
Recent Advances in Pain Management: Relevant Protein Kinases and Their Inhibitors



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Tyrosine Kinase (TK) Group
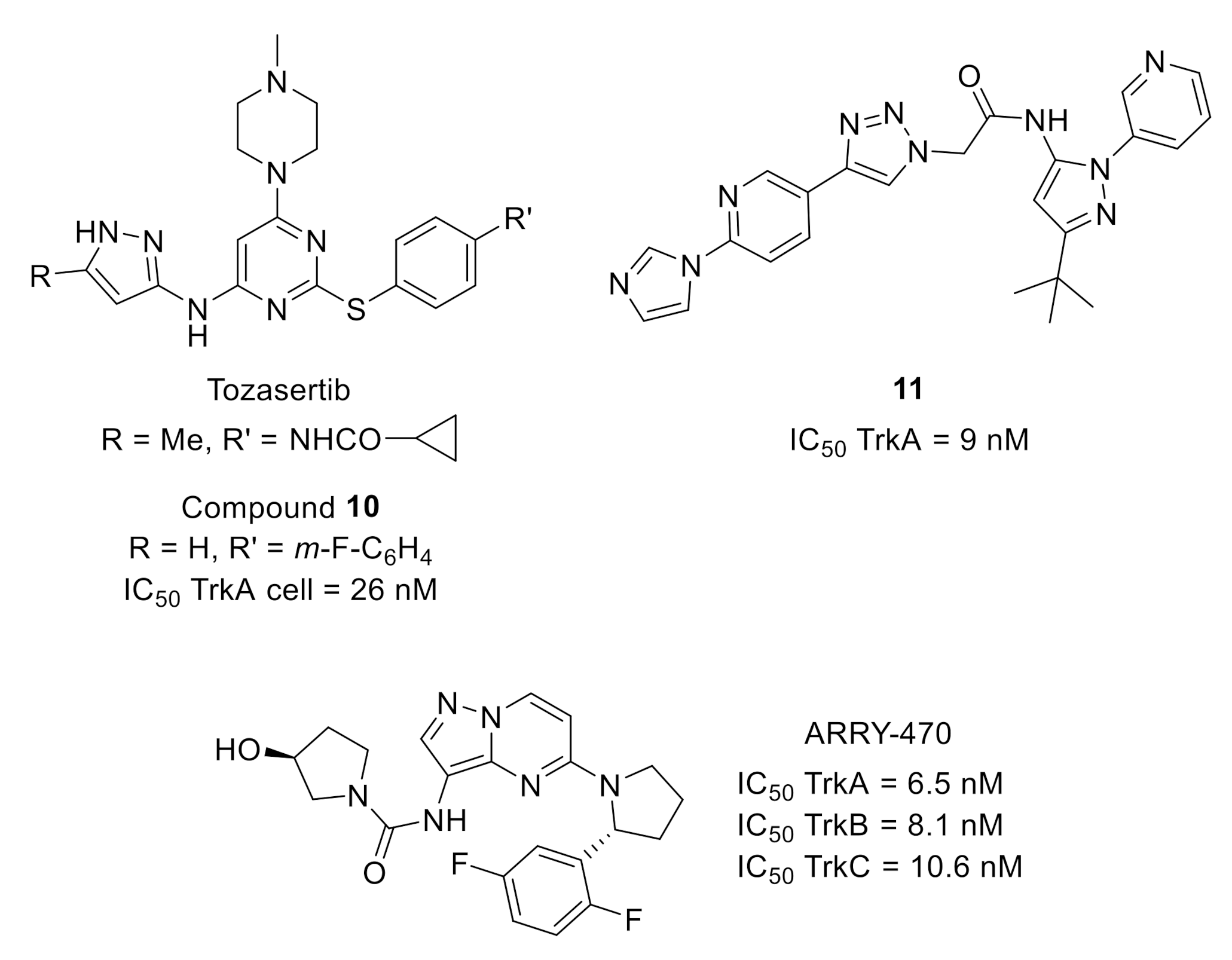
2.1. Tropomyosin-Related Kinases (Trks)
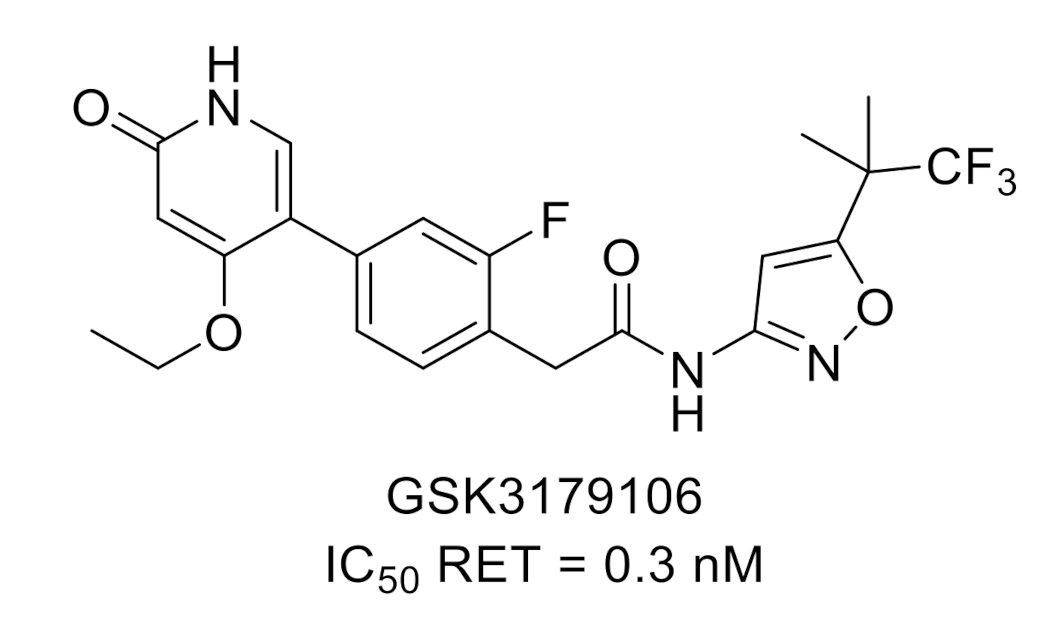
2.2. Rearranged During Transfection (RET)
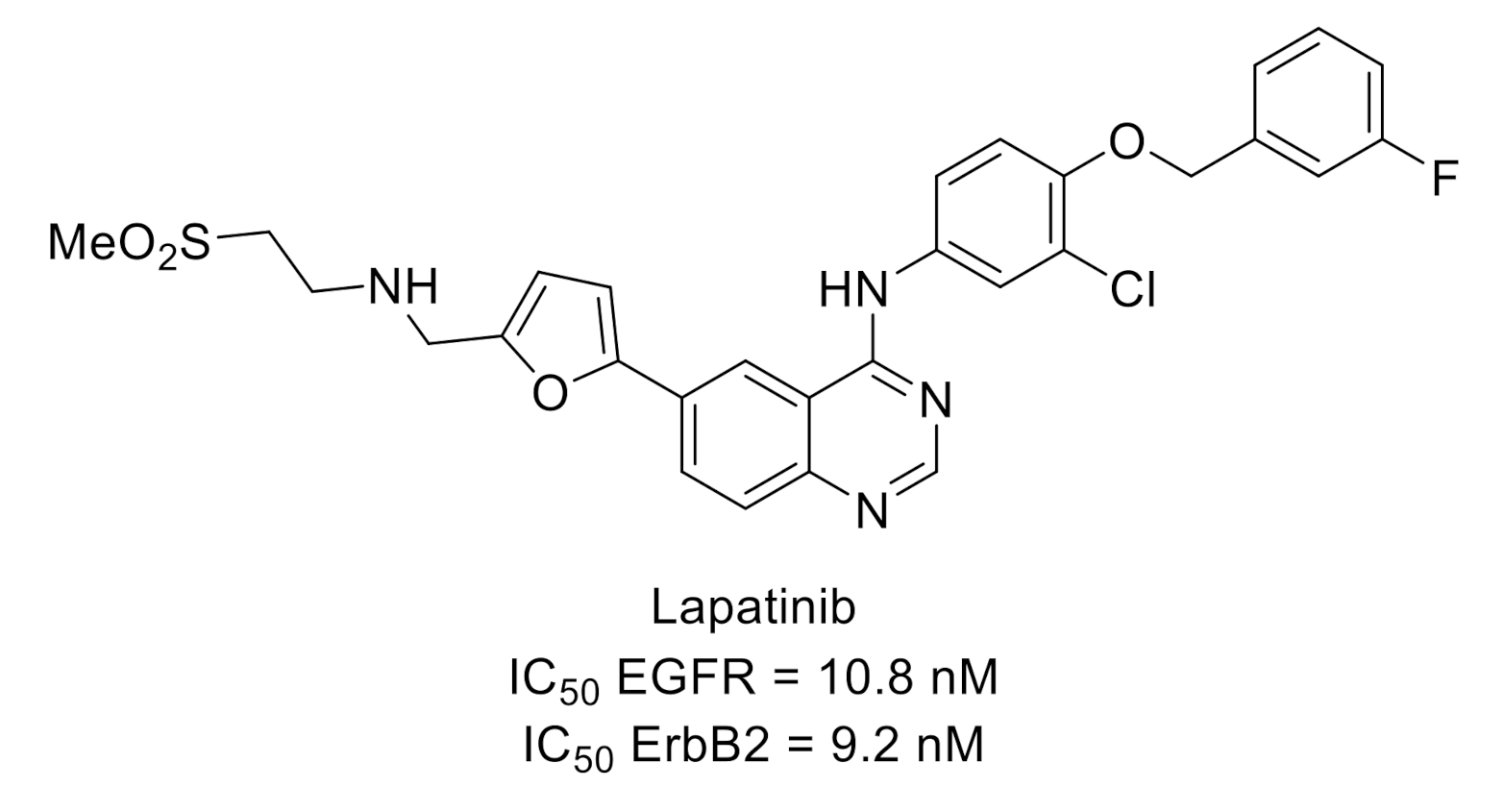
2.3. Epidermal Growth Factor Receptor (EGFR)
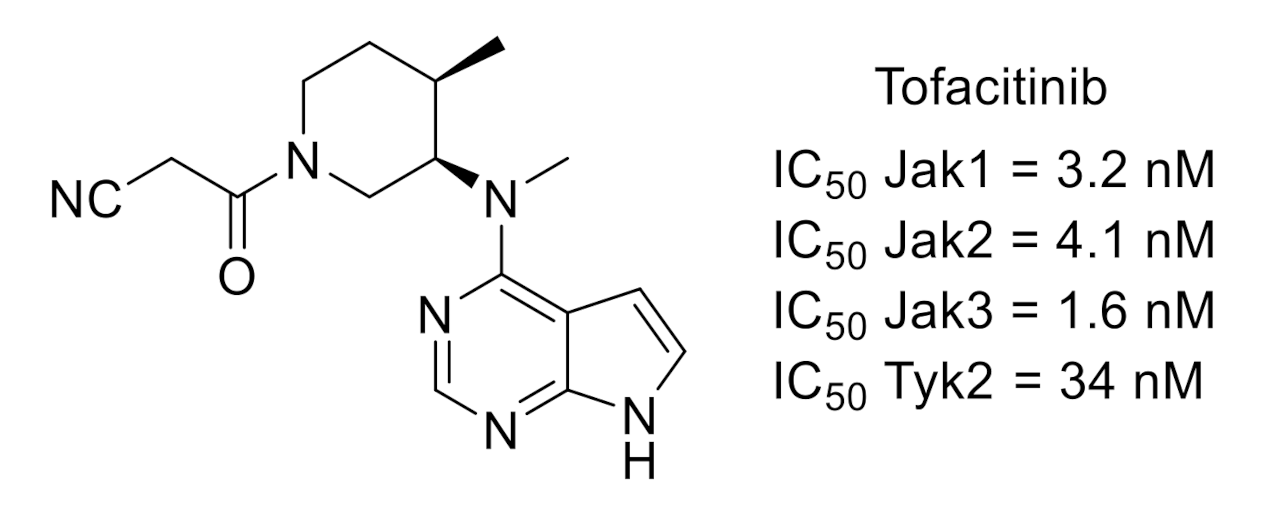
2.4. Janus Kinase (JAK)
2.5. Vascular Endothelial Growth Factor Receptor (VEGFR)
2.6. Src-Family Protein Tyrosine Kinases (SFKs) and Breakpoint Cluster Region (BCR)–Abelson (Abl) Fusion Protein
2.7. FMS-Like Tyrosine Kinase 3 Receptor (FLT3)
3. CMGC Group
3.1. p38 Mitogen-Activated Protein Kinases (p38 MAPKs)
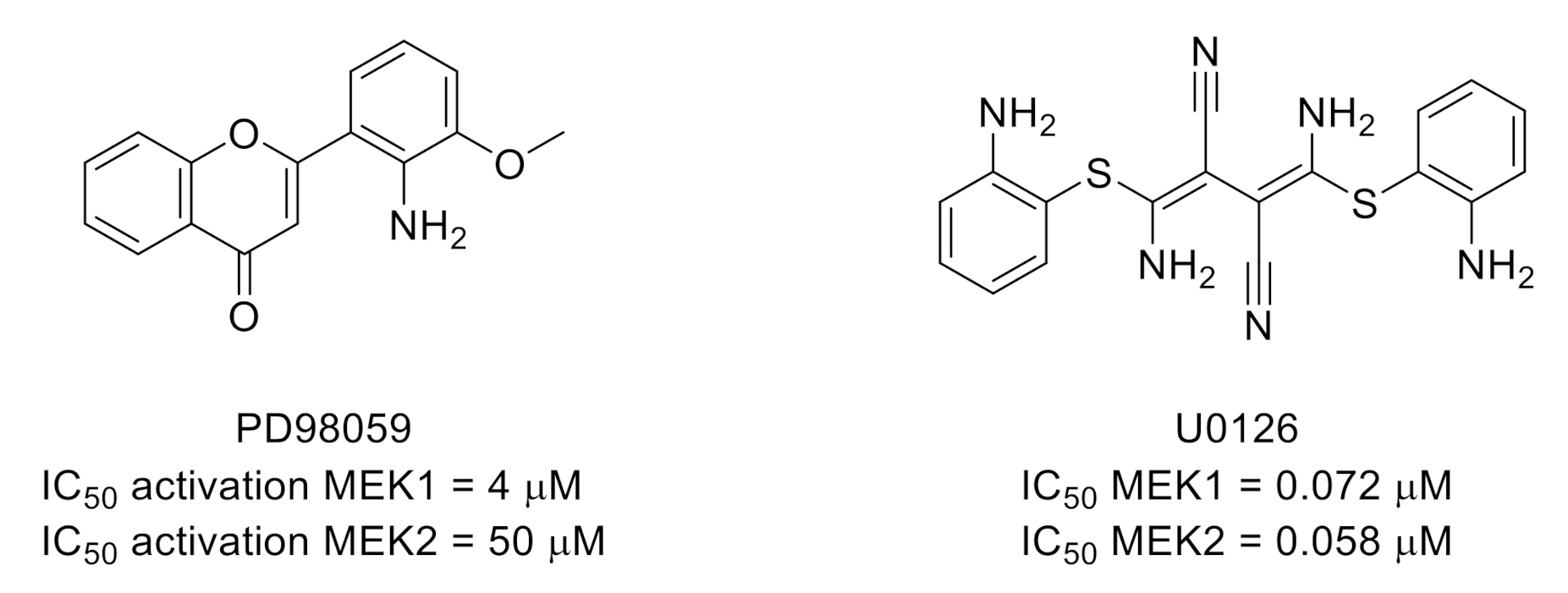
3.2. Mitogen-Activated Protein Kinase Kinases (MEKs)/Extracellular Signal-Regulated Kinases (ERKs)
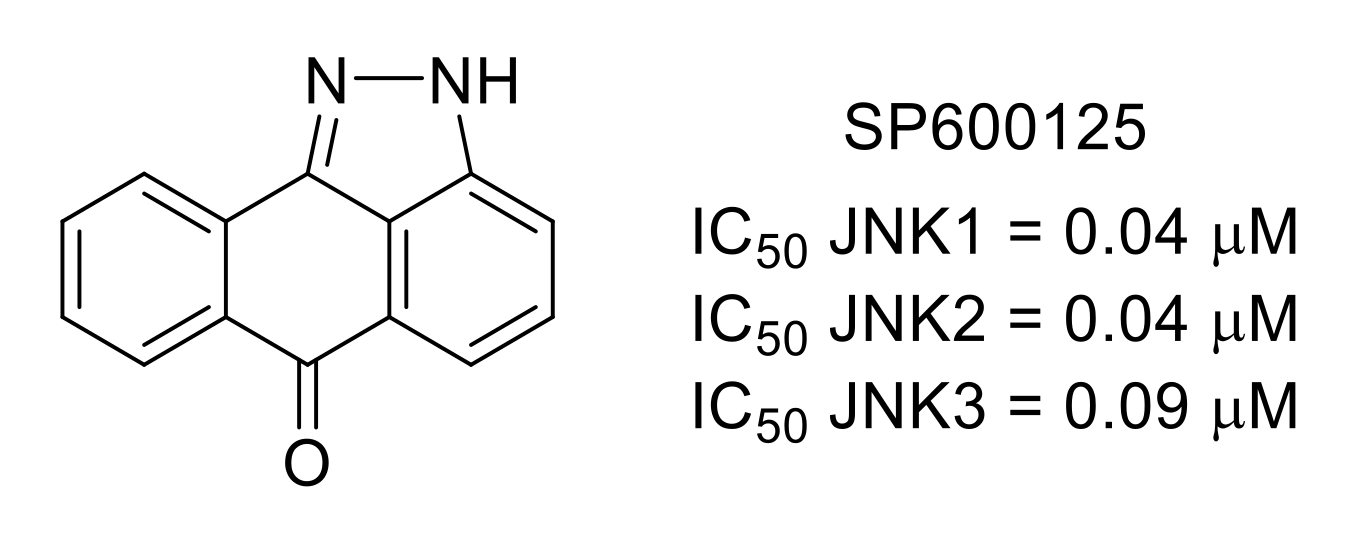
3.3. c-Jun N-Terminal Kinases (JNKs)
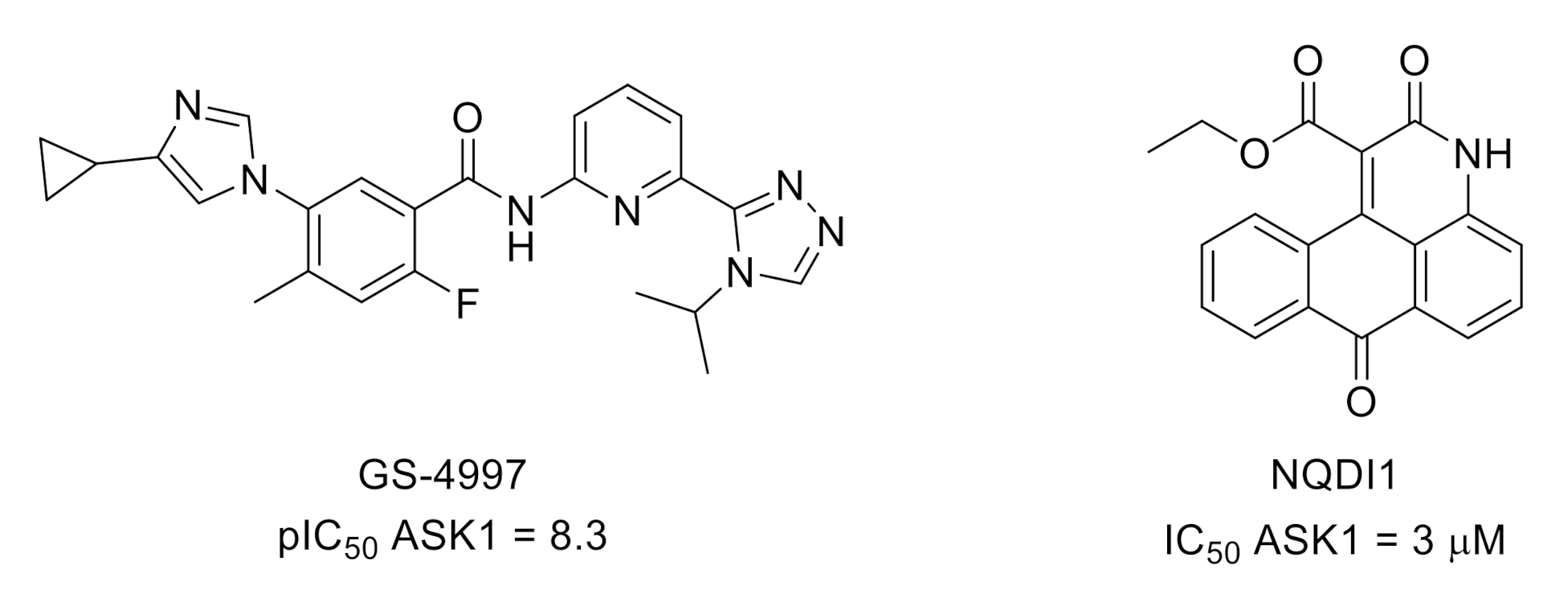
3.4. Apoptosis Signal-Regulating Kinase 1 (ASK1)
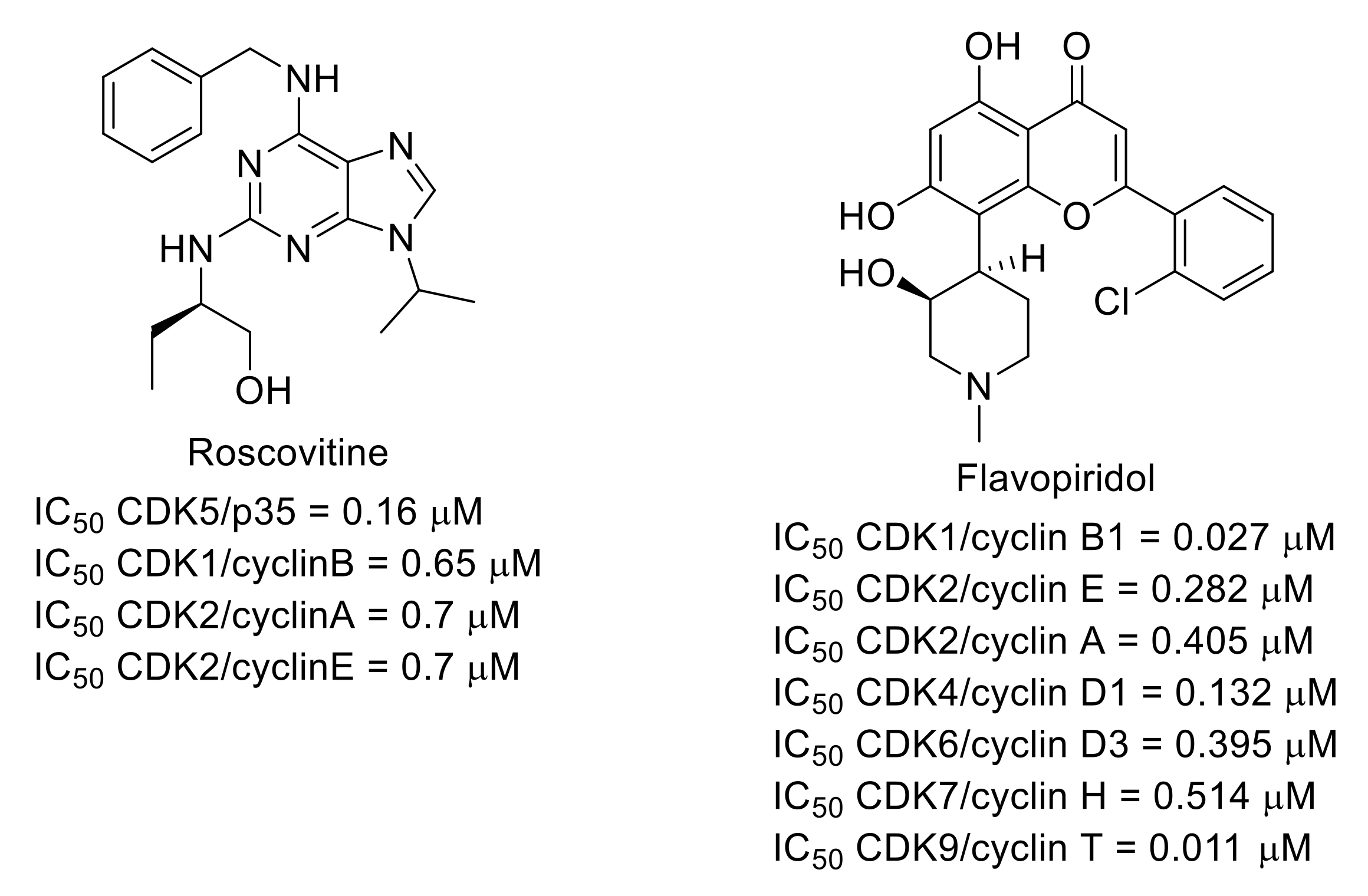
3.5. Cyclin-Dependent Kinases (CDKs)
3.6. CDC-Like Kinase 2 (CLK2)/Dual-Specificity Tyrosine Phosphorylation-Regulated Kinase 1A (DYRK1A)
3.7. Glycogen Synthase Kinase 3 (GSK3)
3.8. Casein Kinase 2 (CK2)
4. Protein Kinases A, G and C (AGC) Group
4.1. PKA, PKB, PKC, PKMζ and PKG
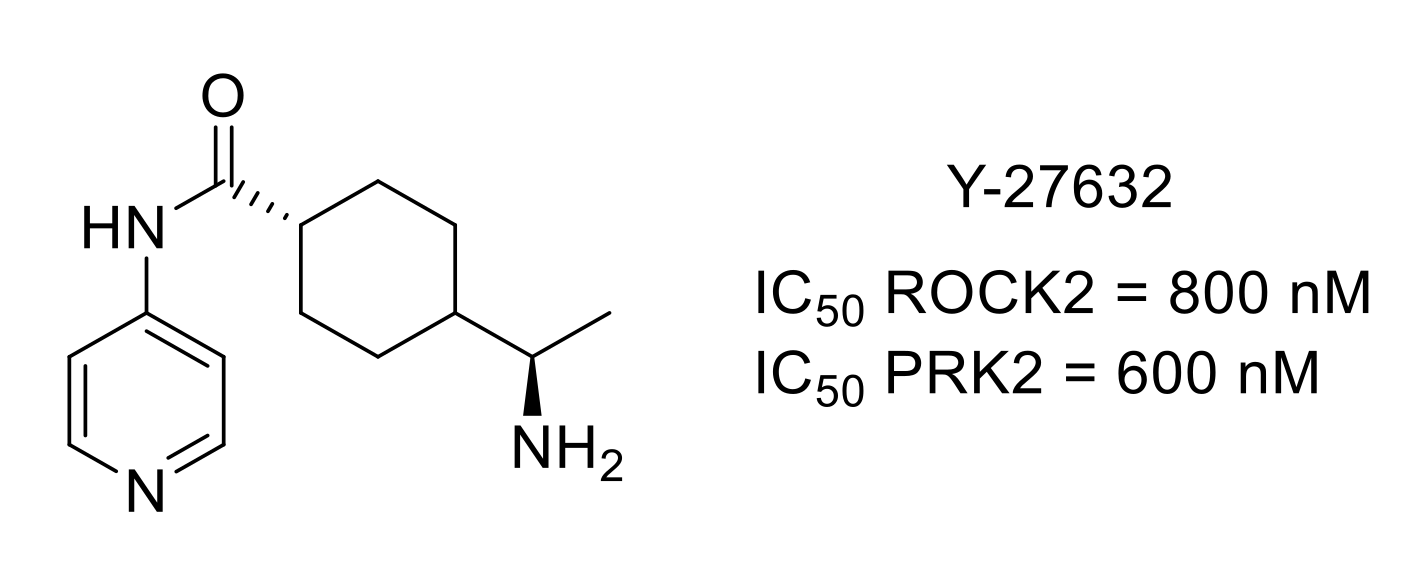
4.2. Rho-Associated Coiled-Coil-Containing Kinase (ROCK)
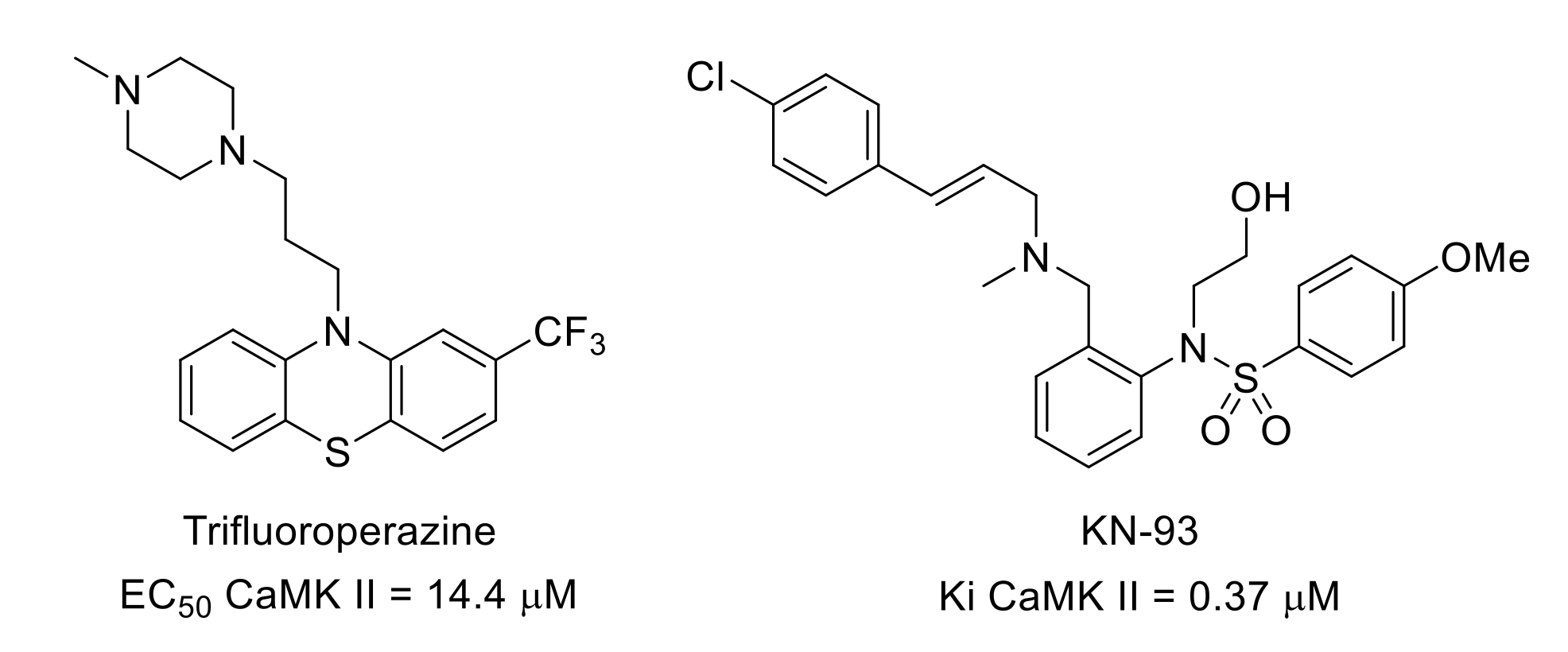
5. Ca2+/Calmodulin-Dependent Protein Kinase (CAMK) Group
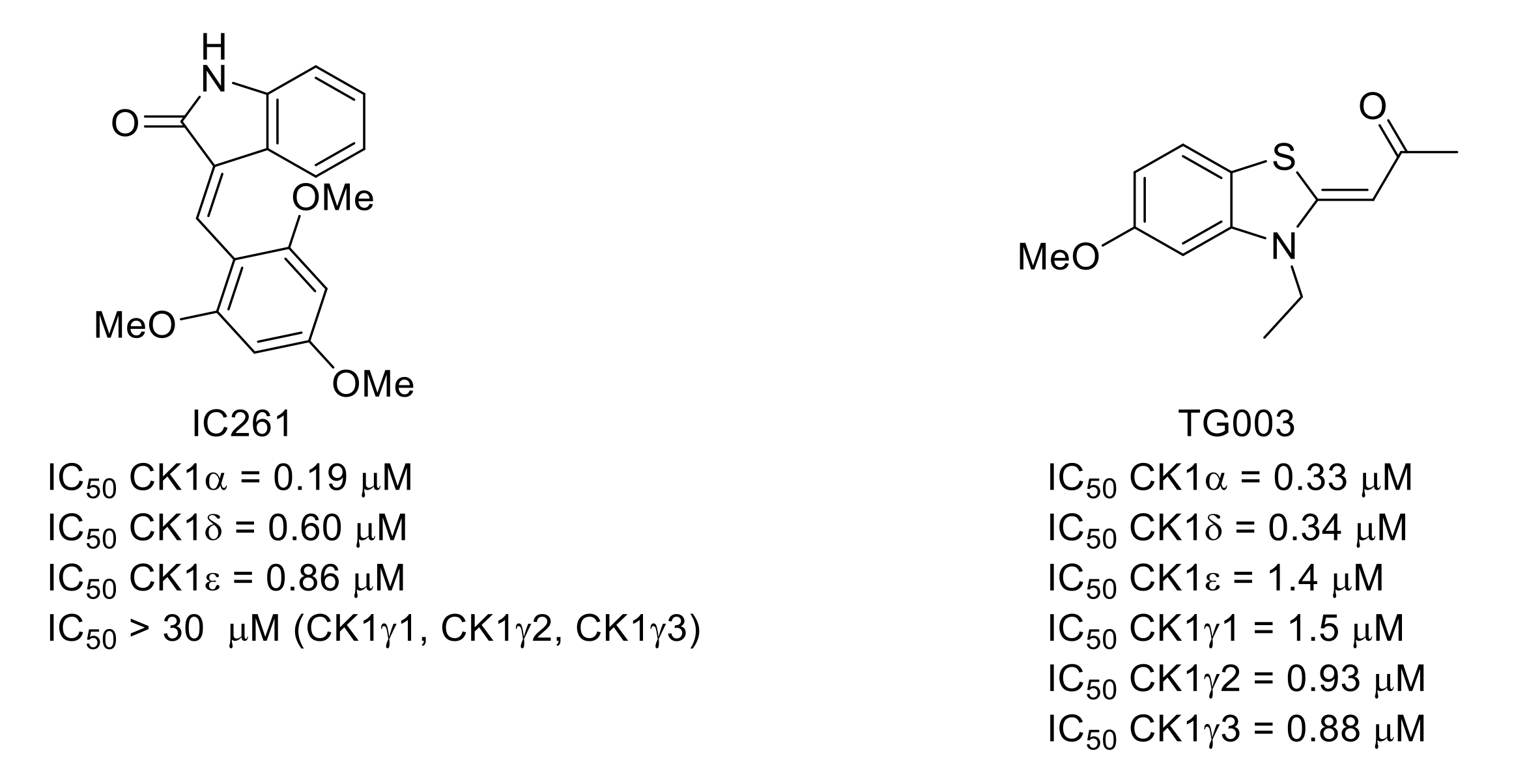
6. Casein Kinase 1 (CK) Group
7. Atypical and Other Protein Kinase Groups
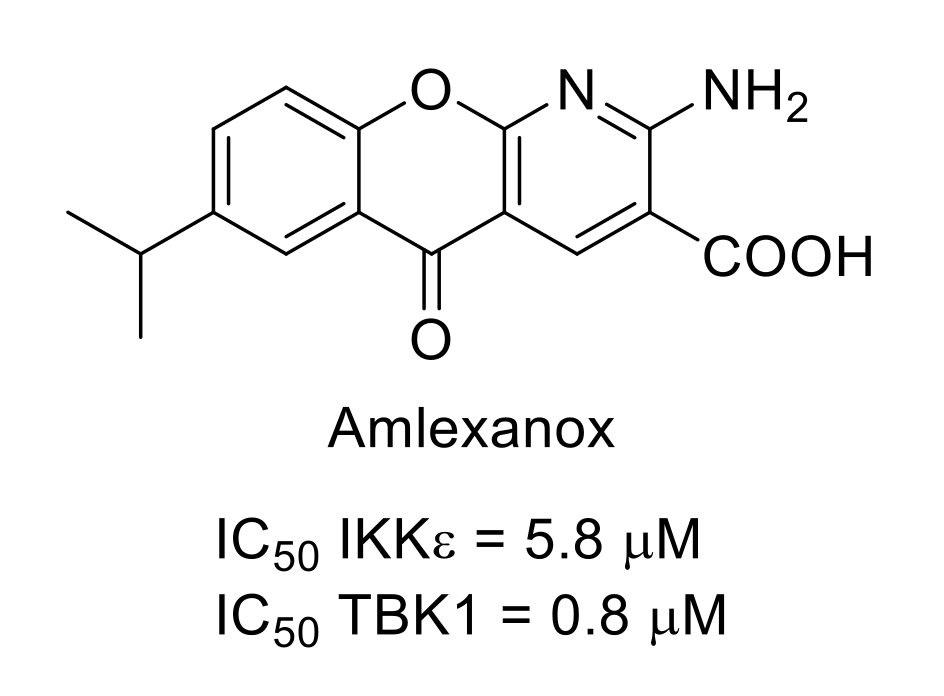
7.1. IκB Kinases (IKKs)
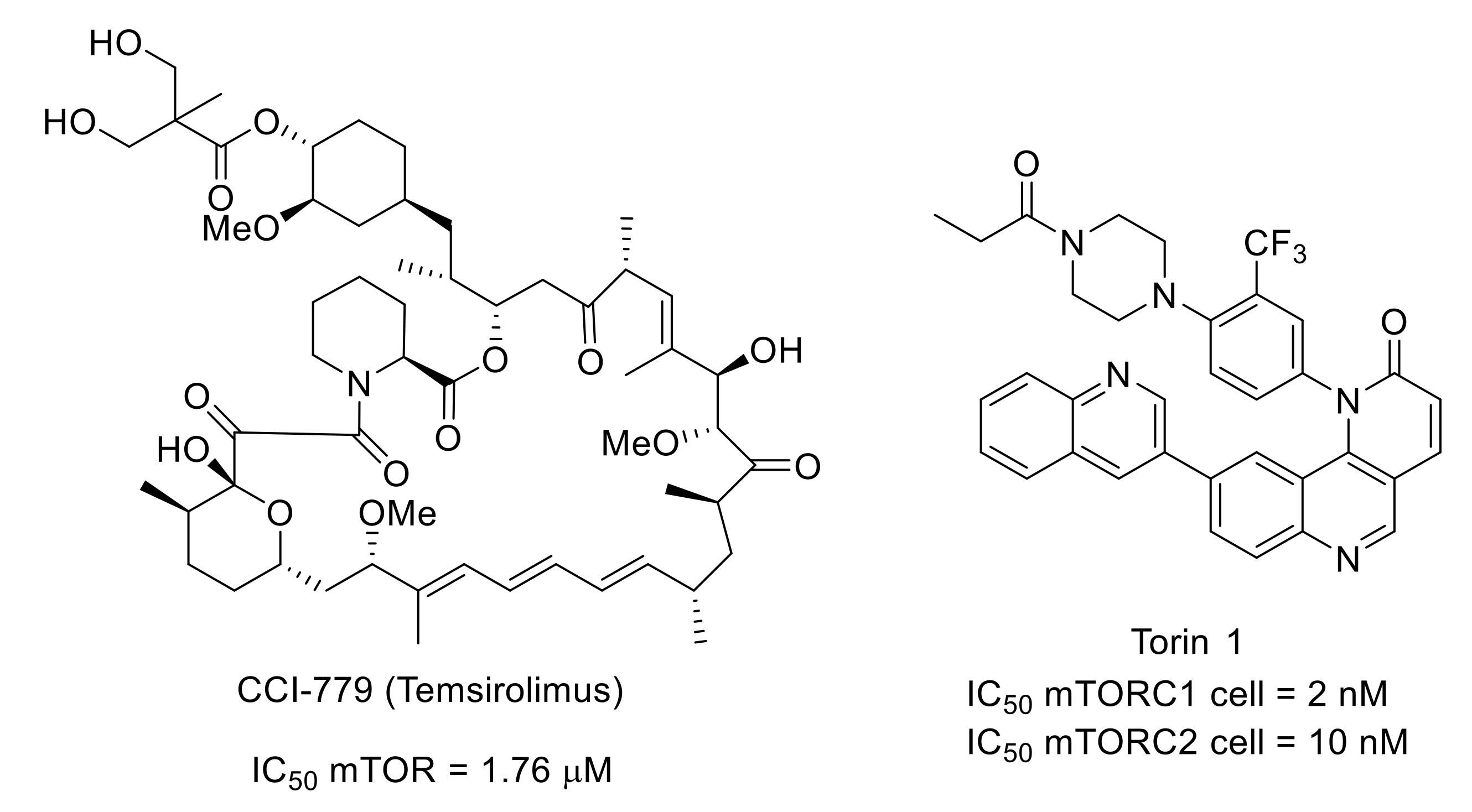
7.2. Mammalian Target of Rapamycin (mTOR)
8. Conclusions
Author Contributions
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Breivik, H.; Eisenberg, E.; O’Brien, T. The individual and societal burden of chronic pain in Europe: The case for strategic prioritisation and action to improve knowledge and availability of appropriate care. BMC Public Health 2013, 24, 1229. [Google Scholar] [CrossRef] [PubMed]
- Bán, E.G.; Brassai, A.; Vizi, E.S. The role of the endogenous neurotransmitters associated with neuropathic pain and in the opioid crisis: The innate pain-relieving system. Brain Res. Bull. 2020, 155, 129–136. [Google Scholar] [CrossRef] [PubMed]
- Micó, J.A.; Ardid, D.; Berrocoso, E.; Eschalier, A. Antidepressants and pain. Trends Pharmacol. Sci. 2006, 27, 348–354. [Google Scholar] [CrossRef] [PubMed]
- Ford, A.C.; Quigley, E.M.M.; Lacy, B.E.; Lembo, A.J.; Saito, Y.A.; Schiller, L.R.; Soffer, E.E.; Spiegel, B.M.R.; Moayyedi, P. Effect of antidepressants and psychological therapies, including hypnotherapy, in irritable bowel syndrome: Systematic review and meta-analysis. Am. J. Gastroenterol. 2014, 109, 1350–1365. [Google Scholar] [CrossRef]
- Jackson, J.L.; Shimeall, W.; Sessums, L.; DeZee, K.J.; Becher, D.; Diemer, M.; Berbano, E.; O’Malley, P.G. Tricyclic antidepressants and headaches: Systematic review and meta-analysis. Brit. Med. J. 2010, 341, c5222. [Google Scholar] [CrossRef]
- Xie, Z.; Yang, X.; Duan, Y.; Han, J.; Liao, C. Small-molecule kinase inhibitors for the treatment of nononcologic diseases. J. Med. Chem. 2021, 64, 1283–1345. [Google Scholar] [CrossRef]
- Indiana, M.; De Souza, F.H.V.; Eduardo, J.; Nascimento, P.G.B.D. Protein kinases and pain. In Protein Kinases; IntechOpen: London, UK, 2012. [Google Scholar] [CrossRef]
- Yousuf, M.S.; Shiers, S.I.; Sahn, J.J.; Price, T.J. Pharmacological manipulation of translation as a therapeutic target for chronic pain. Pharmacol. Rev. 2021, 73, 59–88. [Google Scholar] [CrossRef]
- Mantyh, P.W.; Koltzenburg, M.; Mendell, L.M.; Tive, L.; Sheldon, D.L. Antagonism of nerve growth factor-TrkA signaling and the relief of pain. Anesthesiology 2011, 115, 189–204. [Google Scholar] [CrossRef]
- Cazorla, M.; Jouvenceau, A.; Rose, C.; Guilloux, J.-P.; Pilon, C.; Dranovsky, A.; Prémont, J. Cyclotraxin-B, the first highly potent and selective TrkB inhibitor, has anxiolytic properties in mice. PLoS ONE 2010, 5, e9777. [Google Scholar] [CrossRef]
- Wang, X.; Zhang, L.; Zhan, Y.; Li, D.; Zhang, Y.; Wang, G.; Zhang, M. Contribution of BDNF/TrkB signalling in the rACC to the development of pain-related aversion via activation of ERK in rats with spared nerve injury. Brain Res. 2017, 1671, 111–120. [Google Scholar] [CrossRef]
