
Molecular and Functional Imaging Studies of Psychedelic Drug Action in Animals and Humans

 , , ,
, , , 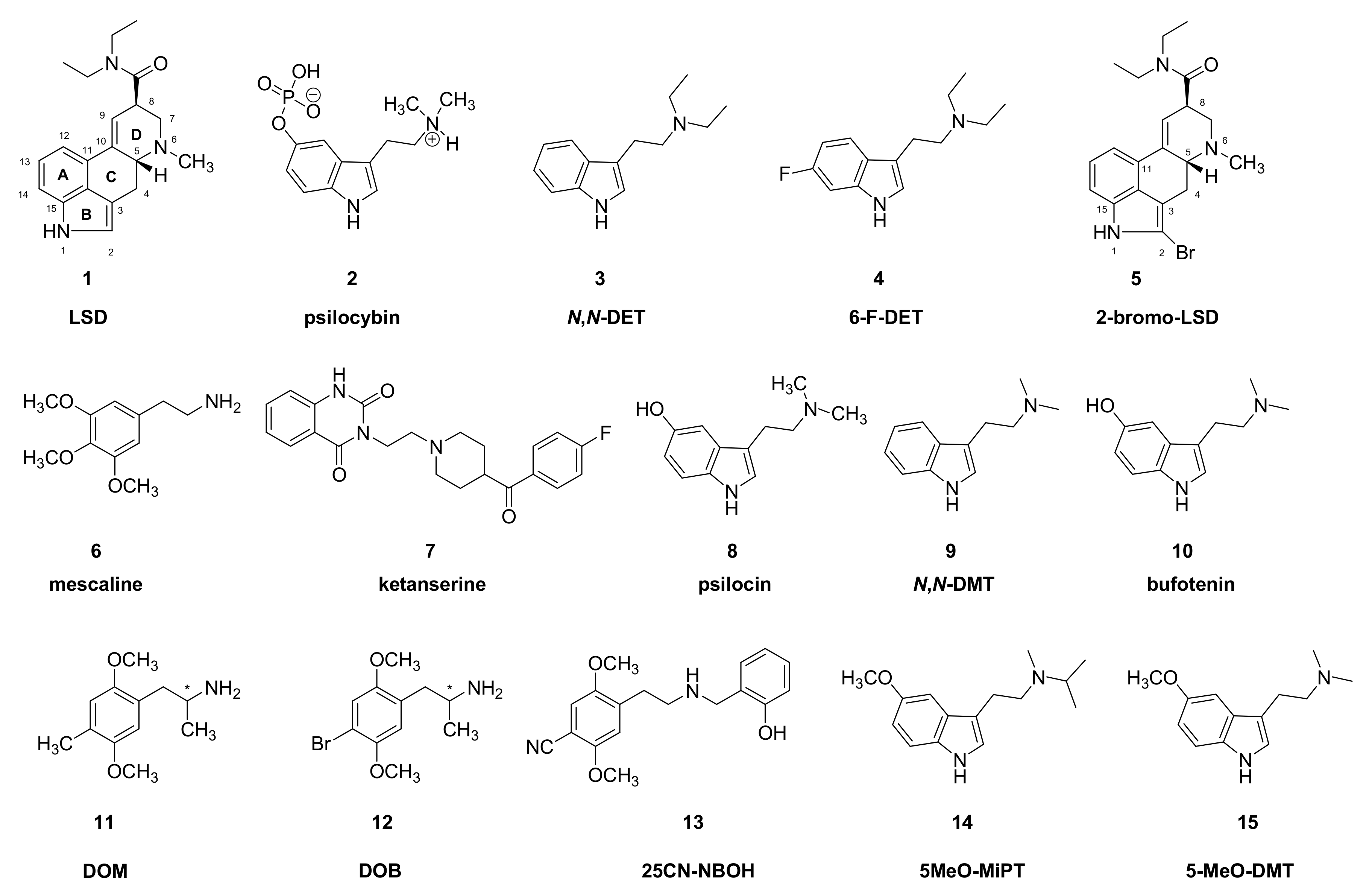{kind=link}
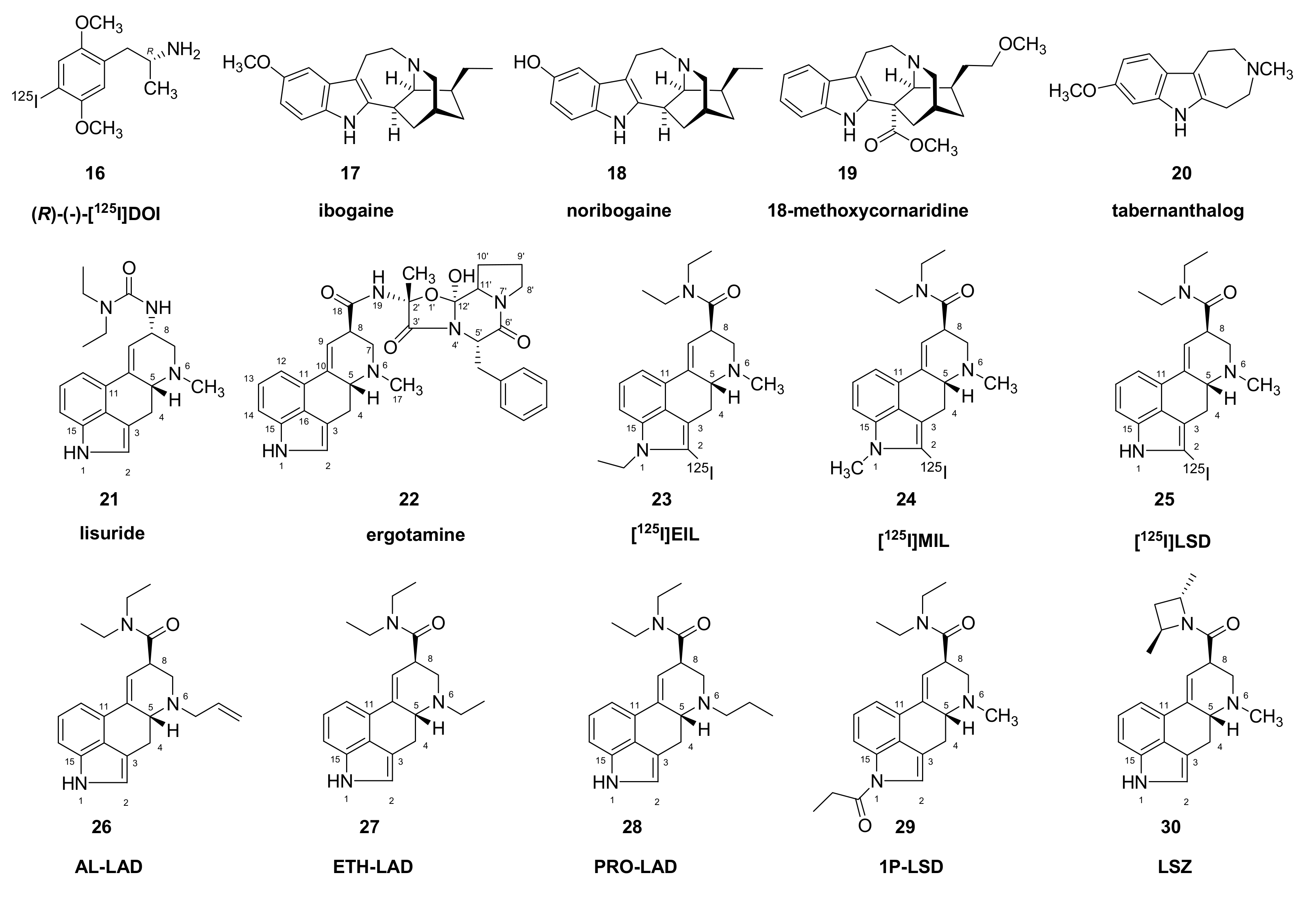{kind=link}
