
Effectiveness of Bioactive Compound as Antibacterial and Anti-Quorum Sensing Agent from Myrmecodia pendans: An In Silico Study

 ,
,
Abstract
1. Introduction
2. Results
2.1. Bioactivity Prediction of the M. pendans Compound via Prediction of Activity Spectra for Substances (PASS) Online Analysis
2.2. Prediction of Bioavailability and Antibacterial Activity of M. pendans through Molecular Interaction with Targeted Proteins
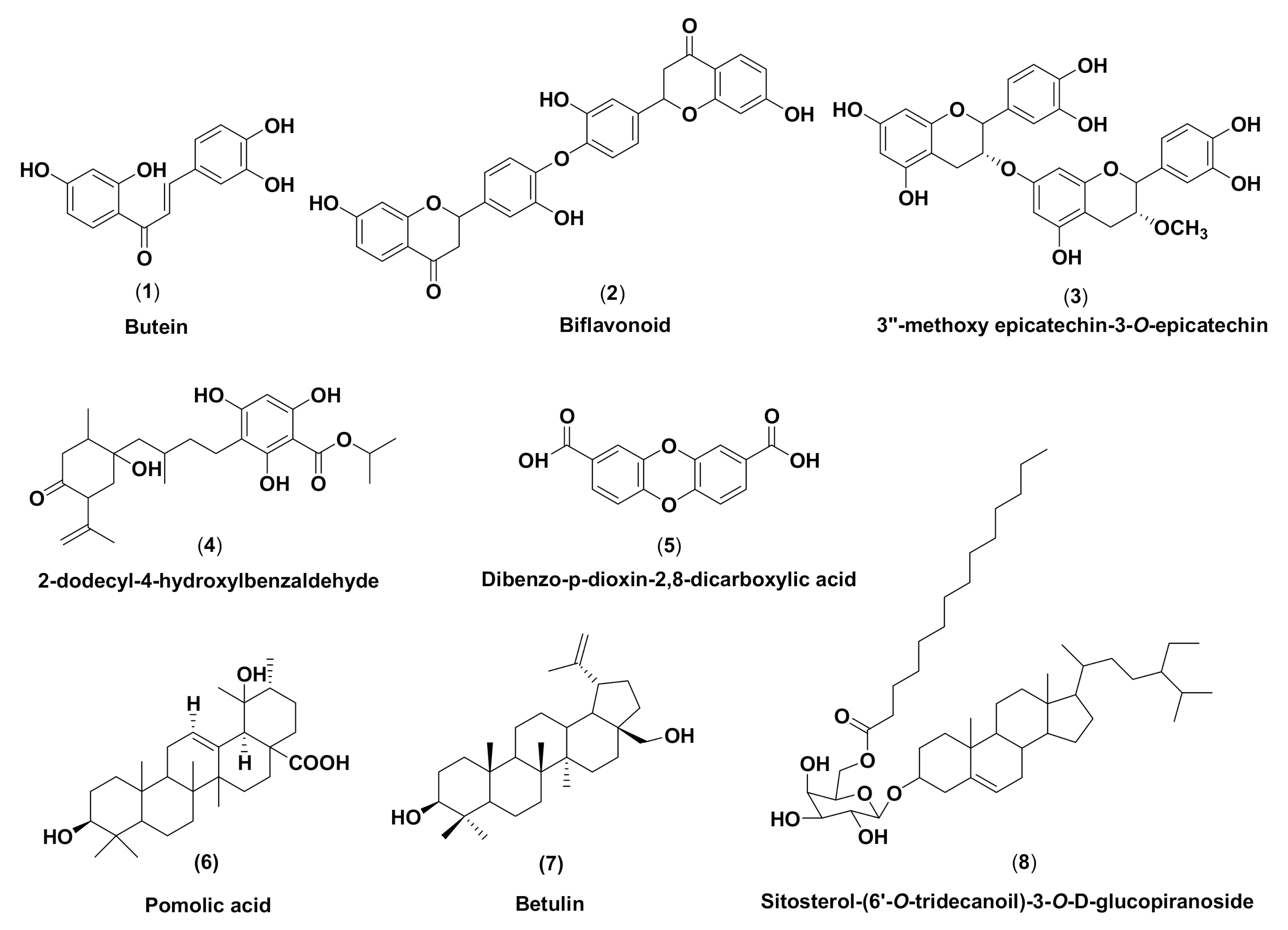
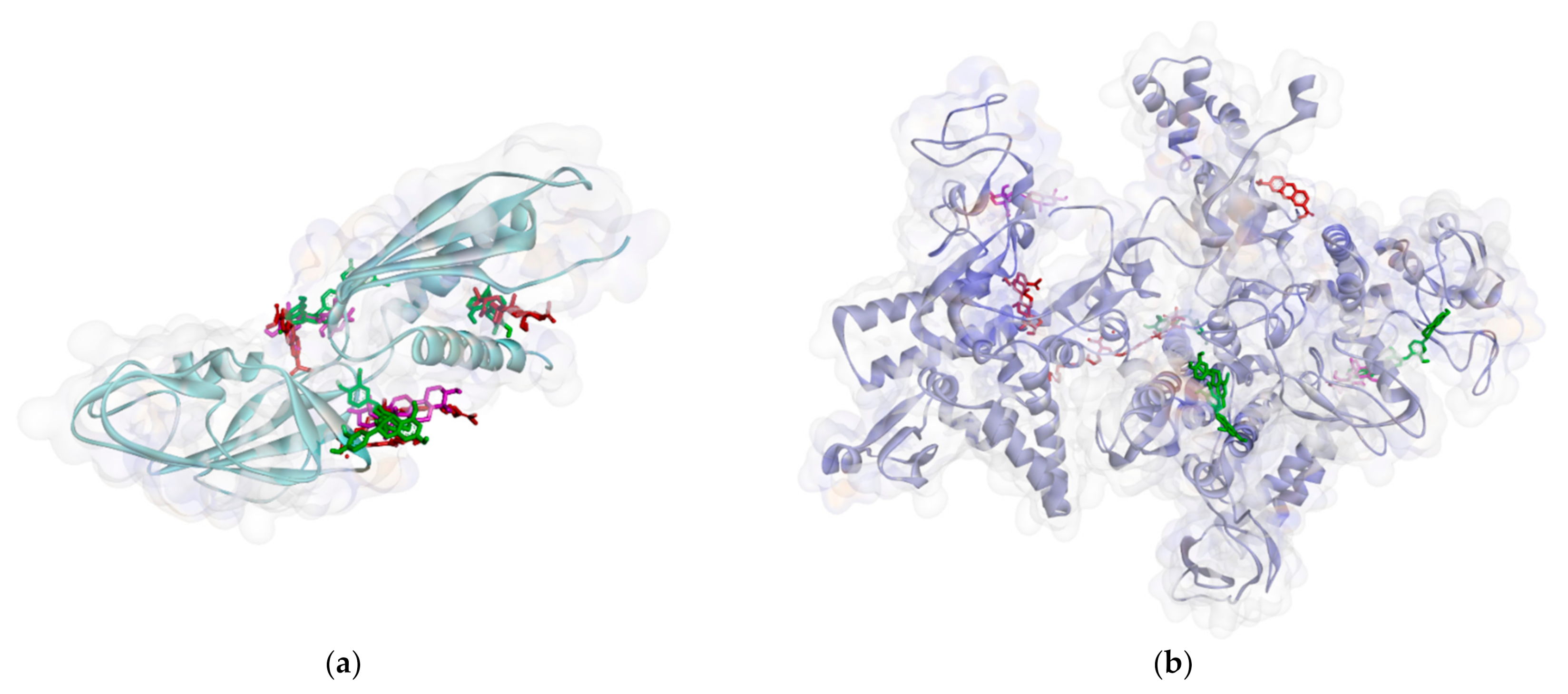
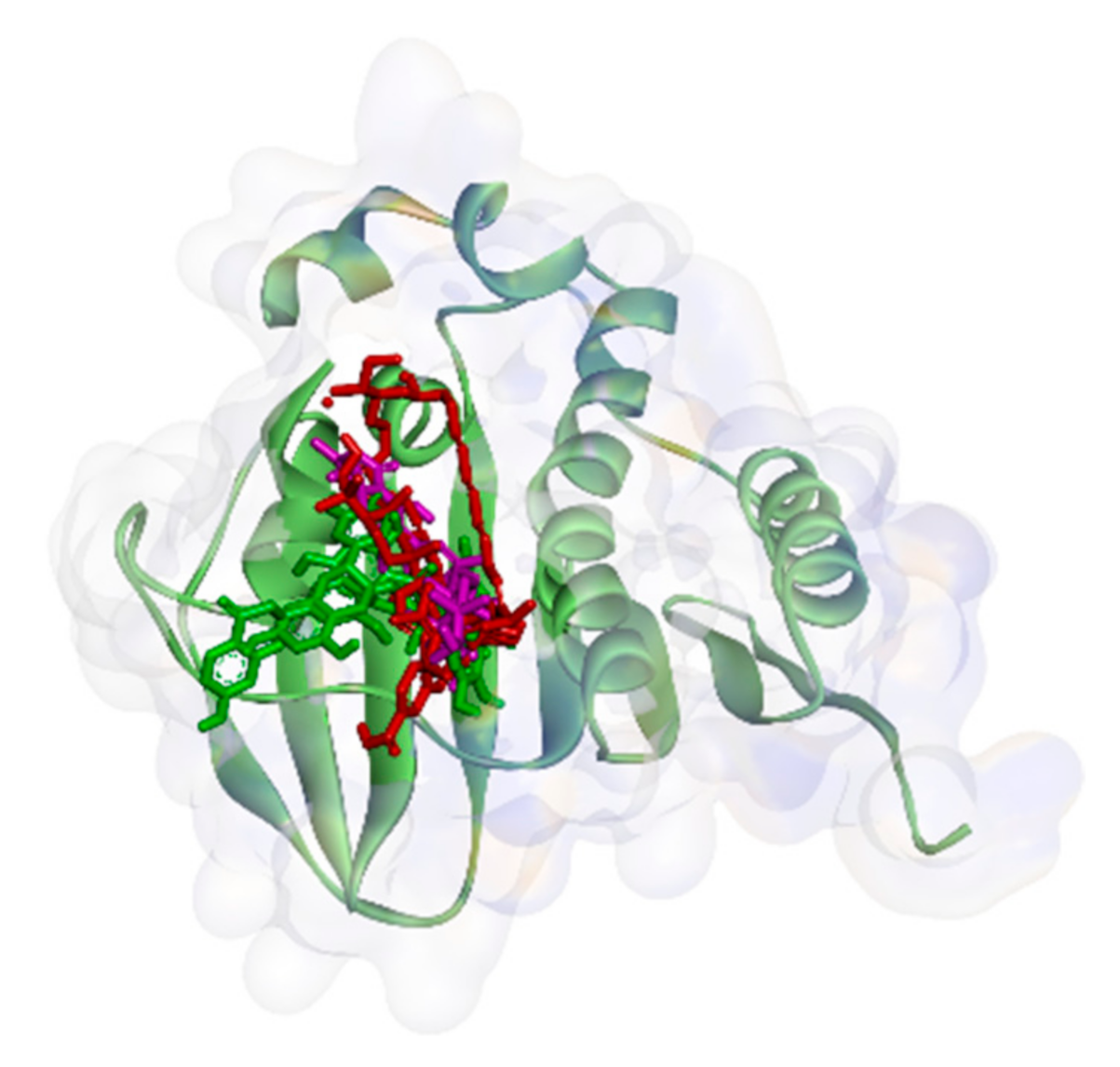
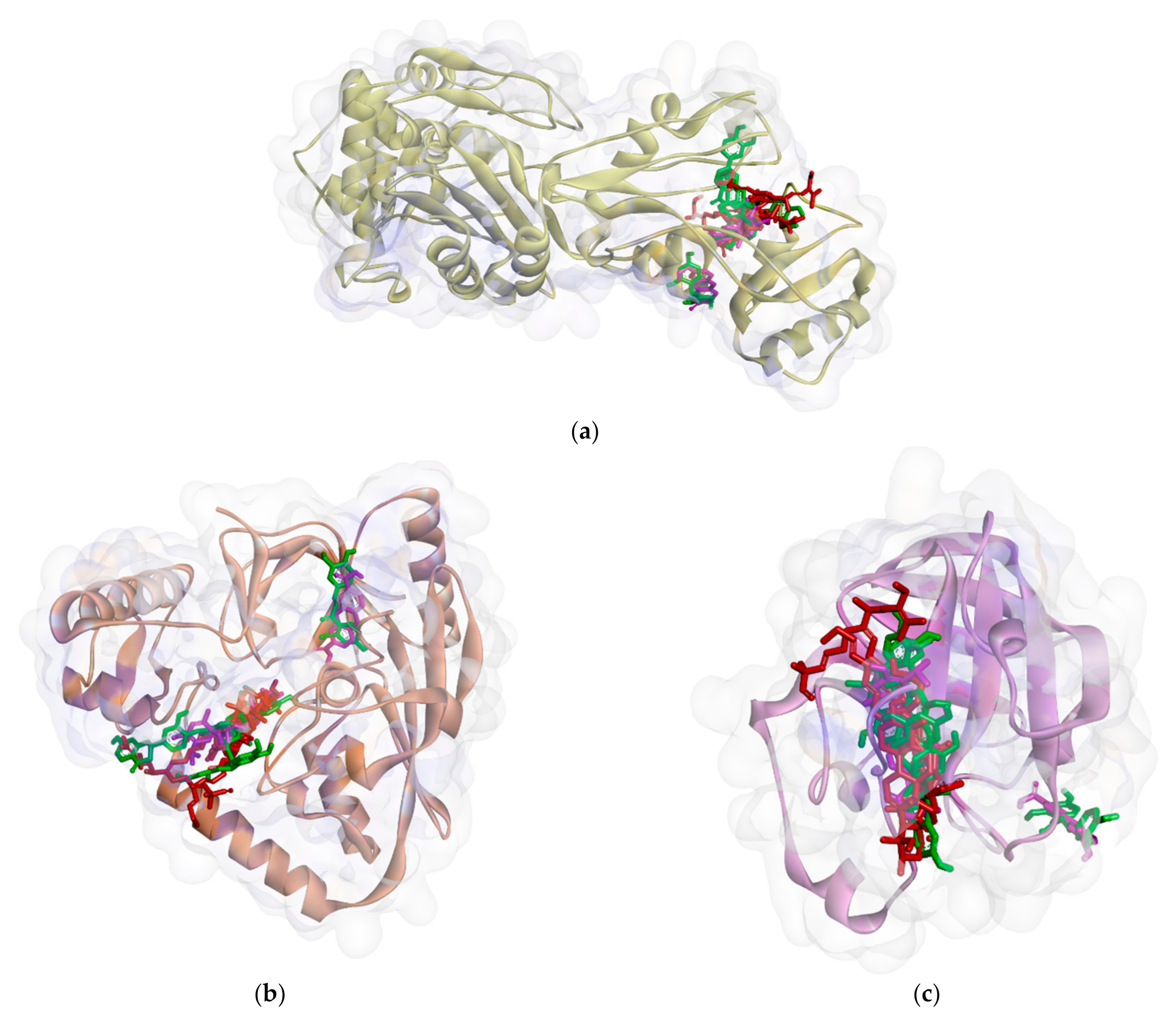
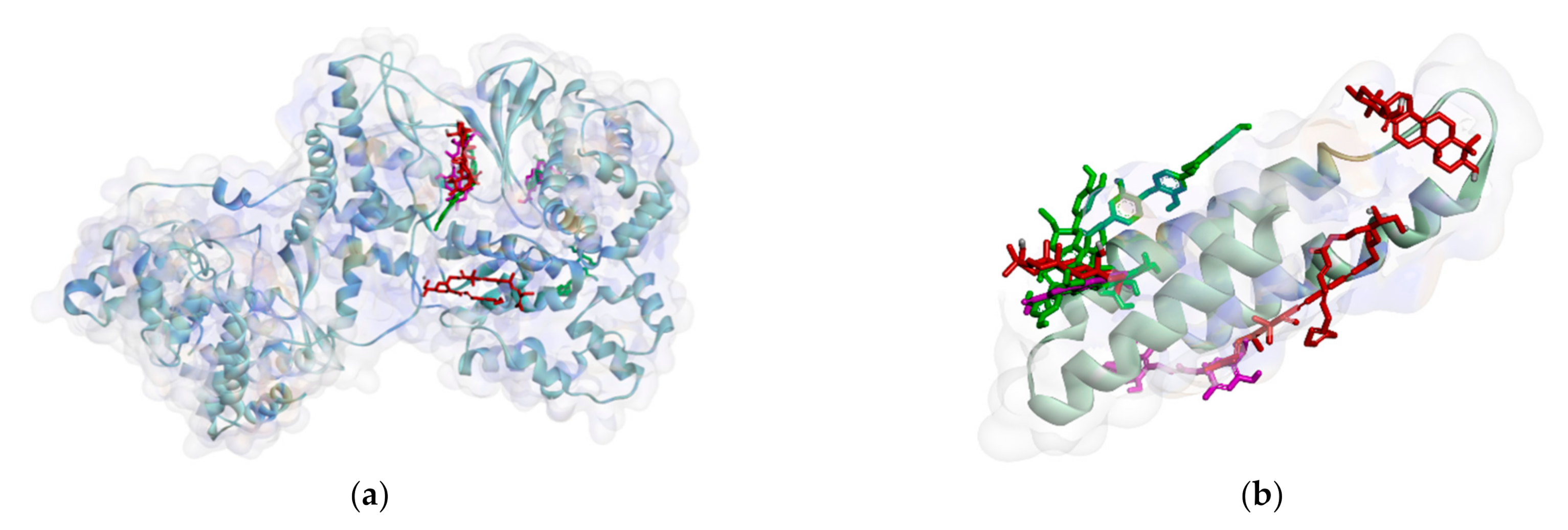
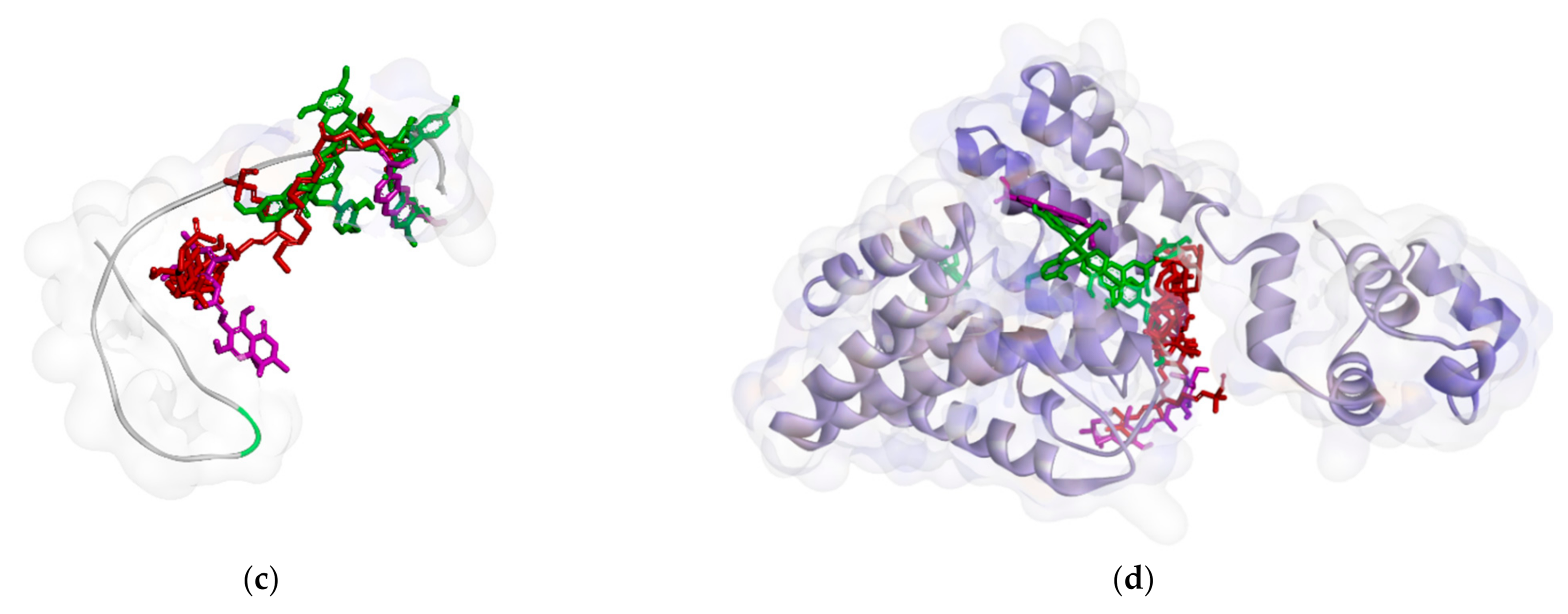
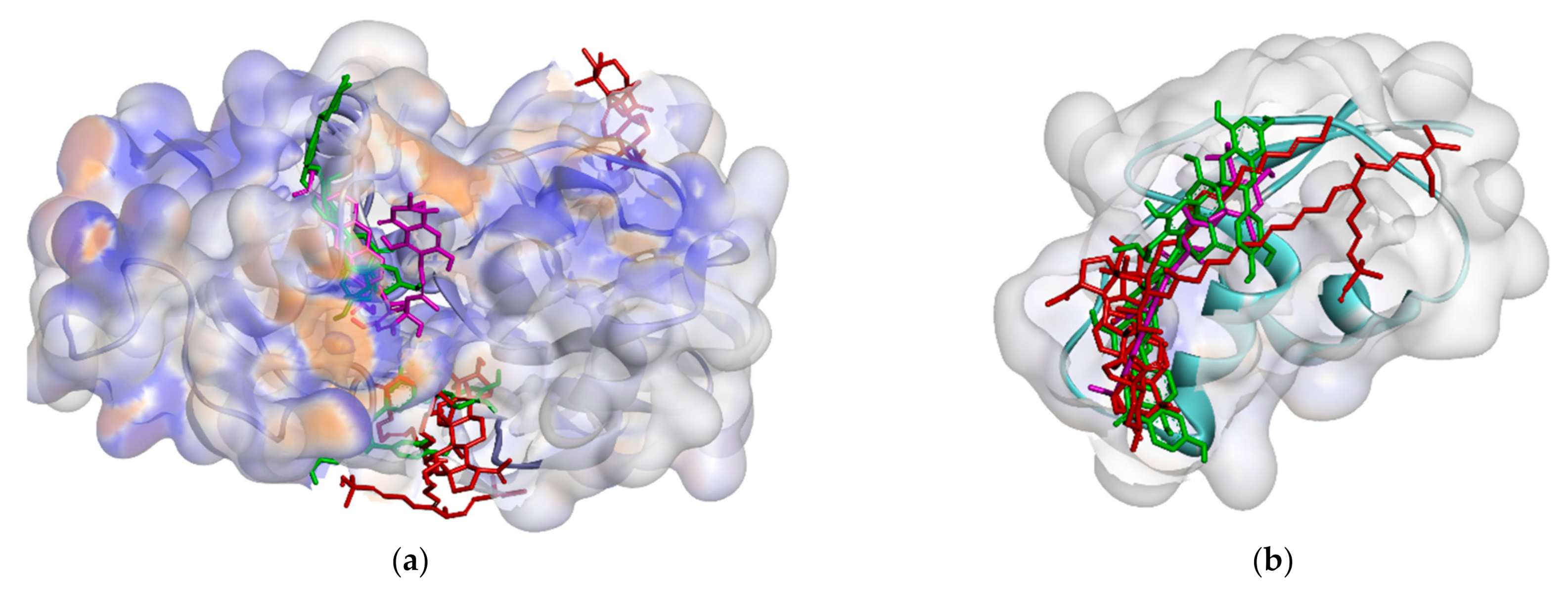
2.2.1. Binding Affinity Analysis of Compounds to Proteins
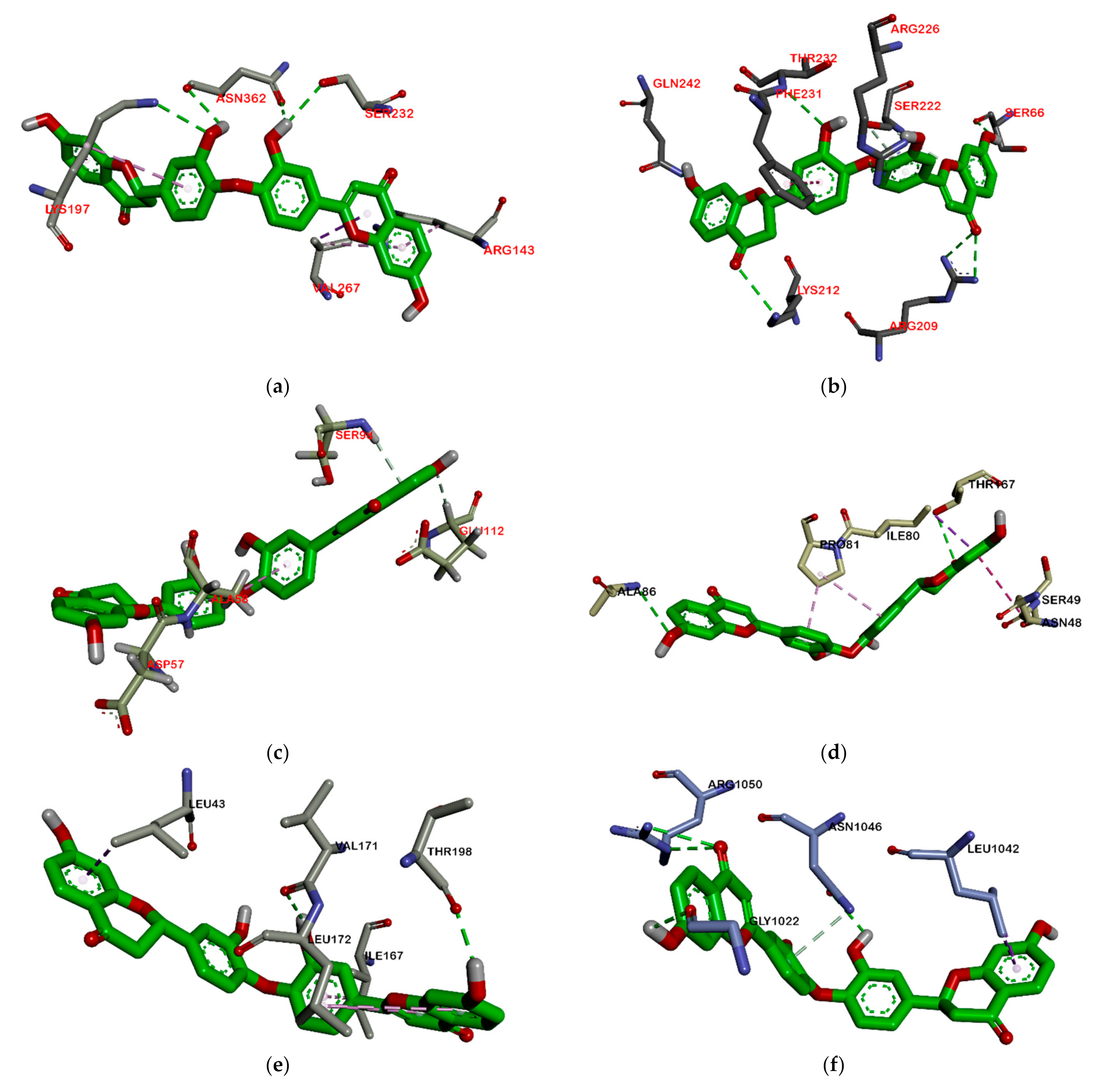
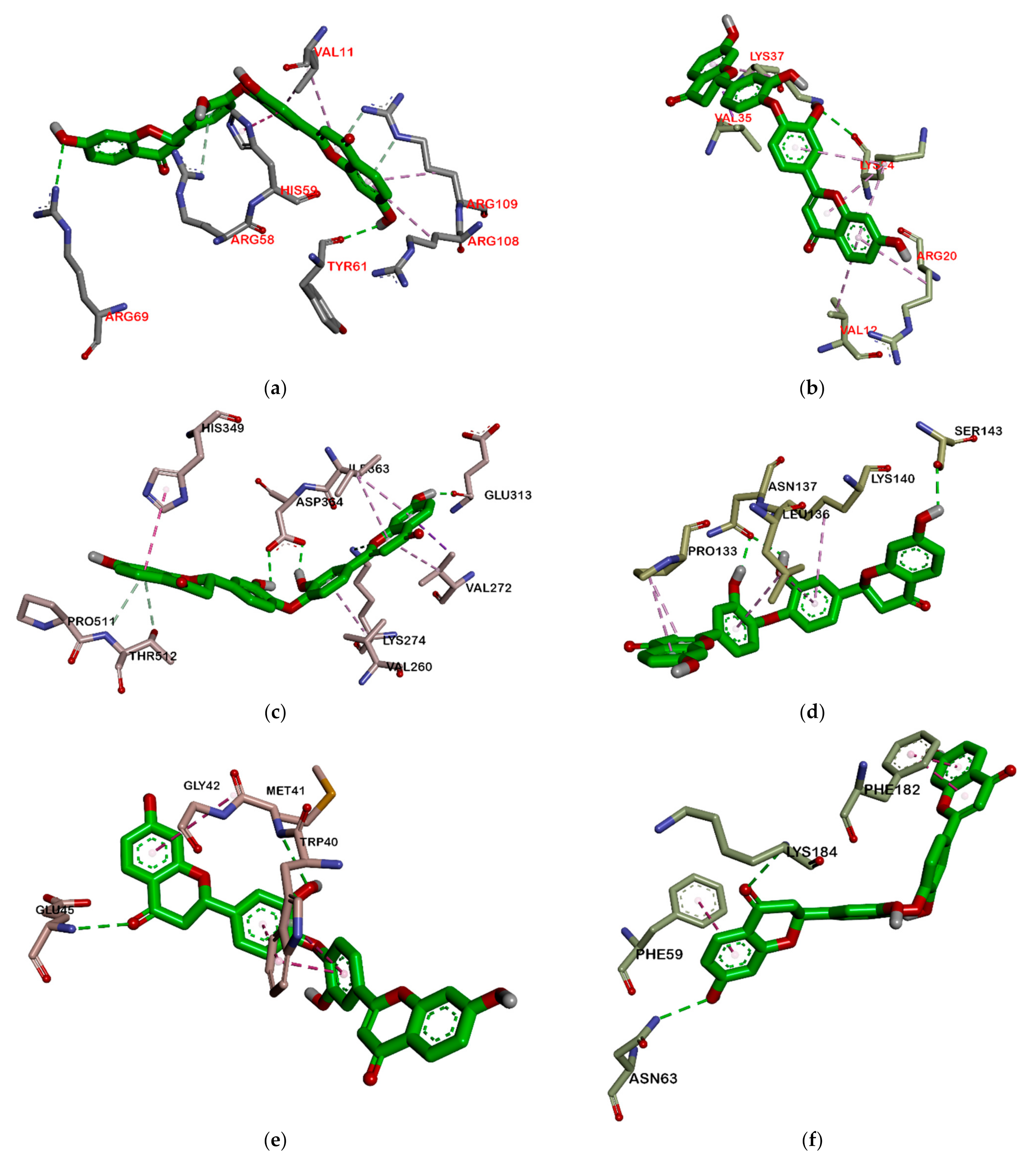
2.2.2. Hydrogen Bond and Hydrophobic Contact Analysis of Compounds to Proteins
2.2.3. Prediction of Lipinski’s Rule
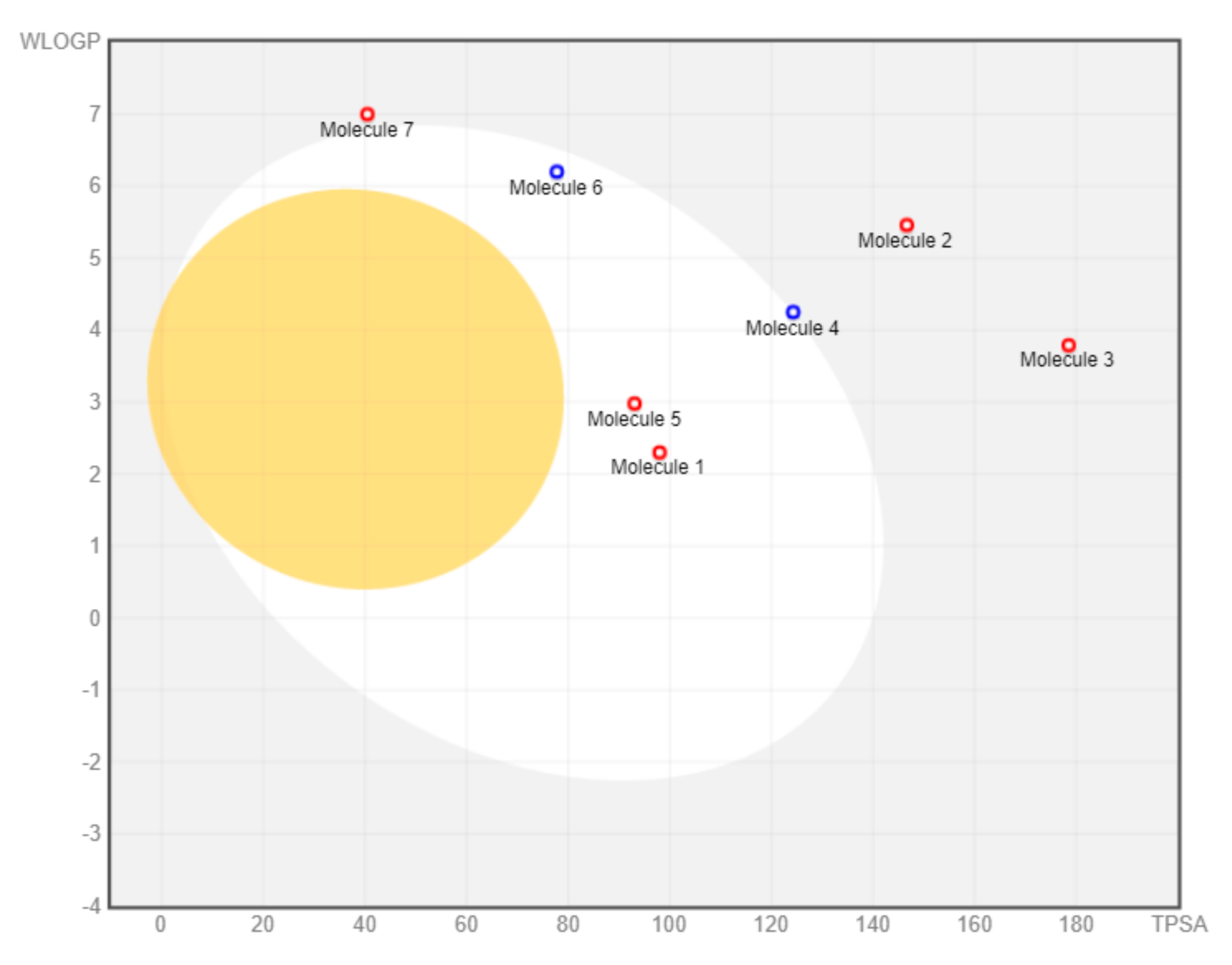
2.2.4. Drug-Likeness Analysis of M. pendans Compounds
2.2.5. Pharmacokinetic Prediction of M. pendans Compounds
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Methods
4.2.1. In Silico Characterization of the M. Pendans Compounds
4.2.2. PASS Online Analysis of M. pendans Compounds
4.2.3. Molecular Docking between Target Protein and M. Pendans Compounds
4.2.4. Complex Protein-Ligand Visualization and Analysis
4.2.5. Analysis of Lipinski Prediction
4.2.6. Analysis of ADMET and Drug-Likeness Prediction
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Bloom, D.E.; Cadarette, D. Infectious disease threats in the twenty-first century: Strengthening the global response. Front. Immunol. 2019, 10, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Chen, H. Increased Enterococcus faecalis infection is associated with clinically active Crohn disease. Medicine 2016, 95, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Friedman, N.D.; Temkin, E. The negative impact of antibiotic resistance. Clin. Microbiol. Infect. 2016, 22, 416–422. [Google Scholar] [CrossRef] [PubMed]
- Jukič, M.; Gobec, S. Reaching toward underexplored targets in antibacterial drug design. Drug Dev. Res. 2019, 80, 6–10. [Google Scholar] [CrossRef]
- Moon, T.M.; D’Andrea, E.D. The structures of penicillin-binding protein 4 (PBP4) and PBP5 from Enterococci provide structural insights into -lactam resistance. J. Biol. Chem. 2018, 293, 18574–18585. [Google Scholar] [CrossRef]
- Selvaraj, C.; Sivakamavalli, J. Structural elucidation of srta enzyme in Enterococcus faecalis: An emphasis on screening of potential inhibitors against the biofilm formation. Mol. Biosyst. 2014, 10, 1775–1789. [Google Scholar] [CrossRef]
- Giuliodori, A.M.; Spurio, R. Antibiotics Targeting the 30S Ribosomal Subunit: A Lesson from Nature to Find and Develop New Drugs. Curr. Top. Med. Chem. 2018, 18, 2080–2096. [Google Scholar] [CrossRef]
