
Development of Novel Potential Pleiotropic Compounds of Interest in Alzheimer’s Disease Treatment through Rigidification Strategy

 ,
,  and
and
Abstract
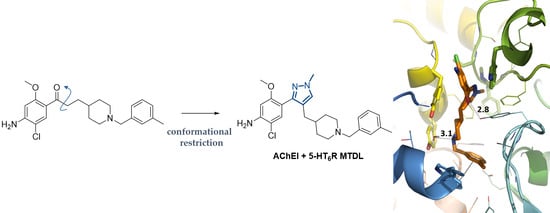
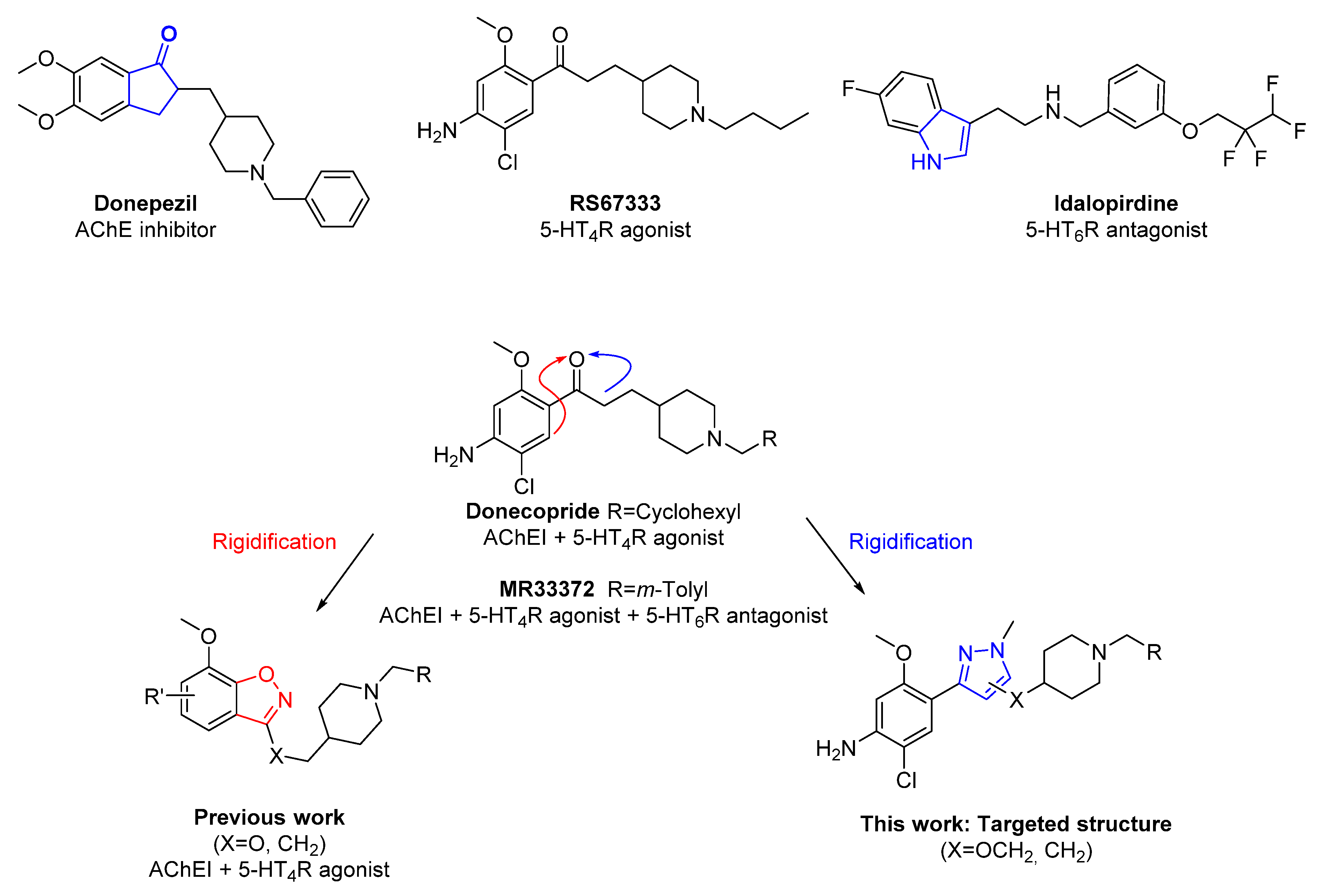
1. Introduction
2. Results
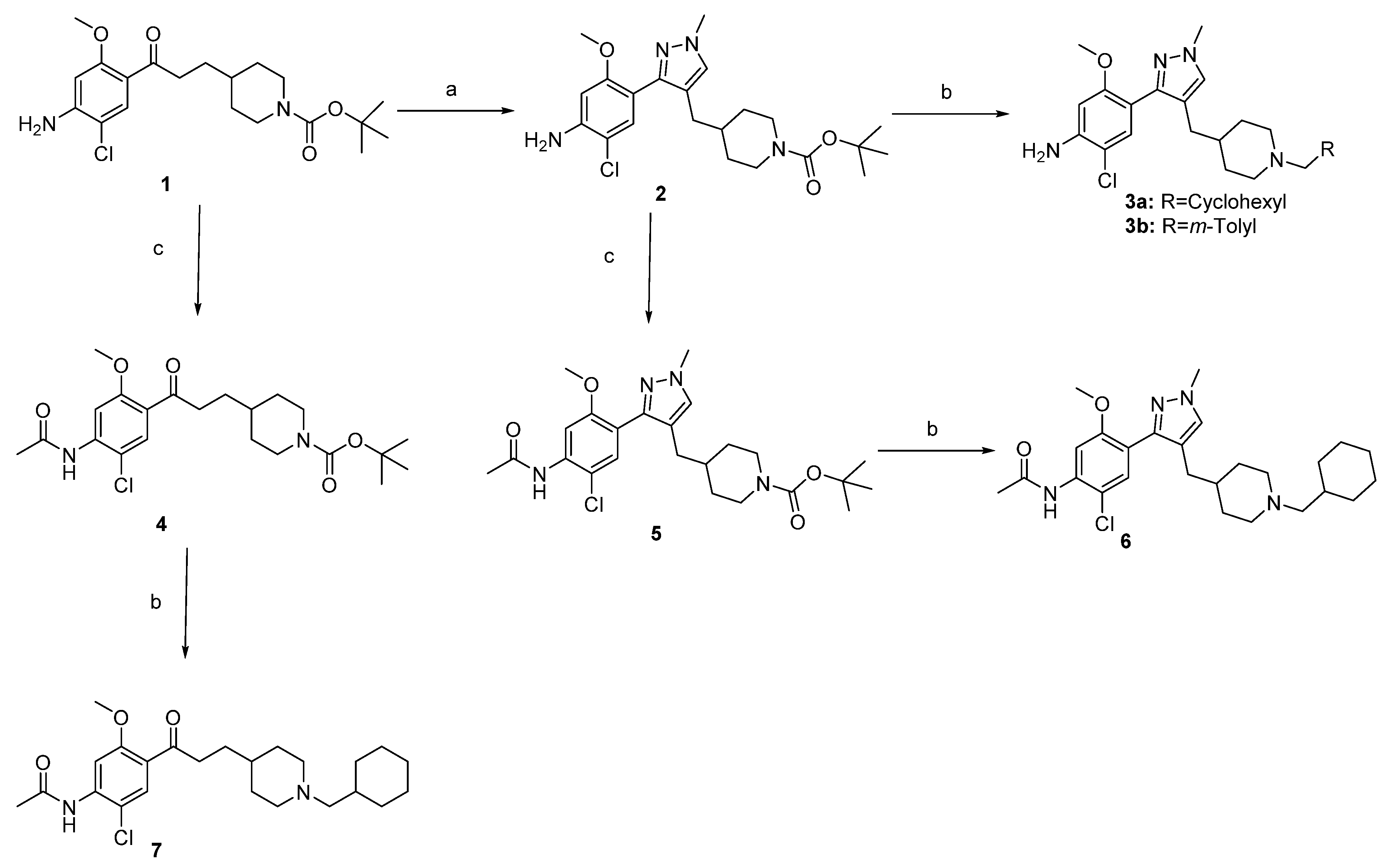
2.1. Chemistry
2.2. In Vitro Results
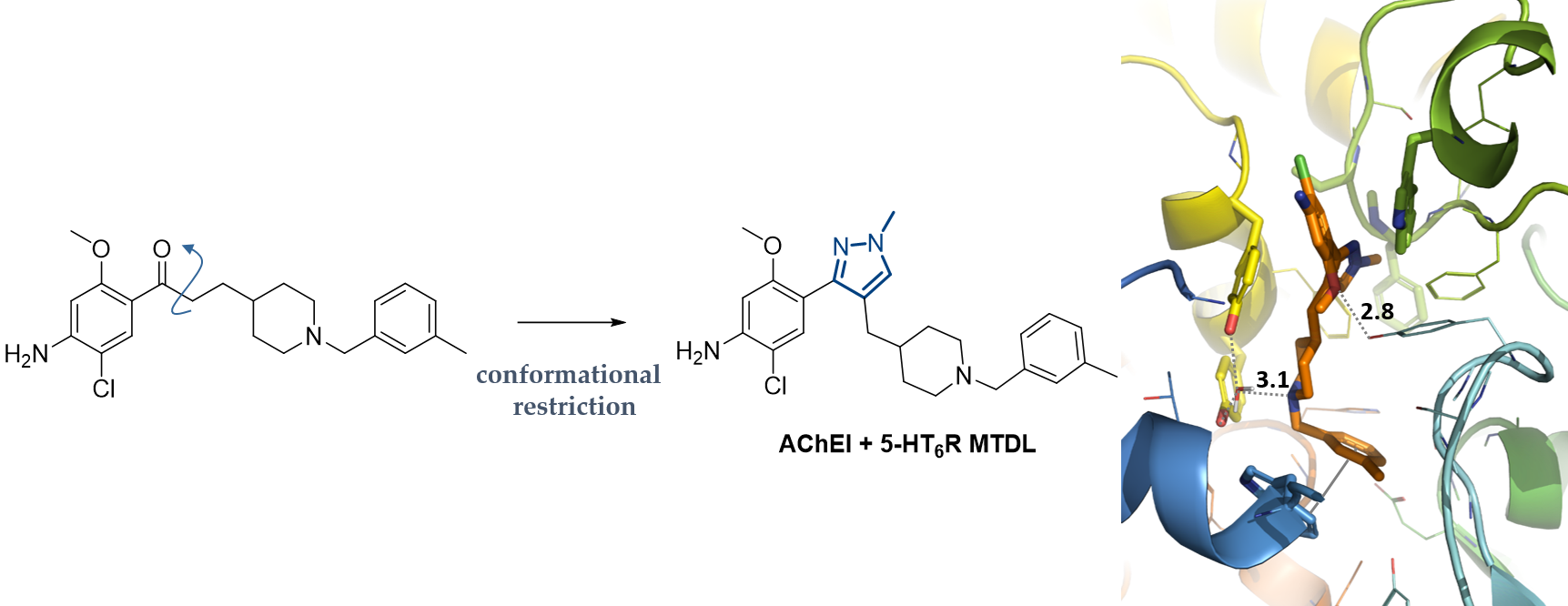
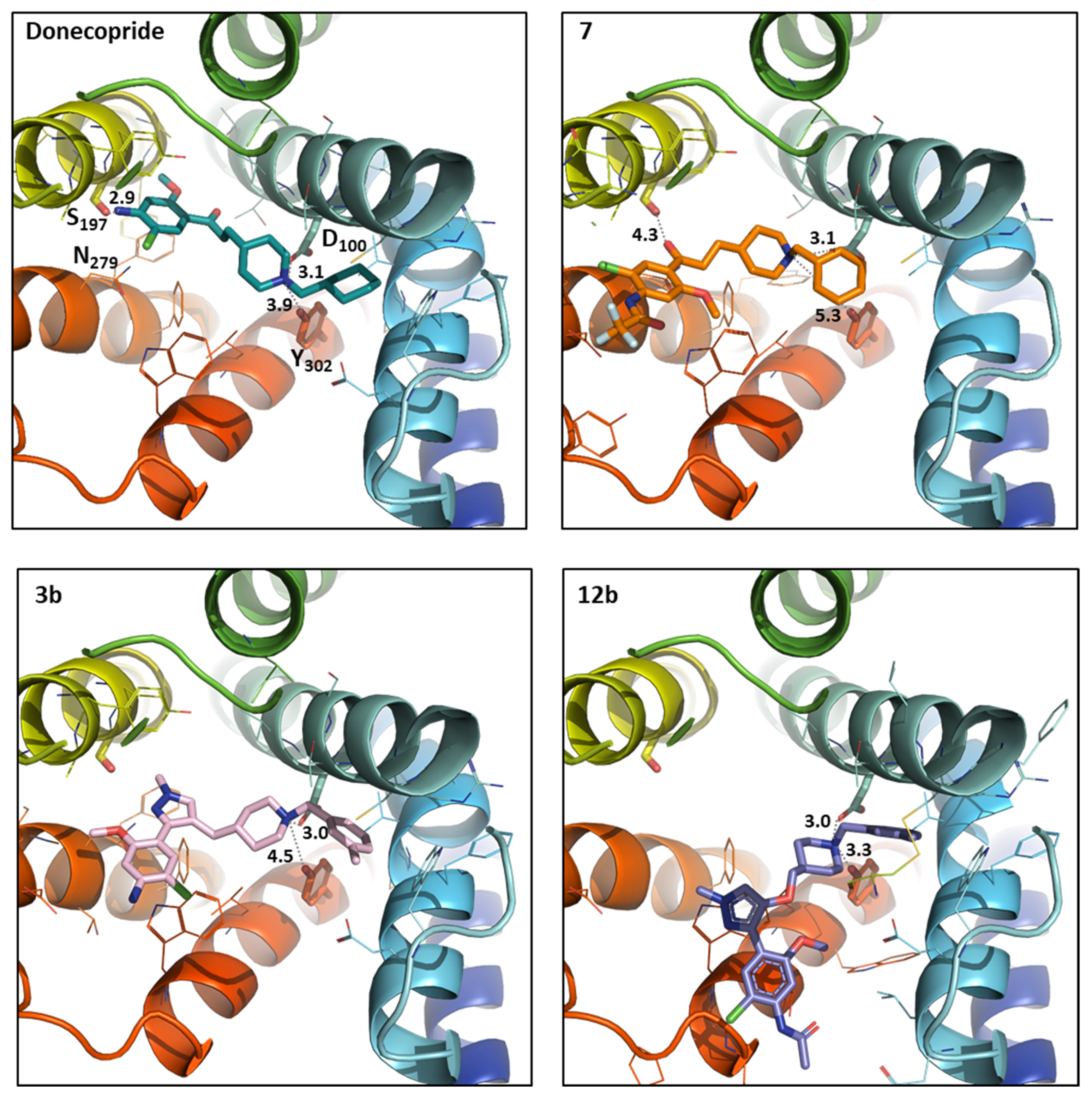
2.3. In Silico Results
3. Experimental
3.1. Chemistry
3.1.1. General Methods
3.1.2. Synthesis of Compounds (2–7, 9–12)
- tert-butyl 4-((3-(4-amino-5-chloro-2-methoxyphenyl)-1-methyl-1H-pyrazol-4-yl)methyl)piperidine-1-carboxylate2. An amount of 375 mg of compound 1 (0.947 mmol) were refluxed in 5 mL of DMF.DMA overnight. After evaporation under reduced pressure, the residue was dissolved in 10 mL of EtOH and 26 mL of methylhydrazine (3.788 mmol, 4 eq) were added. The mixture was refluxed for 4 h. After concentration, the new residue was dissolved in 20 mL of EtOH and 645 mg of ZnCl2 (4.735 mmol, 5 eq) were added. The new reactional mixture was refluxed for 2 h and then concentrated. The residue was dissolved in DCM and washed 3 times with water. The organic phase was dried on MgSO4, concentrated and purified on neutral Al2O3 to afford 155 mg of product 2 as a brown solid. Yield = 38%; m.p. = 67–69 °C; 1H-NMR (CDCl3, 400 MHz): 7.17 (s, 1H), 7.15 (s, 1H), 6.34 (s, 1H), 4.10 (br s, 2H), 3.99 (br s, 2H), 3.87 (s, 3H), 3.71 (s, 3H), 