
Methoxy-Substituted Tyramine Derivatives Synthesis, Computational Studies and Tyrosinase Inhibitory Kinetics


 ,
, 

Abstract

1. Introduction
2. Results and Discussion
2.1. Synthesis of 4-Methoxyphenyl Ethyl Chloroacetamide (2)
2.1.1. In Vitro Tyrosinase Inhibition Assay
2.1.2. Kinetics Mechanism
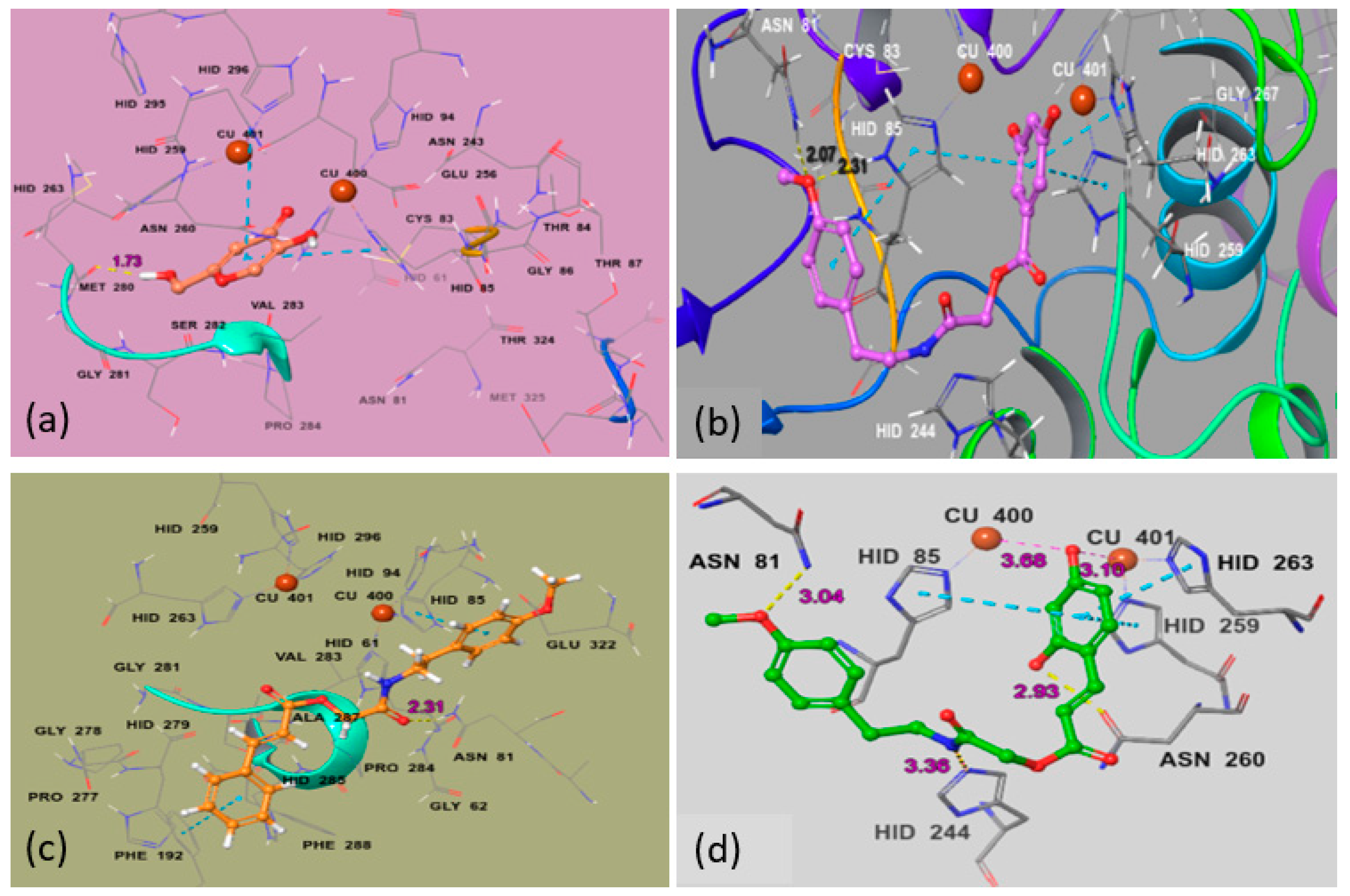
2.1.3. Molecular Docking Studies
3. Materials and Methods
3.1. Chemistry
3.2. General Procedure for the Synthesis of Title Compounds (Ph1–Ph5) and (Ph6–Ph10)
3.3. Spectral Characterization of Synthesized Compounds (Ph1–Ph10)
3.4. Mushroom Tyrosinase Inhibition Assay
3.5. Human Tyrosinase Inhibition Assay
3.5.1. Cell Culture and Preparation of Tyrosinase
3.5.2. Human Tyrosinase Inhibition Assay
3.6. Kinetic Analysis of the Inhibition of Tyrosinase
3.7. Molecular Docking Studies
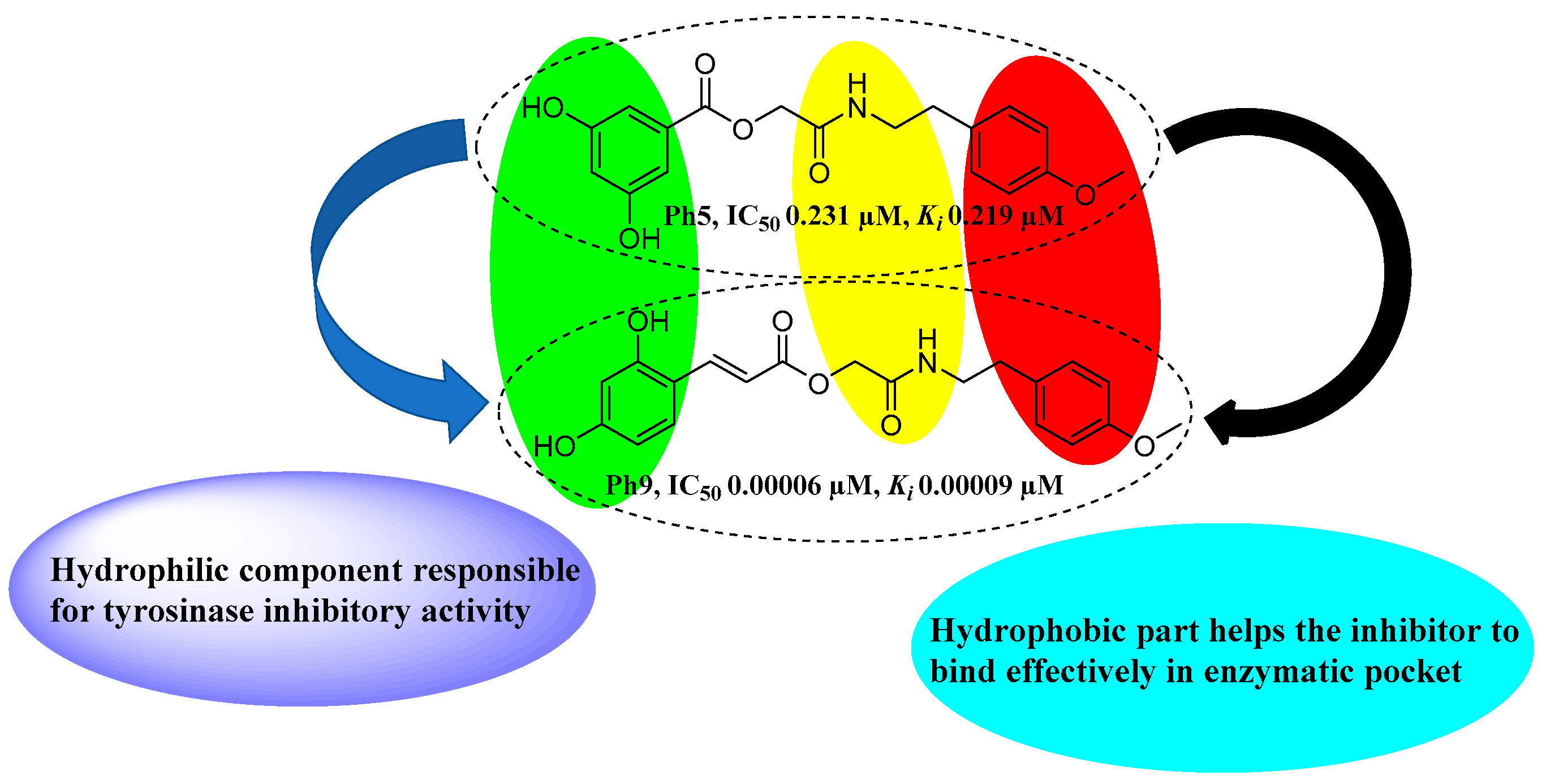
3.8. Structure–Activity Relationship (SAR)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Solomon, E.I.; Sundaram, U.M.; Machonkin, T.E. Multicopper oxidases and oxygenases. Chem. Rev. 1996, 96, 2563–2606. [Google Scholar] [CrossRef]
- Sánchez-Ferrer, Á.; Neptuno Rodríguez-López, J.; García-Cánovas, F.; García-Carmona, F. Tyrosinase: A comprehensive review of its mechanism. Biochim. Biophys. Acta (BBA)/Protein Struct. Mol. 1995, 1247, 1–11. [Google Scholar] [CrossRef]
- Van Holde, K.E.; Miller, K.I.; Decker, H. Hemocyanins and invertebrate evolution. J. Biol. Chem. 2001, 276, 15563–15566. [Google Scholar] [CrossRef]
- Slominski, A.; Costantino, R. Molecular mechanism of tyrosinase regulation by l-DOPA in hamster melanoma cells. Life Sci. 1991, 48, 2075–2079. [Google Scholar] [CrossRef]
- Slominski, A.; Costantino, R. L-tyrosine induces tyrosinase expression via a posttranscriptional mechanism. Experientia 1991, 47, 721–724. [Google Scholar] [CrossRef]
- Slominski, A.; Paus, R. Are l-tyrosine and l-dopa hormone-like bioregulators? J. Theor. Biol. 1990, 143, 123–138. [Google Scholar] [CrossRef]
- Prota, G. Melanins and Melanogenesis; Academic Press: Cambridge, MA, USA, 2012; pp. 34–62. [Google Scholar] [CrossRef]