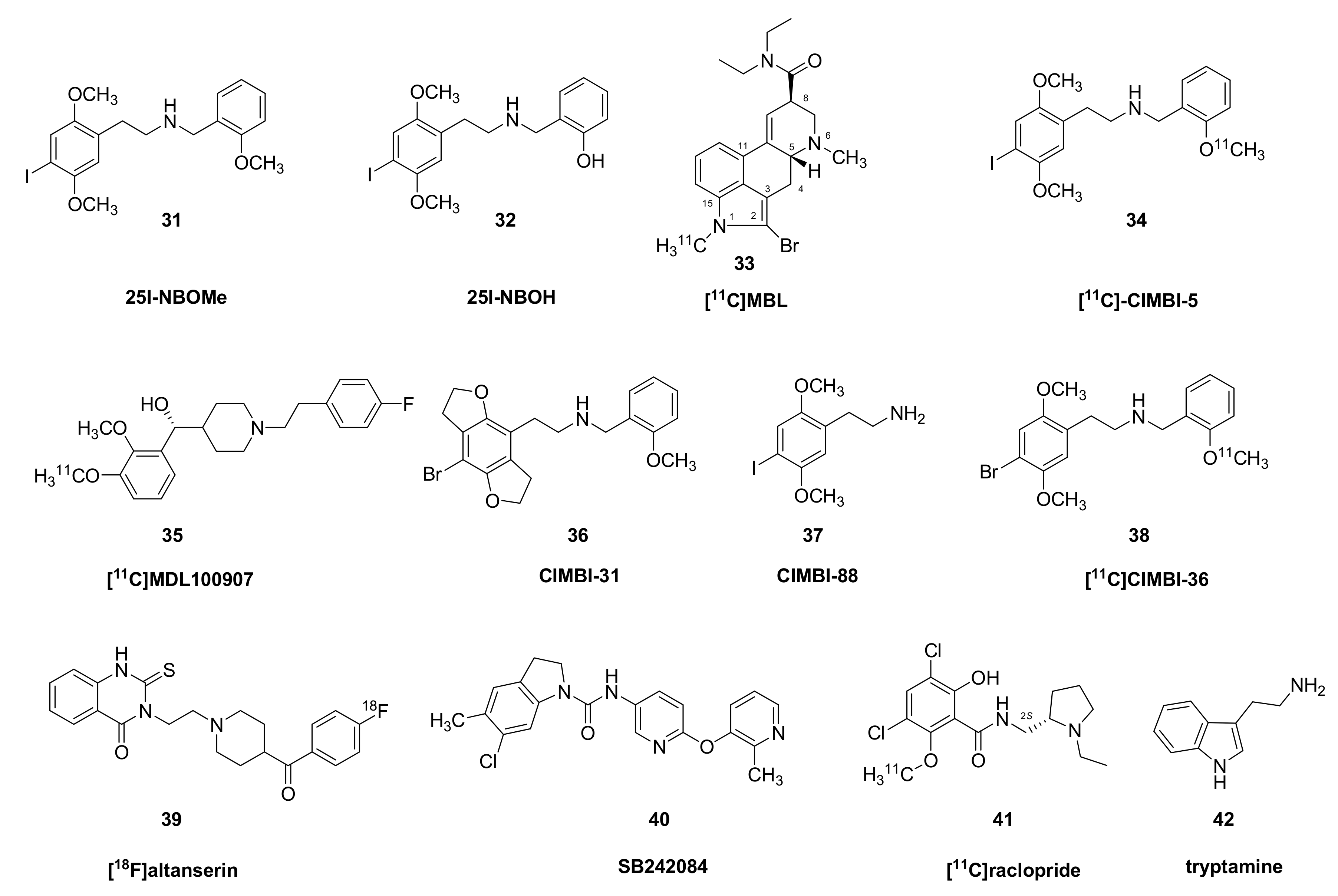{kind=link}
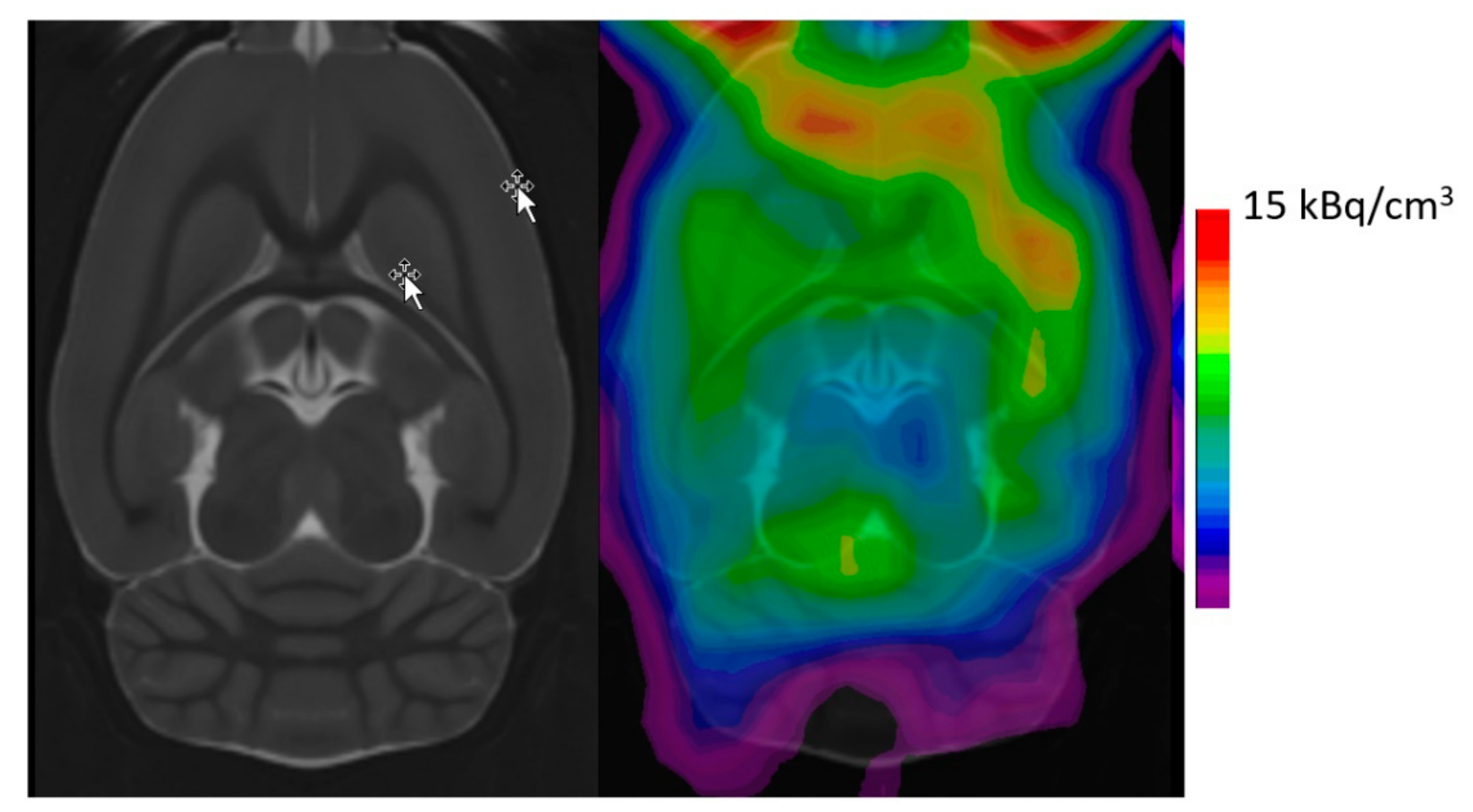{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
- Introduction
- Binding Sites of Hallucinogens in Vitro
- 2.1.
- The Nature of Agonist-Receptor Interactions
- 2.2.
- Affinities of LSD at Neuroreceptors in Vitro
- 2.3.
- Affinities of Hallucinogenic Phenylethylamines in Vitro
- 2.4.
