
Synthesis of a Ni Complex Chelated by a [2.2]Paracyclophane-Functionalized Diimine Ligand and Its Catalytic Activity for Olefin Oligomerization


Abstract
1. Introduction
2. Results and Discussion
2.1. Preparation and Structure of Ni Complexes



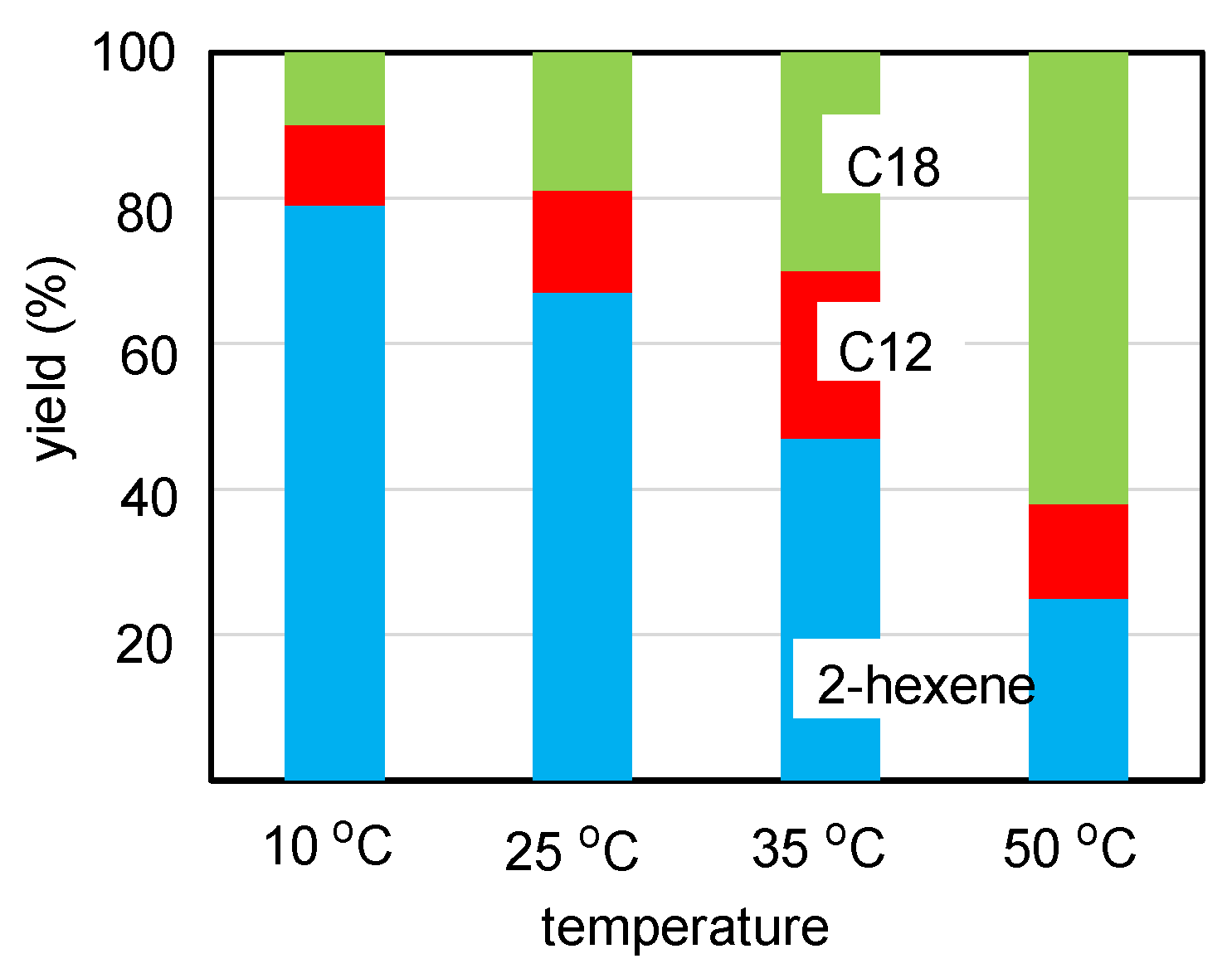
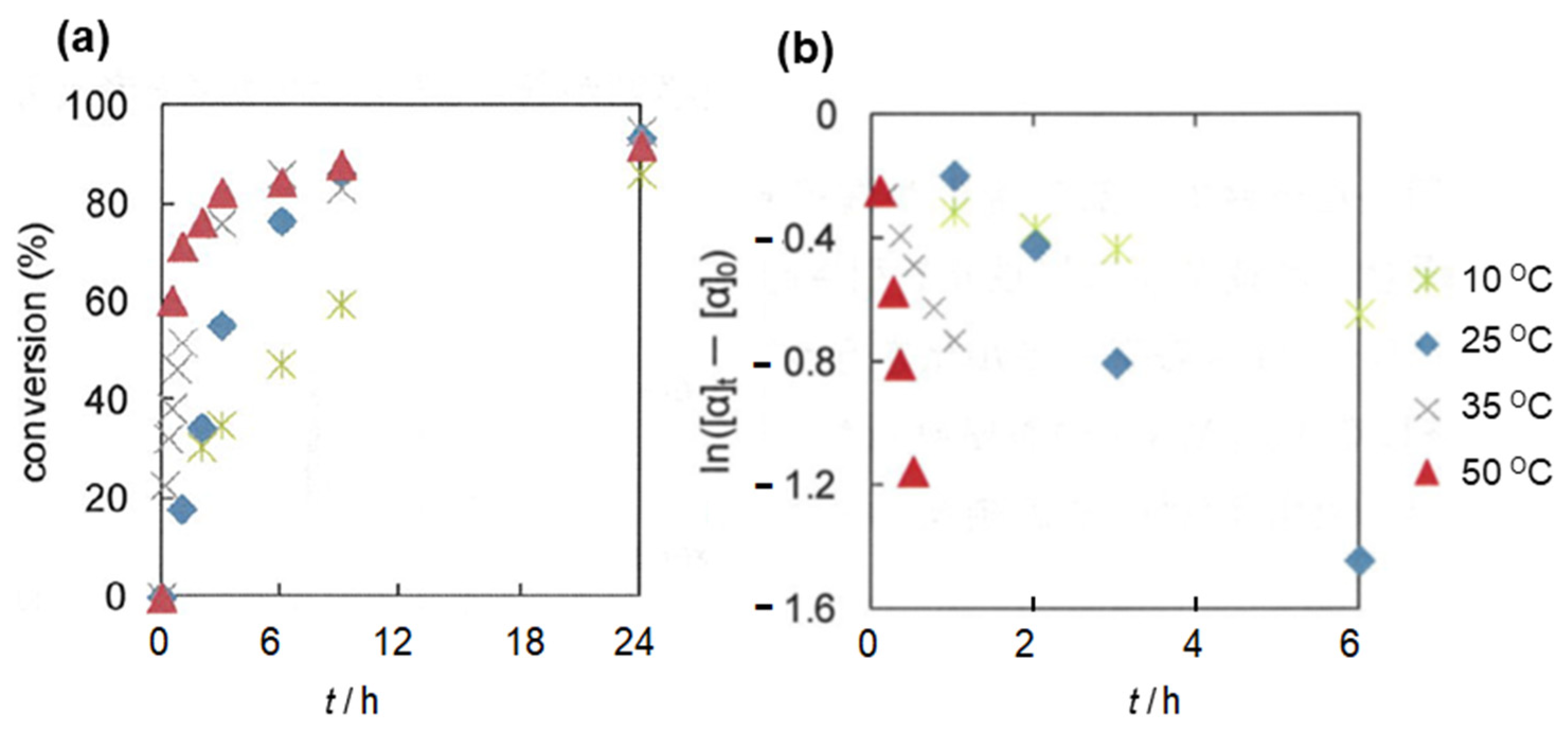
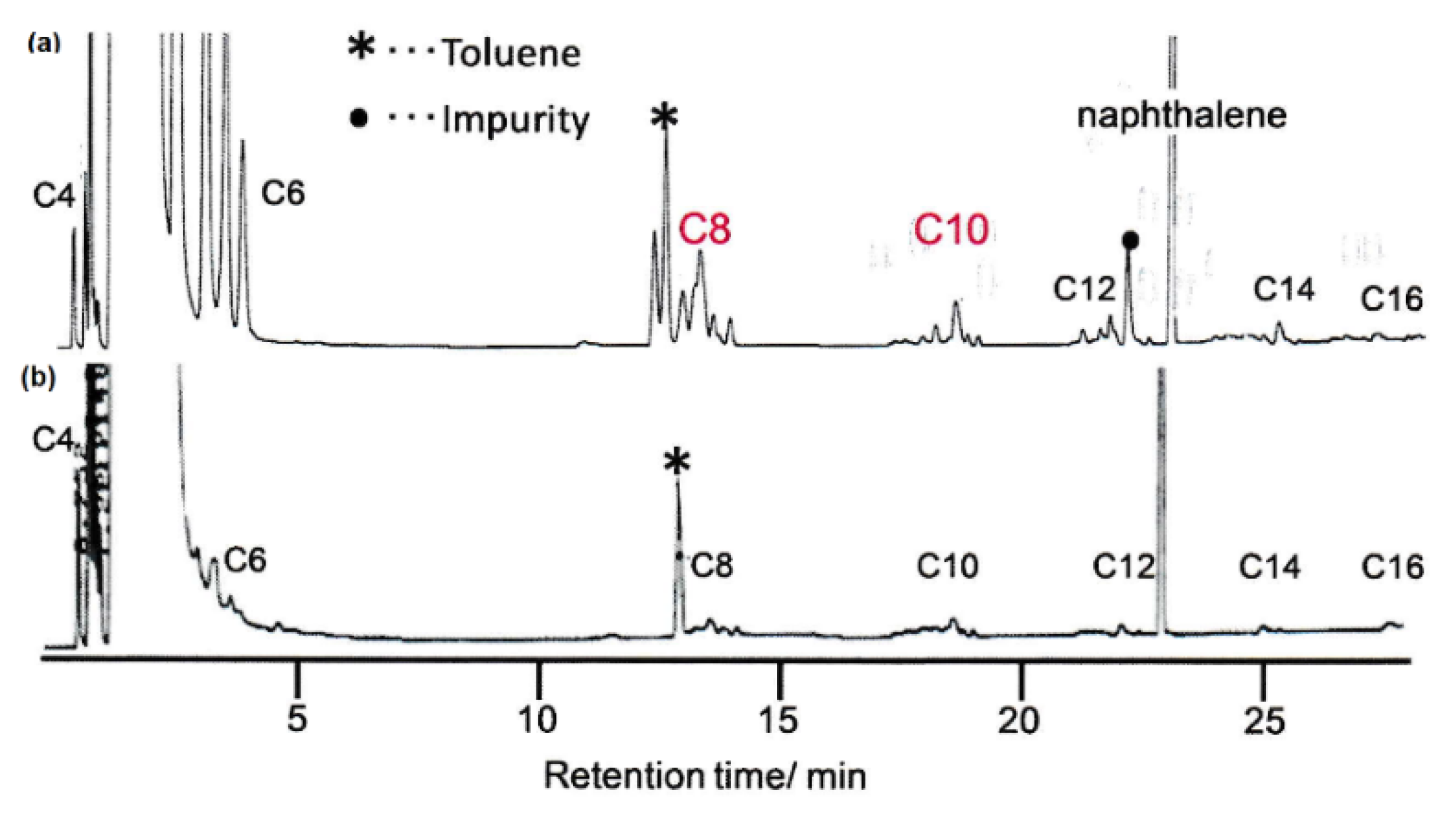
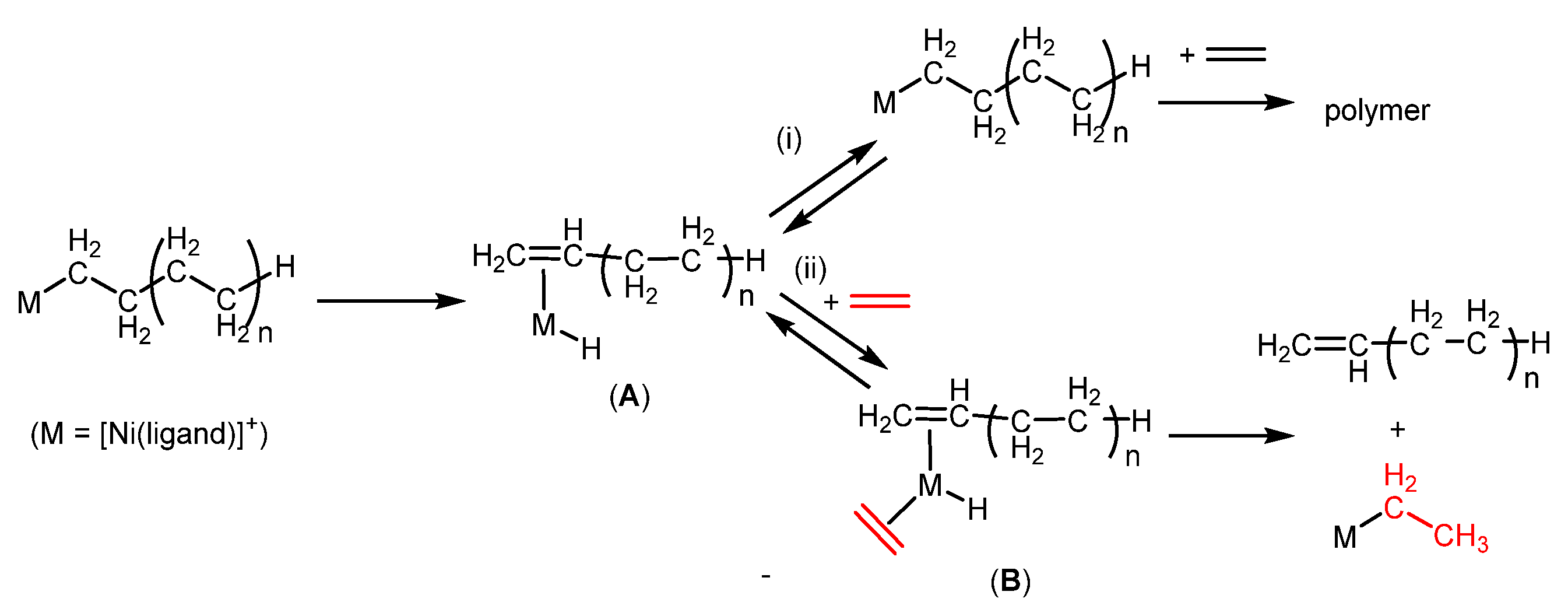
2.2. Olefin Oligomerization Catalyzed by Ni Complexes
3. Conclusions
4. Experimental Section
4.1. General
4.2. Preparation of Racemic Ligand L1
4.3. Preparation of Optically Active Ligand L1
4.4. Preparation of NiBr2(L1)
4.5. X-ray Crystallography of NiBr2(L1)
4.6. Preparation of NiBr2(L2)
4.7. Oligomerization
4.7.1. Oligomerization of Ethylene
4.7.2. Oligomerization of 1-Hexene
4.7.3. Co-Dimerization of Ethylene and 1-Hexene
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Skupinska, J. Oligomerization of α-Olefins to Higher Oligomers. Chem. Rev. 1991, 91, 613–648. [Google Scholar] [CrossRef]
- Dixon, J.T.; Green, M.J.; Hess, F.M.; Morgan, D.H. Advances in Selective Ethylene Trimerisation—A Critical Overview. J. Organomet. Chem. 2004, 689, 3641–3668. [Google Scholar] [CrossRef]
- Speiser, F.; Braustein, P.; Saussine, L. Catalytic Ethylene Dimerization and Oligomerization: Recent Developments with Nickel Complexes Containing P, N-Chelating Ligands. Acc. Chem. Res. 2005, 38, 784–793. [Google Scholar] [CrossRef] [PubMed]
- Bianchini, C.; Giambastiani, G.; Rios, I.G.; Mantovani, G.; Meli, A.; Segarra, A.M. Ethylene Oligomerization, Homopolymerization and Copolymerization by Iron and Cobalt Catalysts with 2,6-(Bis-organylimino)pyridyl Ligands. Coord. Chem. Rev. 2006, 250, 1391–1418. [Google Scholar] [CrossRef]
- Kuhn, P.; Sémeril, D.; Matt, D.; Chetcuti, M.J.; Lutz, P. Structure–Reactivity Relationships in SHOP-Type Complexes: Tunable Catalysts for the Oligomerisation and Polymerisation of Ethylene. Dalton Trans. 2007, 515–528. [Google Scholar] [CrossRef]
- Wass, D.F. Chromium-Catalysed Ethylene Trimerization and Tetramerisation–Breaking the Rules in Olefin Oligomerisation. Dalton Trans. 2007, 816–819. [Google Scholar] [CrossRef] [PubMed]
- Belov, G.P. Selective Dimerization, Oligomerization, Homopolymerization and Copolymerization of Olefins, with Complex Organometallic Catalysts. Russ. J. Appl. Chem. 2008, 81, 1655–1666. [Google Scholar] [CrossRef]
