
Neuroprotective Effects of Curcumin in Methamphetamine-Induced Toxicity

 , , ,
, , , 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. Effects of CUR on PC12 Cell Viability
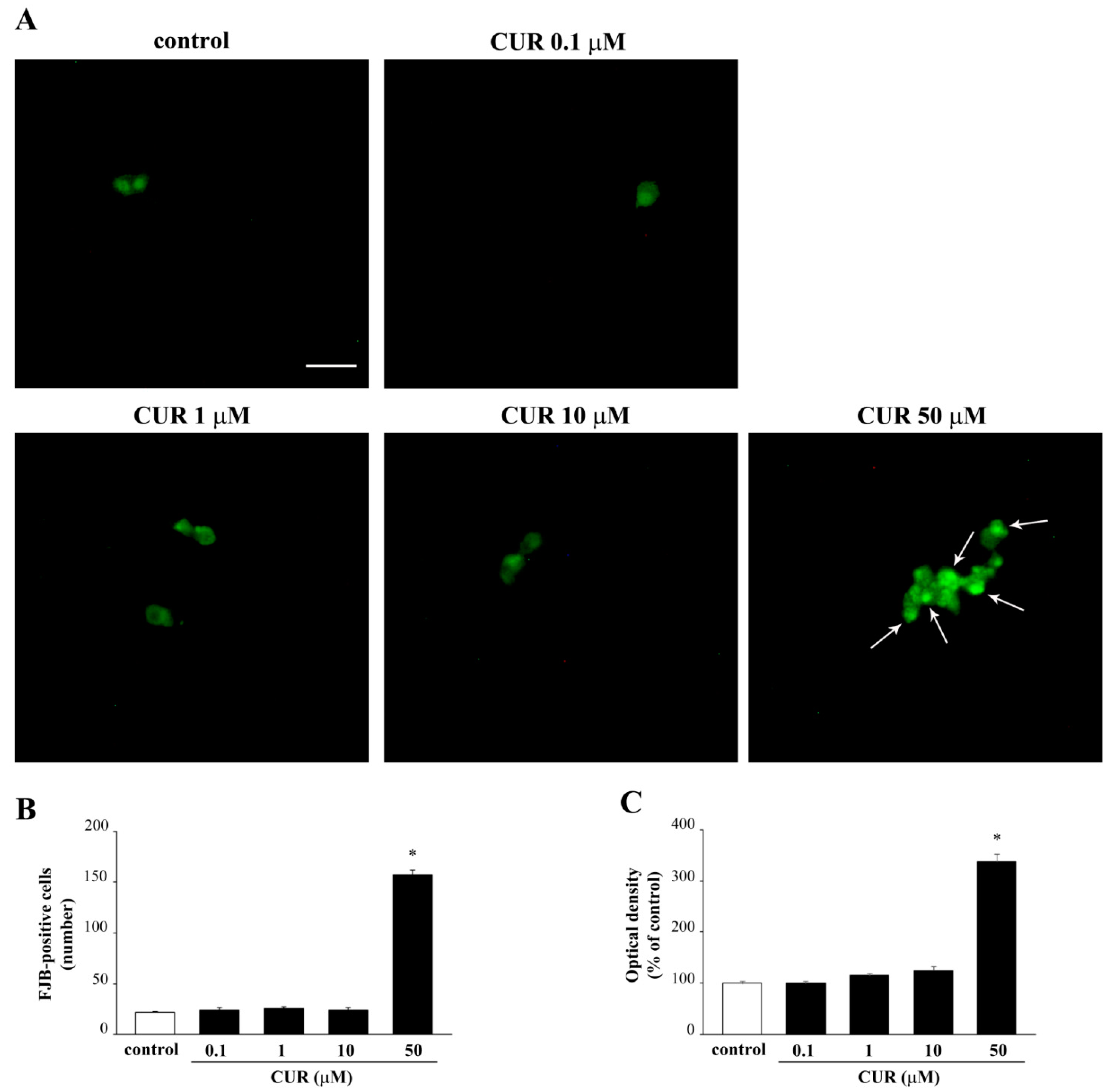
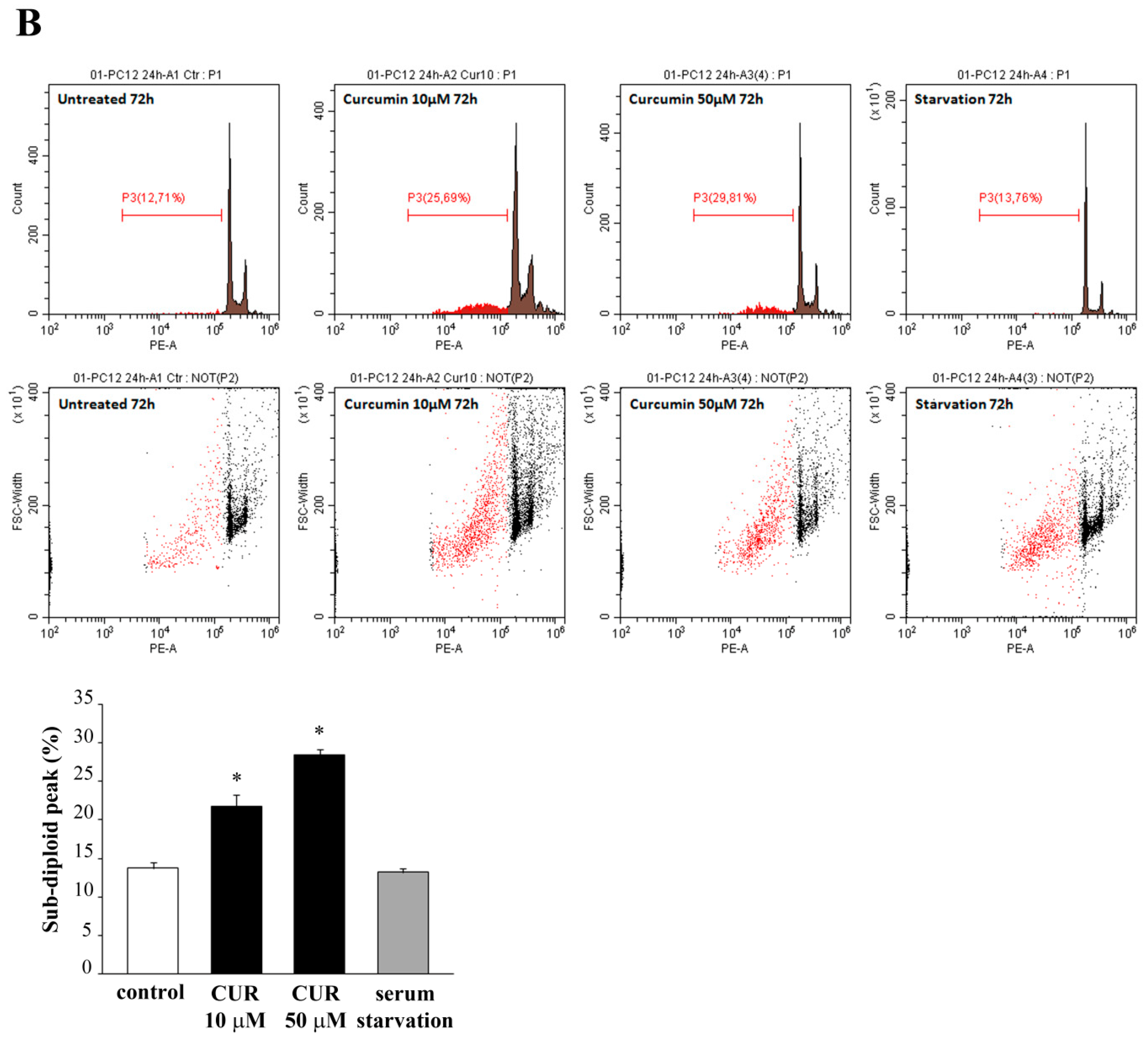
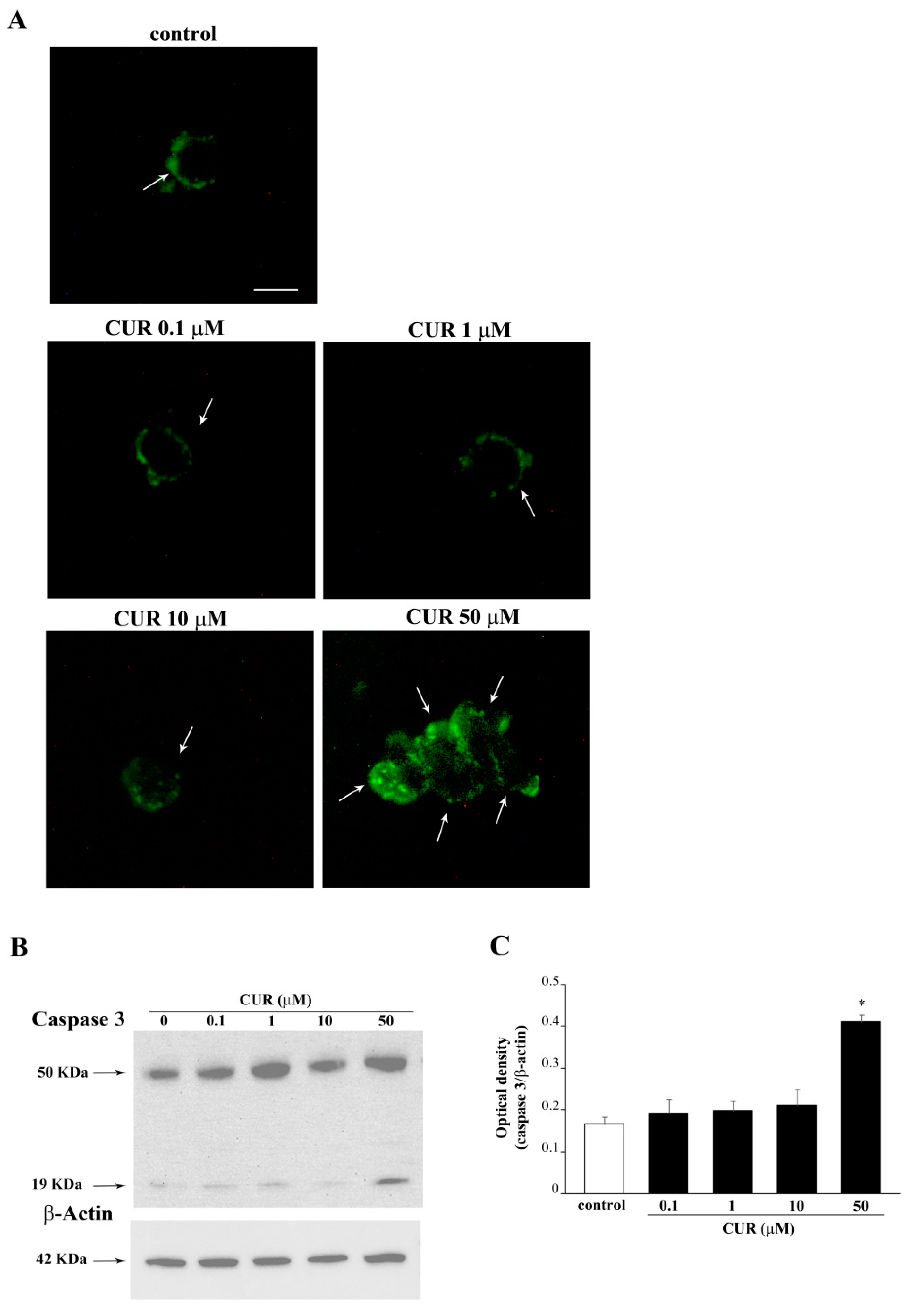
2.2. Apoptotic Effects of CUR
2.3. CUR-Induced Phenotypic Switch
2.4. Dose-Response Curve of METH on PC12 Cell Viability
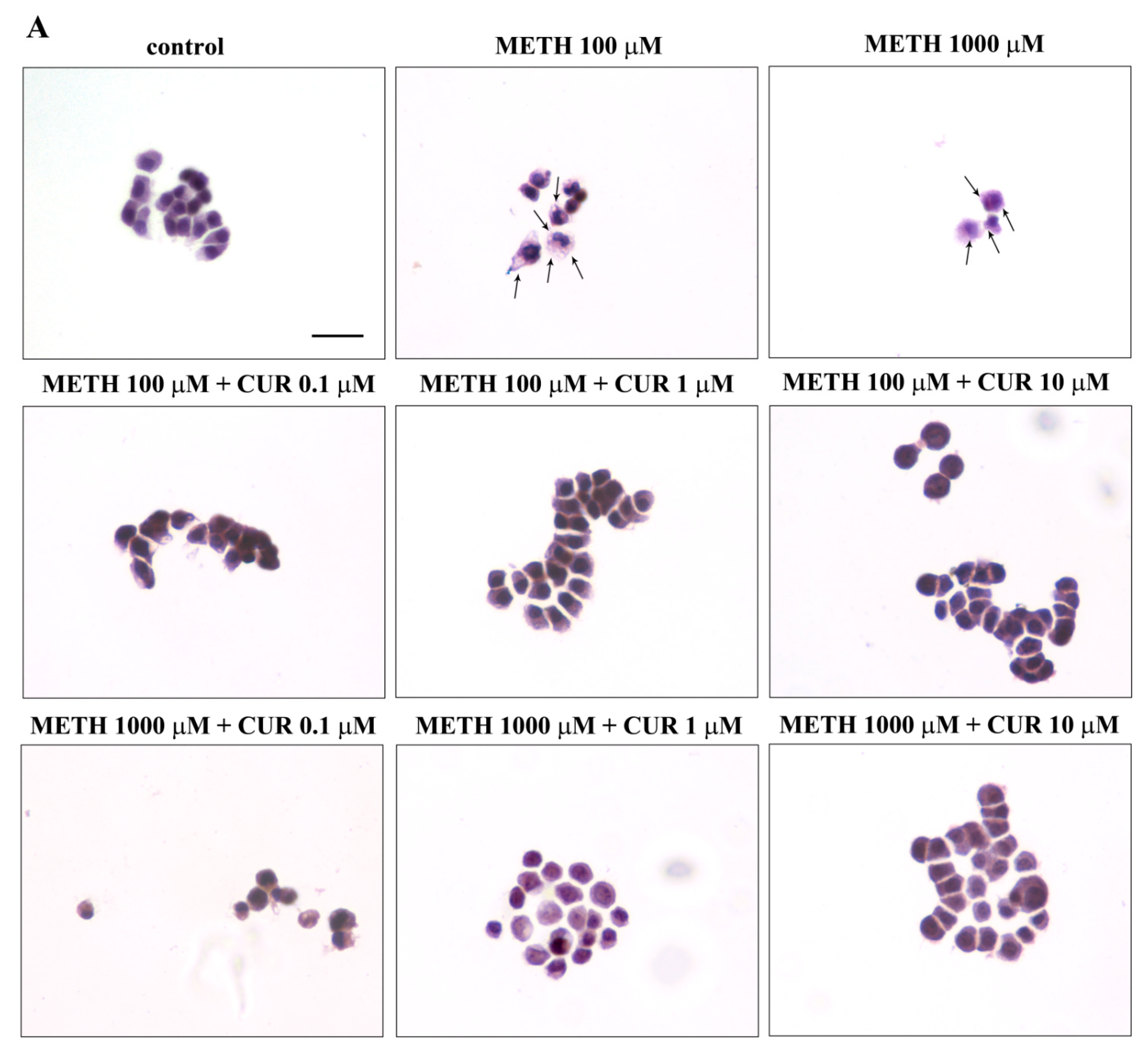
2.5. Phenotypic Effects of METH
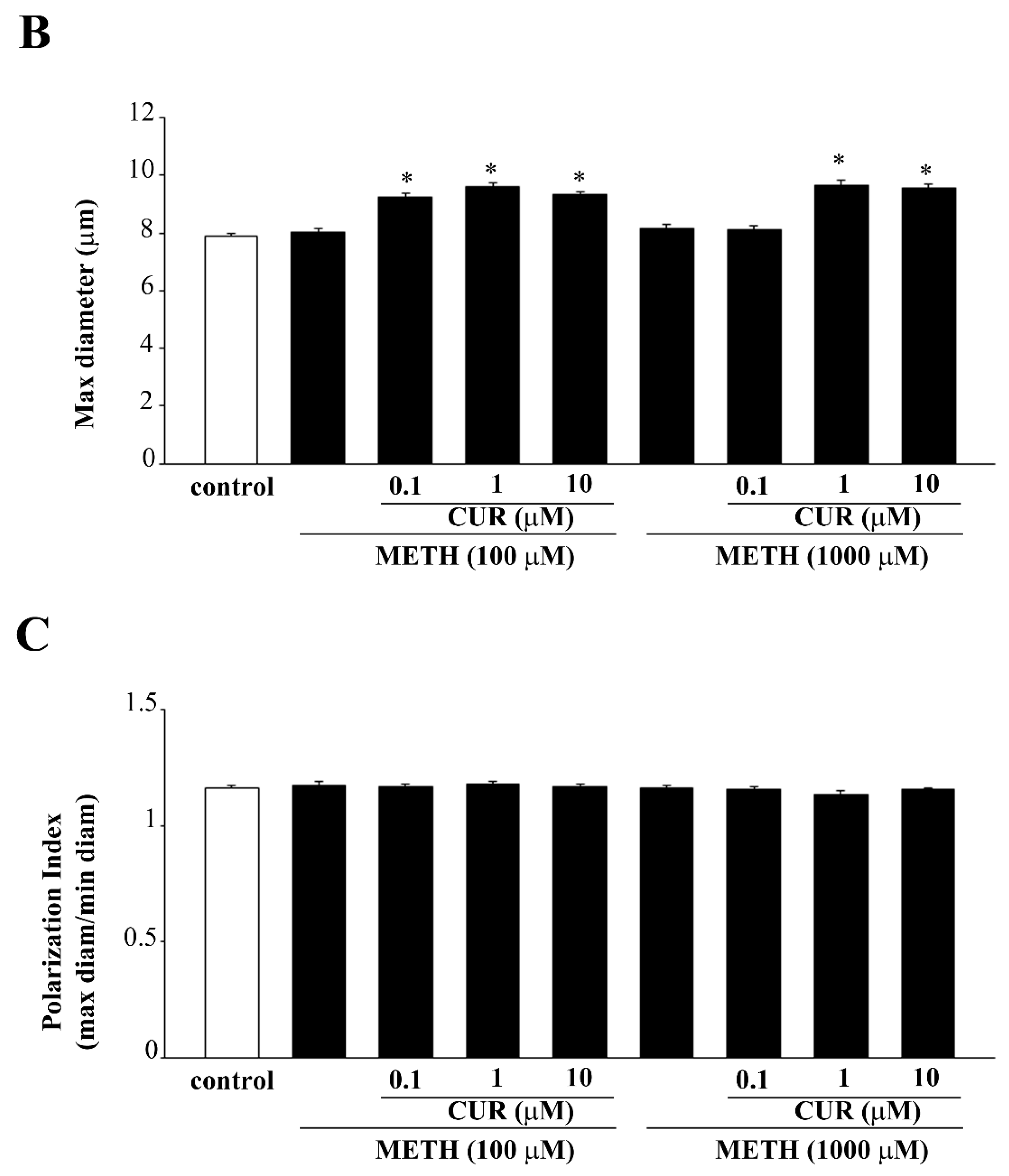
2.6. Dose-Dependent CUR-Induced Protection against METH Toxicity
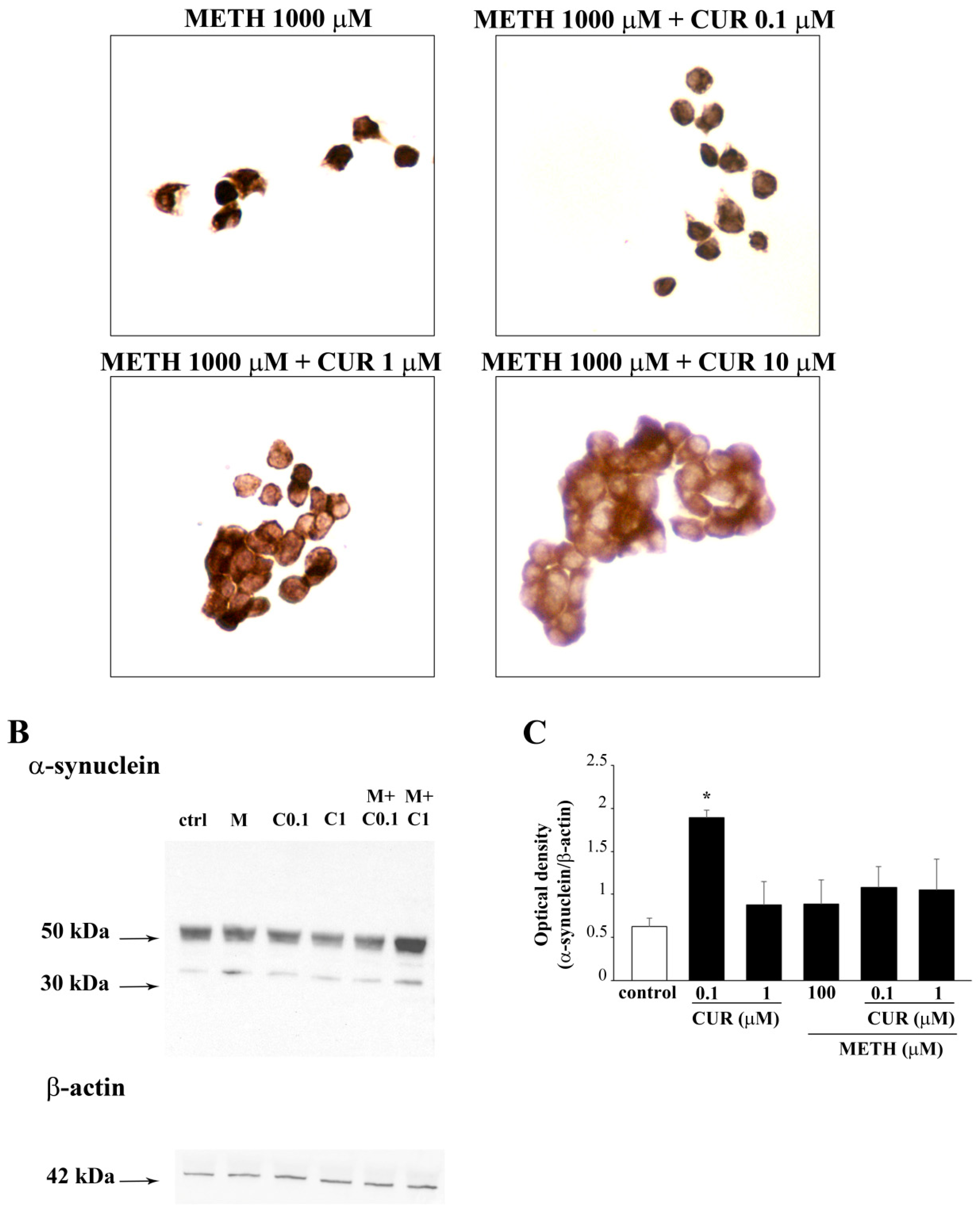
2.7. Reversal by CUR of METH-Induced Cellular Alterations within Spared Cells
2.8. Dose-Dependent Effects of CUR on the Expression of α-Synuclein in Control Cells and Following METH Administration
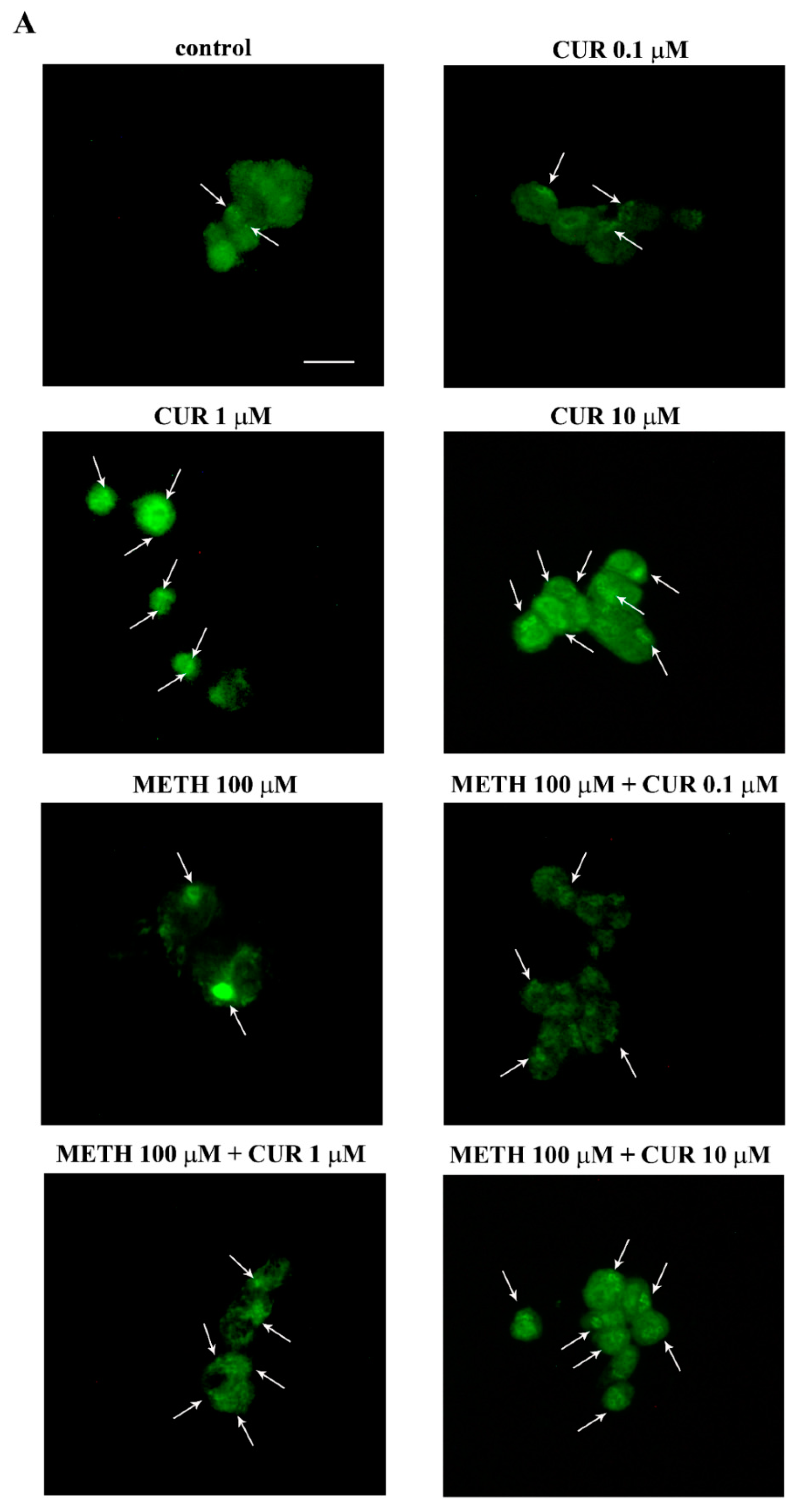
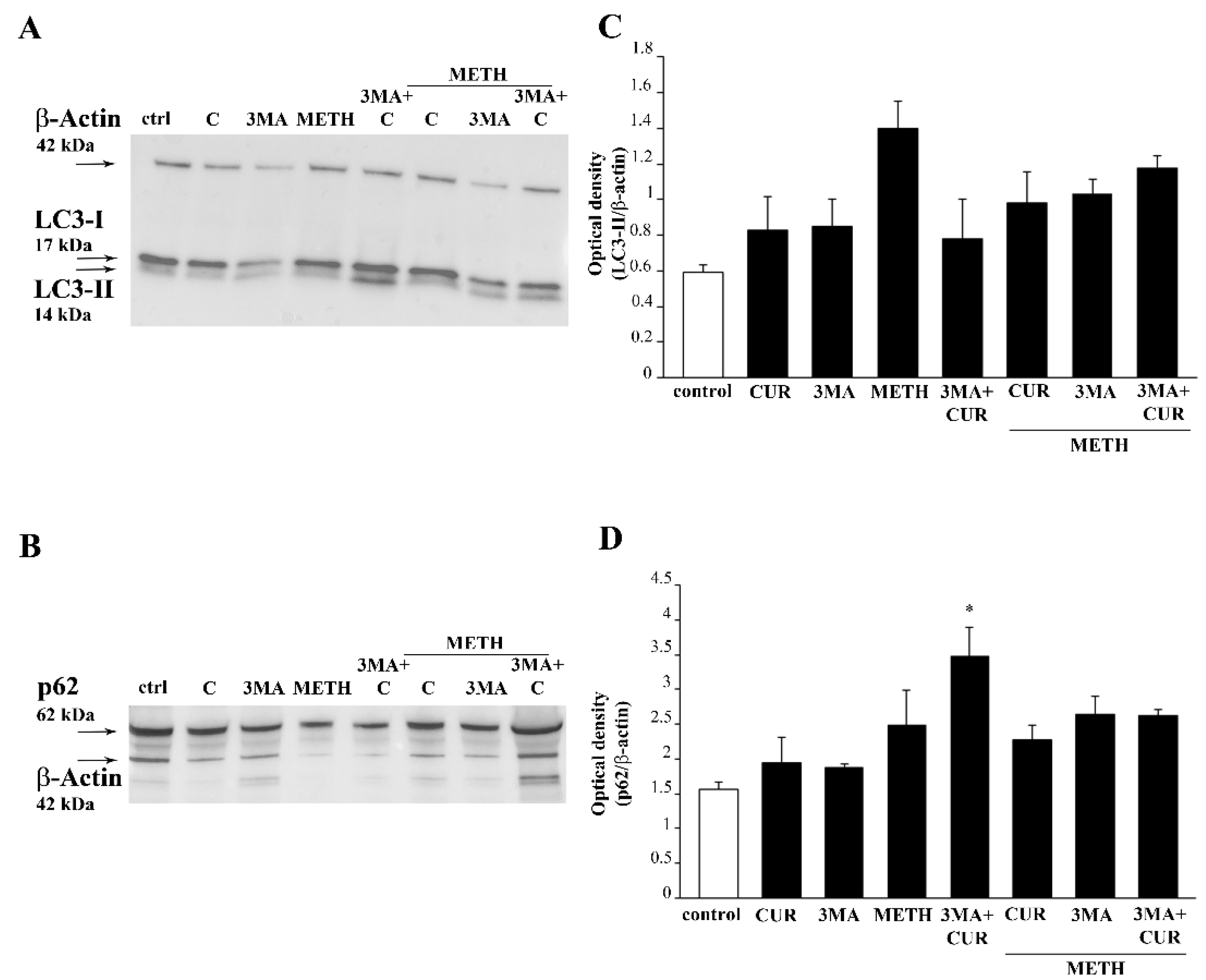
2.9. CUR Increases LC3 and Autophagy Vacuoles
2.10. CUR Counteracts METH-Induced Depolarization of LC3 from Autophagy Vacuoles
2.11. Autophagy Inhibition Occludes the Protective Effect of CUR against METH Toxicity
3. Discussion
4. Materials and Methods
4.1. Experimental Design
4.2. PC12 Cell Culture
4.3. Cell Treatments
4.4. WST-1 Assay
4.5. Trypan Blue Staining
4.6. Fluoro Jade B
4.7. CytofluorimetricAanalysis of Subdiploid (subG1) DNA Peak
4.8. Haematoxylin and Eosin Histochemistry
4.9. Immunocytochemistry at Light Microscopy
4.10. Western Blotting
4.11. Transmission Electron Microscopy
4.12. Postembedding Immunocytochemistry
4.13. Ultrastructural Morphometry
4.14. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Aggarwal, B.B.; Harikumar, K.B. Potential therapeutic effects of curcumin, the anti-inflammatory agent, against neurodegenerative, cardiovascular, pulmonary, metabolic, autoimmune and neoplastic diseases. Int. J. Biochem. Cell Biol. 2009, 41, 40–59. [Google Scholar] [CrossRef]
- Lim, G.P.; Chu, T.; Yang, F.; Beech, W.; Frautschy, S.A.; Cole, G.M. The curry spice curcumin reduces oxidative damage and amyloid pathology in an Alzheimer transgenic mouse. J. Neurosci. 2001, 21, 8370–8377. [Google Scholar] [CrossRef]
- Menon, V.P.; Sudheer, A.R. Antioxidant and anti-inflammatory properties of curcumin. Adv. Exp. Med. Biol. 2007, 595, 105–125. [Google Scholar]