- Affinities of Hallucinogenic Tryptamines in Vitro
- 2.5.
- The Strange Case of Ibogaine
- Ex vivo/In vitro Binding Studies with Hallucinogens
- 3.1.
- LSD Derivatives
- 3.2.
- Phenyethylamine Derivatives
- Molecular Imaging Studies in Vivo with Hallucinogens
- 4.1.
- LSD Derivatives
- 4.2.
- Phenylethylamine Derivatives
- 4.3.
- Tryptamine Derivatives
- 4.4.
- Competition from Hallucinogens at Dopamine Receptors in Vivo
- 4.5.
- Competition from Hallucinogens at Serotonin Receptors in Vivo
- Metabolism of Hallucinogenic Compounds and Tracers
- 4.1.
- LSD
- 5.2.
- Phenylethylamine Derivatives
- 5.3.
- Tryptamine Derivatives
- Ayahuasca and Pharmahuasca
- Effects of Hallucinogens on Energy Metabolism and Perfusion
- 7.1.
- Cerebral Glucose Metabolic Rate
- 7.2.
- Cerebral Blood Flow
- General Conclusions
1. Introduction
2. Binding Sites of Hallucinogens In Vitro
2.1. The Nature of Agonist-Receptor Interactions
2.2. Affinities of LSD at Neuroreceptors In Vitro
2.3. Affinities of Hallucinogenic Phenylethylamines In Vitro
2.4. Affinities of Hallucinogenic Tryptamines In Vitro
2.5. The Strange Case of Ibogaine
3. Ex Vivo/In Vitro Binding Studies with Hallucinogens
3.1. LSD Derivatives
3.2. Phenylethylamine Derivatives
4. Molecular Imaging Studies In Vivo with Hallucinogens
4.1. LSD Derivatives
4.2. Phenylethylamine Derivatives
4.3. Tryptamine Derivatives
4.4. Competition from Hallucinogens at Dopamine Receptors In Vivo
4.5. Competition from Hallucinogens at Serotonin Receptors In Vivo
5. Metabolism of Hallucinogenic Compounds
5.1. LSD
5.2. Phenylethylamines
5.3. Tryptamimes
6. Ayahuasca and Pharmahuasca
7. Effects of Hallucinogens on Energy Metabolism and Cerebral Blood Flow
7.1. Cerebral Glucose Metabolic Rate
7.2. Cerebral Perfusion
8. General Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Muller, F.; Liechti, M.E.; Lang, U.E.; Borgwardt, S. Advances and challenges in neuroimaging studies on the effects of serotonergic hallucinogens: Contributions of the resting brain. Prog. Brain Res. 2018, 242, 159–177. [Google Scholar] [CrossRef]
- Petranker, R.; Anderson, T.; Farb, N. Psychedelic Research and the Need for Transparency: Polishing Alice’s Looking Glass. Front. Psychol. 2020, 11, 1681. [Google Scholar] [CrossRef] [PubMed]
- Doblin, R.E.; Christiansen, M.; Jerome, L.; Burge, B. The Past and Future of Psychedelic Science: An Introduction to This Issue. J. Psychoact. Drugs 2019, 51, 93–97. [Google Scholar] [CrossRef]
- Lawrence, D.W.; Sharma, B.; Griffiths, R.R.; Carhart-Harris, R. Trends in the Top-Cited Articles on Classic Psychedelics. J. Psychoact. Drugs 2021, 1–16. [Google Scholar] [CrossRef]
- Tullis, P. How ecstasy and psilocybin are shaking up psychiatry. Nature 2021, 589, 506–509. [Google Scholar] [CrossRef]
- Dos Santos, R.G.; Bouso, J.C.; Rocha, J.M.; Rossi, G.N.; Hallak, J.E. The Use of Classic Hallucinogens/Psychedelics in a Therapeutic Context: Healthcare Policy Opportunities and Challenges. Risk Manag. Healthc. Policy 2021, 14, 901–910. [Google Scholar] [CrossRef] [PubMed]
- Dos Santos, R.G.; Hallak, J.E.; Baker, G.; Dursun, S. Hallucinogenic/psychedelic 5HT2A receptor agonists as rapid antidepressant therapeutics: Evidence and mechanisms of action. J. Psychopharmacol. 2021. [Google Scholar] [CrossRef]
- Dos Santos, R.G.; Osorio, F.L.; Crippa, J.A.S.; Hallak, J.E.C. Classical hallucinogens and neuroimaging: A systematic review of human studies: Hallucinogens and neuroimaging. Neurosci. Biobehav. Rev. 2016, 71, 715–728. [Google Scholar] [CrossRef]
- Rickli, A.; Moning, O.D.; Hoener, M.C.; Liechti, M.E. Receptor interaction profiles of novel psychoactive tryptamines compared with classic hallucinogens. Eur. Neuropsychopharmacol. J. Eur. Coll. Neuropsychopharmacol. 2016, 26, 1327–1337. [Google Scholar] [CrossRef]
- McKenna, D.J.; Peroutka, S.J. Differentiation of 5-hydroxytryptamine2 receptor subtypes using 125I-R-(-)2,5-dimethoxy-4-iodo-phenylisopropylamine and 3H-ketanserin. J. Neurosci. 1989, 9, 3482–3490. [Google Scholar] [CrossRef] [PubMed]
