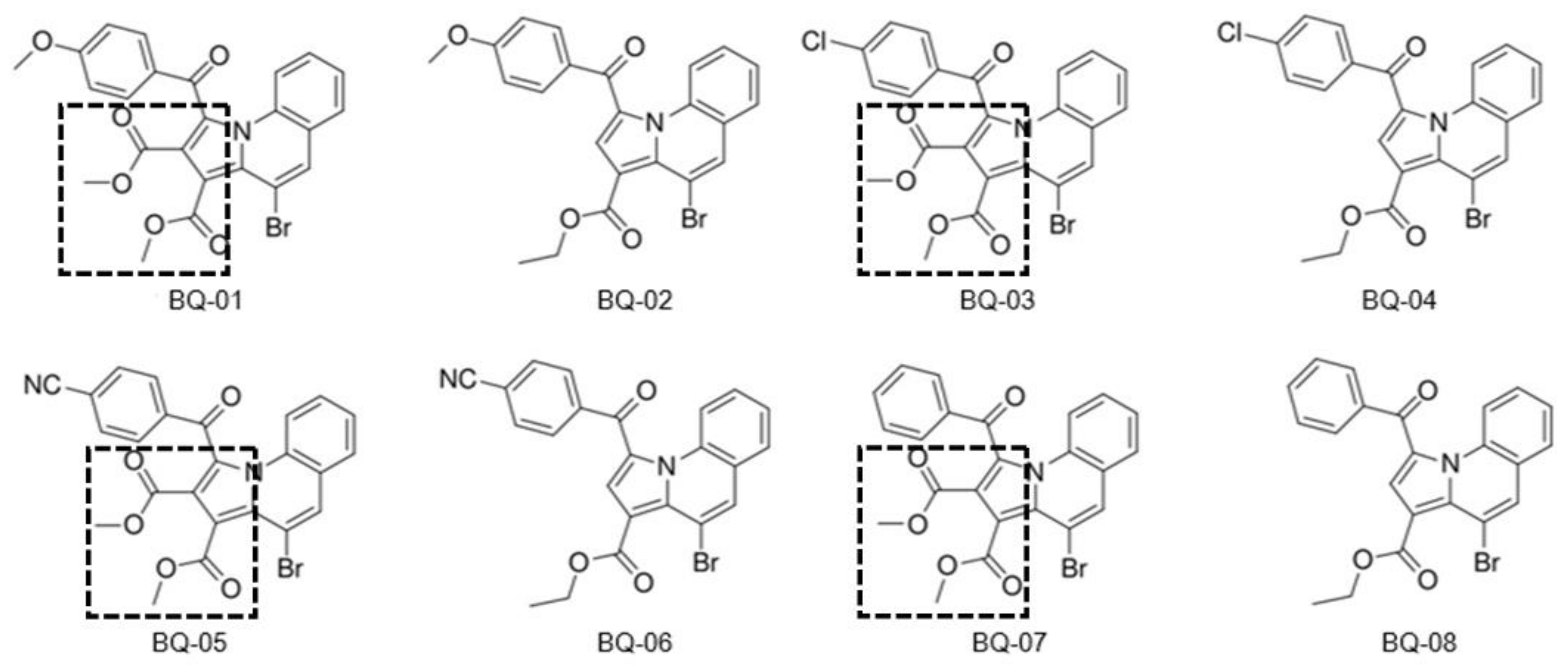
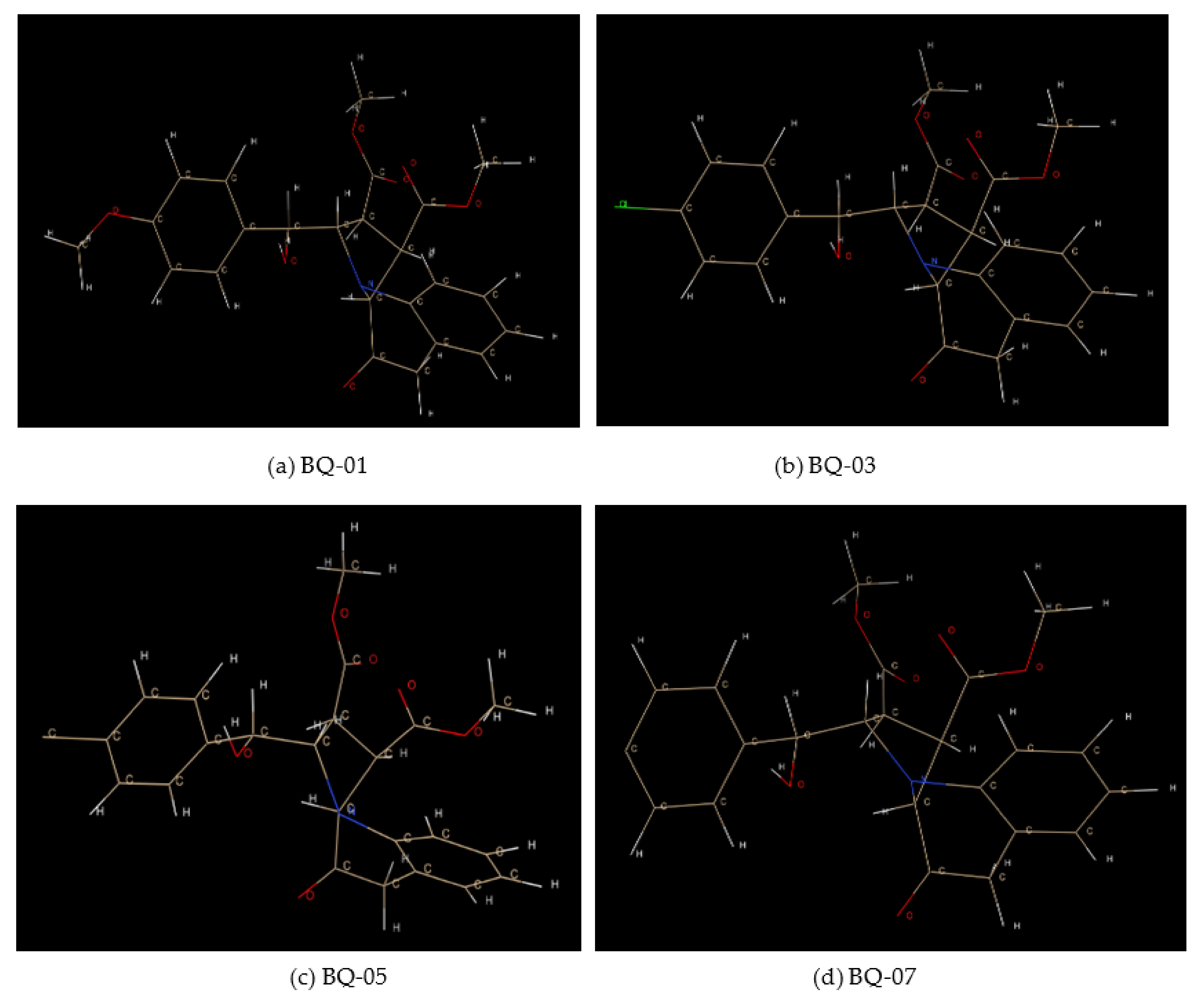
2.1. Analysis of MIC against C. albicans
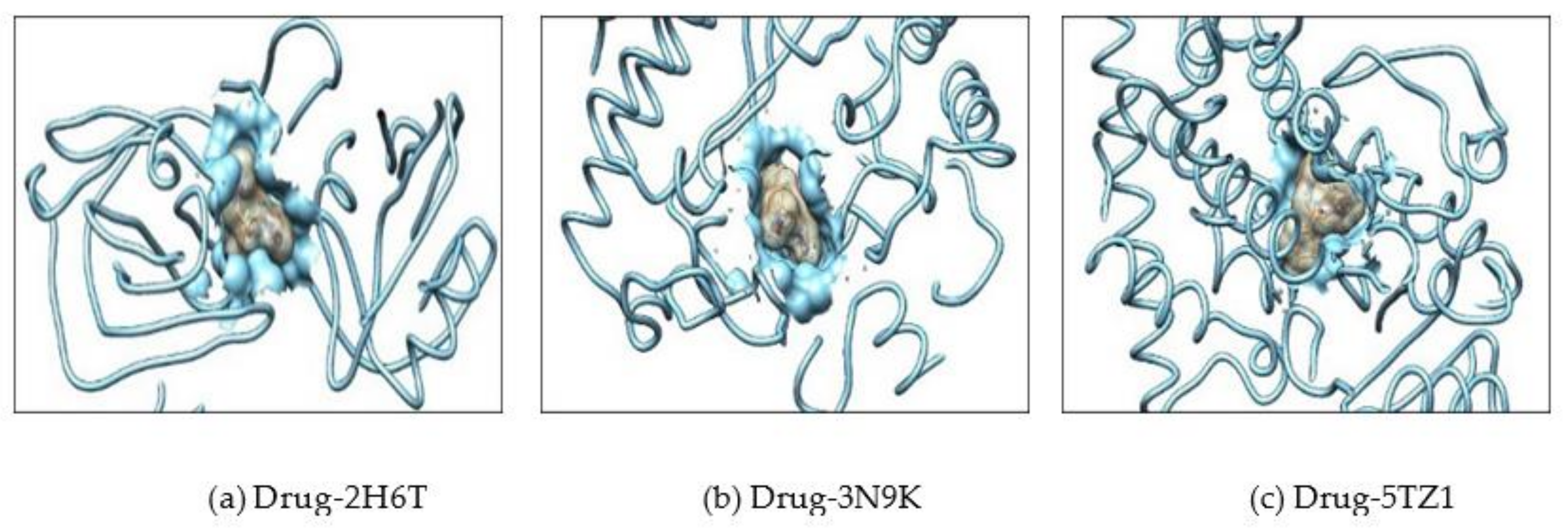
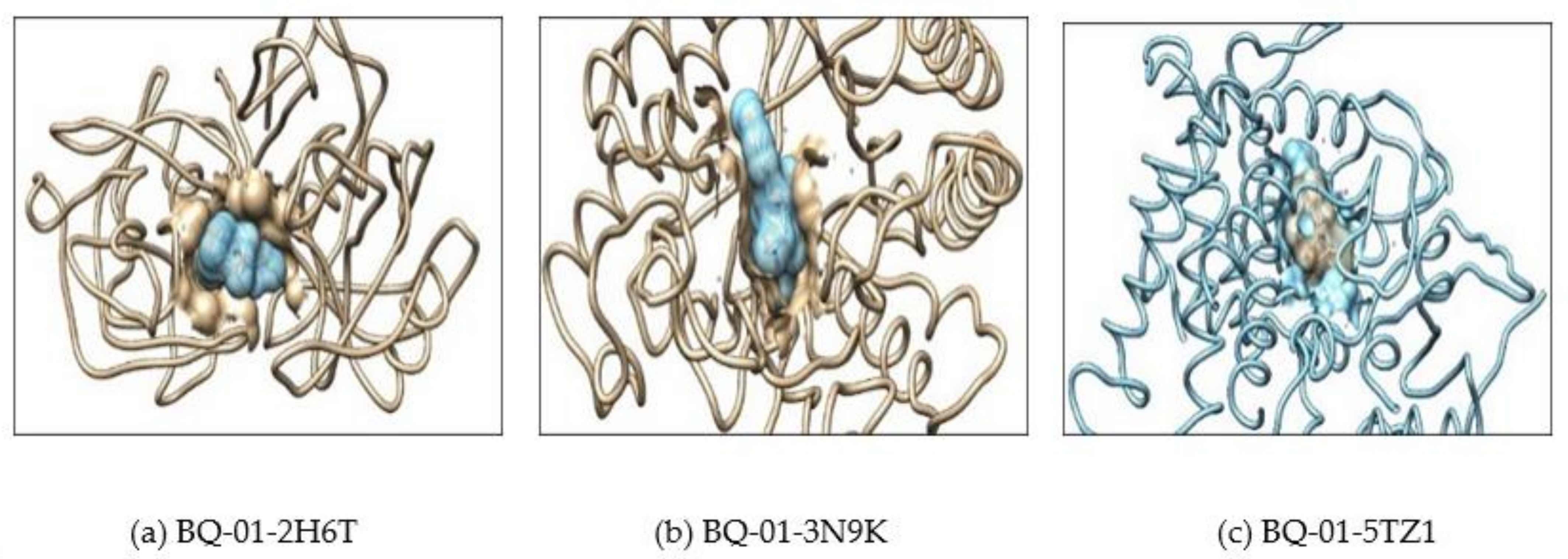
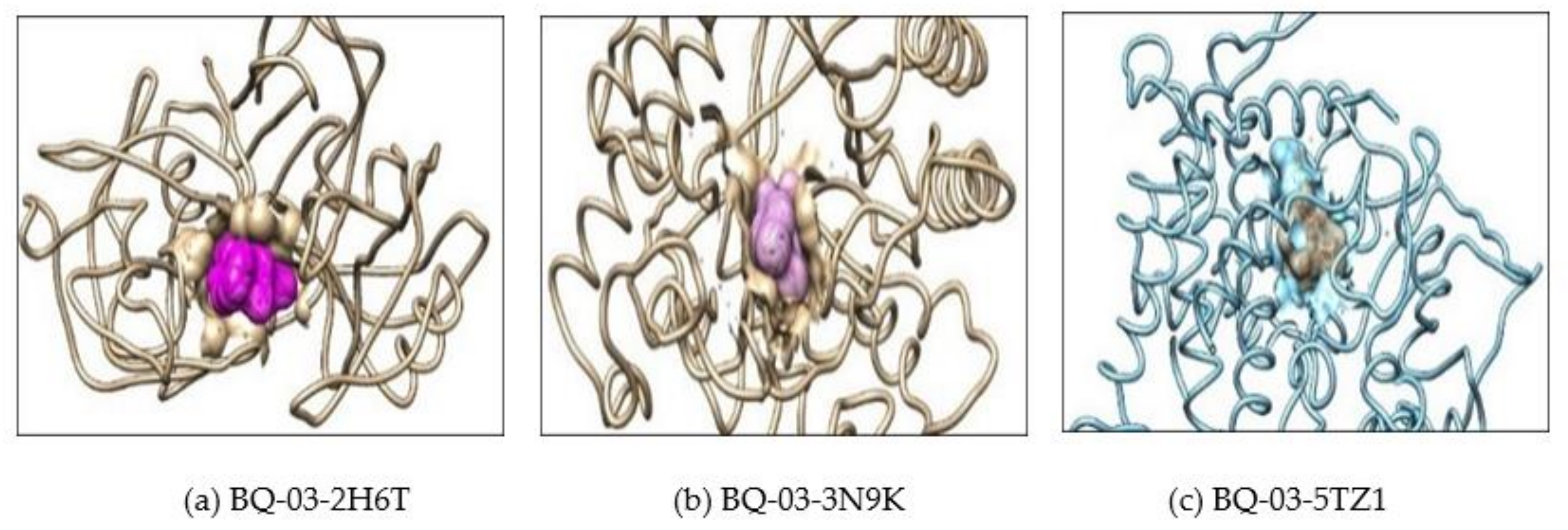
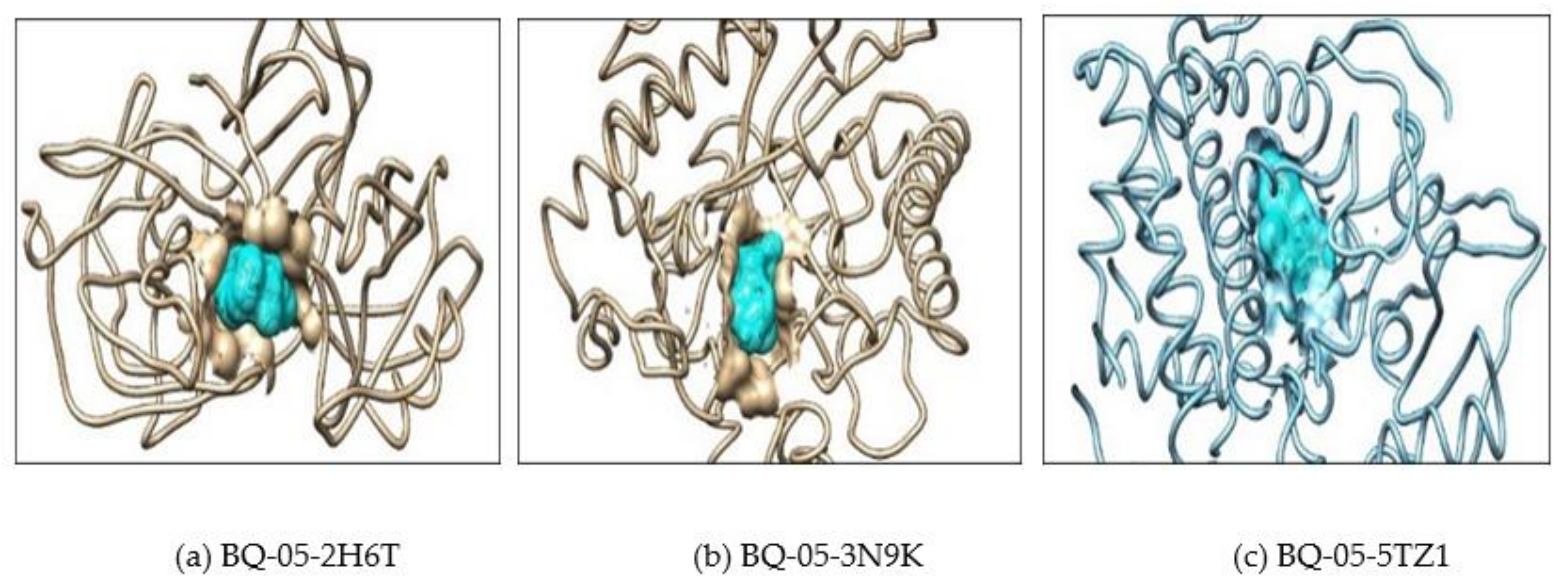
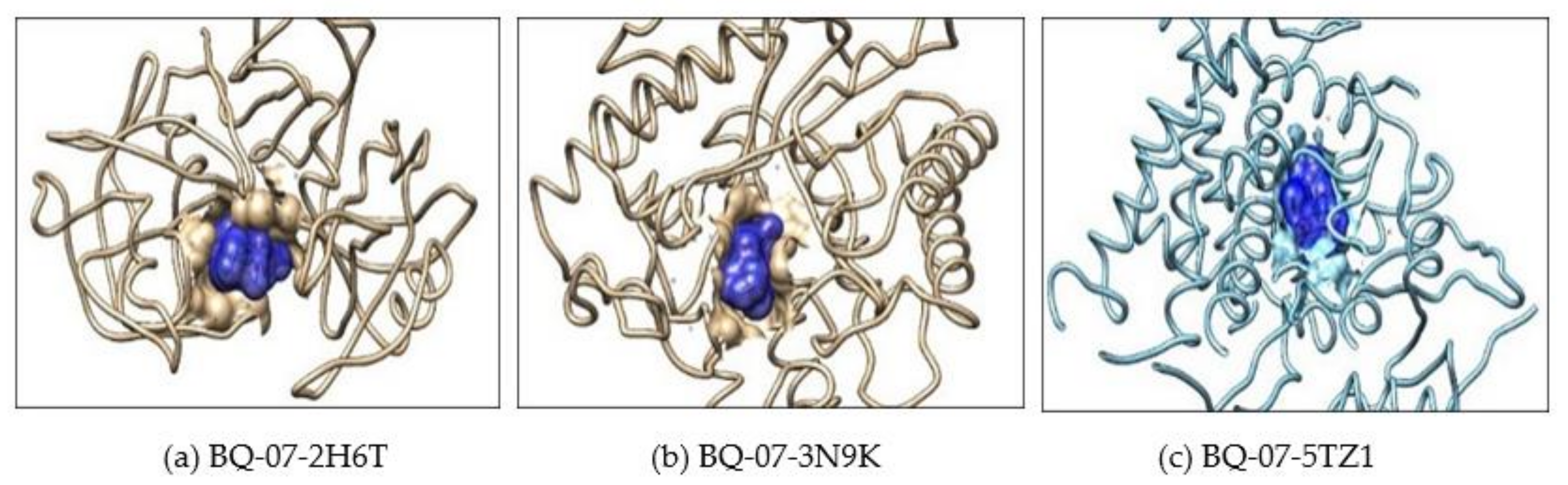
2.2. Molecular Docking
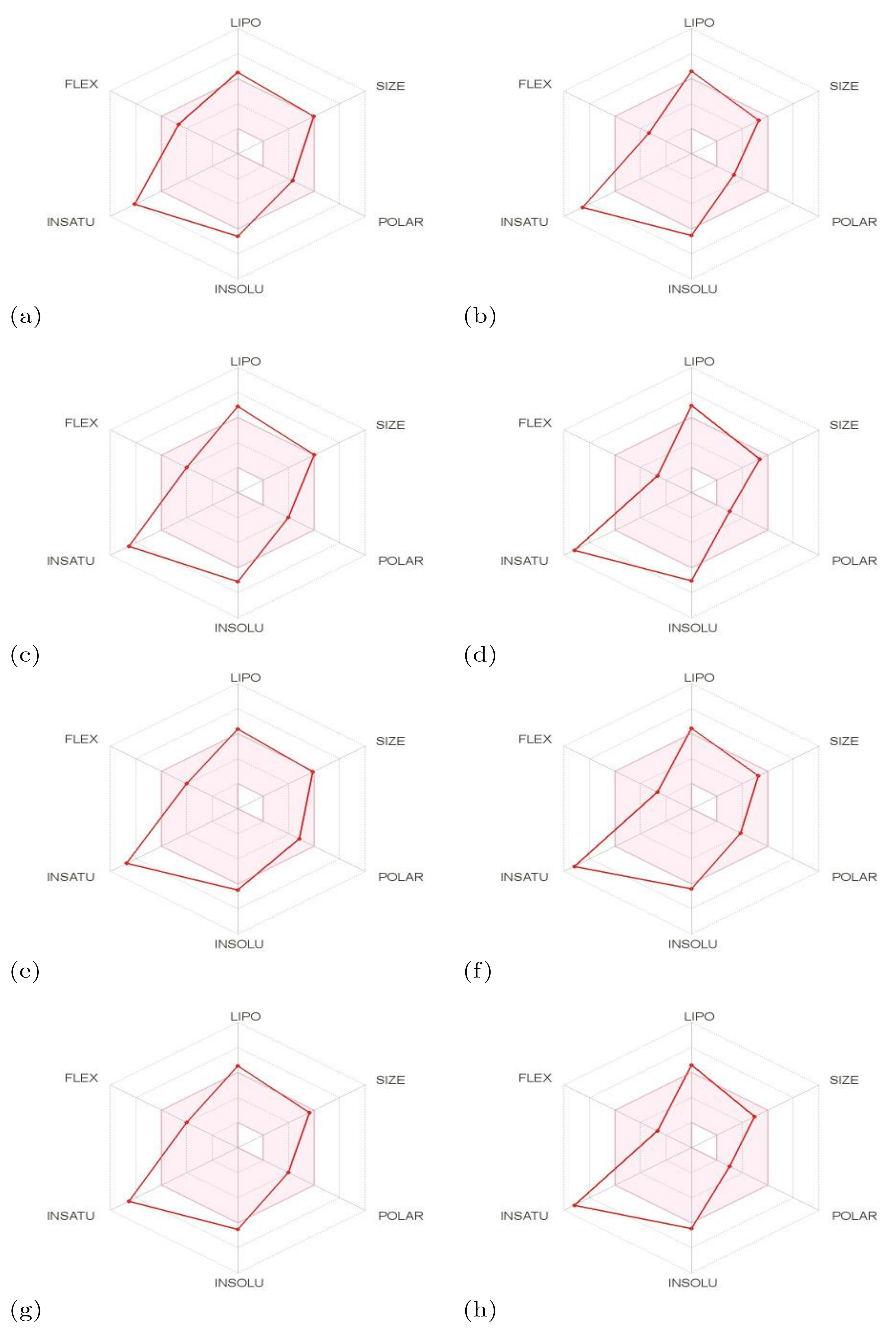
2.3. Determination of the Conceptual DFT Reactivity Descriptors of the Molecules and their Related Pharmacokinetics
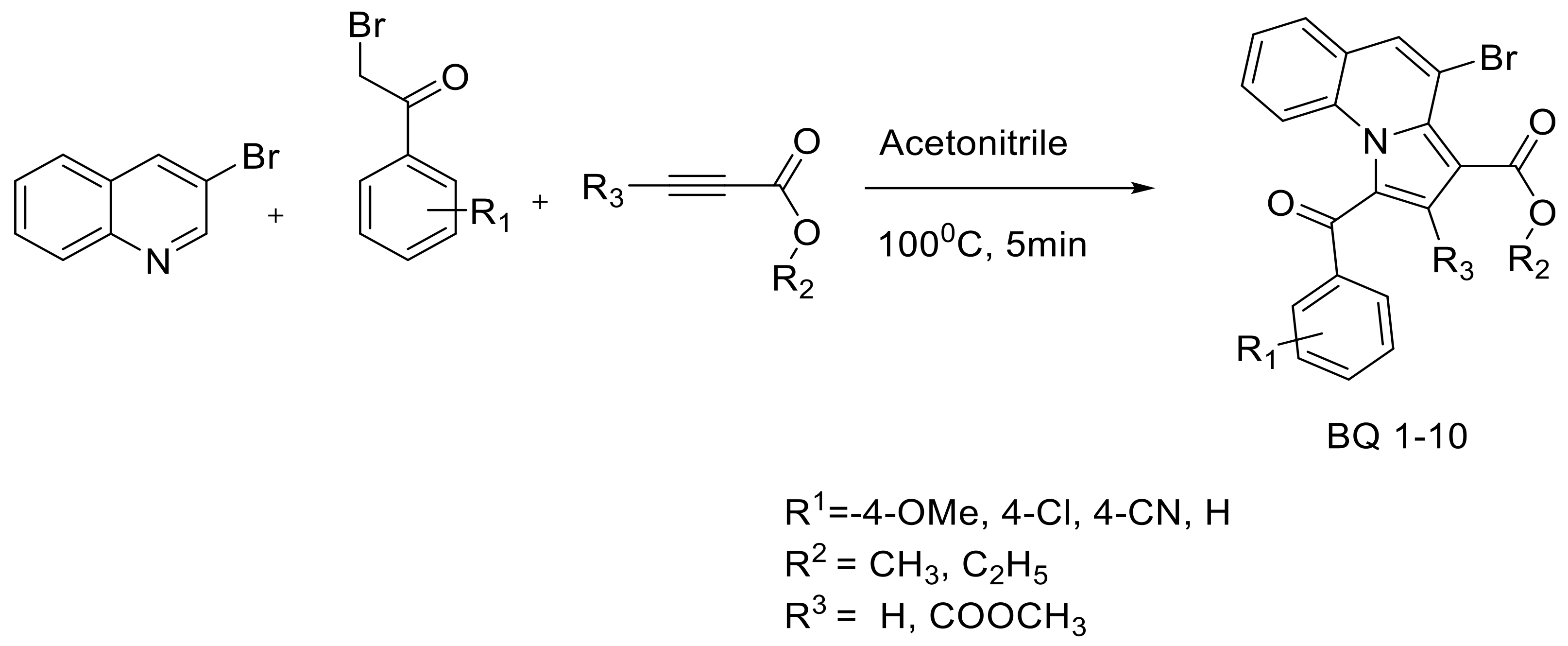
3. Materials and Methods
3.1. Analysis of Minimum Inhibitory Concentration against C. albicans
3.2. Selection of Protein
3.3. Small Molecule Optimization
3.4. In Silico Molecular Interaction and Docking
3.5. Conceptual DFT Reactivity Descriptors of the Molecules and Their Related Pharmacokinetics
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Gudlaugsson, O.; Gillespie, S.; Lee, K.; Berg, J.V.; Hu, J.; Messer, S.; Herwaldt, L.; Pfaller, M.; Diekema, D. Attributable mortality of nosocomial candidemia, revisited. Clin. Infect. Dis. 2003, 37, 1172–1177. [Google Scholar] [CrossRef]
- Antinori, S.; Milazzo, L.; Sollima, S.; Galli, M.; Corbellino, M. Candidemia and invasive candidiasis in adults: A narrative review. Eur. J. Intern. Med. 2016, 34, 21–28. [Google Scholar] [CrossRef]
- Sudbery, P.E. Growth of Candida albicans hyphae. Nat. Rev. Genet. 2011, 9, 737–748. [Google Scholar] [CrossRef]
- Gow, N.A.R.; Van De Veerdonk, F.L.; Brown, A.J.P.; Netea, M.G. Candida albicans morphogenesis and host defence: Discriminating invasion from colonization. Nat. Rev. Genet. 2011, 10, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Su, C.; Liu, H. Candida albicans hyphal initiation and elongation. Trends Microbiol. 2014, 22, 707–714. [Google Scholar] [CrossRef]
- Gow, N.A.R.; Yadav, B. Microbe Profile: Candida albicans: A shape-changing, opportunistic pathogenic fungus of humans. Microbiology 2017, 163, 1145–1147. [Google Scholar] [CrossRef] [PubMed]
- Sardi, J.C.O.; Scorzoni, L.; Bernardi, T.; Fusco-Almeida, A.M.; Giannini, M.J.S.M. Candida species: Current epidemiology, pathogenicity, biofilm formation, natural antifungal products and new therapeutic options. J. Med. Microbiol. 2013, 62, 10–24. [Google Scholar] [CrossRef]
- Dantas, A.D.S.; Lee, K.K.; Raziunaite, I.; Schaefer, K.; Wagener, J.; Yadav, B.; Gow, N.A. Cell biology of Candida albicans–host interactions. Curr. Opin. Microbiol. 2016, 34, 111–118. [Google Scholar] [CrossRef]
- Sanguinetti, M.; Posteraro, B.; Lass-Flörl, C. Antifungal drug resistance among Candidaspecies: Mechanisms and clinical impact. Mycoses 2015, 58, 2–13. [Google Scholar] [CrossRef] [PubMed]
- Cui, J.; Ren, B.; Tong, Y.; Dai, H.; Zhang, L. Synergistic combinations of antifungals and anti-virulence agents to fight againstCandida albicans. Virulence 2015, 6, 362–371. [Google Scholar] [CrossRef] [PubMed]
- Perfect, J.R. The antifungal pipeline: A reality check. Nat. Rev. Drug Discov. 2017, 16, 603–616. [Google Scholar] [CrossRef] [PubMed]
- Qin, Y.; Zhang, L.; Xu, Z.; Zhang, J.; Jiang, Y.-Y.; Cao, Y.; Yan, T. Innate immune cell response uponCandida albicansinfection. Virulence 2016, 7, 512–526. [Google Scholar] [CrossRef]
- Silva, D.R.; Sardi, J.D.C.O.; Freires, I.A.; Silva, A.C.B.; Rosalen, P.L. In silico approaches for screening molecular targets in Candida albicans: A proteomic insight into drug discovery and development. Eur. J. Pharm. 2019, 842, 64–69. [Google Scholar] [CrossRef]
- Howard, K.C.; Dennis, E.K.; Watt, D.S.; Garneau-Tsodikova, S. A comprehensive overview of the medicinal chemistry of antifungal drugs: Perspectives and promise. Chem. Soc. Rev. 2020, 49, 2426–2480. [Google Scholar] [CrossRef]
- Whaley, S.G.; Berkow, E.L.; Rybak, J.M.; Nishimoto, A.T.; Barker, K.S.; Rogers, P.D. Azole antifungal resistance in candida albicans and emerging non-albicans candida species. Front. Microbiol. 2017, 7, 2173. [Google Scholar] [CrossRef] [PubMed]