- McGuinness, D. Alkene Oligomerisation and Polymerisation with Metal-NHC Based Catalysts. Dalton Trans. 2009, 6915–6923. [Google Scholar] [CrossRef]
- Takeuchi, D.; Osakada, K. Oligomerization of Olefins. In Organometallic Reactions and Polymerization; Springer: Berlin/Heidelberg, Germany, 2010; pp. 169–215. [Google Scholar]
- Fujita, T.; Kawai, K. FI Catalysts for Olefin Oligomerization and Polymerization: Production of Useful Olefin-Based Materials by Unique Catalysis. Top. Catal. 2014, 57, 852–877. [Google Scholar] [CrossRef]
- Bianchini, C.; Giambastiani, G.; Luconi, L.; Meli, A. Olefin Oligomerization, Homopolymerization and Copolymerization by Late Transition Metals Supported by (Imino)pyridine Ligands. Coord. Chem. Rev. 2010, 254, 431–455. [Google Scholar] [CrossRef]
- Agapie, T. Selective Ethylene Oligomerization: Recent Advances in Chromium Catalysis and Mechanistic Investigations. Coord. Chem. Rev. 2011, 255, 861–880. [Google Scholar] [CrossRef]
- van Leeuwen, P.W.N.M.; Clément, N.D.; Tschan, M.J.-L. New Processes for the Selective Production of 1-Octene. Coord. Chem. Rev. 2011, 255, 1499–1517. [Google Scholar] [CrossRef]
- Zhang, W.; Sun, W.-H.; Redshaw, C. Tailoring Iron Complexes for Ethylene Oligomerization and/or Polymerization. Dalton Trans. 2013, 42, 8988–8997. [Google Scholar] [CrossRef] [PubMed]
- Olivier-Bourbigou, H.; Breuil, P.A.R.; Magna, L.; Michel, T.; Pastor, M.F.E.; Delcroix, D. Nickel Catalyzed Olefin Oligomerization and Dimerization. Chem. Rev. 2020, 120, 7919–7983. [Google Scholar] [CrossRef]
- Ishii, S.; Nakano, T.; Kawamura, K.; Kinoshita, S.; Ichikawa, S.; Fujita, T. Development of New Selective Ethylene Trimerization Catalysts Based on Highly Active Ethylene Polymerization Catalysts. Catal. Today 2018, 303, 263–270. [Google Scholar] [CrossRef]
- Parfenova, L.V.; Kovyazin, P.V.; Bikmeeva, A.K. Bimetallic Zr, Zr–Hydride Complexes in Zirconocene Catalyzed Alkene Dimerization. Molecules 2020, 25, 2216. [Google Scholar] [CrossRef]
- Guo, J.; Chen, Q.; Zhang, W.; Liang, T.; Sun, W.-H. The Benzhydryl-modified 2-Imino-1,10-Phenanthryliron Precatalyst in Ethylene Oligomerization. J. Organomet. Chem. 2021, 936, 121713. [Google Scholar] [CrossRef]
- Goetjen, T.A.; Zhang, X.; Liu, J.; Hupp, J.T.; Farha, O.K. Metal–Organic Framework Supported Single Site Chromium(III) Catalyst for Ethylene Oligomerization at Low Pressure and Temperature. ACS Sustainable Chem. Eng 2019, 7, 2553–2557. [Google Scholar] [CrossRef]