- Maguire, B.A. Inhibition of Bacterial Ribosome Assembly: A Suitable Drug Target? Microbiol. Mol. Biol. Rev. 2009, 73, 22–35. [Google Scholar] [CrossRef]
- Ma, C.; Yang, X. Bacterial Transcription as a Target for Antibacterial Drug Development. Microbiol. Mol. Biol. Rev. 2016, 80, 139–160. [Google Scholar] [CrossRef]
- Khan, T.; Sankhe, K. DNA gyrase inhibitors: Progress and synthesis of potent compounds as antibacterial agents. Biomed. Pharmacother. 2018, 103, 923–938. [Google Scholar] [CrossRef]
- Gajdács, M.; Spengler, G. The role of drug repurposing in the development of novel antimicrobial drugs: Non-antibiotic pharmacological agents as quorum sensing-inhibitors. Antibiotics 2019, 8, 270. [Google Scholar] [CrossRef]
- Fernandes, M.; Esper, L.M.R. R Quorum sensing in Enterococcus faecium, Enterococcus faecalis and Bacillus cereus strains isolated from ricotta processing. Food Technol. 2016, 48, 17. [Google Scholar] [CrossRef]
- Verbeke, F.; Craemer, S.D. Peptides as quorum sensing molecules: Measurement techniques and obtained levels in vitro and in vivo. Front. Neurosci. 2017, 11, 1–18. [Google Scholar] [CrossRef]
- Rutherford, S.T.; Bassler, B.L. Bacterial quorum sensing: Its role in virulence and possibilities for its control. Cold Spring Harb. Perspect. Med. 2012, 2, 1–26. [Google Scholar] [CrossRef]
- Paluch, E.; Rewak-Soroczyńska, J. Prevention of biofilm formation by quorum quenching. Appl. Microbiol. Biotechnol. 2020, 104, 1871–1881. [Google Scholar] [CrossRef]
- Cook, L.C.; Federle, M.J. Peptide pheromone signaling in Streptococcus and Enterococcus. FEMS Microbiol. Rev. 2014, 38, 473–492. [Google Scholar] [CrossRef]
- Shi, K.; Brown, C.K. Structure of peptide sex pheromone receptor PrgX and PrgX/pheromone complexes and regulation of conjugation in Enterococcus faecalis. Proc. Natl. Acad. Sci. USA 2005, 102, 18596–18601. [Google Scholar] [CrossRef]
- Mylonakis, E.; Engelbert, M. The Enterococcus faecalis fsrB gene, a key component of the fsr quorum-sensing system, is associated with virulence in the rabbit endophthalmitis model. Infect. Immun. 2002, 70, 4678–4681. [Google Scholar] [CrossRef]
- Nakayama, J.; Ca, Y. Gelatinase biosynthesis-activating pheromone: A peptide lactone that mediates a quorum sensing in Enterococcus faecalis. Mol. Microbiol. 2001, 41, 145–154. [Google Scholar] [CrossRef]
- Littlewood, S.; Tattersall, H. The gelatinase biosynthesis-activating pheromone binds and stabilises the FsrB membrane protein in Enterococcus faecalis quorum sensing. FEBS Lett. 2020, 594, 553–563. [Google Scholar] [CrossRef]
- Haas, W.; Shepard, B.D. Two-component regulator of Enterococcus faecalis cytolysin responds to quorum-sensing autoinduction. Nature 2002, 415, 84–87. [Google Scholar] [CrossRef] [PubMed]