- Venugopala, K.N.; Chandrashekharappa, S.; Alwassil, O.I.; Harsha, S.; Mlisana, K.; Odhav, B.; Pillay, M.; Bhandary, S.; Kandeel, M.; Mahomoodally, F.M.; et al. Synthesis and structural elucidation of novel benzothiazole derivatives as anti-tubercular agents: In-silico screening for possible target identification. Med. Chem. 2019, 15, 311–326. [Google Scholar] [CrossRef] [PubMed]
- Siddesh, M.B.; Padmashali, B.; Thriveni, K.S.; Sandeep, C. Synthesis of thiophene-linked pyrimidopyrimidines as pharmaceutical leads. J. Chem. Sci. 2014, 126, 821–826. [Google Scholar] [CrossRef]
- Thriveni, K.S.; Padmashali, B.; Siddesh, M.B.; Sandeep, C. Synthesis of pyrimidine incorporated piperazine derivatives and their antimicrobial activity. Indian J. Pharm. Sci. 2014, 76, 332–338. [Google Scholar]
- Nagesh, K.B.; Padmashali, C.; Sandeep, T.C.M.; Yuvaraj, M.B.; Mallikarjuna, S.M. Synthesis and antimicrobial AC-tivity of benzothiophene substituted coumarins, pyrimidines and pyrazole as new scaffold. Int. J. Pharm. Sci. Rev. Res. 2014, 28, 6–10. [Google Scholar]
- Nagesh, K.B.; Padmashali, C.; Sandeep, T.E.; Lokesh, M. Synthesis and characterization of novel benzothiophene substituted oxadiazole derivatives and their antimicrobial activity. Der Pharm. Chem. 2015, 7, 129–136. [Google Scholar]
- Venugopala, K.N.; Chandrashekharappa, S.; Bhandary, S.; Chopra, D.; Khedr, M.A.; Aldhubiab, B.E.; Attimarad, M.; Odhav, B. Efficient synthesis and characterization of novel substituted 3-benzoylindolizine analogues via the cyclization of aromatic cycloimmoniumylides with electrondeficient alkenes. Curr. Org. Synth. 2018, 15, 388–395. [Google Scholar] [CrossRef]
- Chandrashekharappa, S.; Venugopala, K.N.; Nayak, S.K.; Gleiser, R.M.; García, D.A.; Kumalo, H.M.; Kulkarni, R.S.; Mahomoodally, F.M.; Venugopala, R.; Mohan, M.K.; et al. One-pot microwave assisted synthesis and structural elucidation of novel ethyl 3-substituted-7-methylindolizine-1-carboxylates with larvicidal activity against Anopheles arabiensis. J. Mol. Struct. 2018, 1156, 377–384. [Google Scholar] [CrossRef]
- Chandrashekharappa, S.; Venugopala, K.N.; Tratrat, C.; Mahomoodally, F.M.; Al-Dhubiab, B.E.; Haroun, M.; Venugopala, R.; Mohan, M.K.; Kulkarni, R.S.; Attimarad, M.V.; et al. Efficient synthesis and characterization of novel indolizines: Exploration of in vitro COX-2 inhibitory activity and molecular modelling studies. New J. Chem. 2018, 42, 4893–4901. [Google Scholar] [CrossRef]
- Mallikarjuna, S.M.; Padmashali, M.S.M.B.; Chandrashekharappa, S.; Sandeep, C. Synthesis, anticancer and antituberculosis studies for [1-(4-chlorophenyl) Cyclopropyl] (Piperazine-yl) Methanone derivates. Int. J. Pharm. Pharm. Sci. 2014, 6, 423–427. [Google Scholar]
- Mallikarjuna, S.M.; Chandrashekharappa, S.P.B. Acid amine coupling of (1h-indole-6-yl) Ppiperazin-1-yl) Methanone with substituted acids using hatu coupling reagent and their antimicrobial and antioxidant activity. Int. J. Pharm. Sci. Res. 2017, 8, 2879–2885. [Google Scholar] [CrossRef]
- Sandeep, C.; Padmashali, B.; Kulkarni, R.S.; Mallikarjuna, S.M.; Siddesh, M.B.; Nagesh, H.K.; Thriveni, K.S. Synthesis of substituted 5-acetyl-3- benzoylindolizine-1-carboxylate from substituted 2-acetyl pyridinium bromides. Heterocycl. Lett. 2014, 4, 371–376. [Google Scholar]
- Sandeep, C.; Venugopala, K.N.; Khedr, M.A.; Padmashali, B.; Kulkarni, R.S.; Venugopala, R.; Odhav, B. Design and synthesis of novel indolizine analogues as cox-2 inhibitors: Computational perspective and in vitro screening. Indian J. Pharm. Educ. Res. 2017, 51, 452–460. [Google Scholar] [CrossRef]
- Sandeep, C.; Padmashali, B.; Venugopala, K.N.; Kulkarni, R.S.; Venugopala, R.; Odhav, B. Synthesis and characterization of ethyl 7-acetyl-2-substituted 3-(Substituted benzoyl) Indolizine-1-carboxylates for in vitro anticancer activity. Asian J. Chem. 2016, 28, 1043–1048. [Google Scholar] [CrossRef]
- Sandeep, C.; Venugopala, K.N.; Gleiser, R.M.; Chetram, A.; Padmashali, B.; Kulkarni, R.S.; Venugopala, R.; Odhav, B. Greener synthesis of indolizine analogues using water as a base and solvent: Study for larvicidal activity againstAnopheles arabiensis. Chem. Biol. Drug Des. 2016, 88, 899–904. [Google Scholar] [CrossRef]