- Wang, W.; Xu, J. Curcumin Attenuates Cerebral Ischemia-reperfusion Injury Through Regulating Mitophagy and Preserving Mitochondrial Function. Curr. Neurovasc. Res. 2020, 17, 113–122. [Google Scholar] [CrossRef]
- Song, J.X.; Sun, Y.R.; Peluso, I.; Zeng, Y.; Yu, X.; Lu, J.H.; Xu, Z.; Wang, M.Z.; Liu, L.F.; Huang, Y.Y.; et al. A novel curcumin analog binds to and activates TFEB in vitro and in vivo independent of MTOR inhibition. Autophagy 2016, 12, 1372–1389. [Google Scholar] [CrossRef]
- Zhang, J.; Wang, J.; Xu, J.; Lu, Y.; Jiang, J.; Wang, L.; Shen, H.M.; Xia, D. Curcumin targets the TFEB-lysosome pathway for induction of autophagy. Oncotarget 2016, 7, 75659–75671. [Google Scholar] [CrossRef]
- He, X.J.; Uchida, K.; Megumi, C.; Tsuge, N.; Nakayama, H. Dietary curcumin supplementation attenuates 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) neurotoxicity in C57BL mice. J. Toxicol. Pathol. 2015, 28, 197–206. [Google Scholar] [CrossRef]
- Xia, X.J.; Lian, Y.G.; Zhao, H.Y.; Xu, Q.L. Curcumin protects from oxidative stress and inhibits α-synuclein aggregation in MPTP induced parkinsonian mice. Int. J. Clin. Exp. Med. 2016, 9, 2654–2665. [Google Scholar]
- Wang, J.; Du, X.X.; Jiang, H.; Xie, J.X. Curcumin attenuates 6-hydroxydopamine-induced cytotoxicity by anti-oxidation and nuclear factor-kappa B modulation in MES23.5 cells. Biochem. Pharmacol. 2009, 78, 178–183. [Google Scholar] [CrossRef]
- Jaisin, Y.; Thampithak, A.; Meesarapee, B.; Ratanachamnong, P.; Suksamrarn, A.; Phivthong-Ngam, L.; Phumala-Morales, N.; Chongthammakun, S.; Govitrapong, P.; Sanvarinda, Y. Curcumin I protects the dopaminergic cell lines SH-SY5Y from 6-hydroxydopamine-induced neurotoxicity through attenuation of p53-mediated apoptosis. Neurosci. Lett. 2011, 489, 192–196. [Google Scholar] [CrossRef]
- Maiti, P.; Dunbar, G.L. Use of Curcumin, a Natural Polyphenol for Targeting Molecular Pathways in Treating Age-Related Neurodegenerative Diseases. Int. J. Mol. Sci. 2018, 19, 1637. [Google Scholar] [CrossRef]
- Callaghan, R.C.; Cunningham, J.K.; Sykes, J.; Kish, S.J. Increased risk of Parkinson’s disease in individuals hospitalized with conditions related to the use of methamphetamine or other amphetamine-type drugs. Drug Alcohol Depend. 2012, 120, 35–40. [Google Scholar] [CrossRef]