- Katayama, H.; Yari, H.; Tanaka, M.; Ozawa, F. (Z)-Selective Cross-Dimerization of Arylacetylenes with Silylacetylenes Catalyzed by Vinylidenruthenium Complexes. Chem. Commun. 2005, 4336–4338. [Google Scholar] [CrossRef]
- Xu, H.-D.; Zhang, R.-W.; Li, X.; Huang, S.; Tang, W.; Hu, W.-H. Rhodium-Catalyzed Chemo- and Regioselective Cross-Dimerization of Two Terminal Alkynes. Org. Lett. 2013, 15, 840–843. [Google Scholar] [CrossRef] [PubMed]
- Hirano, M.; Komiya, S. Oxidative Coupling Reactions at Rutheium(0) and Their Applications to Catalytic Homo- and Cross-Dimerizations. Coord. Chem. Rev. 2016, 314, 182–200. [Google Scholar] [CrossRef]
- Kiyota, S.; In, S.; Komine, N.; Hirano, M. Regioselectivity Control by Added MeCN in Ru(0)-catalyzed Cross-dimerization of Internal Alkynes with Methyl Methacrylate. Chem. Lett. 2017, 46, 1040–1043. [Google Scholar] [CrossRef]
- Ueda, Y.; Tsurugi, H.; Mashima, K. Cobalt–Catalyzed E-Selective Cross–Dimerization of Terminal Alkynes: A Mechanism Involving Cobalt (0/II) Redox Cycles. Angew. Chem. Int. Ed. 2020, 59, 1552–1556. [Google Scholar] [CrossRef] [PubMed]
- Brookhart, M.S.; Hauptman, E.M. Cross-Dimerization of Olefins. U.S. Patent US5892101A, 6 April 1999. [Google Scholar]
- Nomura, N.; Jin, J.; Park, H.; RajanBabu, T.V. The Hydrovinylation Reaction: A New Highly Selective Protocol Amenable to Asymmetric Catalysis. J. Am. Chem. Soc. 1998, 120, 459–460. [Google Scholar] [CrossRef]
- RajanBabu, T.V.; Nomura, N.; Jin, J.; Nandi, M.; Park, H.; Sun, X. Heterodimerization of Olefins. 1. Hydrovinylation Reactions of Olefins That Are Amenable to Asymmetric Catalysis. J. Org. Chem. 2003, 68, 8431–8446. [Google Scholar] [CrossRef]
- RajanBabu, T.V. Asymmetric Hydrovinylation Reaction. Chem. Rev. 2003, 103, 2845–2860. [Google Scholar] [CrossRef]
- Saha, B.; RajanBabu, T.V. Syntheses and Applications of 2-Phosphino-2′-alkoxy-1,1′-binaphthyl Ligands. Development of a Working Model for Asymmetric Induction in Hydrovinylation Reactions. J. Org. Chem. 2007, 72, 2357–2363. [Google Scholar] [CrossRef]
- Fassina, V.; Ramminger, C.; Seferin, M.; Monteiro, A.L. Nickel Catalyzed Hydrovinylation of Arylethylenes: General Method of Synthesis of α-Arylpropionic Acids Intermediates. Tetrahedron 2000, 56, 7403–7409. [Google Scholar] [CrossRef]
- Yi, C.S.; He, Z.; Lee, D.W. Hydrovinylation of Alkenes Catalyzed by the Ruthenium–Hydride Complex Formed in Situ from (PCy3)2(CO)RuHCl and HBF4·OEt2. Organometallics 2001, 20, 802–804. [Google Scholar] [CrossRef]