2.57 (t, J = 13.0 Hz, 2H), 2.29 (d, J = 7.0 Hz, 2H), 1.54 (d, J = 13.1 Hz, 2H), 1.45−1.38 (m, 10H), 0.97 (qd, J = 12.2 Hz, J’ = 4.2 Hz, 2H); 13C-NMR (CDCl3, 100 MHz): 156.8, 154.9, 147.4, 143.5, 131.7, 129.3, 118.7, 114.4, 110.7, 98.7, 79.2, 55.6, 44.0 (2C), 38.9, 37.0, 32.0 (2C), 31.1, 28.5 (3C); MS [M + H]+ = 434.86; IR (KBr, cm−1): 3468, 3351, 2931, 1686, 1624, 1461, 1444, 1426, 1365, 1247, 1211, 1173, 1211.
General Procedure for Acetylation
- tert-butyl 4-(3-(4-acetamido-5-chloro-2-methoxyphenyl)-3-oxopropyl)piperidine-1-carboxylate4. From 150 mg of compound 1 (0.379 mmol), 129 mg of compound 4 were obtained as a white solid (gradient of elution: DCM to DCM/EtOAc 9/1). Yield = 78%; m.p. = 123–125 °C; 1H-NMR (CDCl3, 400 MHz): 8.26 (s, 1H), 7.80 (br s, 1H), 7.77 (s, 1H), 4.07 (br s, 2H), 3.90 (s, 3H), 2.94 (m, 2H), 2.65 (m, 2H), 2.26 (s, 3H), 1.66–1.56 (m, 4H), 1.43 (s, 9H), 1.42 (m, 1H), 1.08 (m, 2H); 13C-NMR (CDCl3, 100 MHz): 199.6, 168.7, 158.5, 154.9, 138.8, 130.6, 123.4, 113.7, 104.0, 79.2, 56.0, 44.0 (2C), 40.9, 35.7, 32.1 (2C), 30.9, 28.5 (3C), 25.2; MS [M + H]+ = 438.84; IR (KBr, cm−1): 3323, 2975, 2930, 2853, 1690, 1674, 1599, 1578, 1513, 1451, 1402, 1242, 1163, 1017. HRMS [M + H + Na]+calcd for C22H31ClN2NaO5, 461.1819, exp 461.1813.