- Venugopala, K.N.; Al-Attraqchi, O.H.; Girish, M.B.; Chandrashekharappa, S.; Alwassil, O.I.; Odhav, B.; Tratrat, C.; Nayak, S.K.; Morsy, M.A.; Aldhubiab, B.E.; et al. Novel series of methyl 3-(Substituted benzoyl)-7-substituted-2-phenylindolizine-1-carboxylates as promising anti-inflammatory agents: Molecular modeling studies. Biomolecules 2019, 9, 661. [Google Scholar] [CrossRef] [PubMed]
- Venugopala, K.N.; Tratrat, C.; Haroun, M.; Odhav, B.; Chandrashekharappa, S.; Attimarad, M.; Sreeharsha, N.; Nair, A.; Pottathil, S.; Venugopala, R.; et al. Anti-tubercular potency and computationallyassessed drug-likeness and toxicology of diversely substituted indolizines. Indian J. Pharm. Educ. Res. 2019, 53, 545–552. [Google Scholar] [CrossRef]
- Venugopala, K.N.; Chandrashekharappa, S.; Sreeharsha, N.; Morsy, M.A.; Pottathil, S.; Venugopala, R.; Odhav, B.; Mlisana, K.; Pillay, M.; Abdallah, H.H.; et al. Computational, crystallographic studies, cytotoxicity and anti-tubercular activity of substituted 7-methoxy-indolizine analogues. PLoS ONE 2019, 14, e0217270. [Google Scholar] [CrossRef] [PubMed]
- Venugopala, K.N.; Tratrat, C.; Nair, A.B.; Sreeharsha, N.; Venugopala, R.; Chandrashekharappa, S.; Alwassil, O.I.; Odhav, B.; Pillay, M.; Mahomoodally, F.M.; et al. Anti-tubercular activity of substituted 7-methyl and 7-formylindolizines and in silico study for prospective molecular target identification. Antibiotics 2019, 8, 247. [Google Scholar] [CrossRef] [PubMed]
- Venugopala, K.N.; Tratrat, C.; Pillay, M.; Chandrashekharappa, S.; Al-Attraqchi, O.H.A.E.; Aldhubiab, B.; Attimarad, M.I.; Alwassil, O.; Nair, A.B.; Sreeharsha, N.; et al. In silico design and synthesis of tetrahydropyrimidinones and tetrahydropyrimidinethiones as potential thymidylate kinase inhibitors exerting anti-TB activity against mycobacterium tuberculosis. Drug Des. Dev. Ther. 2020, ume 14, 1027–1039. [Google Scholar] [CrossRef]
- Venugopala, K.N.; Uppar, V.; Chandrashekharappa, S.; Abdallah, H.H.; Pillay, M.; Deb, P.K.; Morsy, M.A.; Aldhubiab, B.E.; Attimarad, M.; Nair, A.B.; et al. Cytotoxicity and Antimycobacterial Properties of Pyrrolo[1,2-a]quinoline Derivatives: Molecular Target Identification and Molecular Docking Studies. Antibiotics 2020, 9, 233. [Google Scholar] [CrossRef] [PubMed]
- Hasija, A.; Bhandary, S.; Venugopala, K.N.; Chandrashekharappa, S.; Chopra, D. Structural investigation of methyl 3-(4-fluorobenzoyl)-7-methyl-2-phenylindolizine-1-carboxylate, an inhibitory drug towards Mycobacterium tuberculosis. Acta Cryst. Sect. E Cryst. Commun. 2020, 76, 567–571. [Google Scholar] [CrossRef] [PubMed]
- Khedr, M.A.; Pillay, M.; Chandrashekharappa, S.; Chopra, D.; Aldhubiab, B.E.; Attimarad, M.; Alwassil, O.I.; Mlisana, K.; Odhav, B.; Venugopala, K.N. Molecular modeling studies and anti-TB activity of trisubstituted indolizine analogues; molecular docking and dynamic inputs. J. Biomol. Struct. Dyn. 2018, 36, 2163–2178. [Google Scholar] [CrossRef] [PubMed]
- Sandeep, C.; Venugopala, K.N.; Khedr, M.A.; Attimarad, M.; Padmashali, B.; Kulkarni, R.S.; Venugopala, R.; Odhav, B. Review on chemistry of natural and synthetic indolizines with their chemical and pharmacological properties. J. Basic Clin. Pharm. 2017, 8, 49–60. [Google Scholar]
- Chandrashekharappa, S.; Venugopala, K.N.; Venugopala, R.; Padmashali, B. Qualitative anti-tubercular activity of synthetic ethyl 7-acetyl2-substituted-3-(4-substituted benzoyl) indolizine-1-carboxylate analogues. J. Appl. Pharm. Sci. 2019, 9, 124–128. [Google Scholar] [CrossRef]
- Sandeep, C.; Padmashali, B.; Kulkarni, R.S. Efficient synthesis of indolizines and new imidazo[1,2-a]pyridines via the expected cyclization of aromatic cycloimmonium ylides with electron deficient alkynes and ethyl cyanoformate. Tetrahedron Lett. 2013, 54, 6411–6414. [Google Scholar] [CrossRef]
- Sandeep, C.; Basavaraj, P.; Rashmi, S.K. Synthesis of isomeric subtituted 6-acetyl-3-benzoylindolizine- 1-carboxylate and 8-acetyl-3-benzoylindolizine-1-carboxylate from subtituteded 3-acetyl pyri-dinium bromides and their antimicrobial activity. J. Appl. Chem. 2013, 2, 1049–1052. [Google Scholar]