- Shirahashi, H.; Toriihaa, E.; Suenaga, Y.; Yoshida, H.; Akaogi, K.; Endou, Y.; Wakabayashi, M.; Takashima, M. The discovery of novel 3-aryl-indazole derivatives as peripherally restricted pan-Trk inhibitors for the treatment of pain. Bioorg. Med. Chem. Lett. 2019, 29, 2320–2326. [Google Scholar] [CrossRef]
- Park, H.; Chi, O.; Kim, J.; Hong, S. Identification of novel inhibitors of tropomyosin-related kinase A through the structure-based virtual screening with homology-modeled protein structure. J. Chem. Inf. Model. 2011, 51, 2986–2993. [Google Scholar] [CrossRef]
- Skerratt, S.E.; Andrews, M.; Bagal, S.K.; Bilsland, J.; Brown, D.; Bungay, P.J.; Cole, S.; Gibson, K.R.; Jones, R.; Morao, I.; et al. The discovery of a potent, selective, and peripherally restricted pan-Trk inhibibitor (PF-06273340) for the treatment of pain. J. Med. Chem. 2016, 59, 10084–10099. [Google Scholar] [CrossRef]
- Wan, S.; Bhati, A.P.; Skeratt, S.; Omoto, K.; Shanmugasundaram, V.; Bagal, S.K.; Coveney, P.V. Evaluation and characterization of Trk kinase inhibitors for the treatment of pain: Reliable binding affinity predictions from theory and computation. J. Chem. Inf. Model. 2017, 57, 897–909. [Google Scholar] [CrossRef]
- Bagal, S.K.; Andrews, M.; Bechle, B.M.; Bian, J.; Bilsland, J.; Blakemore, D.C.; Braganza, J.F.; Bungay, P.J.; Corbett, M.S.; Cronin, C.N.; et al. Discovery of potent, selective, and peripherally restricted pan-Trk kinase inhibitors for the treatment of pain. J. Med. Chem. 2018, 61, 6779–6800. [Google Scholar] [CrossRef]
- Bagal, S.K.; Omoto, K.; Blakemore, D.C.; Bungay, P.J.; Bilsland, J.G.; Clarke, P.J.; Corbett, M.S.; Cronin, C.N.; Cui, J.J.; Dias, R.; et al. Discovery of allosteric, potent, subtype selective, and peripherally restricted TrkA kinase inhibitors. J. Med. Chem. 2019, 62, 247–265. [Google Scholar] [CrossRef]
- Su, H.-P.; Rickert, K.; Burlein, C.; Narayan, K.; Bukhtiyarova, M.; Hurzy, D.M.; Stump, C.A.; Zhang, X.; Reid, J.; Krasowska-Zoladek, A.; et al. Structural characterization of nonactive site, TrkA-selective kinase inhibitors. Proc. Natl. Acad. Sci. USA 2017, 114, E297–E306. [Google Scholar] [CrossRef]
- Hurzy, D.M.; Henze, D.A.; Cabalu, T.D.; Narayan, K.; Heller, A.; Cooke, A.J. Design, synthesis and SAR of substituted indoles as selective TrkA inhibitors. Bioorg. Med. Chem. Lett. 2017, 27, 2695–2701. [Google Scholar] [CrossRef]
- Turk, S.; Merget, B.; Eid, S.; Fulle, S. From cancer to pain target by automated selectivity inversion of a clinical candidate. J. Med. Chem. 2018, 61, 4851–4859. [Google Scholar] [CrossRef]
- Stachel, S.J.; Sanders, J.M.; Henze, D.A.; Rudd, M.T.; Su, H.-P.; Li, Y.; Nanda, K.K.; Egbertson, M.S.; Manley, P.J.; Jones, K.L.G.; et al. Maximizing diversity from a kinase screen: Identification of novel and selective pan-Trk inhibitors for chronic pain. J. Med. Chem. 2014, 57, 5800–5816. [Google Scholar] [CrossRef]
- Ghilardi, J.R.; Freeman, K.T.; Jimenez-Andrade, J.M.; Mantyh, W.G.; Bloom, A.P.; Kuskowski, M.A.; Mantyh, P.W. Administration of a tropomyosin receptor kinase inhibitor attenuates sarcoma-induced nerve sprouting, neuroma formation and bone cancer pain. Mol. Pain 2010, 6, 87. [Google Scholar] [CrossRef]
- Yoon, H.; Shin, I.; Nam, Y.; Kim, N.D.; Lee, K.-B.; Sim, T. Identification of a novel 5-amino-3-(5-cyclopropylisoxazol-3-yl)-1-isopropyl-1H-pyrazole-4-carboxamide as a specific RET kinase inhibitor. Eur. J. Med. Chem. 2017, 125, 1145–1155. [Google Scholar] [CrossRef]
- Yang, J.; Chen, K.; Zhang, G.; Yang, Q.-Y.; Li, Y.-S.; Huang, S.-Z.; Wang, Y.-L.; Yang, W.; Jiang, X.-J.; Yan, H.-X.; et al. Structural optimization and structure-activity relationship studies of N-phenyl-7,8-dihydro-6H-pyrimido[5,4-b][1,4]oxazin-4-amine derivatives as a new class of inhibitors of RET and its drug resistance mutants. Eur. J. Med. Chem. 2018, 143, 1148–1164. [Google Scholar] [CrossRef]
- Moccia, M.; Liu, Q.; Guida, T.; Federico, G.; Brescia, A.; Zhao, Z.; Choi, H.G.; Deng, X.; Tan, L.; Wang, J.; et al. Identification of novel small molecule inhibitors of oncogenic RET kinase. PLoS ONE 2015, 10, e0128364. [Google Scholar] [CrossRef]
- Eidam, H.S.; Russel, J.; Raha, K.; DeMartino, M.; Qin, D.; Guan, H.A.; Zhang, Z.; Zhen, G.; Yu, H.; Wu, C.; et al. Discovery of a first-in-class gut-restricted RET kinase inhibitor as a clinical candidate for the treatment of IBS. ACS Med. Chem. Lett. 2018, 9, 623–628. [Google Scholar] [CrossRef]