- Kalir, A.; Szara, S. Synthesis and Pharmacological Activity of Fluorinated Tryptamine Derivatives. J. Med. Chem. 1963, 6, 716–719. [Google Scholar] [CrossRef]
- Karst, M.; Halpern, J.H.; Bernateck, M.; Passie, T. The non-hallucinogen 2-bromo-lysergic acid diethylamide as preventative treatment for cluster headache: An open, non-randomized case series. Cephalalgia Int. J. Headache 2010, 30, 1140–1144. [Google Scholar] [CrossRef] [PubMed]
- Tfelt-Hansen, P. Is BOL-148 hallucinogenic? Cephalalgia Int. J. Headache 2011, 31, 635–636. [Google Scholar] [CrossRef] [PubMed]
- Blair, J.B.; Kurrasch-Orbaugh, D.; Marona-Lewicka, D.; Cumbay, M.G.; Watts, V.J.; Barker, E.L.; Nichols, D.E. Effect of ring fluorination on the pharmacology of hallucinogenic tryptamines. J. Med. Chem. 2000, 43, 4701–4710. [Google Scholar] [CrossRef]
- Bressloff, P.C.; Cowan, J.D.; Golubitsky, M.; Thomas, P.J.; Wiener, M.C. What geometric visual hallucinations tell us about the visual cortex. Neural Comput. 2002, 14, 473–491. [Google Scholar] [CrossRef] [PubMed]
- Schartner, M.M.; Timmermann, C. Neural network models for DMT-induced visual hallucinations. Neurosci. Conscious. 2020, 2020, niaa024. [Google Scholar] [CrossRef]
- Shulgin, A.T.; Shulgin, A. THIKAL, The Continuation; Transform Press: Berkely, CA, USA, 1997; pp. 337–338. [Google Scholar] [CrossRef]
- Carter, O.L.; Hasler, F.; Pettigrew, J.D.; Wallis, G.M.; Liu, G.B.; Vollenweider, F.X. Psilocybin links binocular rivalry switch rate to attention and subjective arousal levels in humans. Psychopharmacology 2007, 195, 415–424. [Google Scholar] [CrossRef]
- Carter, O.L.; Pettigrew, J.D.; Hasler, F.; Wallis, G.M.; Liu, G.B.; Hell, D.; Vollenweider, F.X. Modulating the rate and rhythmicity of perceptual rivalry alternations with the mixed 5-HT2A and 5-HT1A agonist psilocybin. Neuropsychopharmacology 2005, 30, 1154–1162. [Google Scholar] [CrossRef]
- Zamberlan, F.; Sanz, C.; Martinez Vivot, R.; Pallavicini, C.; Erowid, F.; Erowid, E.; Tagliazucchi, E. The Varieties of the Psychedelic Experience: A Preliminary Study of the Association Between the Reported Subjective Effects and the Binding Affinity Profiles of Substituted Phenethylamines and Tryptamines. Front. Integr. Neurosci. 2018, 12, 54. [Google Scholar] [CrossRef]
- Orsolini, L.; Papanti, G.D.; De Berardis, D.; Guirguis, A.; Corkery, J.M.; Schifano, F. The “Endless Trip” among the NPS Users: Psychopathology and Psychopharmacology in the Hallucinogen-Persisting Perception Disorder. A Systematic Review. Front. Psychiatry 2017, 8, 240. [Google Scholar] [CrossRef]
- Martinotti, G.; Santacroce, R.; Pettorruso, M.; Montemitro, C.; Spano, M.C.; Lorusso, M.; di Giannantonio, M.; Lerner, A.G. Hallucinogen Persisting Perception Disorder: Etiology, Clinical Features, and Therapeutic Perspectives. Brain Sci. 2018, 8. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, B.J.; Scheffel, U.; Lever, J.R.; Karpa, M.D.; Hartig, P.R. N1-methyl-2-125I-lysergic acid diethylamide, a preferred ligand for in vitro and in vivo characterization of serotonin receptors. J. Neurochem. 1987, 48, 115–124. [Google Scholar] [CrossRef] [PubMed]
- Son, H.; Jang, K.; Lee, H.; Kim, S.E.; Kang, K.W.; Lee, H. Use of Molecular Imaging in Clinical Drug Development: A Systematic Review. Nucl. Med. Mol. Imaging 2019, 53, 208–215. [Google Scholar] [CrossRef]
- Cumming, P.; Abi-Dargham, A.; Grunder, G. Molecular imaging of schizophrenia: Neurochemical findings in a heterogeneous and evolving disorder. Behav. Brain Res. 2021, 398, 113004. [Google Scholar] [CrossRef]
- Dumuis, A.; Sebben, M.; Bockaert, J. Pharmacology of 5-hydroxytryptamine-1A receptors which inhibit cAMP production in hippocampal and cortical neurons in primary culture. Mol. Pharmacol. 1988, 33, 178–186. [Google Scholar]
- Nonaka, R.; Nagai, F.; Ogata, A.; Satoh, K. In vitro screening of psychoactive drugs by [(35)S]GTPgammaS binding in rat brain membranes. Biol. Pharm. Bull. 2007, 30, 2328–2333. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Halberstadt, A.L.; Chatha, M.; Klein, A.K.; Wallach, J.; Brandt, S.D. Correlation between the potency of hallucinogens in the mouse head-twitch response assay and their behavioral and subjective effects in other species. Neuropharmacology 2020, 167, 107933. [Google Scholar] [CrossRef]