- Prota, G. The chemistry of melanins and melanogenesis. Fortschr. Chem. Org. Naturst. 1995, 64, 93–148. [Google Scholar] [CrossRef]
- Kadekaro, A.L.; Kanto, H.; Kavanagh, R.; Abdel-Malek, Z.A. Significance of the melanocortin 1 receptor in regulating human melanocyte pigmentation, proliferation, and survival. Ann. N. Y. Acad. Sci. 2003, 994, 359–365. [Google Scholar] [CrossRef]
- Petit, L.; Pierard, G.E. Skin-lightening products revisited. Int. J. Cosmet. Sci. 2003, 25, 169–181. [Google Scholar] [CrossRef]
- Riley, P.A. Melanogenesis and melanoma. Pig. Cell Res. 2003, 16, 548–552. [Google Scholar] [CrossRef]
- Uong, A.; Zon, L.I. Melanocytes in development and cancer. J. Cell. Physiol. 2010, 222, 38–41. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, Y.; Hearing, V.J. Melanocytes and their diseases. Cold Spring Harb. Perspect. Med. 2014, 4, a017046. [Google Scholar] [CrossRef]
- Torihara, M.; Tamai, Y.; Shiono, M.; Tasaka, K. Skin Depigmental Agent. US Patent 4959393, 25 September 1990. [Google Scholar]
- Hu, L. Resorcinol derivatives. In Resorcinol; Springer: Berlin/Heidelberg, Germany, 2005. [Google Scholar] [CrossRef]
- Shore, L.J.; Rocha, S.A.; McKinney, M.D. Skin Lightening Agents, Compositions and Methods. US Patent 7270805, 18 September 2007. [Google Scholar]
- Joo, Y.H.; Baek, H.S.; Lee, C.S.; Choi, S.J.; Rho, H.S.; Park, M.Y.; Shin, S.S.; Lim, K.M.; Park, Y.H. Benzoic Acid Amide Compound. WO Patent 2016159644, 6 October 2016. [Google Scholar]
- Okombi, S.; Rival, D.; Boumendjel, A.; Mariotte, A.; Perrier, E. Para-coumaric Acid or Para-hydroxycinnamic Acid Derivatives and Their Use in Cosmetic or Dermatological Compositions. US Patent 9089499B2, 28 July 2015. [Google Scholar]
- Kaur, S.; Southall, M.D.; Zivin, R.A. Topical Application of 1-hydroxyl-3, 5-BIS (4′ hydroxy styryl) benzene. US Patent 8846013B2, 30 September 2014. [Google Scholar]
- Vanni, A.; Gastaldi, D.; Giunta, G. Kinetic investigations on the double enzymic activity of the tyrosinase mushroom. Ann. Chim. 1990, 80, 35–60. [Google Scholar]