- Østby, O.B.; Dalhus, B.; Gundersen, L.-L.; Rise, F.; Bast, A.; Haenen, G.R.M.M. Synthesis of 1-substituted 7-cyano-2,3-diphenylindolizines and evaluation of antioxidant properties. Eur. J. Org. Chem. 2000, 2000, 3763–3770. [Google Scholar] [CrossRef]
- Baidya, M.; Kumar, J.; Srivastava, A.K.; Jyothi, C.; Gupta, A.; Das, A.K. Synthesis and pharmacological screening of some in-dolizinamido glutamine amino acid derivatives. Indian J. Heterocycl. Chem. 2004, 14, 81–82. [Google Scholar]
- Uppar, V.; Mudnakudu-Nagaraju, K.K.; Basarikatti, A.I.; Chougala, M.; Chandrashekharappa, S.; Mohan, M.K.; Banuprakash, G.; Venugopala, K.N.; Ningegowda, R.; Padmashali, B. Microwave induced synthesis, and pharmacological properties of novel 1-benzoyl-4-bromopyrrolo[1,2-a]quinoline-3-carboxylate analogues. Chem. Data Collect. 2020, 25, 100316. [Google Scholar] [CrossRef]
- Das, A.K.; Mukherjee, I. Synthesis and antimicrobial activity of mannich bases of indolizine analogues. Orient. J. Chem. 2006, 22, 339–342. [Google Scholar]
- Das, A.K.; Mukherjee, I. Synthesis and evaluation of some 3-benzoylindolizine-1-carboxamides as possible anti-inflammatory and analgesic agents. Orient. J. Chem. 2006, 22, 415–420. [Google Scholar]
- Kallay, K.R.; Doerge, R.F. P-substituted 1,2-diphenylindolizines as anti-inflammatory agents. J. Pharm. Sci. 1972, 61, 949–951. [Google Scholar] [CrossRef] [PubMed]
- James, D.A.; Koya, K.; Li, H.; Liang, G.; Xia, Z.; Ying, W.; Wu, Y.; Sun, L. Indole- and indolizine-glyoxylamides displaying cytotoxicity against multidrug resistant cancer cell lines. Bioorg. Med. Chem. Lett. 2008, 18, 1784–1787. [Google Scholar] [CrossRef] [PubMed]
- Morita, K.; Yamamoto, I.H. Synthesis of indolizine derivatives. Biochem. Pharmacol. 1973, 22, 111–115. [Google Scholar]
- Lingala, S.; Nerella, R.; Cherukupally, R.; Das, A.K. Synthesis and comparative anti-tubercular activity of indolizine deriva-tives of isoniazid/Pyrazinamide/Ethionamide. Int. J. Pharm. Sci. Rev. Res. 2011, 6, 128–131. [Google Scholar]
- Liu, Y.; Zhang, Y.; Shen, Y.-M.; Hu, H.-W.; Xu, J.-H. Regioselective synthesis of 3-acylindolizines and benzo- analogues via 1,3-dipolar cycloadditions of N-ylides with maleic anhydride. Org. Biomol. Chem. 2010, 8, 2449–2456. [Google Scholar] [CrossRef]
- Sharma, V.; Kumar, V. Indolizine: A biologically active moiety. Med. Chem. Res. 2014, 23, 3593–3606. [Google Scholar] [CrossRef]
- Gómez, C.M.M.; Kouznetsov, V.V.; Sortino, M.A.; Álvarez, S.L.; Zacchino, S.A. In vitro antifungal activity of polyfunctionalized 2-(hetero)arylquinolines prepared through imino Diels–Alder reactions. Bioorg. Med. Chem. 2008, 16, 7908–7920. [Google Scholar] [CrossRef] [PubMed]
- Vemula, V.R.; Vurukonda, S.; Bairi, C.K. Indolizine derivatives: Recent advances and potential pharmacological activities. Int. J. Pharm. Sci. Rev. Res. 2011, 11, 159–163. [Google Scholar]
- Uppar, V.; Chandrashekharappa, S.; Basarikatti, A.I.; Banuprakash, G.; Mohan, M.; Chougala, M.; Mudnakudu-Nagaraju, K.K.; Ningegowda, R.; Padmashali, B. Synthesis, antibacterial, and antioxidant studies of 7-amino-3-(4-fluorobenzoyl)indolizine-1-carboxylate derivatives. J. Appl. Pharm. Sci. 2020, 10, 77–85. [Google Scholar] [CrossRef]
- Uppar, V.; Chandrashekharappa, S.; Venugopala, K.N.; Deb, P.K.; Kar, S.; Alwassil, O.I.; Gleiser, R.M.; Garcia, D.; Odhav, B.; Mohan, M.K.; et al. Synthesis and characterization of pyrrolo[1,2-a]quinoline derivatives for their larvicidal activity against Anopheles arabiensis. Struct. Chem. 2020, 31, 1533–1543. [Google Scholar] [CrossRef]
- Dillard, R.D.; Pavey, D.E.; Benslay, D.N. Synthesis and antiinflammatory activity of some 2,2-dimethyl-1,2-dihydroquinolines. J. Med. Chem. 1973, 16, 251–253. [Google Scholar] [CrossRef]
- Sechi, M.; Rizzi, G.; Bacchi, A.; Carcelli, M.; Rogolino, D.; Pala, N.; Sanchez, T.W.; Taheri, L.; Dayam, R.; Neamati, N. Design and synthesis of novel dihydroquinoline-3-carboxylic acids as HIV-1 integrase inhibitors. Bioorg. Med. Chem. 2009, 17, 2925–2935. [Google Scholar] [CrossRef]