- Sanchez, R.P., Jr.; Connell, B.T. A Ruthenium-Based Catalyst System for Hydrovinylation at Room Temperature. Organometallics 2008, 27, 2902–2904. [Google Scholar] [CrossRef]
- Kondo, T.; Takagi, D.; Tsujita, H.; Ura, Y.; Wada, K.; Mitsudo, T. Highly Selective Dimerization of Styrenes and Linear Co-dimerization of Styrenes with Ethylene Catalyzed by a Ruthenium Complex. Angew. Chem. Int. Ed. 2007, 46, 5958–5961. [Google Scholar] [CrossRef]
- Gooßen, L.J.; Rodríguez, N. Heterodimerization of Olefins: A Highly Promising Strategy for the Selective Synthesis of Functionalized Alkenes. Angew. Chem. Int. Ed. 2007, 46, 7544–7546. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, D.; Takada, H.; Yamazaki, K.; Osakada, K. Hydrovinylation of Olefins Catalyzed by RuCl2(MeCN)2(cod)/Organoaluminum System. Trans. Mat. Res. Soc. Jpn. 2019, 44, 137–141. [Google Scholar] [CrossRef]
- Yamamoto, Y.; Ohkoshi, N.; Kameda, M.; Itoh, K. Ruthenium–Catalyzed Highly Efficient Intramolecular Olefin Coupling of α,ω-Dienes. Facile and Regioselective Synthesis of exo-Methylenecyclopentanes. J. Org. Chem. 1999, 64, 2178–2179. [Google Scholar] [CrossRef]
- Yamamoto, Y.; Nakag, Y.; Ohkoshi, N.; Itoh, K. Ruthenium(II)–Catalyzed Isomer–Selective Cyclization of 1,6-Dienes Leading to exo-Methylenecyclopentanes: Unprecedented Cycloisomerization Mechanism Involving Ruthenacyclopentane(hydrido) Intermediate. J. Am. Chem. Soc. 2001, 123, 6372–6380. [Google Scholar] [CrossRef] [PubMed]
- Widenhoefer, R.A.; Perch, N.S. Silane–Promoted Cycloisomerization of Functionalized 1,6-Dienes Catalyzed by a Cationic (π-Allyl)palladium Complex. Org. Lett. 1999, 1, 1103–1105. [Google Scholar] [CrossRef]
- Kisanga, P.; Goj, L.A.; Widenhoefer, R.A. Cycloisomerization of Functionalized 1,5- and 1,6-Dienes Catalyzed by Cationic Palladium Phenanthroline Complexes. J. Org. Chem. 2001, 66, 635–637. [Google Scholar] [CrossRef] [PubMed]
- Deckers, P.J.W.; Hessen, B.; Teuben, J.H. Catalytic Trimerization of Ethene with Highly Active Cyclopentadienyl-Arene Titanium Catalysts. Organometallics 2002, 21, 5122–5135. [Google Scholar] [CrossRef]
- Johnson, L.K.; Killian, C.M.; Brookhart, M. New Pd(II)- and Ni(II)-Based Catalyst for Polymerization of Ethylene and α-Olefins. J. Am. Chem. Soc. 1995, 117, 6414–6415. [Google Scholar] [CrossRef]