- Takada, R.; Takada, S.; Takeda, K.; Yasumoto, K.; Watanabe, K.; Udono, T.; Saito, H.; Takahashi, K.; Shibahara, S. Induction of melanocyte-specific microphthalmia-associated transcription factor by Wnt-3a. J. Biol. Chem. 2000, 275, 14013–14016. [Google Scholar] [CrossRef] [PubMed]
- Bellei, B.; Pitisci, A.; Izzo, E.; Picardo, M. Inhibition of melanogenesis by the pyridinyl imidazole class of compounds: Possible involvement of the Wnt/β-catenin signaling pathway. PLoS ONE 2012, 7, e33021. [Google Scholar] [CrossRef] [PubMed]
- Horng, C.-T.; Wu, H.-C.; Chiang, N.-N.; Lee, C.-F.; Huang, Y.-S.; Wang, H.-Y.; Yang, J.-S.; Chen, F.-A. Inhibitory effect of burdock leaves on elastase and tyrosinase activity. Exp. Ther. Med. 2017, 14, 3247–3252. [Google Scholar] [CrossRef][Green Version]
- Hałdys, K.; Goldeman, W.; Anger-Góra, N.; Rossowska, J.; Latajka, R. Monosubstituted acetophenone thiosemicarbazones as potent inhibitors of tyrosinase: Synthesis, inhibitory studies, and molecular docking. Pharmaceuticals 2021, 14, 74. [Google Scholar] [CrossRef] [PubMed]
- Fujita, H.; Menezes, J.C.; Santos, S.M.; Yokota, S.; Kamat, S.P.; Cavaleiro, J.A.S.; Motokawa, T.; Kato, T.; Mochizuki, M.; Fujiwara, T.; et al. Inulavosin and its benzo-derivatives, melanogenesis inhibitors, target the copper loading mechanism to the active site of tyrosinase. Pig. Cell Melan. Res. 2014, 27, 376–386. [Google Scholar] [CrossRef] [PubMed]
- Tai, A.; Sawano, T.; Yazama, F.; Ito, H. Evaluation of antioxidant activity of vanillin by using multiple antioxidant assays. Biochim. Biophys. Acta (BBA)-Gen. Subj. 2011, 1810, 170–177. [Google Scholar] [CrossRef]
- Briganti, S.; Camera, E.; Picardo, M. Chemical and instrumental approaches to treat hyperpigmentation. Pig. Cell Res. 2003, 16, 101–110. [Google Scholar] [CrossRef]
- Shalit, H.; Dyadyuk, A.; Pappo, D. Selective oxidative phenol coupling by iron catalysis. J. Org. Chem. 2019, 84, 1677–1686. [Google Scholar] [CrossRef]