- Russell, J.P.; Mohammadi, E.; Ligon, C.O.; Johnson, A.C.; Gershon, M.D.; Rao, M.; Shen, Y.; Chan, C.-C.; Eidam, H.S.; DeMartino, M.P.; et al. Exploring the potential of RET kinase inhibition for irritable bowel syndrome: A preclinical investigation in rodent models of colonic hypersensitivity. J. Pharmacol. Exp. Ther. 2019, 368, 299–307. [Google Scholar] [CrossRef]
- Ma, F.; Zhang, L.; Westlund, K.N. Trigeminal nerve injury ErbB3/ErbB2 promotes mechanical hypersensitivity. Anesthesiology 2012, 117, 381–388. [Google Scholar] [CrossRef]
- Rusnak, D.W.; Lackey, K.; Affleck, K.; Wood, E.R.; Alligood, K.J.; Rhodes, N.; Keith, B.R.; Murray, D.M.; Knight, W.B.; Mullin, R.J.; et al. The effect of the novel, reversible epidermal growth factor receptor/ErbB-2 tyrosine kinase inhibitor, GW2016, on the growth of human normal and tumor-derived cell lines in vitro and in vivo. Mol. Cancer Ther. 2001, 1, 85–94. [Google Scholar]
- Salaffi, F.; Giacobazzi, G.; Di Carlo, M. Chronic pain in inflammatory arthritis: Mechanisms, metrology, and emerging targets-a focus on the JAK-STAT pathway. Pain Res. Manag. 2018, 8564215. [Google Scholar] [CrossRef]
- Wallenstein, G.V.; Kanik, K.S.; Wilkinson, B.; Cohen, S.; Cutolo, M.; Fleischmann, R.; Genovese, M.C.; Gomez Reino, J.; Gruben, D.; Kremer, J.; et al. Effects of the oral Janus kinase inhibitor tofacitinib on patient-reported outcomes in patients with active rheumatoid arthritis: Results of two phase 2 randomised controlled trials. Clin. Exp. Rheumatol. 2016, 34, 430–442. [Google Scholar]
- Coombs, J.H.; Bloom, B.J.; Breedveld, F.C.; Fletcher, M.P.; Gruben, D.; Kremer, J.M.; Burgos-Vargas, R.; Wilkinson, B.; Zerbini, C.A.F.; Zwillich, S.H. Improved pain, physical functioning and health status in patients with rheumatoid arthritis treated with CP-690,550, an orally active Janus kinase (JAK) inhibitor: Results from a randomised, double-blind, placebo-controlled trial. Ann. Rheum. Dis. 2010, 69, 413–416. [Google Scholar] [CrossRef] [PubMed]
- Meyer, D.M.; Jesson, M.I.; Li, X.; Elrick, M.M.; Funckes-Shippy, C.L.; Warmer, J.D.; Gross, C.J.; Dowty, M.E.; Ramaiah, S.K.; Hirsch, J.L.; et al. Anti-inflammatory activity and neutrophil reductions mediated by the JAK1/JAK3 inhibitor, CP-690,550, in rat adjuvant-induced arthritis. J. Inflamm. 2010, 7, 41. [Google Scholar] [CrossRef]
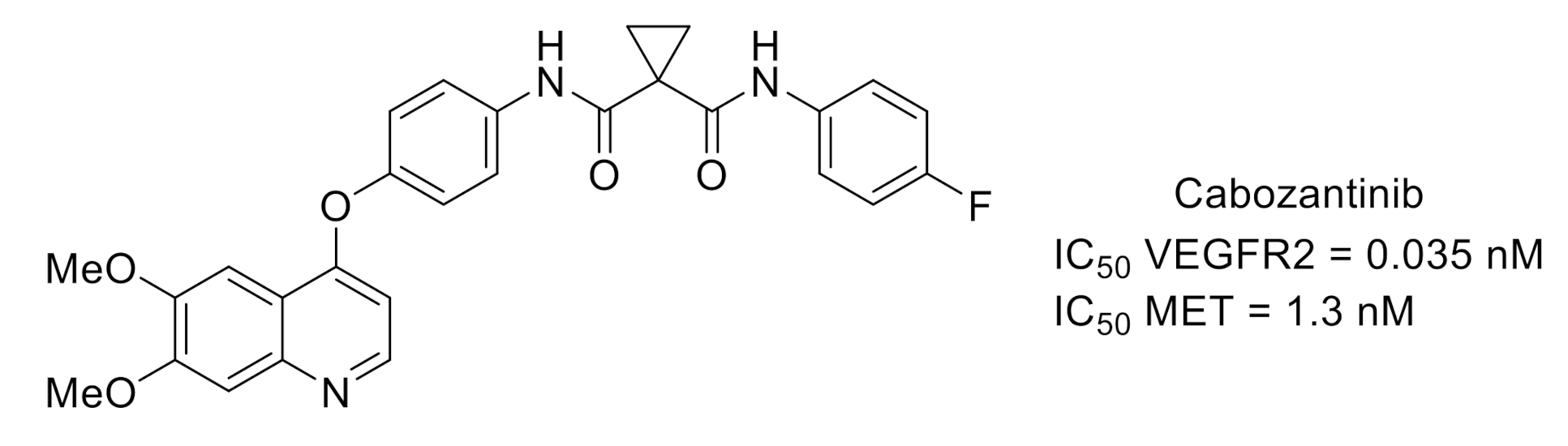
- Yakes, F.M.; Chen, J.; Tan, J.; Yamaguchi, K.; Shi, Y.; Yu, P.; Qian, F.; Chu, F.; Bentzien, F.; Cancilla, B.; et al. Cabozantinib (XL184), a novel MET and VEGFR2 inhibitor, simultaneously suppresses metastasis, angiogenesis, and tumor growth. Mol. Cancer Ther. 2011, 10, 2298–2308. [Google Scholar] [CrossRef] [PubMed]
- Basch, E.; Autio, K.A.; Smith, M.R.; Bennett, A.V.; Weitzman, A.L.; Scheffold, C.; Sweeney, C.; Rathkopf, D.E.; Smith, D.C.; George, D.J.; et al. Effects of cabozantinib on pain and narcotic use in patients with castration-resistant prostate cancer: Results from a phase 2 nonrandomized expansion cohort. Eur. Urol. 2015, 67, 310–318. [Google Scholar] [CrossRef]
- Ge, M.-M.; Zhou, Y.-Q.; Tian, X.-B.; Manyande, A.; Tian, Y.-K.; Ye, D.-W.; Yang, H. Src-family protein tyrosine kinases: A promising target for treating chronic pain. Biomed. Pharmacother. 2020, 125, 110017. [Google Scholar] [CrossRef]
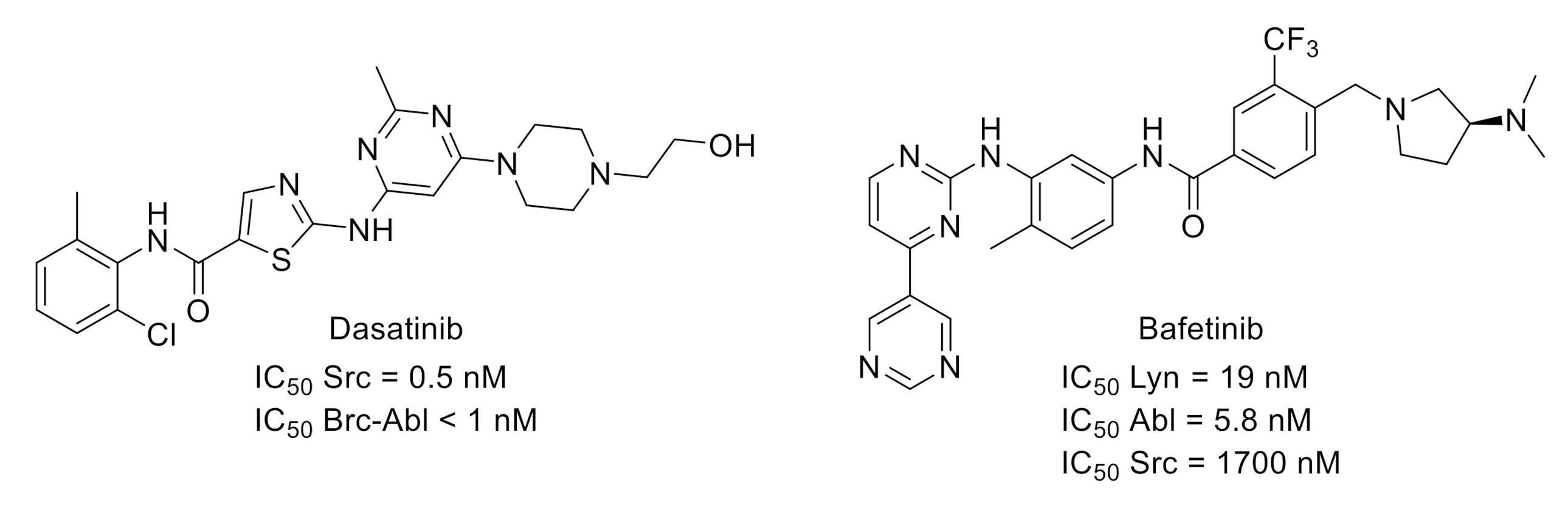
- Lombardo, L.J.; Lee, F.Y.; Chen, P.; Norris, D.; Barrish, J.C.; Behnia, K.; Castaneda, S.; Cornelius, L.A.M.; Das, J.; Doweyko, A.M.; et al. Discovery of N-(2-chloro-6-methylphenyl)-2-(6-(4-(2-hydroxyethyl)-piperazin-1-yl)-2-methylpyrimidin-4-ylamino)thiazole-5-carboxamide (BMS-354825), a dual Src/Abl kinase inhibitor with potent antitumor activity in preclinical assays. J. Med. Chem. 2004, 47, 6658–6661. [Google Scholar] [CrossRef]
- Appel, C.K.; Gallego-Pedersen, S.; Andersen, L.; Blancheflor Kristensen, S.; Ding, M.; Falk, S.; Sayilekshmy, M.; Gabel-Jensen, C.; Heegaard, A.-M. The Src family kinase inhibitor dasatinib delays pain-related behavior and conserves bone in a rat model of cancer-induced bone pain. Sci. Rep. 2017, 7, 4792. [Google Scholar] [CrossRef]
- Grace, M.S.; Lieu, T.; Darby, B.; Abogadie, F.C.; Veldhuis, N.; Bunnett, N.W.; McIntyre, P. The tyrosine kinase inhibitor bafetinib inhibits PAR2-induced activation of TRPV4 channels in vitro and pain in vivo. Br. J. Pharmacol. 2014, 171, 3881–3894. [Google Scholar] [CrossRef]