- Ares-Santos, S.; Granado, N.; Espadas, I.; Martinez-Murillo, R.; Moratalla, R. Methamphetamine causes degeneration of dopamine cell bodies and terminals of the nigrostriatal pathway evidenced by silver staining. Neuropsychopharmacology 2014, 39, 1066–1080. [Google Scholar] [CrossRef]
- Curtin, K.; Fleckenstein, A.E.; Robison, R.J.; Crookston, M.J.; Smith, K.R.; Hanson, G.R. Methamphetamine/amphetamine abuse and risk of Parkinson’s disease in Utah: A population-based assessment. Drug Alcohol Depend. 2015, 146, 30–38. [Google Scholar] [CrossRef]
- Weinshenker, D. Long Road to Ruin: Noradrenergic Dysfunction in Neurodegenerative Disease. Trends Neurosci. 2018, 41, 211–223. [Google Scholar] [CrossRef]
- Fornai, F.; Puglisi-Allegra, S. Autophagy status as a gateway for stress-induced catecholamine interplay in neurodegeneration. Neurosci. Biobehav. Rev. 2021, 123, 238–256. [Google Scholar] [CrossRef]
- Fornai, F.; Giorgi, F.S.; Bassi, L.; Ferrucci, M.; Alessandrì, M.G.; Corsini, G.U. Modulation of dihydroxyphenylacetaldehyde extracellular levels in vivo in the rat striatum after different kinds of pharmacological treatment. Brain Res. 2000, 861, 126–134. [Google Scholar] [CrossRef]
- Gesi, M.; Santinami, A.; Ruffoli, R.; Conti, G.; Fornai, F. Novel aspects of dopamine oxidative metabolism (confounding outcomes take place of certainties). Pharmacol. Toxicol. 2001, 89, 217–224. [Google Scholar] [CrossRef]
- LaVoie, M.J.; Hastings, T.G. Dopamine quinone formation and protein modification associated with the striatal neurotoxicity of methamphetamine: Evidence against a role for extracellular dopamine. J. Neurosci. 1999, 19, 1484–1491. [Google Scholar] [CrossRef]
- Hastings, T.G.; Lewis, D.A.; Zigmond, M.J. Role of oxidation in the neurotoxic effects of intrastriatal dopamine injections. Proc. Natl. Acad. Sci. USA 1996, 93, 1956–1961. [Google Scholar] [CrossRef]
- Conway, K.A.; Rochet, J.-C.; Bieganski, R.M.; Lansbury, P.T. Kinetic stabilization of the α-synuclein protofibril by a dopamine-α-synuclein adduct. Science 2001, 294, 1346–1349. [Google Scholar] [CrossRef]
- Sulzer, D. alpha-synuclein and cytosolic dopamine: Stabilizing a bad situation. Nat. Med. 2001, 7, 1280–1282. [Google Scholar] [CrossRef]
- Fornai, F.; Lenzi, P.; Gesi, M.; Soldani, P.; Ferrucci, M.; Lazzeri, G.; Capobianco, L.; Battaglia, G.; De Blasi, A.; Nicoletti, F.; et al. Methamphetamineproducesneuronalinclusions in the nigrostriatalsystem and in PC12 cells. J. Neurochem. 2004, 88, 114–123. [Google Scholar] [CrossRef]