- Coburn, P.S.; Gilmore, M.S. The Enterococcus faecalis cytolysin: A novel toxin active against eukaryotic and prokaryotic cells. Cell. Microbiol. 2003, 5, 661–669. [Google Scholar] [CrossRef] [PubMed]
- Garsin, D.A. Peptide signals sense and destroy target cells. Microbilogy 2004, 306, 2202–2203. [Google Scholar] [CrossRef] [PubMed]
- Ali, L.; Goraya, M.U. Molecular Mechanism of Quorum-Sensing in Enterococcus faecalis: Its Role in Virulence and Therapeutic Approaches. Int. J. Mol. Sci. 2017, 18, 960–978. [Google Scholar] [CrossRef]
- Asfour, H.Z. Anti—Quorum Sensing Natural Compounds. J. Microsc. Ultrastruct. 2018, 6, 1–10. [Google Scholar] [CrossRef]
- Ramadhany, S.; Achmad, M.H. Formulation of ethanol extract (Myrmecodia pendans) as an antibacterial Streptococcus mutans in chewable lozenges for children with early childhood caries. Syst. Rev. Pharm. 2020, 11, 252–257. [Google Scholar] [CrossRef]
- Binartha, C.T.O.; Suprastiwi, E. Antibacterial effects of sarang semut (Myrmecodia pendans) fractions using three different solvents toward enterococcus faecalis CPS2. Int. J. Appl. Pharm. 2020, 12, 271–275. [Google Scholar] [CrossRef]
- Soviati, N.; Widyarman, A.S. The Effect Ant-Nest Plant (Myrmecodia pendans) Extract on Streptococcus sanguinis and Treponema denticola Biofilms. J. Indones. Dent. Assoc. 2020, 3, 11. [Google Scholar] [CrossRef]
- Soraya, C.; Dharsono, H.D.A. Antibacterial potency of Sarang Semut (Myrmecodia pendans Merr. & Perry) to prevent adhesion and growth of Enterococcus faecalis. Int. J. Sci. Res. 2017, 6, 2694–2698. [Google Scholar] [CrossRef]
- Soraya, C.; Dharsono, H.D.A. Effects of sarang semut (Myrmecodia pendens Merr. & Perry) extracts on Enterococcus faecalis sensitivity. Dent. J. 2016, 49, 175. [Google Scholar] [CrossRef][Green Version]
- Kurnia, D.; Sumiarsa, D. Bioactive compounds isolated from Indonesian epiphytic plant of Sarang Semut and their antibacterial activity against pathogenic oral bacteria. Nat. Prod. Commun. 2017, 12, 1201–1204. [Google Scholar] [CrossRef]
- Gartika, M.; Pramesti, H.T. A terpenoid isolated from sarang semut (Myrmecodia pendans) bulb and its potential for the inhibition and eradication of Streptococcus mutans biofilm. BMC Complement. Altern. Med. 2018, 18, 1–8. [Google Scholar] [CrossRef]
- Kurnia, D.; Apriyanti, E. Antibacterial flavonoids against oral bacteria of Enterococcus faecalis ATCC 29212 from Sarang Semut (Myrmecodia pendans) and its inhibitor activity against enzyme MurA. Curr. Drug Discov. Technol. 2019, 16, 290–296. [Google Scholar] [CrossRef]
- Apriyanti, E.; Kurnia, D. Potential of MurA enzyme and GBAP in Fsr quorum sensing system as antibacterial drugs target: In vitro and in silico study of antibacterial compounds from Myrmecodia pendans. Comb. Chem. Highthroughput Screen. 2020, 23, 1–10. [Google Scholar] [CrossRef]
- Chaudhary, K.K.; Mishra, N.A. Review on molecular docking: Novel tool for drug discovery. JSM Chem. 2016, 4, 1029. [Google Scholar]