- Adams, L.M.; Geyer, M.A. Patterns of exploration in rats distinguish lisuride from lysergic acid diethylamide. Pharmacol. Biochem. Behav. 1985, 23, 461–468. [Google Scholar] [CrossRef]
- Hannon, J.; Hoyer, D. Molecular biology of 5-HT receptors. Behav. Brain Res. 2008, 195, 198–213. [Google Scholar] [CrossRef]
- Parrish, J.C.; Braden, M.R.; Gundy, E.; Nichols, D.E. Differential phospholipase C activation by phenylalkylamine serotonin 5-HT 2A receptor agonists. J. Neurochem. 2005, 95, 1575–1584. [Google Scholar] [CrossRef]
- Berg, K.A.; Maayani, S.; Goldfarb, J.; Scaramellini, C.; Leff, P.; Clarke, W.P. Effector pathway-dependent relative efficacy at serotonin type 2A and 2C receptors: Evidence for agonist-directed trafficking of receptor stimulus. Mol. Pharmacol. 1998, 54, 94–104. [Google Scholar] [CrossRef]
- Gonzalez-Maeso, J.; Weisstaub, N.V.; Zhou, M.; Chan, P.; Ivic, L.; Ang, R.; Lira, A.; Bradley-Moore, M.; Ge, Y.; Zhou, Q.; et al. Hallucinogens recruit specific cortical 5-HT(2A) receptor-mediated signaling pathways to affect behavior. Neuron 2007, 53, 439–452. [Google Scholar] [CrossRef]
- Gonzalez-Maeso, J.; Ang, R.L.; Yuen, T.; Chan, P.; Weisstaub, N.V.; Lopez-Gimenez, J.F.; Zhou, M.; Okawa, Y.; Callado, L.F.; Milligan, G.; et al. Identification of a serotonin/glutamate receptor complex implicated in psychosis. Nature 2008, 452, 93–97. [Google Scholar] [CrossRef] [PubMed]
- Aghajanian, G.K.; Foote, W.E.; Sheard, M.H. Lysergic acid diethylamide: Sensitive neuronal units in the midbrain raphe. Science 1968, 161, 706–708. [Google Scholar] [CrossRef]
- Aghajanian, G.K.; Hailgler, H.J. Hallucinogenic indoleamines: Preferential action upon presynaptic serotonin receptors. Psychopharmacol. Commun. 1975, 1, 619–629. [Google Scholar] [PubMed]
- De Gregorio, D.; Posa, L.; Ochoa-Sanchez, R.; McLaughlin, R.; Maione, S.; Comai, S.; Gobbi, G. The hallucinogen d-lysergic diethylamide (LSD) decreases dopamine firing activity through 5-HT1A, D2 and TAAR1 receptors. Pharmacol. Res. 2016, 113, 81–91. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, K.; Aghajanian, G.K. Effect of hallucinogens on spontaneous and sensory-evoked locus coeruleus unit activity in the rat: Reversal by selective 5-HT2 antagonists. Brain Res. 1986, 385, 395–400. [Google Scholar] [CrossRef]
- Passie, T.; Halpern, J.H.; Stichtenoth, D.O.; Emrich, H.M.; Hintzen, A. The pharmacology of lysergic acid diethylamide: A review. Cns Neurosci. Ther. 2008, 14, 295–314. [Google Scholar] [CrossRef]
- Minuzzi, L.; Cumming, P. Agonist binding fraction of dopamine D2/3 receptors in rat brain: A quantitative autoradiographic study. Neurochem. Int. 2010, 56, 747–752. [Google Scholar] [CrossRef]
- Titeler, M.; Lyon, R.A.; Glennon, R.A. Radioligand binding evidence implicates the brain 5-HT2 receptor as a site of action for LSD and phenylisopropylamine hallucinogens. Psychopharmacology 1988, 94, 213–216. [Google Scholar] [CrossRef]
- Jensen, A.A.; Halberstadt, A.L.; Marcher-Rorsted, E.; Odland, A.U.; Chatha, M.; Speth, N.; Liebscher, G.; Hansen, M.; Brauner-Osborne, H.; Palner, M.; et al. The selective 5-HT2A receptor agonist 25CN-NBOH: Structure-activity relationship, in vivo pharmacology, and in vitro and ex vivo binding characteristics of [(3)H]25CN-NBOH. Biochem. Pharmacol. 2020, 177, 113979. [Google Scholar] [CrossRef]
- Cassels, B.K.; Saez-Briones, P. Dark Classics in Chemical Neuroscience: Mescaline. ACS Chem. Neurosci. 2018, 9, 2448–2458. [Google Scholar] [CrossRef]
- Neff, N.; Rossi, G.V.; Chase, G.D.; Rabinowitz, J.L. Distribution and Metabolism of Mescaline-C14 in the Cat Brain. J. Pharmacol. Exp. Ther. 1964, 144, 1–7. [Google Scholar] [PubMed]
- Lee, H.M.; Roth, B.L. Hallucinogen actions on human brain revealed. Proc. Natl. Acad. Sci. USA 2012, 109, 1820–1821. [Google Scholar] [CrossRef]