- Dubé, D.; Blouin, M.; Brideau, C.; Chan, C.-C.; Desmarais, S.; Ethier, D.; Falgueyret, J.-P.; Friesen, R.W.; Girard, M.; Girard, Y.; et al. Quinolines as potent 5-lipoxygenase inhibitors: Synthesis and biological profile of L-746,530. Bioorg. Med. Chem. Lett. 1998, 8, 1255–1260. [Google Scholar] [CrossRef]
- Testa, M.L.; Lamartina, L.; Mingoia, F. A new entry to the substituted pyrrolo[3,2-c]quinoline derivatives of biological interest by intramolecular heteroannulation of internal imines. Tetrahedron 2004, 60, 5873–5880. [Google Scholar] [CrossRef]
- Martins, C.; Carreiras, M.C.; Leon, R.; Ríos, C.D.L.; Bartolini, M.; Andrisano, V.; Iriepa, I.; Moraleda, I.; Gálvez, E.; Garcia, M.; et al. Synthesis and biological assessment of diversely substituted furo[2,3-b]quinolin-4-amine and pyrrolo[2,3-b]quinolin-4-amine derivatives, as novel tacrine analogues. Eur. J. Med. Chem. 2011, 46, 6119–6130. [Google Scholar] [CrossRef] [PubMed]
- Guha, R.; Bender, A. Computational Approaches in Cheminformatics and Bioinformatics; Wiley: Hoboken, NJ, USA, 2012. [Google Scholar]
- Chakraborty, A.; Pan, S.; Chattaraj, P.K. Biological activity and toxicity: A conceptual dft approach. In Structure and Bonding; Springer International Publishing: Berlin/Heidelberg, Germany, 2013; pp. 143–179. [Google Scholar]
- Pan, S.; Gupta, A.; Subramanian, V.; Chattaraj, P.K. Quantitative structure-activity/Property/Toxicity relationships through conceptual density functional theory-based reactivity descriptors. In Sustainable Nanosystems Development, Properties, and Applications; IGI Global: Hershey, PA, USA, 2015; pp. 123–179. [Google Scholar]
- Pan, S.A.; Gupta, D.; Roy, R.; Sharma, V.; Subramanian, A.; Mitra, P.; Chattaraj., P. Application of conceptual density functional theory in developing qsar models and their usefulness in the prediction of biological activity and toxicity of molecules. In Chemometrics Applications and Research; Apple Academic Press: Palm Bay, FL, USA, 2016; pp. 183–214. [Google Scholar]
- Kon’e, M.G.-R.; N’dri, J.S.; Kodjo, C.G.; Kablan, A.L.C.; Ouattara, L.; Ouattara, O. Combining of dft and qsar results to predict the antibacterial activity of a series of azetidinones derived from dapsone as inhibitors of bacillus subtilis and pseudomonas aeruginosa. SDRP J. Comp. Chem. Mol. Model. 2018, 2, 1–8. [Google Scholar]
- Zermeño-Macías, M.D.L.Á.; González-Chávez, M.M.; Méndez, F.; González-Chávez, R.; Richaud, A. Theoretical reactivity study of indol-4-ones and their correlation with antifungal activity. Molecules 2017, 22, 427. [Google Scholar] [CrossRef] [PubMed]
- Frau, J.; Muñoz, F.; Glossman-Mitnik, D. A molecular electron density theory study of the chemical reactivity of cis- and trans-resveratrol. Molecules 2016, 21, 1650. [Google Scholar] [CrossRef]
- Flores-Holguín, N.; Frau, J.; Glossman-Mitnik, D. Computational peptidology assisted by conceptual density functional theory for the study of five new antifungal tripeptides. ACS Omega 2019, 4, 12555–12560. [Google Scholar] [CrossRef]
- Rajpoot, M.; Bhattacharyya, R.; Sharma, A.K.; Gupta, G.K.; Kumar, V. 2. Drug designing in novel drug discovery: Trends, scope and relevance. In Chemical Drug Design; Springer: Berlin, Germany, 2016; pp. 15–30. [Google Scholar]
- Gore, M.; Jagtap, U.B. Computational Drug Discovery and Design; Springer Science Business Media, LLC.: New York, NY, USA, 2018. [Google Scholar]
- Parr, R.; Yang, W. Density-Functional Theory of Atoms and Molecules; Oxford University Press: New York, NY, USA, 1989. [Google Scholar]
- Chermette, H. Chemical reactivity indexes in density functional theory. J. Comput. Chem. 1999, 20, 129–154. [Google Scholar] [CrossRef]
- Geerlings, P.; De Proft, F.; Langenaeker, W. Conceptual density functional theory. Chem. Rev. 2003, 103, 1793–1873. [Google Scholar] [CrossRef]
- Halgren, T.A. Merck molecular force field: I. Basis, form, scope, parameterization, and performance of mmff94. J. Comput. Chem. 1996, 17, 490–519. [Google Scholar] [CrossRef]