- tert-butyl 4-((3-(4-acetamido-5-chloro-2-methoxyphenyl)-1-methyl-1H-pyrazol-4-yl)methyl)piperidine-1-carboxylate5. From 38 mg of compound 2 (0.088 mmol), 35 mg of compound 5 were obtained as a white solid (gradient of elution: DCM to DCM/EtOAc 6/4). Yield = 83%; m.p. = 147–149 °C; 1H-NMR (CDCl3, 400 MHz): 8.20 (s, 1H), 7.71 (s, 1H), 7.32 (s, 1H), 7.17 (s, 1H), 3.98 (br s, 2H), 3.88 (s, 3H), 3.81 (s, 3H), 2.56 (t, J = 12.8 Hz, 2H), 2.31 (d, J = 7.0 Hz, 2H), 2.26 (s, 3H), 1.53 (d, J = 13.2 Hz, 2H), 1.45−1.38 (m, 10H), 0.96 (qd, J = 12.2 Hz, J’ = 4.3 Hz, 2H); 13C-NMR (CDCl3, 100 MHz): 168.4, 155.7, 154.8, 154.4, 144.7, 134.4, 127.4, 119.3, 113.8, 104.4, 86.0, 79.5, 75.7, 55.9, 43.5 (2C), 36.1, 33.7, 28.6 (2C), 28.5 (3C), 25.1; MS [M + H]+ = 476.82; IR (KBr, cm−1): 3420, 2930, 1688, 1584, 1527, 1450, 1388, 1244, 1161.
- 5-(4-amino-5-chloro-2-methoxyphenyl)-2-methyl-2,4-dihydro-3H-pyrazol-3-one9a. A total of 300 mg of compound 8 (1.107 mmol), 54 µL of methylhydrazine (0.996 mmol, 0.9 eq), and 3 mL of EtOH were stirred under microwaves irradiation at 160 °C for 45 min. The solvent was eliminated under reduced pressure and the residue was purified on silica gel (gradient of elution: DCM to EtOAc) to afford 143 mg of compound 9a as a white solid. Yield = 57%; m.p. = 203–205°C; 13C-NMR (CDCl3, 400 MHz): 7.81 (s, 1H), 6.27 (s, 1H), 4.31 (br s, 2H), 3.78 (s, 3H), 3.68 (s, 2H), 3.36 (s, 3H); 13C-NMR (CDCl3, 100 MHz): 172.5, 157.5, 152.4, 145.6, 128.0, 111.7, 111.4, 98.0, 55.6, 42.1, 31.3; MS [M + H]+ = 253.92; IR (KBr, cm−1): 3448, 3331, 2958, 2229, 1677, 1631, 1603, 1572, 1458, 1348, 1214, 1049; HRMS [M + H]+calcd for C11H13ClN3O2, 254.0696, exp 254.0693.