- Rafiq, M.; Nazir, Y.; Ashraf, Z.; Rafique, H.; Afzal, S.; Mumtaz, A.; Hassan, M.; Ali, A.; Afzal, K.; Yousuf, M.R.; et al. Synthesis, computational studies, tyrosinase inhibitory kinetics and antimelanogenic activity of hydroxy substituted 2-[(4-acetylphenyl) amino]-2-oxoethyl derivatives. J. Enzy. Inhib. Med. Chem. 2019, 34, 1562–1572. [Google Scholar] [CrossRef] [PubMed]
- Nazir, Y.; Saeed, A.; Rafiq, M.; Afzal, S.; Ali, A.; Latif, M.; Zuegg, J.; Hussein, W.M.; Fercher, C.; Barnard, R.T.; et al. Hydroxyl substituted benzoic acid/cinnamic acid derivatives: Tyrosinase inhibitory kinetics, anti-melanogenic activity and molecular docking studies. Bioorg. Med. Chem. Lett. 2020, 30, 126722. [Google Scholar] [CrossRef]
- Parvez, S.; Kang, M.; Chung, H.S.; Bae, H. Naturally occurring tyrosinase inhibitors: Mechanism and applications in skin health, cosmetics and agriculture industries. Phyther. Res. 2007. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Cordoves, C.; Bartolome, B.; Vieira, W.; Virador, V.M. Effects of wine phenolics and sorghum tannins on tyrosinase activity and growth of melanoma cells. J. Agric. Food Chem. 2001, 49, 1620–1624. [Google Scholar] [CrossRef]
- Park, J.B.; Schoene, N. N-Caffeoyltyramine arrests growth of U937 and Jurkat cells by inhibiting protein tyrosine phosphorylation and inducing caspase-3. Cancer Lett. 2003, 202, 161–171. [Google Scholar] [CrossRef]
- Nesterenko, V.; Putt, K.S.; Hergenrother, P.J. Identification from a combinatorial library of a small molecule that selectively induces apoptosis in cancer cells. J. Am. Chem. Soc. 2003, 125, 14672–14673. [Google Scholar] [CrossRef]
- Ley, J.P. Phenolic acid amides of phenolic benzylamines against UVA-induced oxidative stress in skin. Int. J. Cosmet. Sci. 2001, 23, 35–48. [Google Scholar] [CrossRef]
- Khan, K.M.; Maharvi, G.M.; Abbaskhan, A.; Hayat, S.; Khan, M.T.H.; Makhmoor, T.; Choudhary, M.I.; Shaheen, F. Three tyrosinase inhibitors and antioxidant compounds from salsola foetida. Helv. Chim. Acta 2003, 86, 457–464. [Google Scholar] [CrossRef]