- Halgren, T.A. Merck molecular force field. II. Mmff94 van der waals and electrostatic parameters for intermolecular inter-actions. J. Comput. Chem. 1996, 17, 520–552. [Google Scholar] [CrossRef]
- Halgren, T.A. MMFF VI. MMFF94s Option for energy minimization studies. J. Comput. Chem. 1999, 20, 720–729. [Google Scholar] [CrossRef]
- Halgren, T.A.; Nachbar, R.B. Merck molecular force field. IV. Conformational energies and geometries for MMFF94. J. Comput. Chem. 1996, 17, 587–615. [Google Scholar] [CrossRef]
- Halgren, T.A. Merck molecular force field. V. Extension of MMFF94 using experimental data, additional computational data, and empirical rules. J. Comput. Chem. 1996, 17, 616–641. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, G.E.; Scuseria, M.A.; Robb, J.R.; Cheeseman, G.; Scalmani, V.; Barone, B.; Mennucci, G.A.; Petersson, H.; et al. Gaussian 09 Revision E.01; Gaussian Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Peverati, R.; Truhlar, D.G. Screened-exchange density functionals with broad accuracy for chemistry and solid-state physics. Phys. Chem. Chem. Phys. 2012, 14, 16187–16191. [Google Scholar] [CrossRef] [PubMed]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef] [PubMed]
- Weigend, F. Accurate Coulomb-fitting basis sets for H to Rn. Phys. Chem. Chem. Phys. 2006, 8, 1057–1065. [Google Scholar] [CrossRef] [PubMed]
- Flores-Holguín, N.; Frau, J.; Glossman-Mitnik, D. Conceptual dft as a helpful chemoinformatics tool for the study of the cla-vanin family of antimicrobial marine peptides. In Density Functional Theory Calculations; IntechOpen: Rijeka, Croatia, 2019; pp. 1–11. [Google Scholar]
- Frau, J.; Glossman-Mitnik, D. Molecular reactivity and absorption properties of melanoidin blue-g1 through conceptual dft. Molecules 2018, 23, 559. [Google Scholar] [CrossRef]
- Frau, J.; Glossman-Mitnik, D. Conceptual dft study of the local chemical reactivity of the dilysyldipyrrolones a and b in-termediate melanoidins. Chem. Acc. 2018, 137, 1210. [Google Scholar] [CrossRef]
- Frau, J.; Glossman-Mitnik, D. Conceptual dft study of the local chemical reactivity of the colored bisarg melanoidin and its protonated derivative. Front. Chem. 2018, 6, 136. [Google Scholar] [CrossRef]
- Frau, J.; Glossman-Mitnik, D. Computational study of the chemical reactivity of the Blue-M1 intermediate melanoidin. Comput. Chem. 2018, 1134, 22–29. [Google Scholar] [CrossRef]
- Frau, J.; Glossman-Mitnik, D. Chemical reactivity theory applied to the calculation of the local reactivity descriptors of a colored maillard reaction product. Chem. Sci. Int. J. 2018, 22, 1–14. [Google Scholar] [CrossRef]
- Frau, J.; Glossman-Mitnik, D. Blue M2: An intermediate melanoidin studied via conceptual DFT. J. Mol. Model. 2018, 24, 138. [Google Scholar] [CrossRef]
- Marenich, A.V.; Cramer, C.J.; Truhlar, D.G. Universal solvation model based on solute electron density and on a continuum model of the solvent defined by the bulk dielectric constant and atomic surface tensions. J. Phys. Chem. B 2009, 113, 6378–6396. [Google Scholar] [CrossRef] [PubMed]
- Flores-Holguín, N.; Frau, J.; Glossman-Mitnik, D. A fast and simple evaluation of the chemical reactivity properties of the Pristinamycin family of antimicrobial peptides. Chem. Phys. Lett. 2020, 739, 137021. [Google Scholar] [CrossRef]
- Flores-Holguín, N.; Frau, J.; Glossman-Mitnik, D. Conceptual dft- based computational peptidology of marine natural compounds: Dis- codermins a–h. Molecules 2020, 25, 4158. [Google Scholar] [CrossRef]
- Flores-Holguín, N.; Frau, J.; Glossman-Mitnik, D. Virtual screening of marine natural compounds by means of chemoinformatics and cdft-based computational peptidology. Mar. Drugs 2020, 18, 478. [Google Scholar] [CrossRef] [PubMed]
- Frau, J.; Glossman-Mitnik, D. Molecular reactivity of some maillard reaction products studied through conceptual DFT. Contemp. Chem. 2018, 1, 1–14. [Google Scholar]
- Gázquez, J.; Cedillo, A.; Vela, A. Electrodonating and electroaccepting powers. J. Phys. Chem. 2007, 111, 1966–1970. [Google Scholar]