- Kimura, S.; Naito, H.; Segawa, H.; Kuroda, J.; Yuasa, T.; Sato, K.; Yokota, A.; Kamitsuji, Y.; Kawata, E.; Ashihara, E.; et al. NS-187, a potent and selective dual Bcr-Abl/Lyn tyrosine kinase inhibitor, is a novel agent for imatinib-resistant leukemia. Blood 2005, 106, 3948–3954. [Google Scholar] [CrossRef]
- Rivat, C.; Sar, C.; Mechaly, I.; Leyris, J.-P.; Diouloufet, L.; Sonrier, C.; Philipson, Y.; Lucas, O.; Mallié, S.; Jouvenel, A.; et al. Inhibition of neuronal FLT3 receptor tyrosine kinase alleviates peripheral neuropathic pain in mice. Nat. Commun. 2018, 9, 1042. [Google Scholar] [CrossRef]
- Asih, P.R.; Prikas, E.; Stefanoska, K.; Tan, A.R.P.; Ahel, H.I.; Ittner, A. Functions of p38 MAP kinases in the central nervous system. Front. Mol. Neurosci. 2020, 13, 570586. [Google Scholar] [CrossRef]
- Mai, L.; Zhu, X.; Huang, F.; He, H.; Fan, W. p38 mitogen-activated protein kinase and pain. Life Sci. 2020, 256, 117885. [Google Scholar] [CrossRef]
- Madkour, M.M.; Anbar, H.S.; El-Gamal, M.I. Current status and future prospects of p38α/MAPK14 kinase and its inhibitors. Eur. J. Med. Chem. 2021, 213, 113216. [Google Scholar] [CrossRef]
- Lin, X.; Wang, M.; Zhang, J.; Xu, R. p38 MAPK: A potential target of chronic pain. Curr. Med. Chem. 2014, 21, 4405–4418. [Google Scholar] [CrossRef]
- Yasuda, S.; Sugiura, H.; Tanaka, H.; Takigami, S.; Yamagata, K. p38 MAP kinase inhibitors as potential therapeutic drugs for neural diseases. Cent. Nerv. Syst. Agents Med. Chem. 2011, 11, 45–59. [Google Scholar] [CrossRef]
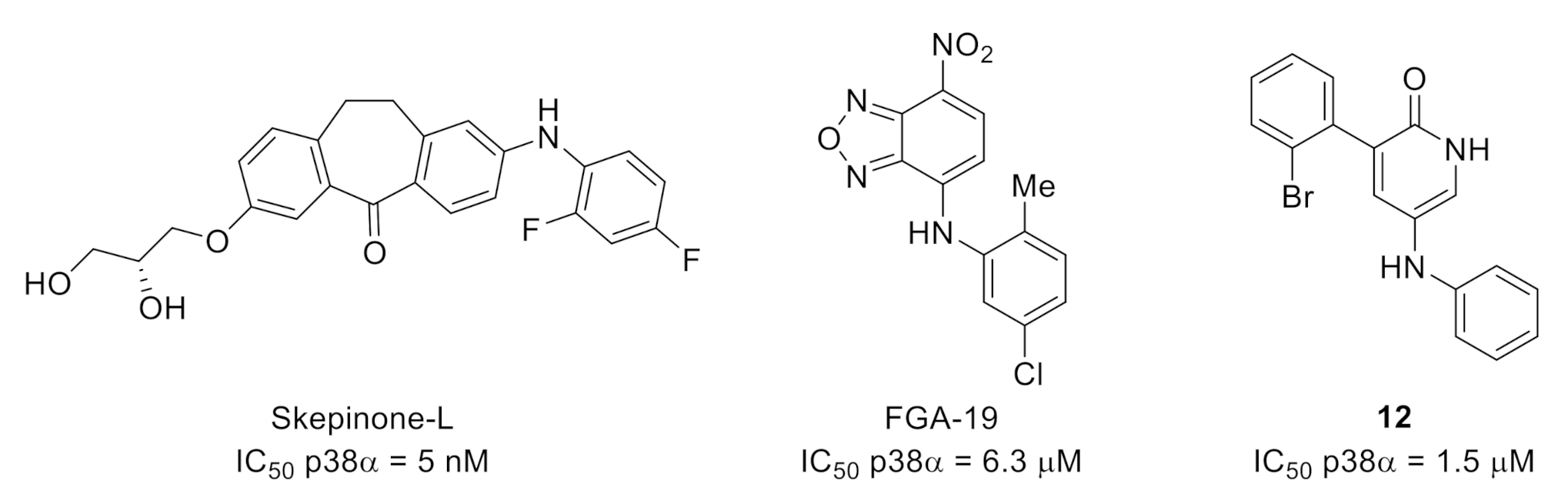
- Koeberle, S.C.; Romir, J.; Fischer, S.; Koeberle, A.; Schattel, V.; Albrecht, W.; Grütter, C.; Werz, O.; Rauh, D.; Stehle, T.; et al. Skepinone-L is a selective p38 mitogen-activated protein kinase inhibitor. Nat. Chem. Biol. 2012, 8, 141–143. [Google Scholar] [CrossRef]
- Taves, S.; Berta, T.; Liu, D.-L.; Gan, S.; Chen, G.; Kim, Y.H.; Van de Ven, T.; Laufer, S.; Ji, R.-R. Spinal inhibition of p38 MAP kinase reduces inflammatory and neuropathic pain in male but not female mice: Sex-dependent microglial signaling in the spinal cord. Brain Behav. Immun. 2016, 55, 70–81. [Google Scholar] [CrossRef]
- Willemen, H.L.D.M.; Campos, P.M.; Lucas, E.; Morreale, A.; Gil-Redondo, R.; Agut, J.; González, F.V.; Ramos, P.; Heijnen, C.; Mayor, F.; et al. A novel p38 MAPK docking-groove-targeted compound is a potent inhibitor of inflammatory hyperalgesia. Biochem, J. 2014, 459, 427–439. [Google Scholar] [CrossRef]
- Visseq, A.; Descheemaeker, A.; Pinto-Pardo, N.; Nauton, L.; Théry, V.; Giraud, F.; Abrunhosa-Thomas, I.; Artola, A.; Anizon, F.; Dallel, R.; et al. Pyridin-2(1H)one derivatives: A possible new class of therapeutics for mechanical allodynia. Eur, J. Med. Chem. 2020, 187, 111917. [Google Scholar] [CrossRef]
- Ji, R.-R.; Gereau, R.W., IV; Malcangio, M.; Strichartz, G.R. MAP kinase and pain. Brain Res. Rev. 2009, 60, 135–148. [Google Scholar] [CrossRef]
- Kondo, M.; Shibuta, I. Extracellular signal-regulated kinases (ERK) 1 and 2 as a key molecule in pain research. J. Oral Sci. 2020, 62, 147–149. [Google Scholar] [CrossRef] [PubMed]
- Alessi, D.R.; Cuenda, A.; Cohen, P.; Dudley, D.T.; Saltiel, A.R. PD 098059 is a specific inhibitor of the activation of mitogen-activated protein kinase kinase in vitro and in vivo. J. Biol. Chem. 1995, 270, 27489–27494. [Google Scholar] [CrossRef] [PubMed]
- Favata, M.F.; Horiuchi, K.Y.; Manos, E.J.; Daulerio, A.J.; Stradley, D.A.; Feeser, W.S.; Van Dyk, D.E.; Pitts, W.J.; Earl, R.A.; Hobbs, F.; et al. Identification of a Novel Inhibitor of Mitogen-activated Protein Kinase Kinase. J. Biol. Chem. 1998, 273, 18623–18632. [Google Scholar] [CrossRef] [PubMed]
- Yamakita, S.; Horii, Y.; Takemura, H.; Matsuoka, Y.; Yamashita, A.; Yamaguchi, Y.; Matsuda, M.; Sawa, T.; Amaya, F. Synergistic activation of ERK1/2 between A-fiber neurons and glial cells in the DRG contributes to pain hypersensitivity after tissue injury. Mol. Pain 2018, 14, 1744806918767508. [Google Scholar] [CrossRef]
- Li, G.; Qi, W.; Li, X.; Zhao, J.; Luo, M.; Chen, J. Recent advances in c-Jun N-terminal kinase (JNK) Inhibitors. Curr. Med. Chem. 2021, 28, 607–627. [Google Scholar] [CrossRef]
- Bennett, B.L.; Sasaki, D.T.; Murray, B.W.; O’Leary, E.O.; Sakata, S.T.; Xu, W.; Leisten, J.C.; Motiwala, A.; Pierce, S.; Satoh, Y.; et al. SP600125, an anthrapyrazolone inhibitor of Jun N-terminal kinase. Proc. Natl. Acad. Sci. USA 2001, 98, 13681–13686. [Google Scholar] [CrossRef]