- Quan, L.; Ishikawa, T.; Michiue, T.; Li, D.R.; Zhao, D.; Oritani, S.; Zhu, B.L.; Maeda, H. Ubiquitin-immunoreactive structures in the midbrain of methamphetamine abusers. Leg Med. 2005, 7, 144–150. [Google Scholar] [CrossRef]
- Fornai, F.; Soldani, P.; Lazzeri, G.; di Poggio, A.B.; Biagioni, F.; Fulceri, F.; Batini, S.; Ruggieri, S.; Paparelli, A. Neuronalinclusions in degenerative disorders. Do they represent static features or a key to understand the dymanics of the disease? Brain Res. Bull. 2005, 65, 275–290. [Google Scholar] [CrossRef]
- Schmued, L.C.; Hopkins, K.J. Fluoro-Jade B: A high affinity fluorescent marker for the localization of neuronal degeneration. Brain Res. 2000, 874, 123–130. [Google Scholar] [CrossRef]
- Schmuck, G.; Kahl, R. The use of Fluoro-Jade in primary neuronal cell cultures. Arch. Toxicol. 2009, 83, 397–403. [Google Scholar] [CrossRef]
- Ullah, I.; Ullah, N.; Naseer, M.I.; Lee, H.Y.; Kim, M.O. Neuroprotection with metformin and thymoquinone against ethanol-induced apoptotic neurodegeneration in prenatal rat cortical neurons. BMC Neurosci. 2012, 13, 11. [Google Scholar] [CrossRef]
- Asanuma, M.; Miyazaki, I.; Higashi, Y.; Diaz-Corrales, F.J.; Shimizu, M.; Miyoshi, K.; Ogawa, N. Suppression of p53-activated gene, PAG608, attenuates methamphetamine-induced neurotoxicity. Neurosci. Lett. 2007, 414, 263–267. [Google Scholar] [CrossRef]
- Castino, R.; Lazzeri, G.; Lenzi, P.; Bellio, N.; Follo, C.; Ferrucci, M.; Fornai, F.; Isidoro, C. Suppression of autophagy precipitates neuronal cell death following low doses of methamphetamine. J. Neurochem. 2008, 106, 1426–1439. [Google Scholar] [CrossRef]
- Wu, J.; Zhu, D.; Zhang, J.; Li, G.; Liu, Z.; Sun, J. Lithium protects against methamphetamine-induced neurotoxicity in PC12 cells via Akt/GSK3β/mTOR pathway. Biochem. Biophys. Res. Commun. 2015, 465, 368–373. [Google Scholar] [CrossRef]
- Lazzeri, G.; Biagioni, F.; Fulceri, F.; Busceti, C.L.; Scavuzzo, M.C.; Ippolito, C.; Salvetti, A.; Lenzi, P.; Fornai, F. mTOR Modulates Methamphetamine-Induced Toxicity through Cell Clearing Systems. Oxid. Med. Cell Longev. 2018, 2018, 6124745. [Google Scholar] [CrossRef]