- Cumming, P.; Wong, D.F.; Gillings, N.; Hilton, J.; Scheffel, U.; Gjedde, A. Specific binding of [(11)C]raclopride and N-[(3)H]propyl-norapomorphine to dopamine receptors in living mouse striatum: Occupancy by endogenous dopamine and guanosine triphosphate-free G protein. J. Cereb. Blood Flow Metab. 2002, 22, 596–604. [Google Scholar] [CrossRef] [PubMed]
- Sadzot, B.; Baraban, J.M.; Glennon, R.A.; Lyon, R.A.; Leonhardt, S.; Jan, C.R.; Titeler, M. Hallucinogenic drug interactions at human brain 5-HT2 receptors: Implications for treating LSD-induced hallucinogenesis. Psychopharmacology 1989, 98, 495–499. [Google Scholar] [CrossRef]
- Harms, A.; Gundisch, D.; Muller, C.E.; Kovar, K.A. Development of a 5-hydroxytryptamine(2A) receptor binding assay for high throughput screening using 96-well microfilter plates. J. Biomol. Screen. 2000, 5, 269–278. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Deliganis, A.V.; Pierce, P.A.; Peroutka, S.J. Differential interactions of dimethyltryptamine (DMT) with 5-HT1A and 5-HT2 receptors. Biochem. Pharmacol. 1991, 41, 1739–1744. [Google Scholar] [CrossRef]
- Smith, R.L.; Canton, H.; Barrett, R.J.; Sanders-Bush, E. Agonist properties of N,N-dimethyltryptamine at serotonin 5-HT2A and 5-HT2C receptors. Pharmacol. Biochem. Behav. 1998, 61, 323–330. [Google Scholar] [CrossRef]
- Valle, M.; Maqueda, A.E.; Rabella, M.; Rodriguez-Pujadas, A.; Antonijoan, R.M.; Romero, S.; Alonso, J.F.; Mananas, M.A.; Barker, S.; Friedlander, P.; et al. Inhibition of alpha oscillations through serotonin-2A receptor activation underlies the visual effects of ayahuasca in humans. Eur. Neuropsychopharmacol. J. Eur. Coll. Neuropsychopharmacol. 2016, 26, 1161–1175. [Google Scholar] [CrossRef]
- Blough, B.E.; Landavazo, A.; Decker, A.M.; Partilla, J.S.; Baumann, M.H.; Rothman, R.B. Interaction of psychoactive tryptamines with biogenic amine transporters and serotonin receptor subtypes. Psychopharmacology 2014, 231, 4135–4144. [Google Scholar] [CrossRef]
- Glennon, R.A.; Dukat, M.; el-Bermawy, M.; Law, H.; De los Angeles, J.; Teitler, M.; King, A.; Herrick-Davis, K. Influence of amine substituents on 5-HT2A versus 5-HT2C binding of phenylalkyl- and indolylalkylamines. J. Med. Chem. 1994, 37, 1929–1935. [Google Scholar] [CrossRef]
- Carbonaro, T.M.; Gatch, M.B. Neuropharmacology of N,N-dimethyltryptamine. Brain Res. Bull. 2016, 126, 74–88. [Google Scholar] [CrossRef] [PubMed]
- Nardai, S.; Laszlo, M.; Szabo, A.; Alpar, A.; Hanics, J.; Zahola, P.; Merkely, B.; Frecska, E.; Nagy, Z. N,N-dimethyltryptamine reduces infarct size and improves functional recovery following transient focal brain ischemia in rats. Exp. Neurol. 2020, 327, 113245. [Google Scholar] [CrossRef] [PubMed]
- Halberstadt, A.L.; Nichols, D.E.; Geyer, M.A. Behavioral effects of alpha,alpha,beta,beta-tetradeutero-5-MeO-DMT in rats: Comparison with 5-MeO-DMT administered in combination with a monoamine oxidase inhibitor. Psychopharmacology 2012, 221, 709–718. [Google Scholar] [CrossRef]
- Strassman, R.J. Human psychopharmacology of N,N-dimethyltryptamine. Behav. Brain Res. 1996, 73, 121–124. [Google Scholar] [CrossRef]
- Maisonneuve, I.M.; Keller, R.W., Jr.; Glick, S.D. Interactions between ibogaine, a potential anti-addictive agent, and morphine: An in vivo microdialysis study. Eur. J. Pharmacol. 1991, 199, 35–42. [Google Scholar] [CrossRef]
- Glick, S.D.; Maisonneuve, I.M.; Szumlinski, K.K. 18-Methoxycoronaridine (18-MC) and ibogaine: Comparison of antiaddictive efficacy, toxicity, and mechanisms of action. Ann. N. Y. Acad. Sci. 2000, 914, 369–386. [Google Scholar] [CrossRef]
- Glick, S.D.; Maisonneuve, I.M.; Szumlinski, K.K. Mechanisms of action of ibogaine: Relevance to putative therapeutic effects and development of a safer iboga alkaloid congener. Alkaloids. Chem. Biol. 2001, 56, 39–53. [Google Scholar] [CrossRef]
- Mash, D.C.; Staley, J.K.; Pablo, J.P.; Holohean, A.M.; Hackman, J.C.; Davidoff, R.A. Properties of ibogaine and its principal metabolite (12-hydroxyibogamine) at the MK-801 binding site of the NMDA receptor complex. Neurosci. Lett. 1995, 192, 53–56. [Google Scholar] [CrossRef]