- Son, S.; Lewis, B.A. Free radical scavenging and antioxidative activity of caffeic acid amide and ester analogues: Structure- activity relationship. J. Agric. Food Chem. 2002, 50, 468–472. [Google Scholar] [CrossRef]
- Okombi, S.; Rival, D.; Bonnet, S.; Mariotte, A.-M.; Perrier, E.; Boumendjel, A. Analogues of N-hydroxycinnamoylphenalkylamides as inhibitors of human melanocyte-tyrosinase. Bioorg. Med. Chem. Lett. 2006, 16, 2252–2255. [Google Scholar] [CrossRef] [PubMed]
- Tamiz, A.P.; Cai, S.X.; Zhou, Z.-L.; Yuen, P.-W.; Schelkun, R.M.; Whittemore, E.R.; Weber, E.; Woodward, R.M.; Keana, J.F.W. Structure-Activity relationship of N-(Phenylalkyl) cinnamides as novel NR2B subtype-selective NMDA receptor antagonists. J. Med. Chem. 1999, 42, 3412–3420. [Google Scholar] [CrossRef] [PubMed]
- Roh, J.S.; Han, J.Y.; Kim, J.H.; Hwang, J.K. Inhibitory effects of active compounds isolated from safflower (Carthamus tinctorius L.) seeds for melanogenesis. Biol. Pharm. Bull. 2004, 27, 1976–1978. [Google Scholar] [CrossRef]
- ElSohly, M.A.; Slade, D. Chemical constituents of marijuana: The complex mixture of natural cannabinoids. Life Sci. 2005, 78, 539–548. [Google Scholar] [CrossRef]
- Manosroi, A.; Chankhampan, C.; Kietthanakorn, B.O.; Ruksiriwanich, W.; Chaikul, P.; Boonpisuttinant, K.; Sainakham, M.; Manosroi, W.; Tangjai, T.; Manosroi, J. Pharmaceutical and cosmeceutical biological activities of hemp (Cannabis sativa L. var. sativa) leaf and seed extracts. Chiang Mai J. Sci 2019, 46, 180–195. [Google Scholar]
- Ruksiriwanich, W.; Sirithunyalug, J.; Khantham, C.; Leksomboon, K.; Jantrawut, P. Skin penetration and stability enhancement of Celastrus paniculatus seed oil by 2-hydroxypropyl-β-cyclodextrin inclusion complex for cosmeceutical applications. Sci. Pharm. 2018, 86, 33. [Google Scholar] [CrossRef] [PubMed]
- Manosroi, A.; Chaikul, P.; Chankhampan, C.; Ruksiriwanich, W.; Manosroi, W.; Manosroi, J. 5-α-Reductase inhibition and melanogenesis induction of the selected Thai plant extracts. Chiang Mai J. Sci. 2018, 45, 220–236. [Google Scholar]
- Wong, K.C.; Teng, Y.E. Volatile components of Mimusops elengi L. flowers. J. Essent. Oil Res. 1994, 6, 453–458. [Google Scholar] [CrossRef]