- Cubells, J.F.; Rayport, S.; Rajendran, G.; Sulzer, D. Methamphetamine neurotoxicity involves vacuolation of endocytic organelles and dopamine-dependent intracellular oxidative stress. J. Neurosci. 1994, 14, 2260–2271. [Google Scholar] [CrossRef]
- Lazzeri, G.; Lenzi, P.; Busceti, C.L.; Ferrucci, M.; Falleni, A.; Bruno, V.; Paparelli, A.; Fornai, F. Mechanisms involved in the formation of dopamine-induced intracellular bodies within striatal neurons. J. Neurochem. 2007, 101, 1414–1427. [Google Scholar] [CrossRef] [PubMed]
- Fornai, F.; Lenzi, P.; Lazzeri, G.; Ferrucci, M.; Fulceri, F.; Giorgi, F.S.; Falleni, A.; Ruggieri, S.; Paparelli, A. Fine ultrastructure and biochemistry of PC12 cells: A comparative approach to understand neurotoxicity. Brain Res. 2007, 1129, 174–190. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, A.; Tagawa, Y.; Yoshimori, T.; Moriyama, Y.; Masaki, R.; Tashiro, Y. Bafilomycin A1 prevents maturation of autophagic vacuoles byinhibiting fusion between autophagosomes and lysosomes in rat hepatomacell line, H-4-II-E cells. Cell Struct. Funct. 1998, 23, 33–42. [Google Scholar] [CrossRef]
- Boya, P.; González-Polo, R.A.; Casares, N.; Perfettini, J.L.; Dessen, P.; Larochette, N.; Métivier, D.; Meley, D.; Souquere, S.; Yoshimori, T.; et al. Inhibition of macroautophagy triggers apoptosis. Mol. Cell Biol. 2005, 25, 1025–1040. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Yoshimori, T. How to interprete LC3 immunoblotting. Autophagy 2007, 3, 542–545. [Google Scholar] [CrossRef] [PubMed]
- Seglen, P.O.; Gordon, P.B. 3-Methyladenine: Specific inhibitor of autophagic/lysosomal protein degradation in isolated rat hepatocytes. Proc. Natl. Acad. Sci. USA 1982, 79, 1889–1892. [Google Scholar] [CrossRef]
- Blommaart, E.F.; Krause, U.; Schellens, J.P. The phosphatidylinositol 3-kinase inhibitors wortmannin and LY294002 inhibit autophagy in isolated rat hepatocytes. Eur. J. Biochem. 1997, 243, 240–246. [Google Scholar] [CrossRef]
- Bjørkøy, G.; Lamark, T.; Pankiv, S.; Øvervatn, A.; Brech, A.; Johansen, T. Monitoring autophagic degradation of p62/SQSTM1. Methods Enzymol. 2009, 452, 181–197. [Google Scholar] [CrossRef]
- Kim, D.S.; Park, S.Y.; Kim, J.K. Curcuminoids from Curcuma longa L. (Zingiberaceae) that protect PC12 rat pheochromocytoma and normal human umbilical vein endothelial cells from betaA(1-42) insult. Neurosci. Lett. 2001, 303, 57–61. [Google Scholar] [CrossRef]