- Ni, H.-D.; Xu, L.S.; Wang, Y.; Li, H.; An, K.; Liu, M.; Liu, Q.; Deng, H.; He, Q.; Huang, B.; et al. Astrocyte activation in the periaqueductal gray promotes descending facilitation to cancer-induced bone pain through the JNK MAPK signaling pathway. Mol. Pain 2019, 15, 1744806919831909. [Google Scholar] [CrossRef]
- Sanna, M.D.; Ghelardini, C.; Galeotti, N. Blockade of the spinal BDNF-activated JNK pathway prevents the development of antiretroviral-induced neuropathic pain. Neuropharmacology 2016, 105, 543–552. [Google Scholar] [CrossRef]
- Ogier, J.M.; Nayagam, B.A.; Lockhart, P.J. ASK1 inhibition: A therapeutic strategy with multi-system benefits. J. Mol. Med. 2020, 98, 335–348. [Google Scholar] [CrossRef]
- Brys, R.; Gibson, K.; Poljak, T.; Van der Plas, S.; Amantini, D. Discovery and development of ASK1 inhibitors. Prog. Med. Chem. 2020, 59, 101–179. [Google Scholar] [CrossRef]
- Lanier, M.; Pickens, J.; Bigi, S.V.; Bradshaw-Pierce, E.L.; Chambers, A.; Cheruvallath, Z.S.; Cole, D.; Dougan, D.R.; Ermolieff, J.; Gibson, T.; et al. Structure-based design of ASK1 inhibitors as potential agents for heart failure. ACS Med. Chem. Lett. 2017, 8, 316–320. [Google Scholar] [CrossRef]
- Volynets, G.P.; Chekanov, M.O.; Synyugin, A.R.; Golub, A.G.; Kukharenko, O.P.; Bdzhola, V.G.; Yarmoluk, S.M. Identification of 3H-naphtho[1,2,3-de]quinoline-2,7-diones as inhibitors of apoptosis signal-regulating kinase 1 (ASK1). J. Med. Chem. 2011, 54, 2680–2686. [Google Scholar] [CrossRef]
- Dai, W.-L.; Bao, Y.-N.; Fan, J.-F.; Li, S.-S.; Zhao, W.-L.; Yu, B.-Y.; Liu, J.-H. Levo-corydalmine attenuates microglia activation and neuropathic pain by suppressing ASK1-p38 MAPK/NF-κB signaling pathways in rat spinal cord. Reg. Anesth. Pain Med. 2020, 45, 219–229. [Google Scholar] [CrossRef]
- Zhou, D.; Zhang, S.; Hu, L.; Gu, Y.-F.; Cai, Y.; Wu, D.; Liu, W.-T.; Jiang, C.-Y.; Kong, X.; Zhang, G.-Q. Inhibition of apoptosis signal-regulating kinase by paeoniflorin attenuates neuroinflammation and ameliorates neuropathic pain. J. Neuroinflammation 2019, 16, 83. [Google Scholar] [CrossRef]
- Pareek, T.K.; Kulkarni, A.B. Cdk5: A new player in pain signaling. Cell Cycle 2006, 5, 585–588. [Google Scholar] [CrossRef]
- Pareek, T.K.; Keller, J.; Kesavapany, S.; Pant, H.C.; Iadarola, M.J.; Brady, R.O.; Kulkarni, A.B. Cyclin-dependent kinase 5 activity regulates pain signaling. Proc. Natl. Acad. Sci. USA 2006, 103, 791–796. [Google Scholar] [CrossRef]
- Utreras, E.; Futatsugi, A.; Pareek, T.K.; Kulkarni, A.B. Molecular roles of Cdk5 in pain signaling. Drug Discov. Today Ther. Strateg. 2009, 6, 105–111. [Google Scholar] [CrossRef]
- Gomez, K.; Vallecillo, T.G.M.; Moutal, A.; Perez-Miller, S.; Delgado-Lezama, R.; Felix, R.; Khanna, R. The role of cyclin-dependent kinase 5 in neuropathic pain. Pain 2020, 161, 2674–2689. [Google Scholar] [CrossRef]
- Meijer, L.; Borgne, A.; Mulner, O.; Chong, J.P.; Blow, J.J.; Inagaki, N.; Inagaki, M.; Delcros, J.G.; Moulinoux, J.P. Biochemical and cellular effects of roscovitine, a potent and selective inhibitor of the cyclin-dependent kinases cdc2, cdk2 and cdk5. Eur. J. Biochem. 1997, 243, 527–536. [Google Scholar] [CrossRef]
- Meijer, L.; Nelson, D.J.; Riazanski, V.; Gabdoulkhakova, A.G.; Hery-Arnaud, G.; Le Berre, R.; Loaëc, N.; Oumata, N.; Galons, H.; Nowak, E.; et al. Modulating Innate and Adaptive Immunity by (R)-Roscovitine: Potential Therapeutic Opportunity in Cystic Fibrosis. J. Innate Immun. 2016, 8, 330–349. [Google Scholar] [CrossRef]
- Byth, K.F.; Thomas, A.; Hughes, G.; Forder, C.; McGregor, A.; Geh, C.; Oakes, S.; Green, C.; Walker, M.; Newcombe, N.; et al. AZD5438, a potent oral inhibitor of cyclin-dependent kinases 1, 2, and 9, leads to pharmacodynamic changes and potent antitumor effects in human tumor xenografts. Mol. Cancer Ther. 2009, 1856–1866. [Google Scholar] [CrossRef]
- Wu, J.; Zhao, Z.; Zhu, X.; Renn, C.L.; Dorsey, S.G.; Faden, A.I. Cell cycle inhibition limits development and maintenance of neuropathic pain following spinal cord injury. Pain 2016, 157, 488–503. [Google Scholar] [CrossRef]
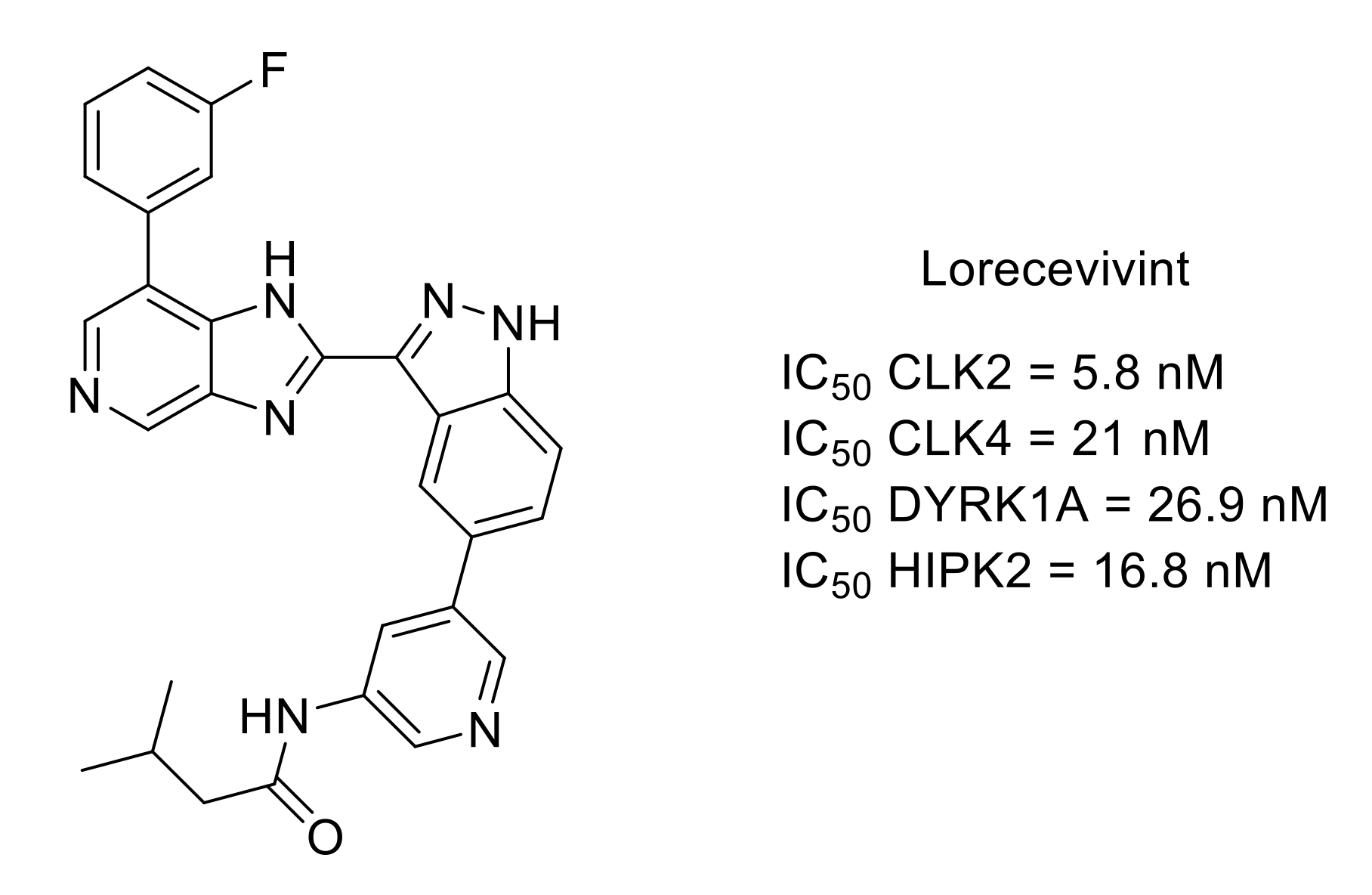
- Deshmukh, V.; O’Green, A.L.; Bossard, C.; Seo, T.; Lamangan, L.; Ibanez, M.; Ghias, A.; Lai, C.; Do, L.; Cho, S.; et al. Modulation of the Wnt pathway through inhibition of CLK2 and DYRK1A by lorecivivint as a novel, potentially disease-modifying approach for knee osteoarthritis treatment. Osteoarthr.Cartil. 2019, 27, 1347–1360. [Google Scholar] [CrossRef]