- Filimonov, D.A.; Lagunin, A.A. Prediction of the biological activity spectra of organic compounds using the pass online web resource. Chem. Heterocycl. Compd. 2014, 50, 444–457. [Google Scholar] [CrossRef]
- Lipinski, C.A.; Lombardo, F. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. 1997, 23, 3–25. [Google Scholar] [CrossRef]
- Nisha, C.M.; Kumar, A. Molecular Docking and In Silico ADMET Study Reveals Acylguanidine 7a as a Potential Inhibitor of β-Secretase. Adv. Bioinform. 2016, 2016, 1–7. [Google Scholar] [CrossRef]
- Subramaniyan, V.; Mathiayalagan, S. Molecular docking and ADME properties of bioactive molecules against human acid-beta-glucosidase enzyme, cause of Gaucher’s disease. In Silico Pharmacol. 2018, 6, 1–11. [Google Scholar] [CrossRef]
- Awaluddin, R.; Muhtadi, W.K. Molecular docking and ADME-toxicity studies of potential compounds of medicinal plants grown in Indonesia as an anti-rheumatoid arthritis. AIP Conf. Proc. 2017, 1823, 20033. [Google Scholar] [CrossRef]
- Boyle, N.M.O.; Banck, M. Open Babel: An open chemical toolbox. J. Cheminform. 2011, 3, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Biasini, M.; Bienert, S. SWISS-MODEL: Modelling protein tertiary and quaternary structure using evolutionary information. Nucleic Acids Res. 2014, 42, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Lagunin, A.; Stepanchikova, A. PASS: Prediction of activity spectra for biologically active substances. Bioinformatics 2000, 16, 747–748. [Google Scholar] [CrossRef] [PubMed]
- Dallakyan, S.; Olson, A.J. Small-Molecule Library Screening by Docking with PyRx. Methods Mol. Biol. 2015, 1263, 243–250. [Google Scholar] [CrossRef]
- Azam, S.S.; Abbas, S.W. Molecular docking studies for the identification of novel melatoninergic inhibitors for acetylserotonin-O-methyltransferase using different docking routines. Theor. Biol. Med. Model. 2013, 10, 1–16. [Google Scholar] [CrossRef]
- Rauf, M.A.; Zubair, S. Ligand docking and binding site analysis with PyMOL and Autodock/Vina. Int. J. Basic Appl. Sci. 2015, 4, 168–177. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 1–13. [Google Scholar] [CrossRef]
- Blake, J.F. Chemoinformatics—Predicting the physicochemical properties of ‘drug-like’ molecules. Curr. Opin. Biotechnol. 2000, 11, 104–107. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No | Bioactivity | Prediction to be Active (Pa) of Compound | |||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | ||
| General | |||||||||
| 1 | Antibacterial | 0.375 | 0.326 | 0.344 | 0.441 | - | - | 0.242 | - |
| 2 | Antibiotic | - | 0.095 | - | 0.251 | - | - | - | - |
| 3 | Antifungal | 0.494 | 0.490 | 0.498 | 0.622 | - | 0.526 | 0.507 | 0.327 |
| 4 | Anti-infective | 0.442 | 0.375 | - | - | - | - | - | - |
| 5 | Antimycobacterial | 0.621 | 0.347 | - | - | - | - | - | - |
| 6 | Antiparasitic | 0.483 | - | - | - | - | - | 0.199 | - |