- Chattaraj, P.K.; Chakraborty, A.; Giri, S. Net electrophilicity. J. Phys. Chem. A 2009, 113, 10068–10074. [Google Scholar] [CrossRef]
- Jaramillo, P.; Domingo, L.R.; Chamorro, E.; Pérez, P. A further exploration of a nucleophilicity index based on the gas-phase ionization potentials. J. Mol. Struct. 2008, 865, 68–72. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. SwissTargetPrediction: Updated data and new features for efficient prediction of protein targets of small molecules. Nucleic Acids Res. 2019, 47, W357–W364. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. Swissadme: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [PubMed]
- Prasad, A.; Govindaraju, S.; Sushma, P.; Jain, A.S.; Chandan, D.; Prasad, M.N.N.; Kollur, S.P.; Srinivasa, C.; Shivamallu, C. Helicobacter pylori infection: A bioinformatic approach. Int. J. Pharm. Sci. Res. 2020, 11, 5469–5483. [Google Scholar]
- O’Boyle, N.M.; Banck, M.A.; James, C.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel: An open chemical toolbox. J. Cheminform. 2011, 3, 33. [Google Scholar] [CrossRef]
- Jain, A.S.; Sushma, P.; Chandan, D.; Beelagi, M.S.; Shashank, P.; Chandan, S.; Prasad, A.; Syed, A.; Marraiki, N.; Prasad, K.S. Effect of flavonoids having anti-inflammatory and antiviral properties on the spike glycoprotein of SARS-CoV-2: An In silico Approach. Saudi J. Biol. Sci. 2020, 28, 1040–1051. [Google Scholar] [CrossRef]
- Prasad, S.K.; Sushma, P.; Chandan, S.; Kollur, S.P.; Syed, A.; Castro, J.O.; Frau, J.; Holguín, N.F.; Mitnik, D.G. Evaluating annona muricata acetogenins as possible anti-sars-cov-2 agents through in silico analysis and theoretical chemistry approaches. Front. Chem. 2021, 8, 624716. [Google Scholar] [CrossRef] [PubMed]
- Dallakyan, S.; Olson, A.J. Small-molecule library screening by docking with PyRx. In NMR-Based Metabolomics; Springer Science and Business Media, LLC.: Berlin/Heidelberg, Germany, 2015; Volume 1263, pp. 243–250. [Google Scholar]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | MIC (µg/mL) |
|---|---|
| BQ-01 | 0.8 |
| BQ-02 | 12.5 |
| BQ-03 | 0.8 |
| BQ-04 | 1.6 |
| BQ-05 | 0.8 |
| BQ-06 | 0.4 |
| BQ-07 | 0.4 |
| BQ-08 | 0.4 |
| Sl No | Macromolecule/Protein Target | Ligand/Small Molecule | Number of Hydrogen Bonds | H-Bond Forming Amino Acid Residues | ∆G (Kcal/mol) |
|---|---|---|---|---|---|
| 1 | 2H6T | Drug | 4 | SER-13, ASP-86, ASP-120 and GLN-121 | −7.5 |
| BQ-01 | 3 | THR-221 and THR-222 | −6.5 | ||
| BQ-03 | 6 | THR-221, THR-222 and GLY-220 | −7.4 | ||
| BQ-05 | 5 | THR-221, THR-222 and GLY-220 | −7.2 | ||
| BQ-07 | 6 | THR-221, THR-222 and VAL-12 | −6.8 | ||
| 2 | 3N9K | Drug | 3 | ASP-145, LEU-304 and ASN-305 | −7.8 |
| BQ-01 | 2 | PHE-144 and ASN-146 | −9.4 | ||
| BQ-03 | 2 | ASN-146 | −9.7 | ||
| BQ-05 | 3 | ASN-146 and TYR-317 | −9.4 | ||
| BQ-07 | 2 | ASN-146 | −9.2 | ||
| 3 | 5TZ1 | Drug | 4 | TYR-132, PHE-463 and ARG-469 | −7.2 |
| BQ-01 | 2 | TYR-132 | −8.9 | ||
| BQ-03 | 2 | TYR-132 | −8.1 | ||
| BQ-05 | 1 | TYR-132 | −8.6 | ||
| BQ-07 | 2 | TYR-132 | −8.7 |
| HOMO | LUMO | SOMO | H-L Gap | JI | JA | JHL | ∆SL | |
|---|---|---|---|---|---|---|---|---|
| BQ-01 | −5.9207 | −2.3551 | −2.5429 | 3.5655 | 0.001 | 0.103 | 0.103 | 0.188 |
| BQ-02 | −5.8513 | −2.2455 | −2.5712 | 3.4058 | 0.008 | 0.061 | 0.062 | 0.126 |
| BQ-03 | −5.9443 | −2.6011 | −2.7470 | 3.3432 | 0.003 | 0.081 | 0.081 | 0.146 |
| BQ-04 | −5.9158 | −2.5666 | −2.6830 | 3.3492 | 0.008 | 0.058 | 0.058 | 0.117 |
| BQ-05 | −5.9634 | −2.9456 | −3.0507 | 3.0177 | 0.003 | 0.056 | 0.056 | 0.105 |
| BQ-06 | −5.9552 | −2.7737 | −2.8771 | 3.1816 | 0.008 | 0.052 | 0.052 | 0.103 |
| BQ-07 | −5.9381 | −2.5225 | −2.6975 | 4.4156 | 0.002 | 0.096 | 0.096 | 0.175 |
| BQ-08 | −5.9073 | −2.5239 | −2.6458 | 3.3835 | 0.008 | 0.060 | 0.061 | 0.122 |
| S | N | |||||||
|---|---|---|---|---|---|---|---|---|
| BQ-01 | 4.1379 | 3.5655 | 2.4011 | 0.2805 | 2.8718 | 7.0940 | 2.9561 | 10.0501 |
| BQ-02 | 4.1484 | 3.4058 | 2.5264 | 0.2936 | 2.9412 | 7.3399 | 3.1916 | 10.5315 |
| BQ-03 | 4.2727 | 3.3432 | 2.7304 | 0.2991 | 2.8482 | 7.8060 | 3.5333 | 11.3393 |