- Yazici, Y.; McAlindon, T.E.; Gibofsky, A.; Lane, N.E.; Clauw, D.; Jones, M.; Bergfeld, J.; Swearingen, C.J.; DiFrancesco, A.; Simsek, I.; et al. Lorecivivint, a novel intraarticular CDC-like kinase 2 and dual specificity tyrosine phosphorylation-regulated kinase 1A inhibitor and Wnt pathway modulator for the treatment of knee osteoarthritis: A phase II randomized trial. Arthritis Rheumatol. 2020, 72, 1694–1706. [Google Scholar] [CrossRef]
- Lories, R.J.; Monteagudo, S. Is Wnt signaling an attractive target for the treatment of osteoarthritis? Rheumatol. Ther. 2020, 7, 259–270. [Google Scholar] [CrossRef]
- Maixner, D.W.; Weng, H.-R. The role of glycogen synthase kinase 3 beta in neuroinflammation and pain. J. Pharm. Pharmacol. (Los Angeles) 2013, 1. [Google Scholar] [CrossRef]
- Noori, T.; Dehpour, A.R.; Sureda, A.; Fakhri, S.; Sobarzo-Sanchez, E.; Farzaei, M.H.; Akkol, E.K.; Khodarahmi, Z.; Hosseini, S.Z.; Alavi, S.D.; et al. The role of glycogen synthase kinase 3 beta in multiple sclerosis. Biomed. Pharmacother. 2020, 132, 110874. [Google Scholar] [CrossRef]
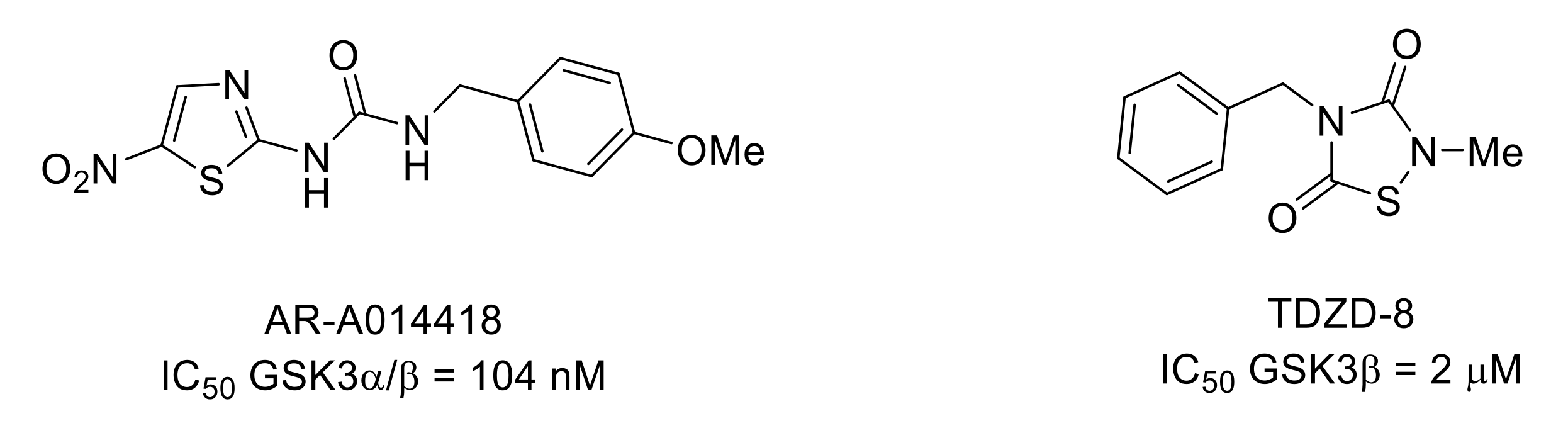
- Martins, D.F.; Rosa, A.O.; Gadotti, V.M.; Mazzardo-Martins, L.; Nascimento, F.P.; Egea, J.; López, M.G.; Santos, A.R.S. The antinociceptive effects of AR-A014418, a selective inhibitor of glycogen synthase kinase-3 beta, in mice. J. Pain 2011, 12, 315–322. [Google Scholar] [CrossRef]
- Mazzardo-Martins, L.; Martins, D.F.; Stramosk, J.; Cidral-Filho, F.J.; Santos, A.R.S. Glycogen synthase kinase 3-specific inhibitor AR-A014418 decreases neuropathic pain in mice: Evidence for the mechanisms of action. NeuroScience 2012, 226, 411–420. [Google Scholar] [CrossRef]
- Li, Y.; Wang, H.; Xie, K.; Wang, C.; Yang, Z.; Yu, Y.; Wang, G. Inhibition of glycogen synthase kinase-3β prevents remifentanil-induced hyperalgesia via regulating the expression and function of spinal N-methyl-D-aspartate receptors in vivo and vitro. PLoS ONE 2013, 8, e77790. [Google Scholar] [CrossRef]
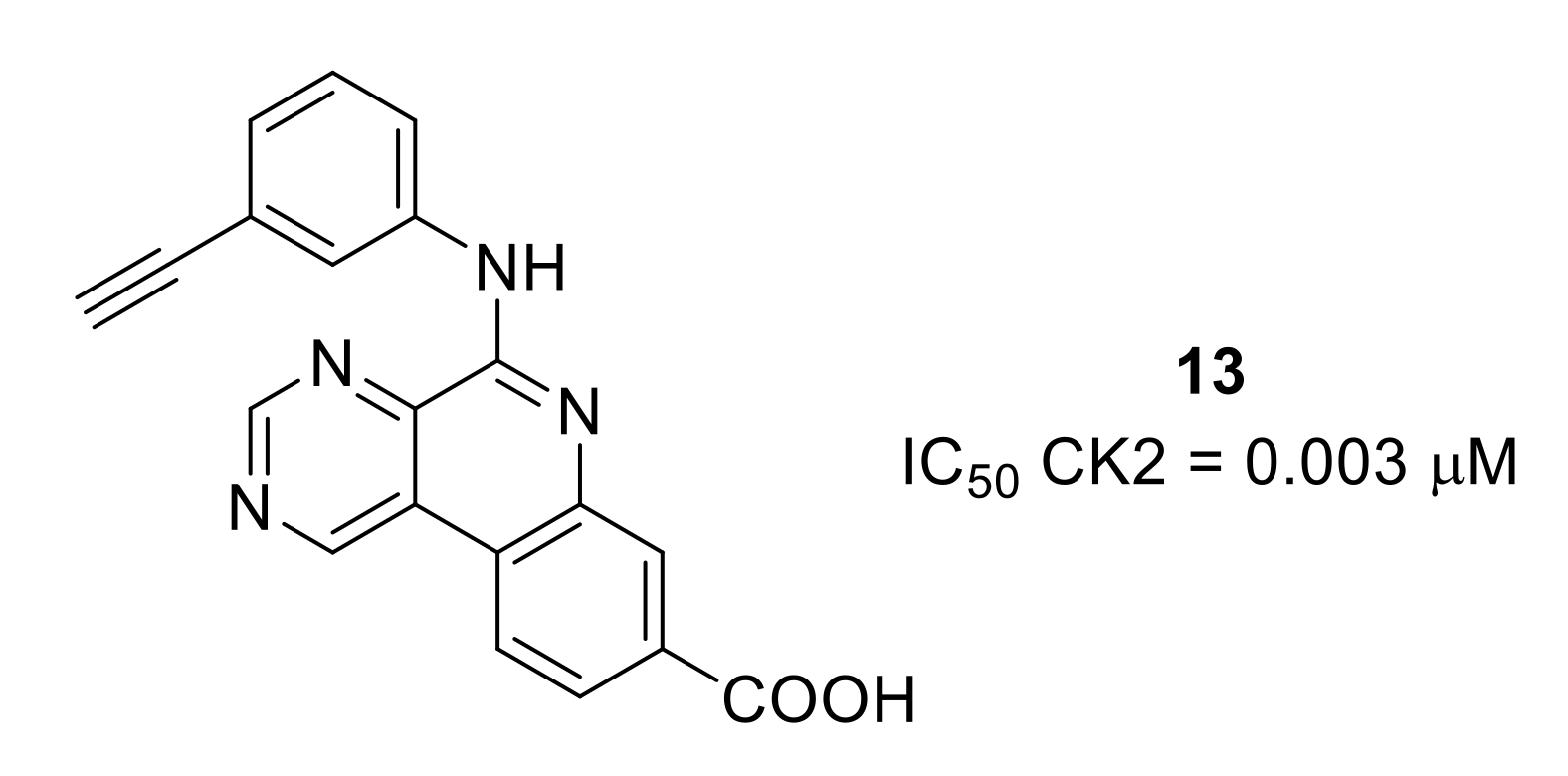
- Li, X.; Shi, X.; Liang, D.-Y.; Clark, J.D. Spinal CK2 regulates nociceptive signaling in models of inflammatory pain. Pain 2005, 115, 182–190. [Google Scholar] [CrossRef] [PubMed]
- Pierre, F.; O’Brien, S.E.; Haddach, M.; Bourdon, P.; Schwaebe, M.K.; Stefan, E.; Darjania, L.; Stansfield, R.; Ho, C.; Siddiqui-Jain, A.; et al. Novel potent pyrimido[4,5-c]quinoline inhibitors of protein kinase CK2: SAR and preliminary assessment of their analgesic and anti-viral properties. Bioorg. Med. Chem. Lett. 2011, 21, 1687–1691. [Google Scholar] [CrossRef]
- Fang, L.; Wu, J.; Lin, Q.; Willis, W.D. Protein kinases regulate the phosphorylation of the GluR1 subunit of AMPA receptors of spinal cord in rats following noxious stimulation. Mol. Brain Res. 2003, 118, 160–165. [Google Scholar] [CrossRef] [PubMed]
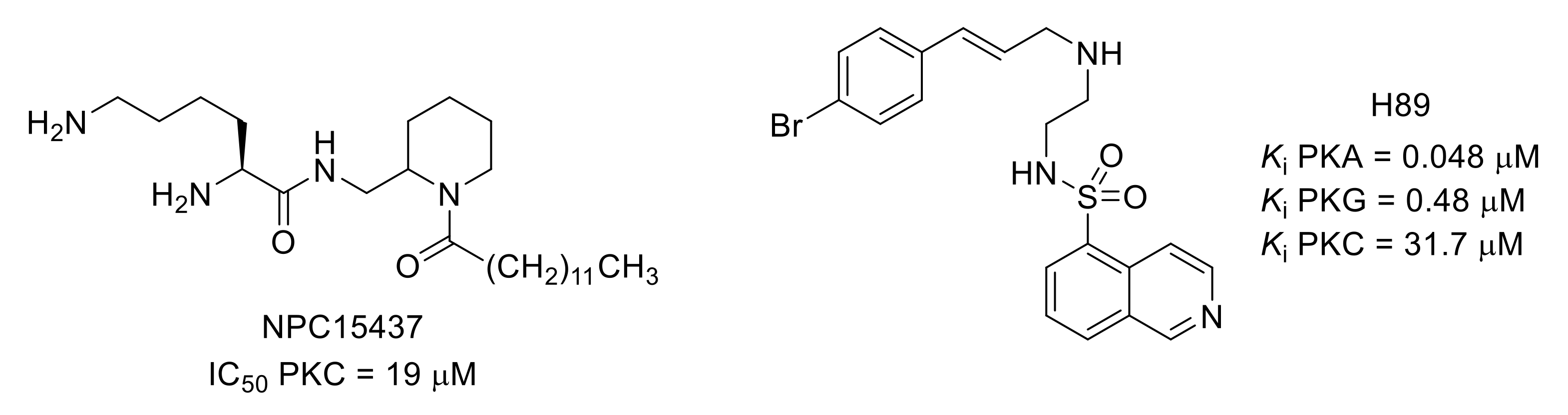
- Sullivan, J.P.; Connor, J.R.; Shearer, B.G.; Burch, R.M. 2,6-diamino-N-([1-(1-oxotridecyl)-2-piperidinyl]methyl)hexanamide (NPC 15437): A selective inhibitor of protein kinase C. Agents Actions 1991, 34, 142–144. [Google Scholar] [CrossRef] [PubMed]
- Zeng, J.; Li, S.; Zhang, C.; Huang, G.; Yu, C. The mechanism of hyperalgesia and anxiety induced by remifentanil: Phosphorylation of GluR1 receptors in the anterior cingulate cortex. J. Mol. Neurosci. 2018, 65, 93–101. [Google Scholar] [CrossRef] [PubMed]
- Chijiwa, T.; Mishima, A.; Hagiwara, M.; Sano, M.; Hayashi, K.; Inoue, T.; Naito, K.; Toshioka, T.; Hidaka, H. Inhibition of forskolin-induced neurite outgrowth and protein phosphorylation by a newly synthesized selective inhibitor of cyclic AMP-dependent protein kinase, N-[2-(p-bromocinnamylamino)ethyl]-5-isoquinolinesulfonamide (H-89), of PC12D pheochromocytoma cells. J. Biol. Chem. 1990, 265, 5267–5272. [Google Scholar] [CrossRef] [PubMed]
- Mochly-Rosen, D.; Das, K.; Grimes, K.V. Protein kinase C, an elusive therapeutic target? Nat. Rev. Drug Discov. 2012, 11, 937–957. [Google Scholar] [CrossRef] [PubMed]
- Churchill, E.N.; Qvit, N.; Mochly-Rosen, D. Rationally designed peptide regulators of protein kinase C. Trends Endocrinol. Metab. 2009, 20, 25–33. [Google Scholar] [CrossRef]
- Pham-Dang, N.; Descheemaeker, A.; Dallel, R.; Artola, A. Activation of medullary dorsal horn γ isoform of protein kinase C interneurons is essential to the development of both static and dynamic facial mechanical allodynia. Eur. J. Neurosci. 2016, 43, 802–810. [Google Scholar] [CrossRef]
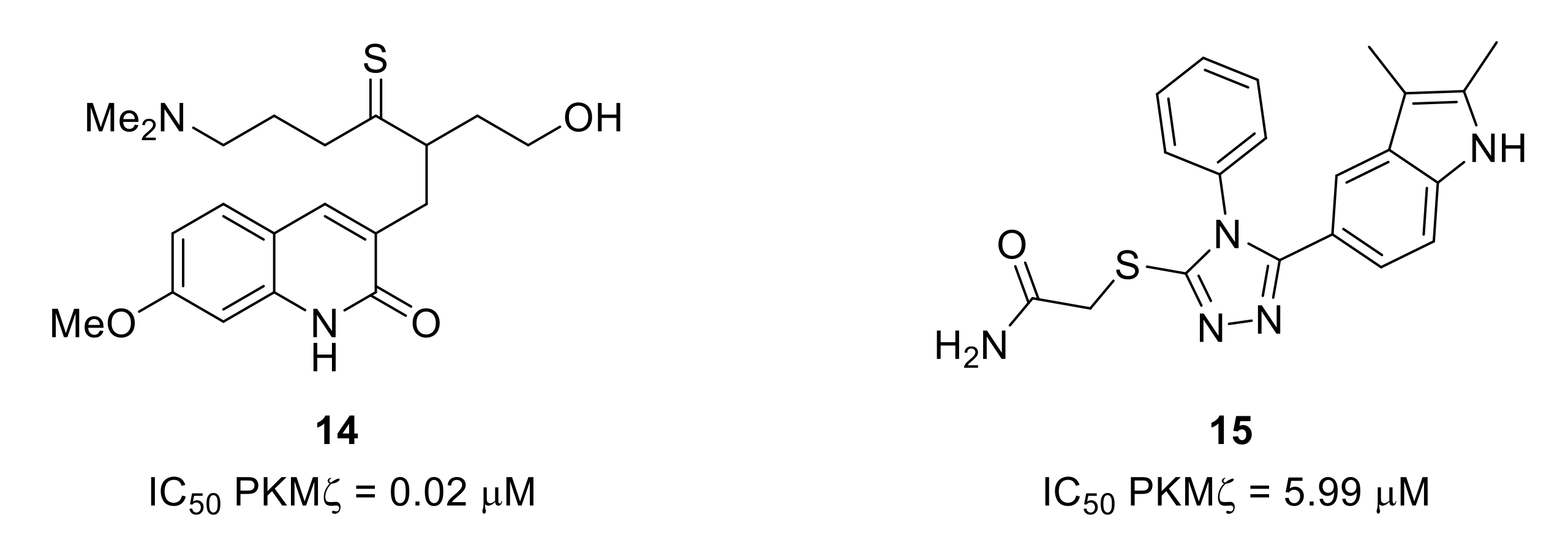