| 7 | Antiseptic | 0.783 | 0.271 | - | 0.277 | 0.751 | - | - | - |
| 8 | Anti-tuberculosis | 0.583 | - | - | - | - | - | - | - |
| DNA Synthesis Pathway | |||||||||
| 1 | DNA synthesis inhibitor | - | - | - | - | - | - | 0.247 | 0.195 |
| 2 | DNA-3-methyladenine glycosylase I inhibitor | 0.255 | - | - | - | 0.725 | - | - | - |
| 3 | DNA-(apurinic or apyrimidinic site) lyase inhibitor | 0.255 | 0.217 | - | - | 0.829 | - | - | - |
| 4 | DNA gyrase inhibitor | - | 0.033 | 0.159 | - | - | - | - | - |
| 5 | DNA ligase (ATP) inhibitor | 0.384 | 0.401 | 0.386 | 0.221 | - | 0.733 | 0.496 | - |
| 6 | DNA polymerase I inhibitor | 0.260 | 0.248 | 0.299 | - | - | - | 0.360 | 0.253 |
| 7 | DNA directed RNA polymerase inhibitor | - | - | 0.137 | 0.164 | - | - | 0.172 | 0.338 |
| 8 | Transcription factor inhibitor | 0.511 | - | - | - | - | - | - | - |
| Protein Synthesis Pathway | |||||||||
| 1 | Protein synthesis inhibitor | - | - | - | 0.322 | - | - | 0.202 | 0.246 |
| 2 | Protein 30S ribosomal subunit inhibitor | - | - | 0.077 | - | - | - | - | - |
| 3 | Tpr proteinase (Porphyromonas gingivalis) inhibitor | - | - | - | - | 0.862 | - | - | - |
| No | Bioactivity (38) | Prediction to be Active (Pa) of Compound | |||||||
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | ||
| Cell Wall Biosynthesis Pathway | |||||||||
| 1 | Cell wall biosynthesis inhibitor | 0.227 | - | 0.204 | - | - | - | - | - |
| 2 | Cell adhesion molecule inhibitor | 0.327 | - | - | - | - | - | - | - |
| 3 | Membrane integrity antagonist | 0.234 | 0.516 | 0.263 | - | - | - | 0.496 | - |
| 4 | Membrane permeability inhibitor | 0.745 | 0.788 | 0.700 | 0.680 | 0.820 | - | 0.411 | - |
| 5 | Peptidoglycan glycosyltransferase inhibitor | - | 0.327 | - | - | - | 0.460 | 0.590 | - |
| 6 | Phospholipid-translocating ATPase inhibitor | - | - | - | - | 0.815 | - | - | - |
| 7 | UDP-glucuronosyltransferase substrate | 0.709 | 0.827 | 0.771 | 0.527 | - | - | 0.554 | 0.183 |
| 8 | UDP-N-acetylglucosamine 4-epimerase inhibitor | 0.559 | - | - | - | 0.924 | - | - | - |
| RNA Synthesis Pathway | |||||||||
| 1 | RNA synthesis inhibitor | 0.313 | 0.326 | 0.328 | 0.261 | - | - | 0.360 | 0.208 |
| 2 | RNA directed DNA polymerase inhibitor | 0.161 | 0.172 | 0.207 | 0.204 | - | - | 0.161 | - |
| 3 | RNA-directed RNA polymerase inhibitor | 0.418 | - | - | - | - | - | 0.276 | - |
| 4 | tRNA nucleotidyltransferase inhibitor | 0.234 | - | - | - | 0.805 | - | - | - |
| 5 | tRNA-pseudouridine synthase I inhibitor | 0.359 | 0.217 | - | - | 0.902 | - | - | - |
| 6 | Alanine-tRNA ligase inhibitor | 0.184 | - | - | - | - | - | - | - |
| 7 | Aspartate-tRNA ligase inhibitor | 0.145 | 0.196 | - | - | - | - | - | - |
| 8 | Asparagine-tRNA ligase inhibitor | 0.124 | 0.151 | - | - | - | - | - | - |
| 9 | Glutamate-tRNA ligase inhibitor | 0.248 | 0.205 | - | - | - | - | - | - |
| No | Bioactivity | Prediction to Be Active (Pi) | |||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | ||