- 3-(4-acetamido-5-chloro-2-methoxyphenyl)-1-methyl-1H-pyrazol-5-yl acetate11. A total of 200 mg of compound 9a (0.80 mmol) were stirred in 2 mL of anhydric acetic at room temperature overnight. Water was added to obtain a white suspension. This latter was filtrated and washed with water. A total of 250 mg of product 11 were then obtained as a white solid. Yield = 94%. m.p. = 191–193°C; 1H-NMR (CDCl3, 400 MHz): 8.21 (s, 1H), 7.92 (s, 1H), 7.68 (s, 1H), 6.61 (s, 1H), 3.89 (s, 3H), 3.73 (s, 3H), 2.35 (s, 3H), 2.25 (s, 3H); 13C-NMR (CDCl3, 100 MHz): 168.4, 166.1, 155.8, 145.1, 144.8, 134.6, 127.3, 118.8, 113.6, 104.3, 95.5, 55.9, 34.7, 25.1, 20.7; MS [M + H]+ = 337.92; IR (KBr, cm−1): 3422, 2945, 1787, 1772, 1699, 1585, 1511, 1483, 1450, 1352, 1243, 1191, 1011. HRMS [M + H]+ calcd for C15H17ClN3O4, 338.0908, exp 338.0914.
- N-(2-chloro-5-methoxy-4-(1-methyl-5-oxo-4,5-dihydro-1H-pyrazol-3-yl)phenyl)acetamide9b. A total of 250 mg of compound 11 (0.742 mmol) and 21 mg of LiOH (0.890 mmol, 1.2 eq) were dissolved in 14 mL of EtOH/water (5/2). The reactional mixture was stirred at room temperature for 1 h 45 min. After evaporation in vacuo, water was added, adjusted at pH 4–5 and extracted 3 times with CHCl3. The organic phases were combined, washed once with water, dried on MgSO4, and concentrated to obtain 176 mg of monoprotected compound 9b and used without further purification. Yield = 76%; m.p. = 219 -221 °C; 1H-NMR (CDCl3, 400 MHz): 8.26 (s, 1H), 7.94 (s, 1H), 7.73 (br s, 1H), 3.88 (s, 3H), 3.74 (s, 2H), 3.39 (s, 3H), 2.27 (s, 3H); 13C-NMR (CDCl3, 100 MHz) 172.6, 168.6, 156.8, 151.6, 136.9, 126.9, 116.2, 114.0, 104.1, 56.0, 42.0, 31.3, 25.2; MS [M + H]+ = 295.85; IR (KBr, cm−1): 3316, 2340, 1681, 1606, 1586, 1547, 1506, 1450, 1392, 1209, 1015. HRMS [M + H]+ calcd for C13H15ClN3O3, 296.0802, exp 296.0803.
General Procedure for the O-Alkylation
- Tert-butyl 4-(((3-(4-acetamido-5-chloro-2-methoxyphenyl)-1-methyl-1H-pyrazol-5-yl)oxy)methyl)piperidine-1-carboxylate10b. From 167 mg of compound 9b (0.379 mmol), 60 mg of compound 10b were obtained as a white solid. Yield = 22%; m.p. = 182–184 °C; 1H-NMR (CDCl3, 400 MHz): 8.19 (s, 1H), 7.89 (s, 1H), 7.69 (br s, 1H), 6.00 (s, 1H), 4.15 (br s, 2H), 3.92 (d, J = 6.3 Hz, 2H), 3.89 (s, 3H), 3.66 (s, 3H), 2.73 (t, J = 13.0 Hz, 2H), 2.23 (s, 3H), 1.98 (m, 1H), 1.78 (d, J = 12.6 Hz, 2H), 1.45 (s, 9H), 1.27 (qd, J = 12.1 Hz, J’ = 4.2 Hz, 2H); 13C-NMR (CDCl3, 100 MHz): 168.4, 155.7, 154.8, 154.4, 144.7, 134.4, 127.4, 119.3, 113.8, 104.4, 86.0, 79.5, 75.7, 55.9, 43.5 (2C), 36.1, 33.7, 28.6 (2C), 28.5 (3C), 25.1; [M + H]+ = 492.76; IR (KBr, cm−1): 3420, 2936, 2853, 1690, 1585, 1557, 1528, 1450, 1436, 1365, 1247, 1173, 1147, 1018.
- Tert-butyl 4-(((1-methyl-3-phenyl-1H-pyrazol-5yl)oxy)methyl)piperidine-1-carboxylate10c. From 218 mg of compound 9c (0.647 mmol), 129 mg of compound 10c were obtained as a yellow solid. Yield = 27%; m.p. = 133–135 °C; 1H-NMR (CDCl3, 400 MHz): 7.73 (m, 2H), 7.37 (m, 2H), 7.27 (m, 1H), 5.80 (s, 1H), 4.16 (br s, 2H), 3.93 (d, J = 6.4 Hz, 2H), 3.68 (s, 3H), 2.75 (t, J = 12.8 Hz, 2H), 2.00 (m, 1H), 1.80 (d, J = 13.0 Hz, 2H), 1.47 (s, 9H), 1.29 (qd, J = 11.8 Hz, J’ = 4.6 Hz, 2H); 13C-NMR (CDCl3, 400 MHz): 155.1, 154.8, 149.3, 133.8, 128.5 (2C), 127.6, 125.2 (2C), 82.0, 79.6, 75.9, 43.5 (2C), 36.0, 33.8, 28.6 (2C), 28.5 (3C); MS [M + H]+ = 372.01; IR (KBr, cm−1): 3426, 2925, 2853, 1742, 1690, 1626, 1558, 1512, 1454, 1422, 1367, 1275.
- Tert-butyl 4-(((3-(3,4-dimethoxyphenyl)-1-methyl-1H-pyrazol-5-yl)oxy)methyl)piperidine-1-carboxylate10d. From 151 mg of compound 9d (0.379 mmol), 80 mg of compound 10d were obtained as a yellow oil. Yield = 21%; 1H-NMR (CDCl3, 400 MHz): 7.33 (d, J = 2.0 Hz, 1H), 7.22 (d, J = 8.2 Hz, J’ = 2.0 Hz, 1H), 6.86 (d, J = 8.3 Hz, 1H), 5.74 (s, 1H), 4.18 (br s, 2H), 3.95 (s, 3H), 3.93 (d, J = 6.4 Hz, 2H), 3.90 (s, 3H), 3.67 (s, 3H), 2.75 (t, J = 13.0 Hz, 2H), 2.00 (m, 1H), 1.79 (d, J = 10.3 Hz, 2H), 1.47 (s, 9H), 1.29 (qd, J = 11.7 Hz, J = 3.9 Hz, 2H); MS [M + H]+ = 431.91;
General Procedure for the N-Alkylation
- 2-chloro-4-(4-((1-(cyclohexylmethyl)piperidin-4-yl)methyl)-1-methyl-1H-pyrazol-3-yl)-5-methoxyaniline3a. From 59 mg of compound 2 (0.136 mmol), 18 mg of compound 3a were obtained as a pale brown gum. Yield = 31%; 1H-NMR (CDCl3, 400 MHz): 7.17 (s, 1H), 7.15 (s, 1H), 6.34 (s, 1H), 4.09 (br s, 2H), 3.88 (br s, 2H), 3.72 (s, 3H), 2.83 (m, 2H), 2.29 (d, J = 6.6 Hz, 2H), 2.10 (m, 2H), 1.78−1.10 (m, 17 H), 0.89−0.80 (m, 2H); 13C-NMR (CDCl3, 100 MHz): 156.8, 147.4, 143.4, 131.8, 129.4, 119.1, 114.6, 110.7, 98.7, 66.0, 55.6, 54.4 (2C), 38.9, 36.9, 35.1, 32.0 (2C), 31.9 (2C), 31.1, 26.7, 26.1 (2C); MS [M + H]+ = 431.02; IR (KBr, cm−1): 3438, 2921, 2849, 1622, 1525, 1461, 1445, 1402, 1211. HRMS [M + H]+ calcd for C24H36ClN4O, 431.2578, exp 431.2576.