- Mendonça, L.M.; da Silva Machado, C.; Teixeira, C.C.; de Freitas, L.A.; de Lourdes Pires Bianch, M.; Antunes, L.M. Curcumin reduces cisplatin-induced neurotoxicity in NGF-differentiated PC12 cells. Neurotoxicology 2013, 34, 205–211. [Google Scholar] [CrossRef]
- Chang, C.H.; Chen, H.X.; Yü, G.; Peng, C.C.; Peng, R.Y. Curcumin-Protected PC12 Cells Against Glutamate-Induced Oxidative Toxicity. Food Technol. Biotechnol. 2014, 52, 468–478. [Google Scholar] [CrossRef] [PubMed]
- Rahaman, M.S.; Banik, S.; Akter, M.; Rahman, M.M.; Sikder, M.T.; Hosokawa, T.; Saito, T.; Kurasaki, M. Curcumin alleviates arsenic-induced toxicity in PC12 cells via modulating autophagy/apoptosis. Ecotoxicol. Environ. Saf. 2020, 200, 110756. [Google Scholar] [CrossRef] [PubMed]
- Siddiqui, M.A.; Kashyap, M.P.; Kumar, V.; Tripathi, V.K.; Khanna, V.K.; Yadav, S.; Pant, A.B. Differential protection of pre-, co- and post-treatment of curcumin against hydrogen peroxide in PC12 cells. Hum. Exp. Toxicol. 2011, 30, 192–198. [Google Scholar] [CrossRef]
- Ao, G.Z.; Chu, X.J.; Ji, Y.Y.; Wang, J.W. Antioxidant properties and PC12 cell protective effects of a novel curcumin analogue (2E,6E)-2,6-bis(3,5- dimethoxybenzylidene)cyclohexanone (MCH). Int. J. Mol. Sci. 2014, 15, 3970–3988. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, S.; Karmenyan, A.; Tsai, J.W.; Chiou, A. Inhibitory effects of curcumin and cyclocurcumin in 1-methyl-4-phenylpyridinium (MPP+) induced neurotoxicity in differentiated PC12 cells. Sci. Rep. 2017, 7, 16977. [Google Scholar] [CrossRef] [PubMed]
- Palikaras, K.; Tavernarakis, N. Mitochondrial homeostasis: The interplay between mitophagy and mitochondrial biogenesis. Exp. Gerontol. 2014, 56, 182–188. [Google Scholar] [CrossRef]
- Ferese, R.; Lenzi, P.; Fulceri, F.; Biagioni, F.; Fabrizi, C.; Gambardella, S.; Familiari, P.; Frati, A.; Limanaqi, F.; Fornai, F. Quantitative Ultrastructural Morphometry and Gene Expression of mTOR-Related Mitochondriogenesis within Glioblastoma Cells. Int. J. Mol. Sci. 2020, 21, 4570. [Google Scholar] [CrossRef] [PubMed]
- Ferrucci, M.; Biagioni, F.; Lenzi, P.; Gambardella, S.; Ferese, R.; Calierno, M.T.; Falleni, A.; Grimaldi, A.; Frati, A.; Esposito, V.; et al. Rapamycin promotes differentiation increasing βIII-tubulin, NeuN, and NeuroD while suppressing nest inexpression in glioblastoma cells. Oncotarget 2017, 8, 29574–29599. [Google Scholar] [CrossRef]