| General | |||||||||
| 1 | Antibacterial | 0.030 | 0.051 | 0.045 | 0.023 | - | - | 0.087 | - |
| 2 | Antibiotic | - | 0.087 | - | 0.020 | - | - | - | - |
| 3 | Antifungal | 0.030 | 0.032 | 0.031 | 0.016 | - | 0.026 | 0.029 | 0.071 |
| 4 | Anti0infective | 0.030 | 0.057 | - | - | - | - | - | - |
| 5 | Antimycobacterial | 0.000 | 0.056 | - | - | - | - | - | - |
| 6 | Antiparasitic | 0.010 | - | - | - | - | - | 0.098 | - |
| 7 | Antiseptic | 0.000 | 0.043 | - | 0.041 | 0.005 | - | - | - |
| 8 | Anti-tuberculosis | 0.000 | - | - | - | - | - | - | - |
| DNA Synthesis Pathway | |||||||||
| 1 | DNA synthesis inhibitor | - | - | - | 0.086 | 0.168 | |||
| 2 | DNA-3-methyladenine glycosylase I inhibitor | 0.007 | - | - | - | 0.003 | - | - | - |
| 3 | DNA-(apurinic or apyrimidinic site) lyase inhibitor | 0.010 | 0.141 | - | - | 0.004 | - | - | - |
| 4 | DNA gyrase inhibitor | - | 0.015 | 0.003 | - | - | - | - | - |
| 5 | DNA ligase (ATP) inhibitor | 0.010 | 0.016 | 0.019 | 0.087 | - | 0.001 | 0.006 | - |
| 6 | DNA polymerase I inhibitor | 0.120 | 0.154 | 0.065 | - | - | - | 0.025 | 0.143 |
| 7 | DNA directed RNA polymerase inhibitor | - | - | 0.104 | 0.065 | - | - | 0.057 | 0.013 |
| 8 | Transcription factor inhibitor | 0.010 | - | - | - | - | - | - | - |
| Protein Synthesis Pathway | |||||||||
| 1 | Protein synthesis inhibitor | - | - | - | 0.027 | - | - | 0.053 | 0.041 |
| 2 | Protein 30S ribosomal subunit inhibitor | - | - | 0.035 | - | - | - | - | - |
| 3 | Tpr proteinase (Porphyromonas gingivalis) inhibitor | - | - | - | - | 0.002 | - | - | - |
| Cell Wall Biosynthesis Pathway | |||||||||
| 1 | Cell wall biosynthesis inhibitor | 0.100 | - | 0.148 | - | - | - | - | - |
| 2 | Cell adhesion molecule inhibitor | 0.060 | - | - | - | - | - | - | - |
| 3 | Membrane integrity antagonist | 0.162 | 0.040 | 0.139 | - | - | - | 0.496 | - |
| 4 | Membrane permeability inhibitor | 0.023 | 0.012 | 0.039 | 0.047 | 0.007 | - | 0.198 | - |
| 5 | Peptidoglycan glycosyltransferase inhibitor | - | 0.091 | - | - | - | 0.032 | 0.009 | - |
| 6 | Phospholipid-translocating ATPase inhibitor | - | - | - | - | 0.004 | - | - | - |
| 7 | UDP-glucuronosyltransferase substrate | 0.014 | 0.005 | 0.009 | 0.025 | - | - | 0.024 | 0.110 |
| 8 | UDP-N-acetylglucosamine 4-epimerase inhibitor | 0.041 | - | - | - | 0.002 | - | - | - |
| RNA Synthesis Pathway | |||||||||
| 1 | RNA synthesis inhibitor | 0.046 | 0.042 | 0.039 | 0.088 | - | - | 0.028 | 0.158 |
| 2 | RNA directed DNA polymerase inhibitor | 0.160 | 0.135 | 0.082 | 0.085 | - | - | 0.159 | - |
| 3 | RNA-directed RNA polymerase inhibitor | 0.041 | - | - | - | - | - | 0.178 | - |
| 4 | tRNA nucleotidyltransferase inhibitor | 0.040 | - | - | - | 0.002 | - | - | - |
| 5 | tRNA-pseudouridine synthase I inhibitor | 0.056 | 0.124 | - | - | 0.002 | - | - | - |