- Zhang, L.; Guo, S.; Zhao, Q.; Li, Y.; Song, C.; Wang, C.; Yu, Y.; Wang, G. Spinal protein kinase M ζ regulates α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor trafficking and dendritic spine plasticity via kalirin-7 in the pathogenesis of remifentanil-induced postincisional hyperalgesia in rats. Anesthesiology 2018, 129, 173–186. [Google Scholar] [CrossRef]
- Li, X.-Y.; Ko, H.-G.; Chen, T.; Descalzi, G.; Koga, K.; Wang, H.; Kim, S.S.; Shang, Y.; Kwak, C.; Park, S.-W.; et al. Alleviating neuropathic pain hypersensitivity by inhibiting PKMζ in the anterior cingulate cortex. Science 2010, 330, 1400–1404. [Google Scholar] [CrossRef]
- An, K.; Zhen, C.; Liu, Z.-H.; Zhao, Q.; Liu, H.-P.; Zhong, X.L.; Huang, W.-Q. Spinal protein kinase Mζ contributes to the maintenance of peripheral inflammation-primed persistent nociceptive sensitization after plantar incision. Eur. J. Pain 2015, 19, 39–47. [Google Scholar] [CrossRef]
- Cha, M.; Um, S.W.; Kwon, M.; Nam, T.S.; Lee, B.H. Repetitive motor cortex stimulation reinforces the pain modulation circuits of peripheral neuropathic pain. Sci. Rep. 2017, 7, 7986. [Google Scholar] [CrossRef]
- Purkayastha, P.; Alokam, R.; Malapati, A.; Sriram, D.; Yogeeswari, P. Structural models for the design of PKMzeta inhibitors with neurobiological indications. Mol. Inf. 2015, 34, 665–678. [Google Scholar] [CrossRef]
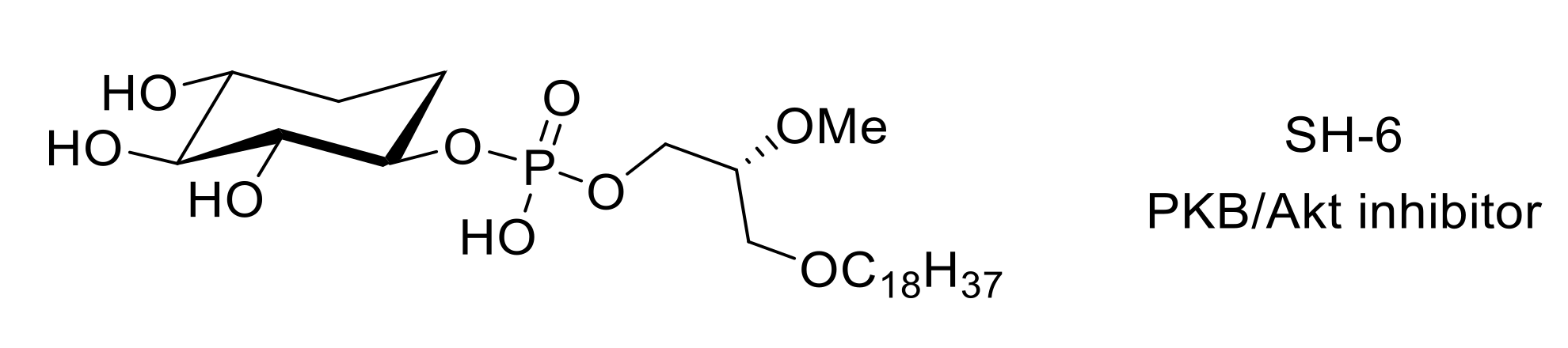
- Kozikowski, A.P.; Sun, H.; Brognard, J.; Dennis, P.A. Novel PI analogues selectively block activation of the pro-survival serine/threonine kinase Akt. J. Am. Chem. Soc. 2003, 125, 1144–1145. [Google Scholar] [CrossRef]
- Sun, R.-Q.; Tu, Y.-J.; Yan, J.-Y.; Willis, W.D. Activation of protein kinase B/Akt signaling pathway contributes to mechanical hypersensitivity induced by capsaicin. Pain 2006, 120, 86–96. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.; Wu, L.-Y. Influence of phosphatidylinositol-3-kinase/protein kinase B-mammalian target of rapamycin signaling pathway on the neuropathic pain complicated by nucleoside reverse transcriptase inhibitors for the treatment of HIV infection. Chin. Med. J. 2018, 131, 1849–1856. [Google Scholar] [CrossRef]
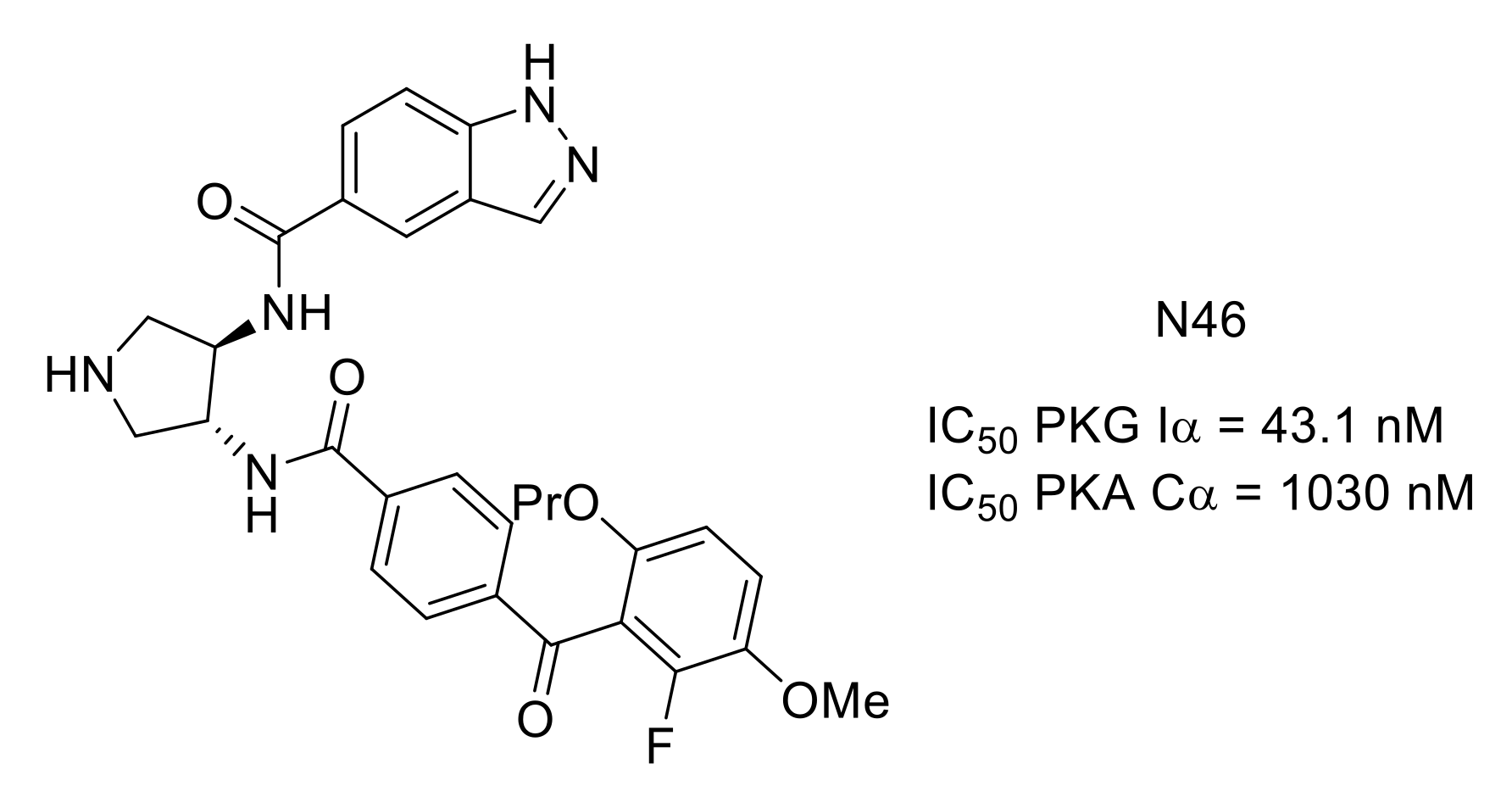
- Qin, L.; Sankaran, B.; Aminzai, S.; Casteel, D.E.; Kim, C. Structural basis for selective inhibition of human PKG Iα by the balanol-like compound N46. J. Biol. Chem. 2018, 293, 10985–10992. [Google Scholar] [CrossRef]
- Sung, Y.-J.; Sofoluke, N.; Nkamany, M.; Deng, S.; Xie, Y.; Greenwood, J.; Farid, R.; Landry, D.W.; Ambron, R.T. A novel inhibitor of active protein kinase G attenuates chronic inflammatory and osteoarthritic pain. Pain 2017, 158, 822–832. [Google Scholar] [CrossRef]
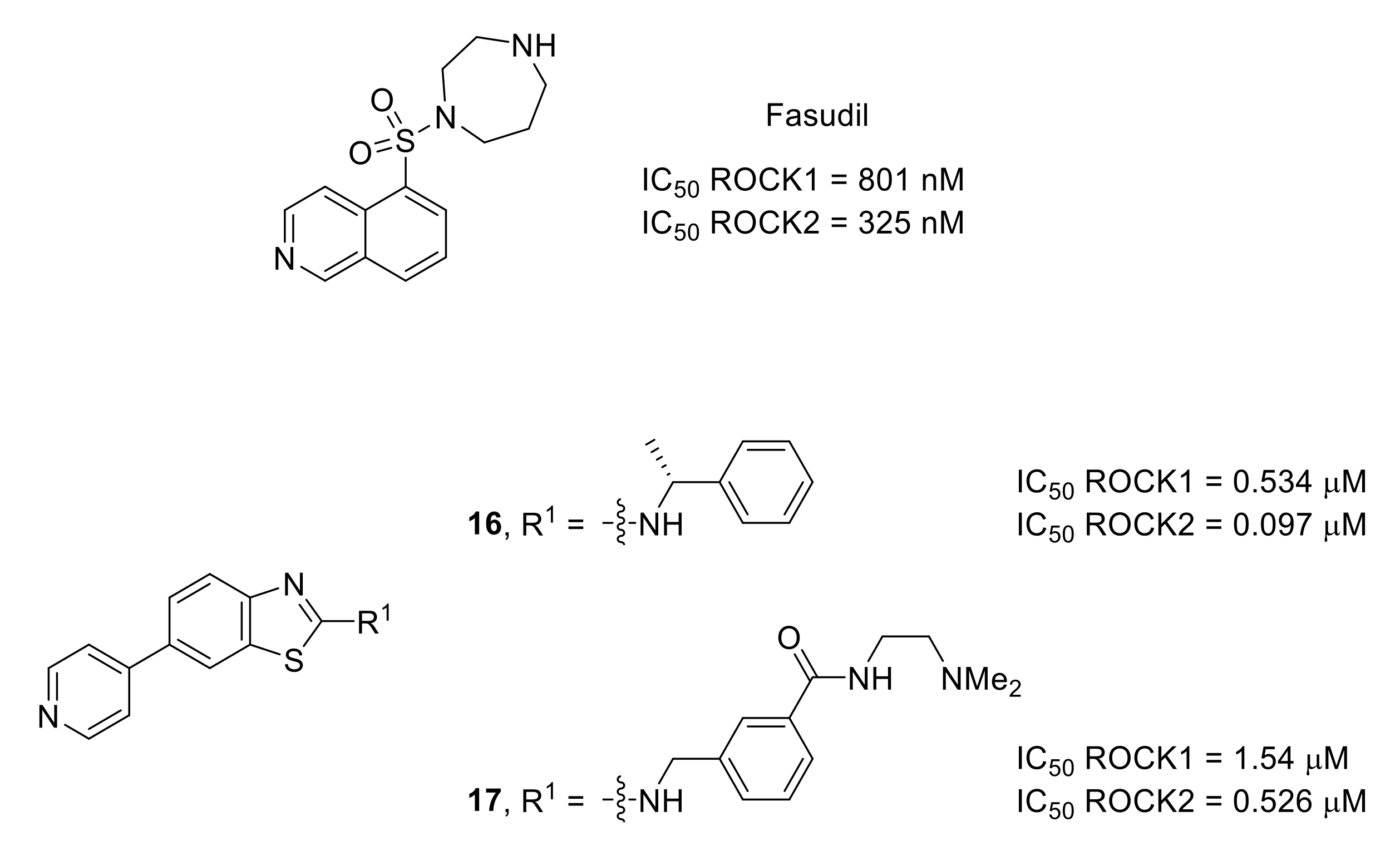
- Mueller, B.K.; Mack, H.; Teusch, N. Rho kinase, a promising drug target for neurological disorders. Nat. Rev. Drug Discov. 2005, 4, 387–398. [Google Scholar] [CrossRef]
- Boyce-Rustay, J.M.; Simler, G.H.; McGaraughty, S.; Chu, K.L.; Wensink, E.J.; Vasudevan, A.; Honore, P. Characterization of fasudil in preclinical models of pain. J. Pain 2010, 11, 941–949. [Google Scholar] [CrossRef] [PubMed]
- Judge, R.A.; Vasudevan, A.; Scott, V.E.; Simler, G.H.; Pratt, S.D.; Namovic, M.T.; Putman, C.B.; Aguirre, A.; Stoll, V.S.; Mamo, M.; et al. Design of aminobenzothiazole inhibitors of Rho kinases 1 and 2 by using protein kinase A as a structure surrogate. ChemBioChem 2018, 19, 613–621. [Google Scholar] [CrossRef] [PubMed]
- Davies, S.P.; Reddy, H.; Caivano, M.; Cohen, P. Specificity and mechanism of action of some commonly used protein kinase inhibitors. Biochem. J. 2000, 351, 95–105. [Google Scholar] [CrossRef]
- Ohsawa, M.; Ishikura, K.-I.; Mutoh, J.; Hisa, H. Involvement of inhibition of RhoA/Rho kinase signaling in simvastatin-induced amelioration of neuropathic pain. Neuroscience 2016, 333, 204–213. [Google Scholar] [CrossRef]
- Molokie, R.E.; Wilkie, D.J.; Wittert, H.; Suarez, M.L.; Yao, Y.; Zhao, Z.; He, Y.; Wang, Z.J. Mechanism-driven phase I translational study of trifluoroperazine in adults with sickle cell disease. Eur. J. Pharmacol. 2014, 723, 419–424. [Google Scholar] [CrossRef]
- Sumi, M.; Kiuchi, K.; Ishikawa, T.; Ishii, A.; Hagiwara, M.; Nagatsu, T.; Hidaka, H. The newly synthesized selective Ca2+/calmodulin dependent protein kinase II inhibitor KN-93 reduces dopamine contents in PC12h cells. Biochem. Biophys. Res. Commun. 1991, 181, 968–975. [Google Scholar] [CrossRef]
- Chen, Y.; Luo, F.; Yang, C.; Kirkmire, C.M.; Wang, Z.J. Acute inhibition of Ca2+/calmodulin-dependent protein kinase II reverses experimental neuropathic pain in mice. J. Pharmacol. Exp. Ther. 2009, 330, 650–659. [Google Scholar] [CrossRef]