- Killian, C.M.; Johnson, L.K.; Brookhart, M. Preparation of Linear α-Olefins Using Cationic Nickel(II) α-Diimine Catalysts. Organometallics 1997, 16, 2005–2007. [Google Scholar] [CrossRef]
- Svejda, S.A.; Brookhart, M. Ethylene Oligomerization and Propylene Dimerization Using Cationic (α-Diimine)nickel(II) Catalysts. Organometallics 1999, 18, 65–74. [Google Scholar] [CrossRef]
- Schmid, M.; Eberhardt, R.; Klinga, M.; Leskelä, M.; Rieger, B. New C2v− and Chiral C2− Symmetric Olefin Polymerization Catalysts Based on Nickel(II) and Pallaium(II) Diimine Complexes Bearing 2,6-Diphenyl Aniline Moieties: Synthesis, Structural Characterization, and First Insight into Polymerization Properties. Organometallics 2001, 20, 2321–2330. [Google Scholar] [CrossRef]
- Camacho, D.H.; Guan, Z. Living Polymerization of α-Olefins at Elevated Temperatures Catalyzed by a Highly Active and Robust Cyclophane-Based Nickel Catalyst. Macromolecules 2005, 38, 2544–2546. [Google Scholar] [CrossRef]
- Cherian, A.E.; Rose, J.M.; Lobkovsky, E.B.; Coates, G.W. A C2−Symmetric, Living α-Diimine Ni(II) Catalyst: Regioblock Copolymers from Propylene. J. Am. Chem. Soc. 2005, 127, 13770–13771. [Google Scholar] [CrossRef]
- Meinhard, D.; Wegner, M.; Kipiani, G.; Hearley, A.; Reuter, P.; Fischer, S.; Marti, O.; Rieger, B. New Nickel(II) Diimine Complexes and the Control of Polyethylene Microstructure by Catalyst Design. J. Am. Chem. Soc. 2007, 129, 9182–9191. [Google Scholar] [CrossRef] [PubMed]
- Meinhard, D.; Rieger, B. Novel Unsymmetric α-Diimine Nickel(II) Complexes: Suitable Catalysts for Copolymerization Reactions. Chem. Asian J. 2007, 2, 386–392. [Google Scholar] [CrossRef] [PubMed]
- Anselment, T.M.J.; Vagin, S.I.; Rieger, B. Activation of Late Transition Metal Catalysts for Olefin Polymerizations and Olefin/CO Copolymerization. Dalton Trans. 2008, 34, 4537–4548. [Google Scholar] [CrossRef]
- Popeney, C.S.; Rheingold, A.L.; Guan, Z. Nickel(II) and Palladium(II) Polymerization Catalysts Bearing a Fuorinated Cyclophane Ligand: Stabilization of the Reactive Intermediate. Organometallics 2009, 28, 4452–4463. [Google Scholar] [CrossRef]
- Camacho, D.H.; Guan, Z. Designing Late-transition Metal Catalysts for Olefin Insertion Polymerization and Copolymerization. Chem. Commun. 2010, 46, 7879–7893. [Google Scholar] [CrossRef] [PubMed]
- Okada, T.; Takeuchi, D.; Shishido, A.; Ikeda, T.; Osakada, K. Isomerization Polymerization of 4-Alkylcyclopentane Catalyzed by Pd Complexes: Hydrocarbon Polymers with Isotactic−Type Stereochemistry and Liquid−Crystalline Properties. J. Am. Chem. Soc. 2009, 131, 10852–10853. [Google Scholar] [CrossRef]