- Kietthanakorn, B.; Ruksiriwanich, W.; Manosroi, W.; Manosroi, J.; Manosroi, A. Biological activities of supercritical carbon dioxide fluid (scCO2) extracts from medicinal flowers. Chiang Mai J. Sci. 2012, 39, 84–96. [Google Scholar]
- Phrutivorapongkul, A.; Kiattisin, K.; Jantrawut, P.; Chansakaow, S.; Vejabhikul, S.; Leelapornpisid, P. Appraisal of biological activities and identification of phenolic compound of African marigold (Tagetes erecta L.) flower extract. Pak. J. Pharm. Sci 2013, 26, 1071–1076. [Google Scholar] [PubMed]
- Lai, X.; Wichers, H.J.; Soler-Lopez, M.; Dijkstra, B.W. Structure and function of human tyrosinase and tyrosinase-related proteins. Chem. Eur. J. 2018, 24, 47–55. [Google Scholar] [CrossRef]
- Baell, J.B.; Nissink, J.W.M. Seven year itch: Pan-assay interference compounds (PAINS) in 2017-utility and limitations. ACS Chem. Biol. 2018, 13, 36–44. [Google Scholar] [CrossRef]
- Pintus, F.; Matos, M.J.; Vilar, S.; Hripcsak, G.; Varela, C.; Uriarte, E.; Santana, L.; Borges, F.; Medda, R.; Di Petrillo, A.; et al. New insights into highly potent tyrosinase inhibitors based on 3-heteroarylcoumarins: Anti-melanogenesis and antioxidant activities, and computational molecular modeling studies. Bioorg. Med. Chem. 2017, 25, 1687–1695. [Google Scholar] [CrossRef]
- Sidhu, G.S.; Sattur, P.B.; Jaleel, S. Synthesis and anticonvulsant activity of some N-phenethylacetamides. J. Pharm. Pharmacol. 1962, 14, 125. [Google Scholar] [CrossRef]
- Seo, W.D.; Ryu, Y.B.; Curtis-Long, M.J.; Lee, C.W.; Ryu, H.W.; Jang, K.C.; Park, K.H. Evaluation of anti-pigmentary effect of synthetic sulfonylamino chalcone. Eur. J. Med. Chem. 2010, 45. [Google Scholar] [CrossRef]
- Sugimoto, K.; Nishimura, T.; Nomura, K.; Sugimoto, K.; Kuriki, T. Syntheses of arbutin-α-glycosides and a comparison of their inhibitory effects with those of α-arbutin and arbutin on human tyrosinase. Chem. Pharm. Bull. 2003, 51, 798–801. [Google Scholar] [CrossRef]