- 2-chloro-5-methoxy-4-(1-methyl-4-((1-(3-methylbenzyl)piperidin-4-yl)methyl)-1H-pyrazol-3-yl)aniline 3b. From 41 mg of compound 2 (0.095 mmol), 19 mg of compound 3b were obtained as a pale yellow gum. Yield = 46%; 1H-NMR (CDCl3, 400 MHz): 7.18 (t, J = 7.6 Hz, 1H), 7.17 (s, 1H), 7.14 (s, 1H), 7.10 (s, 1H), 7.05 (m, 2H), 6.33 (s, 1H), 4.09 (br s, 2H), 3.86 (s, 3H), 3.71 (s, 3H), 3.40 (s, 2H), 2.81 (m, 2H), 2.33 (s, 3H), 2.28 (d, J = 6.8 Hz, 2H), 1.82 (td, J = 11.8 Hz, J’ = 2.4 Hz, 2H), 1.55 (d, J = 12.7 Hz, 2H), 1.28 (m, 1H), 1.15 (qd, J = 12.3 Hz, J’ = 3.6 Hz, 2H). 13C-NMR (CDCl3, 100 MHz): 156.8, 147.4, 143.4, 138.4, 137.7, 131.8, 130.0, 129.3, 128.0, 127.6, 126.4, 119.2, 114.6, 110.7, 98.7, 63.6, 55.6, 53.9 (2C), 38.9, 36.9, 32.2 (2C), 31.1, 21.4; MS [M + H]+ = 439.74; IR (KBr, cm−1): 3434, 2925, 1623, 1525, 1460, 1445, 1402, 1211. HRMS [M + H]+ calcd for C25H32ClN4O, 439.2265, exp 439.2268.
- N-(2-chloro-4-(4-((1-(cyclohexylmethyl)piperidin-4-yl)methyl)-1-methyl-1H-pyrazol-3-yl)-5-methoxyphenyl)acetamide 6. From 60 mg of compound 5 (0.126 mmol), 32 mg of compound 6 were obtained as a beige solid. Yield = 46%; m.p. = 148–150 °C; 1H-NMR (CDCl3, 400 MHz): 8.20 (s, 1H), 7.70 (br s, 1H), 7.31 (s, 1H), 7.16 (s, 1H), 3.87 (s, 3H), 3.80 (s, 3H), 2.77 (d, J = 11.8 Hz, 2H), 2.29 (d, J = 6.8 Hz, 2H), 2.26 (s, 3H), 2.03 (d, J = 7.0 Hz, 2H), 1.74−1.61 (m, 6H), 1.52 (d, J = 12.6 Hz, 2H), 1.43 (m, 1H), 1.28−1.08 (m, 7H), 0.87−0.77 (m, 2H); 13C-NMR (CDCl3, 100 MHz): 168.5, 156.4, 146.8, 135.1, 130.9, 129.6, 119.8, 119.4, 112.9, 103.8, 66.2, 55.8, 54.5 (2C), 38.9, 37.0, 35.3, 32.3 (2C), 32.1 (2C), 31.2, 26.8, 26.2 (2C), 25.2; MS [M + H]+ = 472.92; IR (KBr, cm−1): 3420, 2921, 2849, 1694, 1625, 1584, 1527, 1449, 1388, 1243; HRMS [M + H]+calcd for C26H34N3O3, 473.2683, exp 473.2682.
- N-(2-chloro-4-(3-(1-(cyclohexylmethyl)piperidin-4-yl)propanoyl)-5-methoxyphenyl)acetamide 7. From 81 mg of compound 4 (0.185 mmol), 34 mg of compound 7 were obtained as a beige gum. Yield = 42%; 1H-NMR (CDCl3, 400 MHz): 8.26 (s, 1H), 7.77 (s, 1H), 7.75 (br s, 1H), 3.90 (s, 3H), 2.93 (t, J = 7.6 Hz, 2H), 2.83 (d, J = 11.0 Hz, 2H), 2.25 (s, 3H), 2.05 (d, J = 7.1 Hz, 2H), 1.79–1.45 (m, 10H), 1.23–1.05 (m, 8H), 0.83 (m, 2H); 13C-NMR (CDCl3, 100 MHz): 200.1, 168.6, 158.4, 138.6, 130.6, 123.5, 113.5, 103.9, 66.2, 55.9, 53.3 (2C), 41.2, 35.7, 35.2, 32.2 (2C), 32.1 (2C), 31.0, 26.8, 26.2 (2C), 25.2; MS [M + H]+ = 434.82; IR (KBr, cm−1): 3345, 2921, 2849, 1704, 1672, 1599, 1578, 1512, 1450, 1401, 1263, 1241, 1176, 1014; HRMS [M + H]+calcd for C24H36ClN2O3, 435.2414, exp 435.2415.
- N-(2-chloro-5-methoxy-4-(1-methyl-5-((1-(3-methylbenzyl)piperidin-4-yl)methoxy)-1H-pyrazol-3-yl)phenyl)acetamide 12b. From 70 mg of compound 10b (0.126 mmol), 45 mg of compound 12b were obtained as a beige solid. Yield = 64%; m.p. = 174–176 °C; 1H-NMR (CDCl3, 400 MHz): 8.22 (s, 1H), 7.90 (s, 1H), 7.67 (br s, 1H), 7.21 (t, J = 7.4 Hz, 1H), 7.14−7.06 (m, 3H), 6.00 (s, 1H), 3.93 (d, J = 6.3 Hz, 2H), 3.91 (s, 3H), 3.67 (s, 3H), 3.48 (s, 2H), 2.94 (d, J = 11.2 Hz, 2H), 2.35 (s, 3H), 2.25 (s, 3H), 2.00 (td, J = 11.9 Hz, J’ = 2.5 Hz, 2H), 1.85−1.77 (m 3H), 1.44 (qd, J = 11.9 Hz, J’ = 3.7 Hz, 2H); 13C-NMR (CDCl3, 100 MHz): 168.4, 155.7, 154.7, 144.7, 138.2, 137.8, 134.4, 130.0, 128.1, 127.8, 127.4, 126.3, 119.4, 113.7, 104.3, 86.0, 76.2, 63.5, 56.0, 53.3 (2C), 35.8, 33.7, 28.9 (2C), 25.1, 21.4; MS [M + H]+ = 496.82; IR (KBr, cm−1): 3415, 2937, 1694, 1680, 1585, 1557, 1527, 1449, 1361, 1243; HRMS [M + H]+calcd for C27H34ClN4O3, 497.2319, exp 497.2319.