- Shinojima, N.; Yokoyama, T.; Kondo, Y.; Kondo, S. Roles of the Akt/mTOR/p70S6K and ERK1/2 signaling pathways in curcumin-induced autophagy. Autophagy 2007, 3, 635–637. [Google Scholar] [CrossRef]
- Aoki, H.; Takada, Y.; Kondo, S.; Sawaya, R.; Aggarwal, B.B.; Kondo, Y. Evidence that curcumin suppresses the growth of malignant gliomas in vitro and in vivo through induction of autophagy: Role of Akt and extracellular signal-regulated kinase signaling pathways. Mol. Pharmacol. 2007, 72, 29–39. [Google Scholar] [CrossRef]
- Hasima, N.; Ozpolat, B. Regulation of autophagy by polyphenolic compounds as a potential therapeutic strategy for cancer. Cell Death Dis. 2014, 5, e1509. [Google Scholar] [CrossRef] [PubMed]
- Su, J.; Zhou, X.; Yin, X.; Wang, L.; Zhao, Z.; Hou, Y.; Zheng, N.; Xia, J.; Wang, Z. The effects of curcumin on proliferation, apoptosis, invasion, and NEDD4 expression in pancreatic cancer. Biochem. Pharmacol. 2017, 140, 28–40. [Google Scholar] [CrossRef] [PubMed]
- Shakeri, A.; Cicero, A.F.G.; Panahi, Y.; Mohajeri, M.; Sahebkar, A. Curcumin: A naturally occurring autophagy modulator. J. Cell. Physiol. 2019, 234, 5643–5654. [Google Scholar] [CrossRef]
- Limanaqi, F.; Biagioni, F.; Busceti, C.L.; Ryskalin, L.; Polzella, M.; Frati, A.; Fornai, F. Phytochemicals Bridging Autophagy Induction and Alpha-Synuclein Degradation in Parkinsonism. Int. J. Mol. Sci. 2019, 20, 3274. [Google Scholar] [CrossRef]
- Wu, Y.; Liang, S.; Xu, B.; Zhang, R.; Xu, L. Protective effect of curcumin on dopamine neurons in Parkinson’s disease and its mechanism. Zhejiang Da Xue Xue Bao Yi Xue Ban 2018, 47, 480–486. [Google Scholar]
- Beevers, C.S.; Chen, L.; Liu, L.; Luo, Y.; Webster, N.J.; Huang, S. Curcumin disrupts the Mammalian target of rapamycin-raptor complex. Cancer Res. 2009, 69, 1000–1008. [Google Scholar] [CrossRef]
- Riccardi, C.; Nicoletti, I. Analysis of apoptosis by propidium iodide staining and flow cytometry. Nat. Protoc. 2006, 1, 1458–1461. [Google Scholar] [CrossRef]
- Fabrizi, C.; Pompili, E.; De Vito, S.; Somma, F.; Catizone, A.; Ricci, G.; Lenzi, P.; Fornai, F.; Fumagalli, L. Impairment of the autophagic flux in astrocytes intoxicated by trimethyltin. Neurotoxicology 2016, 52, 12–22. [Google Scholar] [CrossRef] [PubMed]