- Allen, K.E.; Campos, J.; Daugulis, O.; Brookhart, M. Living Polymerization of Ethylene and Copolymerization of Ethylene/Methyl Acrylate Using “Sandwich” Diimine Palladium Catalysts. ACS Catal. 2015, 5, 456–464. [Google Scholar] [CrossRef]
- Chen, Z.; Liu, W.; Daugulis, O.; Brookhart, M. Mechanistic Studies of Pd(II)-Catalyzed Copolymerization of Ethylene and Vinylalkoxysilanes: Evidence for a β-Silyl Elimination Chain Transfer Mechanism. J. Am. Chem. Soc. 2016, 138, 16120–16129. [Google Scholar] [CrossRef] [PubMed]
- Takano, S.; Takeuchi, D.; Osakada, K. Olefin Polymerization Catalyzed by Double-Decker Dipalladium Complexes: Low Branched Poly(α-Olefin)s by Selective Insertion of the Monomer Molecules. Chem. Eur. J. 2015, 21, 16209–16218. [Google Scholar] [CrossRef] [PubMed]
- Mahmood, Q.; Zeng, Y.; Wang, X.; Sun, Y.; Sun, W.-H. Advancing Polyethylene Properties by Incorporating NO2 Moiety in 1,2-Bis(arylimino)acenaphthynickel Precatalysts: Synthesis, Characterization and Ethylene Polymerization. Dalton Trans. 2017, 46, 6934–6947. [Google Scholar] [CrossRef] [PubMed]
- Belokon, Y.; Moscalenko, M.; Ikonnikov, N.; Yashkina, L.; Antonov, D.; Vorontsov, E.; Rozenberg, V. Asymmetric Trimethylsilylcyanation of Benzaldehyde Catalyzed by (salen)Ti(IV) Complexes Derived from (R)- and/or (S)-4-Hydroxy-5-formyl[2.2]paracyclophane and Diamines. Tetrahedron Asymmetry 1997, 8, 3245–3250. [Google Scholar] [CrossRef]
- Duan, W.; Ma, Y.; Xia, H.; Liu, X.; Ma, Q.; Sun, J. Design and Synthesis of Planar Chiral Heterocyclic Carbene Precursors Derived from [2.2]Paracyclophane. J. Org. Chem. 2008, 73, 4330–4333. [Google Scholar] [CrossRef]
- Ma, Q.; Ma, Y.; Liu, X.; Duan, W.; Qu, B.; Song, C. Planar Chiral Imidazolium Salts Based on [2.2]Paracyclophane in the Asymmetric Rhodium-catalyzed 1,2-Addition of Arylboronic Acids to Aldehydes. Tetrahedron Asymmetry 2010, 21, 292–298. [Google Scholar] [CrossRef]
- Göker, V.; Kohl, S.R.; Rominger, F.; Meyer-Eppler, G.; Volbach, L.; Schnakenburg, G.; Lützen, A.; Hashmi, A.S.K. Chiral [2.2]Paracyclophane–Based NAC– and NHC–gold(I) Complexes. J. Organomet. Chem. 2015, 795, 45–52. [Google Scholar] [CrossRef]