- 4-(((1-methyl-3-phenyl-1H-pyrazol-5-yl)oxy)methyl)-1-(3-methylbenzyl)piperidine 12c. From 62 mg of compound 10c (0.126 mmol), 20 mg of compound 12c were obtained as an oil. Yield = 32%; 1H-NMR (CDCl3, 400 MHz): 7.73 (m, 2H), 7.37 (m, 2H), 7.27 (m, 1H), 7.22 (t, J = 7.5 Hz, 1H), 7.15 (s, 1H), 7.13 (d, J = 7.5 Hz, 1H), 7.08 (d, J = 7.5 Hz, 1H), 5.80 (s, 1H), 3.94 (d, J = 6.4 Hz, 2H), 3.68 (s, 3H), 3.52 (s, 2H), 2.98 (d, J = 10.7 Hz, 2H), 2.35 (s, 3H), 2.04 (t, J = 1.6 HZ, 2H), 1.86 (m, 1H), 1.80 (d, J = 13.5 Hz, 2H), 1.48 (m, 2H); 13C-NMR (CDCl3, 100 MHz): 155.3, 149.3, 137.9, 137.6, 133.9, 130.1, 128.5 (2C), 128.1, 127.9, 127.5, 126.5, 125.2 (2C), 82.0, 76.2, 63.4, 53.2 (2C), 35.7, 33.8, 28.7 (2C), 21.4; MS [M + H]+ = 376.00; HRMS [M + H]+calcd for C24H30N3O, 376.2389, exp 376.2392.
- 4-(((3-(3,4-dimethoxyphenyl)-1_methyl-1H-pyrazol-5-yl)oxy)methyl)-1-(3-methylbenzyl)piperidine12d. From 70 mg of compound 10d (0.126 mmol), 35 mg of compound 12d were obtained as a pale brown oil. Yield = 67%; 1H-NMR (CDCl3, 400 MHz): 7.33 (d, J = 2.0 Hz, 1H), 7.24−7.19 (m, 2H), 7.14−7.06 (m, 3H), 6.87 (d, J = 8.4 Hz, 1H), 5.73 (s, 1H), 3.95 (s, 3H), 3.93 (d, J = 6.4 Hz, 2H), 3.90 (s, 3H), 3.67 (s, 3H), 3.49 (s, 2H), 2.95 (d, J = 11.7 Hz, 2H), 2.35 (s, 3H), 2.00 (td, J = 11.7 Hz, J’ = 2.4 Hz, 2H), 1.85 (m, 1H), 1.81 (d, J = 13.0 Hz, 2H), 1.44 (qd, J = 12.3 Hz, J = 3.7 Hz, 2H); 13C-NMR (CDCl3, 100 MHz): 155.3, 149.2, 149.0, 148.7, 138.1, 137.8, 130.0, 128.1, 127.8, 127.1, 126.4, 117.8, 111.0, 108.1, 81.7, 76.3, 63.5, 55.9, 55.9, 53.2 (2C), 35.8, 33.7, 28.8 (2C), 21.4; MS [M + H]+ = 435.94; IR (KBr, cm−1): 2935, 1609, 1589, 1558, 1525, 1466, 1435, 1258, 1234, 1029; HRMS [M + H]+calcd for C26H34N3O3, 436.2600, exp 436.2596.
3.2. Biological Evaluation
3.2.1. In Vitro Tests of AChE Biological Activity
3.2.2. Pharmacological Characterization of Drugs on Human 5-HT4R
3.2.3. Pharmacological Characterization of Drugs on Human 5-HT6R
3.3. In Silico Study
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Cavalli, A.; Bolognesi, M.L.; Minarini, A.; Rosini, M.; Tumiatti, V.; Recanatini, M.; Melchiorre, C. Multi-target-Directed Ligands To Combat Neurodegenerative Diseases. J. Med. Chem. 2008, 51, 347–372. [Google Scholar] [CrossRef]
- Albertini, C.; Salerno, A.; de Sena Murteira Pinheiro, P.; Bolognesi, M.L. From combinations to multitarget-directed ligands: A continuum in Alzheimer’s disease polypharmacology. Med. Res. Rev. 2020, 1–28. [Google Scholar] [CrossRef]
- Lalut, J.; Rochais, C.; Dallemagne, P. Multiple Ligands in Neurodegenerative Diseases. In Drug Selectivity–An Evolving Concept in Drug Discovery, Book Series “Methods and Principles in Medicinal Chemistry; Wiley-VCH Publishing House: Hoboken, NJ, USA, 2017; pp. 477–508. [Google Scholar]
- Rodríguez-Soacha, D.A.; Scheiner, M.; Decker, M. Multi-target-directed-ligands acting as enzyme inhibitors and receptor ligands. Eur. J. Med. Chem. 2019, 180, 690–706. [Google Scholar] [CrossRef]
- Lecoutey, C.; Hedou, D.; Freret, T.; Giannoni, P.; Gaven, F.; Since, M.; Bouet, V.; Ballandonne, C.; Corvaisier, S.; Fréon, A.M.; et al. Design of donecopride, a dual serotonin subtype 4 receptor agonist/acetylcholinesterase inhibitor with potential interest for Alzheimer’s disease treatment. Proc. Natl. Acad. Sci. USA 2014, 111, E3825–E3830. [Google Scholar] [CrossRef]
- Rochais, C.; Lecoutey, C.; Gaven, F.; Giannoni, P.; Hamidouche, K.; Hedou, D.; Dubost, E.; Genest, D.; Yahiaoui, S.; Freret, T.; et al. Novel Multitarget-Directed Ligands (MTDLs) with Acetylcholinesterase (AChE) Inhibitory and Serotonergic Subtype 4 Receptor (5-HT4R) Agonist Activities As Potential Agents against Alzheimer’s Disease: The Design of Donecopride. J. Med. Chem. 2015, 58, 3172–3187. [Google Scholar] [CrossRef] [PubMed]
- Rochais, C.; Lecoutey, C.; Hamidouche, K.; Giannoni, P.; Gaven, F.; Cem, E.; Mignani, S.; Baranger, K.; Freret, T.; Bockaert, J.; et al. Donecopride, a Swiss army knife with potential against Alzheimer’s disease. Br. J. Pharmacol. 2020, 177, 1988–2005. [Google Scholar] [CrossRef] [PubMed]
- Cachard-Chastel, M.; Lezoualc’H, F.; Dewachter, I.; Deloménie, C.; Croes, S.; Devijver, H.; Langlois, M.; Van Leuven, F.; Sicsic, S.; Gardier, A.M. 5-HT4 receptor agonists increase sAPPα levels in the cortex and hippocampus of male C57BL/6j mice. Br. J. Pharmacol. 2007, 150, 883–892. [Google Scholar] [CrossRef]
- Maillet, M.; Robert, S.J.; Cacquevel, M.; Gastineau, M.; Vivien, D.; Bertoglio, J.; Zugaza, J.L.; Fischmeister, R.; Lezoualc’H, F. Crosstalk between Rap1 and Rac regulates secretion of sAPPα. Nat. Cell Biol. 2003, 5, 633–639. [Google Scholar] [CrossRef]
- Egiannoni, P.; Egaven, F.; Bundel, D.E.; Ebaranger, K.; Emarchetti-Gauthier, E.; Roman, F.S.; Evaljent, E.; Emarin, P.; Ebockaert, J.; Erivera, S.; et al. Early administration of RS 67333, a specific 5-HT4 receptor agonist, prevents amyloidogenesis and behavioral deficits in the 5XFAD mouse model of Alzheimer’s disease. Front. Aging Neurosci. 2013, 5, 96. [Google Scholar] [CrossRef]