- Funayama, M.; Nishino, T.; Hirota, A.; Murao, S.; Takenishi, S.; Nakano, H. Enzymatic synthesis of (+) catechin-α-glucoside and its effect on tyrosinase activity. Biosci. Biotechnol. Biochem. 1993, 57, 1666–1669. [Google Scholar] [CrossRef]
- Mendes, E.; Perry, M.d.J.; Francisco, A.P. Design and discovery of mushroom tyrosinase inhibitors and their therapeutic applications. Expert Opin. Drug Discov. 2014, 9, 533–554. [Google Scholar] [CrossRef] [PubMed]
- Chang, T.S. An updated review of tyrosinase inhibitors. Int. J. Mol. Sci. 2009, 10, 2440–2475. [Google Scholar] [CrossRef]
- Shi, Y.; Chen, Q.-X.; Wang, Q.; Song, K.-K.; Qiu, L. Inhibitory effects of cinnamic acid and its derivatives on the diphenolase activity of mushroom (Agaricus bisporus) tyrosinase. Food Chem. 2005, 92, 707–712. [Google Scholar] [CrossRef]
- Kim, K.-D.; Song, M.-H.; Yum, E.-K.; Jeon, O.-S.; Ju, Y.-W.; Chang, M.-S. 2,4-dihydroxycinnamic esters as skin depigmenting agents. Bull. Korean Chem. Soc. 2009, 30, 1619–1621. [Google Scholar] [CrossRef][Green Version]
- Loizzo, M.R.; Tundis, R.; Menichini, F. Natural and synthetic tyrosinase inhibitors as antibrowning agents: An update. Compr. Rev. Food Sci. Food Saf. 2012, 11, 378–398. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

| Compound | Substitution Pattern | Mushroom Tyrosinase IC50 ± SEM (µM) | Human Tyrosinase (% Inhibition) | Yield (%) | |
|---|---|---|---|---|---|
| R1 | R2 | ||||
| Ph1 | 3-OH | 4.065 ± 0.154 | 75.590 ± 5.653 | 78 | |
| Ph2 | 4-OH | 0.463 ± 0.017 | 73.049 ± 5.346 | 82 | |
| Ph3 | 2,4-di-OH | 0.839 ± 0.0381 | 79.710 ± 6.61 | 76 | |
| Ph4 | 3,4-di-OH | 0.329 ± 0.021 | 82.17 ± 7.045 | 71 | |
| Ph5 | 3,5-di-OH | 0.231 ± 0.0078 | 64.8 ± 3.84 | 76 | |
| Ph6 | -H | 0.00204 ± 0.000087 | 92.24 ± 7.985 | 84 | |
| Ph7 | 2-OH | 0.1042 ± 0.0038 | 85.6 ± 6.041 | 75 | |
| Ph8 | 4-OH | 0.923 ± 0.053 | 86.27 ± 6.542 | 78 | |
| Ph9 | 2,4-di-OH | 0.00005950 ± 0.00000225 | 94.59 ± 8.071 | 76 | |
| Ph10 | 4-Cl | 0.4520 ± 0.0171 | 79.23 ± 5.952 | 82 | |
| Kojic Acid | 16.69 ± 2.6 | 72.94 ± 5.125 | |||
| Code | Dose (µM) | Vmax (∆A/s) | Km (mM) | Inhibition Type | Ki (µM) | Ki′ (µM) |