- Crystal and Calculation Data. Crystal size (mm), 0.25 × 0.20 x0.10; Measurement –160 °C, MoKα; Formula, C46H40Br2Cl2N2Ni; Fw 910.25; Crystal System Monoclinic; Space Group Cc; Crystal Lattice a 11.585(4) (Å), b 19.475(6) (Å), c 18.169(5) (Å), β 96.154(5)°, V4076(2) (Å3); Z 4; F(000) 1848.00; dcalcd 1.483 g cm−3; total Independent Reflections, 8793, Reflections>2σ, 6019; Parameters 537; GOF 1.019; R 0.0404, wR 0.0802. Available online: https://www.ccdc.cam.ac.uk/structures/.
- Janeta, M.; Heidlas, J.X.; Daugulis, O.; Brookhart, M. 2,4,6-Triphenylpyridinium: A Bulky, Highly Electron–Withdrawing Substituent That Enhances Properties of Nickel(II) Ethylene Polymerization Catalysts. Angew. Chem. Int. Ed. 2021, 60, 4566–4569. [Google Scholar] [CrossRef]
- Ernst, L.; Wittkowski, L. Diastereomers Composed of Two Planar-Chiral Subunits: Bis([2.2]paracyclophan-4-yl)methane and Analogues. Eur. J. Org. Chem. 1999, 1653–1663. [Google Scholar] [CrossRef]
- Reich, H.J.; Cram, D.J. Macro Rings. XXXVIII. Determination of Positions of Substituents in the [2.2]Paracyclophane Nucleus through Nuclear Magnetic Resonance Spectra. J. Am. Chem Soc. 1969, 91, 3534–3543. [Google Scholar] [CrossRef]
- Ricci, G.; Ruzziconi, R.; Giorgio, E. Atropisomeric (R,R)-2,2′-Bi([2]paracyclo[2](5,8)quinolinophane) and (R,R)-1,1′-Bi([2]paracyclo[2](5,8)isoquinolinophane): Synthesis, Structural Analysis, and Chiropical Properties. J. Org. Chem. 2005, 70, 1011–1018. [Google Scholar] [CrossRef] [PubMed]
- Marchand, A.; Maxwell, A.; Mootoo, B.; Pelter, A.; Reid, A. Oxazoline Mediated Routes to a Unique Amino-acid, 4-Amino-13-Carboxy[2.2]paracyclophane, of Planar Chirality. Tetrahedron 2000, 56, 7331–7338. [Google Scholar] [CrossRef]
- Hitchcock, P.B.; Rowlands, G.J.; Parmer, R. The Synthesis of Enantiomerically Pure 4-Substituted [2.2]Paracyclophane Derivatives by Sulfoxide-metal Exchange. Chem. Commun. 2005, 4219–4221. [Google Scholar] [CrossRef] [PubMed]
- Reich, H.J.; Yelm, K.E. Asymmetric Induction in the Oxidation of [2.2]Paracyclophane-Substituted Selenides. Application of Chirality Transfer in the Selenoxide [2,3] Sigmatropic Rearrangement. J. Org. Chem. 1991, 56, 5672–5679. [Google Scholar] [CrossRef]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A Complete Structure Solution, Refinement and Analysis Program. J. Appl. Cryst. 2009, 42, 339–341. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Catalyst | Conditions | Products/mmol | C4TOF/h−1 b | |||
|---|---|---|---|---|---|---|---|
| Temp/°C | Time/h | 1-Butene | 2-Butene | Hexenes | |||
| 1 | NiBr2(L1) | 10 | 3 | 0.32 | 0.11 | 0.00 | 29 |