- Luo, F.; Yang, C.; Chen, Y.; Shukla, P.; Tang, L.; Wang, L.X.; Wang, Z.J. Reversal of chronic inflammatory pain by acute inhibition of Ca2+/calmodulin-dependent protein kinase II. J. Pharmacol. Exp. Ther. 2008, 325, 267–275. [Google Scholar] [CrossRef]
- Jiang, M.; Zhang, W.; Cheng, C.; Ma, Z.; Gu, X. Intrathecal injection of KN93 attenuates paradoxical remifentanil-induced postoperative hyperalgesia by inhibiting spinal CaMKII phosphorylation in rats. Pharmacol. Biochem. Behav. 2015, 134, 35–41. [Google Scholar] [CrossRef]
- Mashhoon, N.; DeMaggio, A.J.; Tereshko, V.; Bergmeier, S.C.; Egli, M.; Hoekstra, M.F.; Kuret, J. Crystal structure of a conformation-selective casein kinase-1 inhibitor. J. Biol. Chem. 2000, 275, 20052–20060. [Google Scholar] [CrossRef]
- Muraki, M.; Ohkawara, B.; Hosoya, T.; Onogi, H.; Koizumi, J.; Koizumi, T.; Sumi, K.; Yomoda, J.-I.; Murray, M.V.; Kimura, H.; et al. Manipulation of alternative splicing by a newly developed inhibitor of Clks. J. Biol. Chem. 2004, 279, 24246–24254. [Google Scholar] [CrossRef]
- Kurihara, T.; Sakurai, E.; Toyomoto, M.; Kii, I.; Kawamoto, D.; Asada, T.; Tanabe, T.; Yoshimura, M.; Hagiwara, M.; Miyata, A. Alleviation of behavioral hypersensitivity in mouse models of inflammatory pain with two structurally different casein kinase 1 (CK1) inhibitors. Mol. Pain 2014, 10, 17. [Google Scholar] [CrossRef]
- Reilly, S.M.; Chiang, S.-H.; Decker, S.J.; Chang, L.; Uhm, M.; Larsen, M.J.; Rubin, J.R.; Mowers, J.; White, N.M.; Hochberg, I.; et al. An inhibitor of the protein kinases TBK1/IKKε improves obesity related metabolic dysfunctions. Nat. Med. 2013, 19, 313–321. [Google Scholar] [CrossRef]
- Beyett, T.S.; Gan, X.; Reilly, S.M.; Chang, L.; Gomez, A.V.; Saltiel, A.R.; Showalter, H.D.; Tesmer, J.J.G. Carboxylic acid derivatives of amlexanox display enhanced potency towards TBK1 and IKKε and reveal mechanisms for selective inhibition. Mol. Pharmacol. 2018, 94, 1210–1219. [Google Scholar] [CrossRef]
- Möser, C.V.; Möller, M.; Fleck, S.C.; Thomas, D.; Geisslinger, G.; Niederberger, E. Inhibition of the protein kinase IKKepsilon attenuates neuropathic pain in mice. Neuropharmacology 2019, 146, 198–211. [Google Scholar] [CrossRef] [PubMed]
- Asante, C.O.; Wallace, V.C.; Dickenson, A.H. Mammalian target of rapamycin signaling in the spinal cord is required for neuronal plasticity and behavorial hypersensitivity associated with neuropathy in the rat. J. Pain 2010, 11, 1356–1367. [Google Scholar] [CrossRef] [PubMed]
- Shor, B.; Zhang, W.-G.; Toral-Barza, L.; Lucas, J.; Abraham, R.T.; Gibbons, J.J.; Yu, K. A new pharmacologic action of CCI-779 involved FKBP12-independent inhibition of mTOR kinase activity and profound repression of global protein synthesis. Cancer Res. 2008, 68, 2934–2943. [Google Scholar] [CrossRef]
- Liu, Q.; Chang, J.W.; Wang, J.; Kang, S.A.; Thoreen, C.C.; Markhard, A.; Hur, W.; Zhang, J.; Sim, T.; Sabatini, D.M.; et al. Discovery of 1-(4-(4-propionylpiperazin-1-yl)-3-(trifluoromethyl)phenyl)-9-(quinolin-3-yl)benzo[h][1,6]naphthyridin-2(1H)-one as a highly potent, selective mammalian target of rapamycin (mTOR) inhibitor for the treatment of cancer. J. Med. Chem. 2010, 53, 7146–7155. [Google Scholar] [CrossRef]
- Obara, I.; Tochiki, K.K.; Géranton, S.M.; Carr, F.B.; Lumb, B.M.; Liu, Q.; Hunt, S.P. Systemic inhibition of the mammalian target of rapamycin (mTOR) pathway reduces neuropathic pain in mice. Pain 2011, 152, 2582–2595. [Google Scholar] [CrossRef]





























Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Giraud, F.; Pereira, E.; Anizon, F.; Moreau, P. Recent Advances in Pain Management: Relevant Protein Kinases and Their Inhibitors. Molecules 2021, 26, 2696. https://doi.org/10.3390/molecules26092696
Giraud F, Pereira E, Anizon F, Moreau P. Recent Advances in Pain Management: Relevant Protein Kinases and Their Inhibitors. Molecules. 2021; 26(9):2696. https://doi.org/10.3390/molecules26092696
Chicago/Turabian StyleGiraud, Francis, Elisabeth Pereira, Fabrice Anizon, and Pascale Moreau. 2021. "Recent Advances in Pain Management: Relevant Protein Kinases and Their Inhibitors" Molecules 26, no. 9: 2696. https://doi.org/10.3390/molecules26092696
APA StyleGiraud, F., Pereira, E., Anizon, F., & Moreau, P. (2021). Recent Advances in Pain Management: Relevant Protein Kinases and Their Inhibitors. Molecules, 26(9), 2696. https://doi.org/10.3390/molecules26092696