|---|---|---|---|---|---|---|
| Ph5 | 0.0 | 1.251 × 10−5 | 0.181 | Mixed-Inhibition | 0.219 | 0.475 |
| 0.2 | 8.266 × 10−6 | 0.238 | ||||
| 0.4 | 5.242 × 10−6 | 0.263 | ||||
| Ph6 | 0.0 | 1.272 × 10−5 | 0.232 | Non-competitive | 0.0023 | - |
| 0.001 | 7.060 × 10−6 | 0.232 | ||||
| 0.002 | 6.929 × 10−6 | 0.238 | ||||
| Ph9 | 0.0 | 4.848 × 10−6 | 0.192 | Mixed-Inhibition | 0.000093 | - |
| 0.00003 | 4.666 × 10−6 | 0.227 | ||||
| 0.00015 | 4.285 × 10−6 | 0.238 | ||||
| 0.00012 | 2.666 × 10−6 | 0.25 |
| Compound | Docking Energy (Kcal/mol), PDB ID (2Y9X) |
|---|---|
| Ph1 | −5.492 |
| Ph2 | −4.070 |
| Ph3 | −5.850 |
| Ph4 | −6.830 |
| Ph5 | −5.339 |
| Ph6 | −4.543 |
| Ph7 | −5.890 |
| Ph8 | −5.511 |
| Ph9 | −7.930 |
| Ph10 | −5.516 |
| Kojic Acid | −4.149 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nazir, Y.; Rafique, H.; Kausar, N.; Abbas, Q.; Ashraf, Z.; Rachtanapun, P.; Jantanasakulwong, K.; Ruksiriwanich, W. Methoxy-Substituted Tyramine Derivatives Synthesis, Computational Studies and Tyrosinase Inhibitory Kinetics. Molecules 2021, 26, 2477. https://doi.org/10.3390/molecules26092477
Nazir Y, Rafique H, Kausar N, Abbas Q, Ashraf Z, Rachtanapun P, Jantanasakulwong K, Ruksiriwanich W. Methoxy-Substituted Tyramine Derivatives Synthesis, Computational Studies and Tyrosinase Inhibitory Kinetics. Molecules. 2021; 26(9):2477. https://doi.org/10.3390/molecules26092477
Chicago/Turabian StyleNazir, Yasir, Hummera Rafique, Naghmana Kausar, Qamar Abbas, Zaman Ashraf, Pornchai Rachtanapun, Kittisak Jantanasakulwong, and Warintorn Ruksiriwanich. 2021. "Methoxy-Substituted Tyramine Derivatives Synthesis, Computational Studies and Tyrosinase Inhibitory Kinetics" Molecules 26, no. 9: 2477. https://doi.org/10.3390/molecules26092477
APA StyleNazir, Y., Rafique, H., Kausar, N., Abbas, Q., Ashraf, Z., Rachtanapun, P., Jantanasakulwong, K., & Ruksiriwanich, W. (2021). Methoxy-Substituted Tyramine Derivatives Synthesis, Computational Studies and Tyrosinase Inhibitory Kinetics. Molecules, 26(9), 2477. https://doi.org/10.3390/molecules26092477