- Lalut, J.; Karila, D.; Dallemagne, P.; Rochais, C. Modulating 5-HT4and 5-HT6receptors in Alzheimer’s disease treatment. Futur. Med. Chem. 2017, 9, 781–795. [Google Scholar] [CrossRef]
- Barnes, N.M.; Ahern, G.P.; Becamel, C.; Bockaert, J.; Camilleri, M.; Chaumont-Dubel, S.; Claeysen, S.; Cunningham, K.A.; Fone, K.C.; Gershon, M.; et al. International Union of Basic and Clinical Pharmacology. CX. Classification of Receptors for 5-hydroxytryptamine; Pharmacology and Function. Pharmacol. Rev. 2021, 73, 310–520. [Google Scholar] [CrossRef]
- Karila, D.; Freret, T.; Bouet, V.; Boulouard, M.; Dallemagne, P.; Rochais, C. Therapeutic Potential of 5-HT6Receptor Agonists. J. Med. Chem. 2015, 58, 7901–7912. [Google Scholar] [CrossRef] [PubMed]
- Benhamú, B.; Martín-Fontecha, M.; Vázquez-Villa, H.; Pardo, L.; López-Rodríguez, M.L. Serotonin 5-HT6Receptor Antagonists for the Treatment of Cognitive Deficiency in Alzheimer’s Disease. J. Med. Chem. 2014, 57, 7160–7181. [Google Scholar] [CrossRef]
- Arnt, J.; Bang-Andersen, B.; Grayson, B.; Bymaster, F.P.; Cohen, M.P.; Giethlen, B.; Kreilgaard, M.; McKinzie, D.L.; Neill, J.C.; Nielsen, S.M.; et al. Lu AE58054, a 5-HT6 antagonist, reverses cognitive impairment induced by subchronic phencyclidine in a novel object recognition test in rats. Int. J. Neuropsychopharmacol. 2010, 13, 1021–1033. [Google Scholar] [CrossRef]
- Ferris, C.F.; Kulkarni, P.; Yee, J.R.; Nedelman, M.; De Jong, I.E.M. The Serotonin Receptor 6 Antagonist Idalopirdine and Acetylcholinesterase Inhibitor Donepezil Have Synergistic Effects on Brain Activity—A Functional MRI Study in the Awake Rat. Front. Pharmacol. 2017, 8, 279. [Google Scholar] [CrossRef] [PubMed]
- Yahiaoui, S.; Hamidouche, K.; Ballandonne, C.; Davis, A.; Sopkova-de Oliveira Santos, J.; Freret, T.; Boulouard, M.; Rochais, C.; Dallemagne, P. Design, synthesis, and pharmacological evaluation of multitarget-directed ligands with both serotonergic subtype 4 receptor (5-HT4R) partial agonist and 5-HT6R antagonist activities, as potential treatment of Alzheimer’s disease. Eur. J. Med. Chem. 2016, 121, 283–293. [Google Scholar] [CrossRef] [PubMed]
- Hatat, B.; Yahiaoui, S.; Lecoutey, C.; Davis, A.; Freret, T.; Boulouard, M.; Claeysen, S.; Rochais, C.; Dallemagne, P. A Novel in vivo Anti-amnesic Agent, Specially Designed to Express Both Acetylcholinesterase (AChE) Inhibitory, Serotonergic Subtype 4 Receptor (5-HT4R) Agonist and Serotonergic Subtype 6 Receptor (5-HT6R) Inverse Agonist Activities, With a Potential Interest Against Alzheimer’s Disease. Front. Aging Neurosci. 2019, 11, 148. [Google Scholar] [CrossRef]
- Fang, Z.; Song, Y.; Zhan, P.; Zhang, Q.; Liu, X. Conformational restriction: An effective tactic in ’follow-on’-based drug discovery. Futur. Med. Chem. 2014, 6, 885–901. [Google Scholar] [CrossRef] [PubMed]
- Lalut, J.; Payan, H.; Davis, A.; Lecoutey, C.; Legay, R.; Santos, J.S.-D.O.; Claeysen, S.; Dallemagne, P.; Rochais, C. Rational design of novel benzisoxazole derivatives with acetylcholinesterase inhibitory and serotoninergic 5-HT4 receptors activities for the treatment of Alzheimer’s disease. Sci. Rep. 2020, 10, 3014. [Google Scholar] [CrossRef]
- Bredereck, H.; Simchen, G.; Rebsdat, S.; Kantlehner, W.; Horn, P.; Wahl, R.; Hoffmann, H.; Grieshaber, P. Darstellung und Eigenschaften der Amidacetale und Aminalester. Chem. Ber. 1968, 101, 41–50. [Google Scholar] [CrossRef]
- Beria, I.; Ballinari, D.; Bertrand, J.A.; Borghi, D.; Bossi, R.T.; Brasca, M.G.; Cappella, P.; Caruso, M.; Ceccarelli, W.; Ciavolella, A.; et al. Identification of 4,5-Dihydro-1H-pyrazolo[4,3-h]quinazoline Derivatives as a New Class of Orally and Selective Polo-Like Kinase 1 Inhibitors. J. Med. Chem. 2010, 53, 3532–3551. [Google Scholar] [CrossRef]
- Genest, D.; Rochais, C.; Lecoutey, C.; Sopkova Oliveira Santos, J.S.; Ballandonne, C.; Butt-Gueulle, S.; Legay, R.; Since, M.; Dallemagne, P. Design, synthesis and biological evaluation of novel indano- and thiaindano-pyrazoles with potential interest for Alzheimer’s disease. Med. Chem. Comm. 2013, 4, 925. [Google Scholar] [CrossRef]
- Ellman, G.L.; Courtney, K.; Andres, V.; Featherstone, R.M. A new and rapid colorimetric determination of acetylcholinesterase activity. Biochem. Pharmacol. 1961, 7, 88–95. [Google Scholar] [CrossRef]
- Cheung, J.; Rudolph, M.J.; Burshteyn, F.; Cassidy, M.S.; Gary, E.N.; Love, J.; Franklin, M.C.; Height, J.J. Structures of Human Acetylcholinesterase in Complex with Pharmacologically Important Ligands. J. Med. Chem. 2012, 55, 10282–10286. [Google Scholar] [CrossRef]
- Jones, G.; Willett, P.; Glen, R.C. Molecular recognition of receptor sites using a genetic algorithm with a description of desolvation. J. Mol. Biol. 1995, 245, 43–53. [Google Scholar] [CrossRef]
- Jones, G.; Willett, P.; Glen, R.C.; Leach, A.R.; Taylor, R. Development and validation of a genetic algorithm for flexible docking. J. Mol. Biol. 1997, 267, 727–748. [Google Scholar] [CrossRef]
- Dubost, E.; Dumas, N.; Fossey, C.; Magnelli, R.; Butt-Gueulle, S.; Ballandonne, C.; Caignard, D.H.; Dulin, F.; Santos, J.S.D.-O.; Millet, P.; et al. Synthesis and Structure–Affinity Relationships of Selective High-Affinity 5-HT4 Receptor Antagonists: Application to the Design of New Potential Single Photon Emission Computed Tomography Tracers. J. Med. Chem. 2012, 55, 9693–9707. [Google Scholar] [CrossRef] [PubMed]