| 2 | NiBr2(L1) | 10 | 6 | 0.85 | 0.90 | 0.00 | 58 |
| 3 | NiBr2(L1) | 25 | 1 | 0.15 | 0.05 | 0.01 | 40 |
| 4 | NiBr2(L1) | 25 | 3 | 0.67 | 0.63 | 0.10 | 86 |
| 5 | NiBr2(L1) | 25 | 6 | 0.98 | 2.78 | 0.75 | 126 |
| 6 | NiBr2(L1) | 25 | 8 | 0.99 | 3.75 | 1.10 | 124 |
| 7 | NiBr2(L2) | 25 | 1 | – c | – c | – c | – c |
| 8 | NiBr2(L1) | 50 | 3 | 0.12 | 0.18 | 0.00 | 20 |
| 9 | NiBr2(L1) | 50 | 6 | 0.20 | 0.34 | 0.00 | 18 |
| Entry | Co-Catalyst b | Conditions | Products (%) | C12,C18 TOF/h−1 | |||
|---|---|---|---|---|---|---|---|
| Temp/°C | Time/h | 2-Hexene | C12 | C18 | |||
| 1 | MAO (300) | 10 | 6 | 34 | 2.9 | 6.6 | 4.8 |
| 2 | MAO (300) | 10 | 24 | 79 | 9.5 | 8.6 | 2.3 |
| 3 | MAO (50) | 25 | 24 | 66 | 2.6 | 17 | 2.5 |
| 4 | MAO (300) | 25 | 6 | 56 | 7.3 | 13 | 10.2 |
| 5 | MAO (300) | 25 | 24 | 62 | 11 | 18 | 3.6 |
| 6 | MAO (1000) | 25 | 24 | 21 | 2.1 | 18 | 2.5 |
| 7 | MAO (300) | 35 | 0.5 | 33 | 4.6 | 0.0 | 28 |
| 8 | MAO (300) | 35 | 6 | 54 | 16 | 5.6 | 11 |
| 9 | MAO (300) | 35 | 24 | 45 | 22 | 28 | 6.3 |
| 10 | MAO (300) | 50 | 0.5 | 49 | 7.2 | 3.0 | 61 |
| 11 | MAO (300) | 50 | 6 | 40 | 11 | 34 | 23 |
| 12 | MAO (300) | 50 | 24 | 23 | 12 | 57 | 8.6 |
| 13 | MMAO (300) | 25 | 6 | 37 | 6.8 | 18 | 12 |
| 14 | AlMe3 (300) | 50 | 1 | 98 | 0.0 | 0.0 | 0.0 |
| 15 | Et2AlCl (300) | 50 | 1 | 95 | 0.0 | 0.0 | 0.0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Takeuchi, D.; Tojo, Y.-a.; Osakada, K. Synthesis of a Ni Complex Chelated by a [2.2]Paracyclophane-Functionalized Diimine Ligand and Its Catalytic Activity for Olefin Oligomerization. Molecules 2021, 26, 2719. https://doi.org/10.3390/molecules26092719
Takeuchi D, Tojo Y-a, Osakada K. Synthesis of a Ni Complex Chelated by a [2.2]Paracyclophane-Functionalized Diimine Ligand and Its Catalytic Activity for Olefin Oligomerization. Molecules. 2021; 26(9):2719. https://doi.org/10.3390/molecules26092719
Chicago/Turabian StyleTakeuchi, Daisuke, Yoshi-aki Tojo, and Kohtaro Osakada. 2021. "Synthesis of a Ni Complex Chelated by a [2.2]Paracyclophane-Functionalized Diimine Ligand and Its Catalytic Activity for Olefin Oligomerization" Molecules 26, no. 9: 2719. https://doi.org/10.3390/molecules26092719
APA StyleTakeuchi, D., Tojo, Y.-a., & Osakada, K. (2021). Synthesis of a Ni Complex Chelated by a [2.2]Paracyclophane-Functionalized Diimine Ligand and Its Catalytic Activity for Olefin Oligomerization. Molecules, 26(9), 2719. https://doi.org/10.3390/molecules26092719