- Klionsky, D.J.; Abdel-Aziz, A.K.; Abdelfatah, S.; Abdellatif, M.; Abdoli, A.; Abel, S.; Abeliovich, H.; Abildgaard, M.H.; Abudu, Y.P.; Acevedo-Arozena, A.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy (4th edition). Autophagy 2021, 17, 1–382. [Google Scholar] [CrossRef] [PubMed]
- Lenzi, P.; Lazzeri, G.; Biagioni, F.; Busceti, C.L.; Gambardella, S.; Salvetti, A.; Fornai, F. The Autophagoproteasome—A Novel Cell Clearing Organelle in Baseline and Stimulated Conditions. Front. Neuroanat. 2016, 10, 78. [Google Scholar] [CrossRef]
- Bendayan, M.; Zollinger, M. Ultrastructural localization of antigenic sites on osmium-fixed tissues applying the protein A-gold technique. J. Histochem. Cytochem. 1983, 31, 101–109. [Google Scholar] [CrossRef] [PubMed]
- D’Alessandro, D.; Mattii, L.; Moscato, S.; Bernardini, N.; Segnani, C.; Dolfi, A.; Bianchi, F. Immunohistochemical demonstration of the small GTPaseRhoAA on epoxy-resin embedded sections. Micron 2004, 35, 287–296. [Google Scholar] [CrossRef]



























Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ryskalin, L.; Puglisi-Allegra, S.; Lazzeri, G.; Biagioni, F.; Busceti, C.L.; Balestrini, L.; Fornasiero, A.; Leone, S.; Pompili, E.; Ferrucci, M.; et al. Neuroprotective Effects of Curcumin in Methamphetamine-Induced Toxicity. Molecules 2021, 26, 2493. https://doi.org/10.3390/molecules26092493
Ryskalin L, Puglisi-Allegra S, Lazzeri G, Biagioni F, Busceti CL, Balestrini L, Fornasiero A, Leone S, Pompili E, Ferrucci M, et al. Neuroprotective Effects of Curcumin in Methamphetamine-Induced Toxicity. Molecules. 2021; 26(9):2493. https://doi.org/10.3390/molecules26092493
Chicago/Turabian StyleRyskalin, Larisa, Stefano Puglisi-Allegra, Gloria Lazzeri, Francesca Biagioni, Carla L. Busceti, Linda Balestrini, Andrea Fornasiero, Stefano Leone, Elena Pompili, Michela Ferrucci, and et al. 2021. "Neuroprotective Effects of Curcumin in Methamphetamine-Induced Toxicity" Molecules 26, no. 9: 2493. https://doi.org/10.3390/molecules26092493
APA StyleRyskalin, L., Puglisi-Allegra, S., Lazzeri, G., Biagioni, F., Busceti, C. L., Balestrini, L., Fornasiero, A., Leone, S., Pompili, E., Ferrucci, M., & Fornai, F. (2021). Neuroprotective Effects of Curcumin in Methamphetamine-Induced Toxicity. Molecules, 26(9), 2493. https://doi.org/10.3390/molecules26092493