| 6 | Alanine-tRNA ligase inhibitor | 0.099 | - | - | - | - | - | - | - |
| 7 | Aspartate-tRNA ligase inhibitor | 0.070 | 0.046 | - | - | - | - | - | - |
| 8 | Asparagine-tRNA ligase inhibitor | 0.064 | 0.048 | - | - | - | - | - | - |
| 9 | Glutamate-tRNA ligase inhibitor | 0.103 | 0.160 | - | - | - | - | - | - |
| No. | Compound | PBP | MurB | SrtA | DNA Gyrase | RNA Polymerase | Ribosomal | ClyM | FsrB | GBAP | PgrX | ||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Alpha | Beta | 30S | 50S | ||||||||||
| 1 | Compound 1 | −6.9 | −7.2 | −5.6 | −6.6 | −6.6 | −6.7 | −7.5 | −5.6 | −8.3 | −5.8 | −5.5 | −6.6 |
| 2 | Compound 2 | −11.2 | −11.5 | −7.6 | −8.6 | −8.6 | −9.0 | −9.4 | −6.8 | −10.4 | −7.7 | −6.9 | −8.5 |
| 3 | Compound 3 | −10.5 | −9.5 | −7.0 | −8.3 | −8.4 | −9.0 | −7.4 | −5.8 | −9.4 | −7.1 | −6.3 | −9.0 |
| 4 | Compound 4 | −8.0 | −7.9 | −6.5 | −7.1 | −5.6 | −6.7 | −6.1 | −4.7 | −7.8 | −6.0 | −5.3 | −6.3 |
| 5 | Compound 5 | −7.5 | −8.4 | −6.4 | −7.2 | −6.3 | −7.3 | −7.5 | −5.4 | −8.5 | −5.8 | −5.2 | −6.7 |
| 6 | Compound 6 | −10.1 | −8.8 | −7.1 | −7.5 | −7.9 | −8.2 | −7.6 | −6.2 | −8.9 | −6.6 | −6.0 | −7.4 |
| 7 | Compound 7 | −7.5 | −6.4 | −6.7 | −8.1 | −7.0 | −7.5 | −7.0 | −6.1 | −7.9 | −6.1 | −5.3 | −6.4 |
| 8 | Compound 8 | −8.1 | −6.8 | −5.4 | −6.0 | −5.7 | −6.6 | −5.9 | −5.3 | −8.3 | −6.7 | −4.6 | −6.4 |
| Average binding affinity | −8.7 | −8.3 | −6.5 | −7.3 | −7.0 | −7.6 | −7.3 | −5.7 | −8.7 | −6.5 | −5.6 | −7.2 | |
| 9 | Penicillin | −7.5 | NT | NT | NT | NT | NT | NT | NT | NT | NT | NT | NT |
| 10 | Carbapenems | −8.5 | NT | NT | NT | NT | NT | NT | NT | NT | NT | NT | NT |
| 11 | Glycopeptides | NT | −7.4 | NT | NT | NT | NT | NT | NT | NT | NT | NT | NT |
| 12 | Quercetin | NT | −8.1 | NT | NT | NT | NT | NT | NT | NT | NT | NT | NT |
| 13 | Amoxicillin | NT | NT | −5.8 | NT | NT | NT | NT | NT | NT | NT | NT | −6.3 |
| 14 | Cefixime | NT | NT | −5.2 | NT | NT | NT | NT | NT | NT | NT | NT | −6.9 |
| 15 | Curcumin | NT | NT | −5.7 | NT | NT | NT | NT | NT | NT | NT | NT | NT |
| 16 | Sitafloxacin | NT | NT | NT | −6.0 | NT | NT | NT | NT | NT | NT | NT | NT |
| 17 | Rifamycin | NT | NT | NT | NT | −6.7 | −7.7 | NT | NT | NT | NT | NT | NT |
| 18 | Tetracycline | NT | NT | NT | NT | NT | NT | −7.4 | NT | NT | NT | NT | NT |
| 19 | Chloramphenicol | NT | NT | NT | NT | NT | NT | NT | −4.9 | NT | NT | NT | NT |
| 20 | (+)-AMP | NT | NT | NT | NT | NT | NT | NT | NT | −8 | NT | NT | NT |
| 21 | Ambuic acid | NT | NT | NT | NT | NT | NT | NT | NT | NT | −5.3 | −5.3 | NT |
| Ligand | Lipinski’s Rule of Five | Drug-Likeness | ||||
|---|---|---|---|---|---|---|
| Molecular Mass (Dalton) | Hydrogen Bond Donor | Hydrogen Bond Acceptors | Log P | Molar Refractivity | ||
| Less Than 500 Dalton | Less Than 5 | Less Than 10 | Less Than 5 | 40–130 | Lipinski’s Rule Follows | |
| M1 | 272 | 4 | 5 | 2.405 | 72.908 | Yes |
| M2 | 524 | 4 | 9 | 5.714 | 137.134 | No |