| BQ-04 | 4.2412 | 3.3492 | 2.6854 | 0.2986 | 2.8767 | 7.7006 | 3.4595 | 11.1601 |
| BQ-05 | 4.4545 | 3.0177 | 3.2877 | 0.3314 | 2.8291 | 8.9912 | 4.5367 | 13.5279 |
| BQ-06 | 4.3644 | 3.1816 | 2.9936 | 0.3143 | 2.8373 | 8.3682 | 4.0037 | 12.3719 |
| BQ-07 | 4.2303 | 3.4156 | 2.6197 | 0.2928 | 2.8544 | 7.5679 | 3.3377 | 10.9056 |
| BQ-08 | 4.2156 | 3.3835 | 2.6262 | 0.2956 | 2.8852 | 7.5716 | 3.3560 | 10.9277 |
| ∆G of Solvation | MW | log P | TPSA | Molecular Volume | |
|---|---|---|---|---|---|
| BQ-01 | −12.66 | 496.31 | 5.07 | 83.33 | 380.28 |
| BQ-02 | −9.69 | 438.28 | 5.31 | 57.02 | 335.75 |
| BQ-03 | −10.91 | 500.73 | 5.69 | 74.10 | 368.27 |
| BQ-04 | −8.46 | 442.70 | 5.93 | 47.79 | 323.74 |
| BQ-05 | −12.81 | 491.30 | 4.76 | 97.89 | 371.59 |
| BQ-06 | −10.64 | 433.26 | 5.00 | 71.58 | 327.06 |
| BQ-07 | −11.22 | 466.29 | 5.01 | 74.10 | 354.73 |
| BQ-08 | −8.45 | 408.25 | 5.25 | 47.79 | 310.20 |
| BQ-01 | BQ-02 | BQ-03 | BQ-04 | |
|---|---|---|---|---|
| GI absorption | HIGH | HIGH | HIGH | HIGH |
| BBB permeant | NO | YES | NO | YES |
| P-gp substrate | NO | NO | NO | NO |
| CYP1A2 inhibitor | NO | YES | NO | YES |
| CYP2C19 inhibitor | YES | YES | YES | YES |
| CYP2C9 inhibitor | YES | YES | YES | YES |
| CYP2D6 inhibitor | NO | NO | NO | NO |
| CYP3A4 inhibitor | NO | NO | NO | NO |
| Log Kp/cm s−1 (skin permeation) | −5.15 | −4.69 | −4.71 | −4.26 |
| BQ-05 | BQ-06 | BQ-07 | BQ-08 | |
| GI absorption | HIGH | HIGH | HIGH | HIGH |
| BBB permeant | NO | NO | NO | YES |
| P-gp substrate | NO | NO | NO | NO |
| CYP1A2 inhibitor | NO | YES | YES | YES |
| CYP2C19 inhibitor | YES | YES | YES | YES |
| CYP2C9 inhibitor | YES | YES | YES | YES |
| CYP2D6 inhibitor | NO | NO | NO | NO |
| CYP3A4 inhibitor | NO | NO | NO | NO |
| Log Kp/cm s−1 (skin permeation) | −5.30 | −4.85 | −4.95 | −4.49 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Uppar, V.; Chandrashekharappa, S.; Shivamallu, C.; P, S.; Kollur, S.P.; Ortega-Castro, J.; Frau, J.; Flores-Holguín, N.; Basarikatti, A.I.; Chougala, M.; et al. Investigation of Antifungal Properties of Synthetic Dimethyl-4-Bromo-1-(Substituted Benzoyl) Pyrrolo[1,2-a] Quinoline-2,3-Dicarboxylates Analogues: Molecular Docking Studies and Conceptual DFT-Based Chemical Reactivity Descriptors and Pharmacokinetics Evaluation. Molecules 2021, 26, 2722. https://doi.org/10.3390/molecules26092722
Uppar V, Chandrashekharappa S, Shivamallu C, P S, Kollur SP, Ortega-Castro J, Frau J, Flores-Holguín N, Basarikatti AI, Chougala M, et al. Investigation of Antifungal Properties of Synthetic Dimethyl-4-Bromo-1-(Substituted Benzoyl) Pyrrolo[1,2-a] Quinoline-2,3-Dicarboxylates Analogues: Molecular Docking Studies and Conceptual DFT-Based Chemical Reactivity Descriptors and Pharmacokinetics Evaluation. Molecules. 2021; 26(9):2722. https://doi.org/10.3390/molecules26092722
Chicago/Turabian StyleUppar, Vijayakumar, Sandeep Chandrashekharappa, Chandan Shivamallu, Sushma P, Shiva Prasad Kollur, Joaquín Ortega-Castro, Juan Frau, Norma Flores-Holguín, Atiyaparveen I. Basarikatti, Mallikarjun Chougala, and et al. 2021. "Investigation of Antifungal Properties of Synthetic Dimethyl-4-Bromo-1-(Substituted Benzoyl) Pyrrolo[1,2-a] Quinoline-2,3-Dicarboxylates Analogues: Molecular Docking Studies and Conceptual DFT-Based Chemical Reactivity Descriptors and Pharmacokinetics Evaluation" Molecules 26, no. 9: 2722. https://doi.org/10.3390/molecules26092722
APA StyleUppar, V., Chandrashekharappa, S., Shivamallu, C., P, S., Kollur, S. P., Ortega-Castro, J., Frau, J., Flores-Holguín, N., Basarikatti, A. I., Chougala, M., Mohan M, M., Banuprakash, G., Jayadev, Venugopala, K. N., Nandeshwarappa, B. P., Veerapur, R., Al-Kheraif, A. A., Elgorban, A. M., Syed, A., ... Glossman-Mitnik, D. (2021). Investigation of Antifungal Properties of Synthetic Dimethyl-4-Bromo-1-(Substituted Benzoyl) Pyrrolo[1,2-a] Quinoline-2,3-Dicarboxylates Analogues: Molecular Docking Studies and Conceptual DFT-Based Chemical Reactivity Descriptors and Pharmacokinetics Evaluation. Molecules, 26(9), 2722. https://doi.org/10.3390/molecules26092722