- Cherezov, V.; Rosenbaum, D.M.; Hanson, M.A.; Rasmussen, S.G.F.; Thian, F.S.; Kobilka, T.S.; Choi, H.-J.; Kuhn, P.; Weis, W.I.; Kobilka, B.K.; et al. High-Resolution Crystal Structure of an Engineered Human 2-Adrenergic G Protein-Coupled Receptor. Science 2007, 318, 1258–1265. [Google Scholar] [CrossRef]
- Rivail, L.; Giner, M.; Gastineau, M.; Berthouze, M.; Soulier, J.-L.; Fischmeister, R.; Lezoualc’H, F.; Maigret, B.; Sicsic, S.; Berque-Bestel, I. New insights into the human 5-HT4 receptor binding site: Exploration of a hydrophobic pocket. Br. J. Pharmacol. 2004, 143, 361–370. [Google Scholar] [CrossRef] [PubMed]
- Jain, E.; Bairoch, A.; Duvaud, S.; Phan, I.; Redaschi, N.; Suzek, B.E.; Martin, M.J.; McGarvey, P.; Gasteiger, E. Infrastructure for the life sciences: Design and implementation of the UniProt website. BMC Bioinform. 2009, 10, 1–19. [Google Scholar] [CrossRef]
- Shia, J.; Blundell, T.L.; Mizuguchia, K. FUGUE: Sequence-structure homology recognition using environment-specific substitution tables and structure-dependent gap penalties11Edited by B. Honig. J. Mol. Biol. 2001, 310, 243–257. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Zhou, Y. Fold recognition by combining sequence profiles derived from evolution and from depth-dependent structural alignment of fragments. Proteins Struct. Funct. Bioinform. 2004, 58, 321–328. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Madden, T.L.; Schäffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef]
- Schäaffer, A.A.; Aravind, L.; Madden, T.L.; Shavirin, S.; Spouge, J.L.; Wolf, Y.I.; Koonin, E.V.; Altschul, S.F. Improving the accuracy of PSI-BLAST protein database searches with composition-based statistics and other refinements. Nucleic Acids Res. 2001, 29, 2994–3005. [Google Scholar] [CrossRef] [PubMed]
- Söding, J. Protein homology detection by HMM-HMM comparison. Bioinformatics 2004, 21, 951–960. [Google Scholar] [CrossRef]
- Pons, J.-L.; Labesse, G. @TOME-2: A new pipeline for comparative modeling of protein-ligand complexes. Nucleic Acids Res. 2009, 37, W485–W491. [Google Scholar] [CrossRef] [PubMed]
- Eswar, N.; Eramian, D.; Webb, B.; Shen, M.; Sali, A. Protein structure modeling with MODELLER. Methods Mol. Biol. 2008, 426, 145–159. [Google Scholar] [CrossRef]
- Eisenberg, D.; Lüthy, R.; Bowie, J.U. VERIFY3D: Assessment of protein models with three-dimensional profiles. Methods Enzymol. 1997, 277, 396–404. [Google Scholar] [CrossRef]
- Gracy, J.; Chiche, L.; Sallantin, J. Improved alignment of weakly homologous protein sequences using structural information. Protein Eng. Des. Sel. 1993, 6, 821–829. [Google Scholar] [CrossRef]
- Robert, X.; Gouet, P. Deciphering key features in protein structures with the new ENDscript server. Nucl. Acids Res. 2014, 42, 320–324. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| hAChE | h5-HT4R | h5-HT6R | ||||
|---|---|---|---|---|---|---|
| Compound | % Inhibition at 10−6 M | IC50 (nM) (n = 3) | % Inhibition at 10−6 M | Ki (nM) (n = 3) | % Inhibition at 10−6M | Ki (nM) (n = 3) |
| Donepezil | 98% | 7.7 ± 1.3 | − | − | − | − |
| RS67333 | − | − | 100 | 5.1 ± 0.51 | − | − |
| Idalopirdine | − | − | − | − | 106 | 7.05 ± 0.61 |
| Donecopride | 96% | 16 ± 5 | 100% | 7.05 ± 0.61 | 30% | ND |
| MR33372 | 89% | (16 ± 4) × 10 | 98% | (17 ± 1) × 10 | 80% | (23 ± 5) × 10 |
| 7 | 98% | 34 ± 5 | 94% | (3 ± 1) × 102 | − | ND |
| 3a | 34% | ND | 34% | ND | 2% | ND |
| 3b | 76% | 110 ± 4 | 20% | ND | 68% | (47 ± 9) × 10 |
| 6 | 31% | ND | 16% | ND | 0% | ND |
| 12b | 15% | ND | 9% | ND | 0% | ND |
| 12c | 15% | ND | 10% | ND | 6% | ND |
| 12d | 6% | ND | 9% | ND | 9% | ND |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lecoutey, C.; Legay, R.; Davis, A.; Sopková-de Oliveira Santos, J.; Dallemagne, P.; Rochais, C. Development of Novel Potential Pleiotropic Compounds of Interest in Alzheimer’s Disease Treatment through Rigidification Strategy. Molecules 2021, 26, 2536. https://doi.org/10.3390/molecules26092536
Lecoutey C, Legay R, Davis A, Sopková-de Oliveira Santos J, Dallemagne P, Rochais C. Development of Novel Potential Pleiotropic Compounds of Interest in Alzheimer’s Disease Treatment through Rigidification Strategy. Molecules. 2021; 26(9):2536. https://doi.org/10.3390/molecules26092536
Chicago/Turabian StyleLecoutey, Cédric, Rémi Legay, Audrey Davis, Jana Sopková-de Oliveira Santos, Patrick Dallemagne, and Christophe Rochais. 2021. "Development of Novel Potential Pleiotropic Compounds of Interest in Alzheimer’s Disease Treatment through Rigidification Strategy" Molecules 26, no. 9: 2536. https://doi.org/10.3390/molecules26092536
APA StyleLecoutey, C., Legay, R., Davis, A., Sopková-de Oliveira Santos, J., Dallemagne, P., & Rochais, C. (2021). Development of Novel Potential Pleiotropic Compounds of Interest in Alzheimer’s Disease Treatment through Rigidification Strategy. Molecules, 26(9), 2536. https://doi.org/10.3390/molecules26092536