| M3 | 576 | - | 11 | 4.441 | 146.781 | No |
| M4 | 448 | 4 | 7 | 4.249 | 120.748 | Yes |
| M5 | 272 | 2 | 6 | 3.138 | 68.719 | Yes |
| M6 | 472 | 3 | 4 | 6.204 | 134.071 | No |
| M7 | 442 | 2 | 2 | 6.997 | 132.062 | No |
| M8 | 772 | 3 | 7 | 10.711 | 221.185 | No |
| Ligand | clogP | Solubility | TPSA | Mutagenic | Tumorigenic | Irritant | Reproductive Effective |
|---|---|---|---|---|---|---|---|
| M1 | 1.92 | −2.66 | 97.99 | High risk | low risk | Medium risk | low risk |
| M2 | 4.92 | −6.77 | 142.7 | low risk | low risk | low risk | low risk |
| M3 | 4.6 | −3.41 | 178.5 | low risk | low risk | low risk | low risk |
| M4 | 4.87 | −4.2 | 124.2 | low risk | low risk | low risk | low risk |
| M5 | 2.67 | −5.15 | 93.06 | Medium risk | low risk | low risk | low risk |
| M6 | 5.18 | −5.66 | 77.76 | low risk | low risk | low risk | low risk |
| M7 | 6.72 | −6.3 | 40.46 | low risk | low risk | low risk | low risk |
| M8 | 11.5 | −9.75 | 105.4 | low risk | low risk | low risk | low risk |
| Ligand | GI Absorption | BBB Permeant | Pgp Substrate | Inhibitor of | Bioavailability Score | ||||
|---|---|---|---|---|---|---|---|---|---|
| CYP1A2 | CYP2C19 | CYP2C9 | CYP2D6 | CYP3A4 | |||||
| M1 | High | No | No | Yes | No | Yes | No | Yes | 0.55 |
| M2 | Low | No | No | No | No | Yes | No | No | 0.55 |
| M3 | Low | No | No | No | No | No | No | Yes | 0.17 |
| M4 | Low | No | Yes | No | No | No | No | Yes | 0.55 |
| M5 | High | No | No | Yes | No | No | No | No | 0.56 |
| M6 | High | No | Yes | No | No | No | No | No | 0.56 |
| M7 | Low | No | No | No | No | No | No | No | 0.55 |
| M8 | Low | No | Yes | No | No | No | No | Yes | 0.17 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Satari, M.H.; Apriyanti, E.; Dharsono, H.D.A.; Nurdin, D.; Gartika, M.; Kurnia, D. Effectiveness of Bioactive Compound as Antibacterial and Anti-Quorum Sensing Agent from Myrmecodia pendans: An In Silico Study. Molecules 2021, 26, 2465. https://doi.org/10.3390/molecules26092465
Satari MH, Apriyanti E, Dharsono HDA, Nurdin D, Gartika M, Kurnia D. Effectiveness of Bioactive Compound as Antibacterial and Anti-Quorum Sensing Agent from Myrmecodia pendans: An In Silico Study. Molecules. 2021; 26(9):2465. https://doi.org/10.3390/molecules26092465
Chicago/Turabian StyleSatari, Mieke Hemiawati, Eti Apriyanti, Hendra Dian Adhita Dharsono, Denny Nurdin, Meirina Gartika, and Dikdik Kurnia. 2021. "Effectiveness of Bioactive Compound as Antibacterial and Anti-Quorum Sensing Agent from Myrmecodia pendans: An In Silico Study" Molecules 26, no. 9: 2465. https://doi.org/10.3390/molecules26092465
APA StyleSatari, M. H., Apriyanti, E., Dharsono, H. D. A., Nurdin, D., Gartika, M., & Kurnia, D. (2021). Effectiveness of Bioactive Compound as Antibacterial and Anti-Quorum Sensing Agent from Myrmecodia pendans: An In Silico Study. Molecules, 26(9), 2465. https://doi.org/10.3390/molecules26092465