- Asjad, H.M.M.; Kasture, A.; El-Kasaby, A.; Sackel, M.; Hummel, T.; Freissmuth, M.; Sucic, S. Pharmacochaperoning in a Drosophila model system rescues human dopamine transporter variants associated with infantile/juvenile parkinsonism. J. Biol. Chem. 2017, 292, 19250–19265. [Google Scholar] [CrossRef]
- Rabin, R.A.; Winter, J.C. Ibogaine and noribogaine potentiate the inhibition of adenylyl cyclase activity by opioid and 5-HT receptors. Eur. J. Pharmacol. 1996, 316, 343–348. [Google Scholar] [CrossRef]
- Glick, S.D.; Maisonneuve, I.M.; Kitchen, B.A.; Fleck, M.W. Antagonism of alpha 3 beta 4 nicotinic receptors as a strategy to reduce opioid and stimulant self-administration. Eur. J. Pharmacol. 2002, 438, 99–105. [Google Scholar] [CrossRef]
- Cameron, L.P.; Tombari, R.J.; Lu, J.; Pell, A.J.; Hurley, Z.Q.; Ehinger, Y.; Vargas, M.V.; McCarroll, M.N.; Taylor, J.C.; Myers-Turnbull, D.; et al. A non-hallucinogenic psychedelic analogue with therapeutic potential. Nature 2021, 589, 474–479. [Google Scholar] [CrossRef] [PubMed]
- Pottie, E.; Kupriyanova, O.V.; Brandt, A.L.; Laprairie, R.B.; Shevryn, V.A.; Stove, C.P. Serotonin 2A receptor (5-HT2AR) activation by 25H-NBOMe positional isomers; in vitro functional evaluation and molecular docking. Pharmacol. Transl. Sci. 2021, 4, 479–487. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.P.; Singh, P.; Bindal, M.C. QSAR studies on hallucinogens. Chem. Rev. 1983, 83, 633–649. [Google Scholar] [CrossRef]
- Scheffel, U.; Lever, J.R.; Stathis, M.; Ricaurte, G.A. Repeated administration of MDMA causes transient down-regulation of serotonin 5-HT2 receptors. Neuropharmacology 1992, 31, 881–893. [Google Scholar] [CrossRef]
- Lever, J.R.; Scheffel, U.A.; Musachio, J.L.; Stathis, M.; Wagner, H.N., Jr. Radioiodinated D-(+)-N1-ethyl-2-iodolysergic acid diethylamide: A ligand for in vitro and in vivo studies of serotonin receptors. Life Sci. 1991, 48, PL73–PL78. [Google Scholar] [CrossRef]
- Bennett, J.P., Jr.; snyder, S.H. Stereospecific binding of D-lysergic acid diethylamide (LSD) to brain membranes: Relationship to serotonin receptors. Brain Res. 1975, 94, 523–544. [Google Scholar] [CrossRef]
- McKenna, D.J.; Saavedra, J.M. Autoradiography of LSD and 2,5-dimethoxyphenylisopropylamine psychotomimetics demonstrates regional, specific cross-displacement in the rat brain. Eur. J. Pharmacol. 1987, 142, 313–315. [Google Scholar] [CrossRef]
- McKenna, D.J.; Mathis, C.A.; Shulgin, A.T.; Sargent, T., 3rd; Saavedra, J.M. Autoradiographic localization of binding sites for 125I-DOI, a new psychotomimetic radioligand, in the rat brain. Eur. J. Pharmacol. 1987, 137, 289–290. [Google Scholar] [CrossRef]
- Nakada, M.T.; Wieczorek, C.M.; Rainbow, T.C. Localization and characterization by quantitative autoradiography of [125I]LSD binding sites in rat brain. Neurosci. Lett. 1984, 49, 13–18. [Google Scholar] [CrossRef]
- Altar, C.A.; Boyar, W.C.; Marien, M.R. 125I-LSD autoradiography confirms the preferential localization of caudate-putamen S2 receptors to the caudal (peripallidal) region. Brain Res. 1986, 372, 130–136. [Google Scholar] [CrossRef]
- Hopf, A.; Eckert, H. Autoradiographic studies on the distribution of psychoactive drugs in the rat brain. 3. 14C-psilocin. Psychopharmacologia 1969, 16, 201–222. [Google Scholar] [CrossRef]
- Korr, H. Autoradiographic studies on the distribution of 3H-mescaline in the brain of the marmoset, Callithrix jacchus. Psychopharmacologia 1976, 46, 115–117. [Google Scholar] [CrossRef] [PubMed]
- Korr, H.; Seiler, N. Autoradiographic studies on the distribution of 3H-2,3,4-trimethoxy-beta-phenylethylamine in the mouse. Psychopharmacologia 1976, 46, 53–58. [Google Scholar] [CrossRef] [PubMed]
- Palenicek, T.; Balikova, M.; Bubenikova-Valesova, V.; Horacek, J. Mescaline effects on rat behavior and its time profile in serum and brain tissue after a single subcutaneous dose. Psychopharmacology 2008, 196, 51–62. [Google Scholar] [CrossRef]
- Glennon, R.A.; Raghupathi, R.; Bartyzel, P.; Teitler, M.; Leonhardt, S. Binding of phenylalkylamine derivatives at 5-HT1C and 5-HT2 serotonin receptors: Evidence for a lack of selectivity. J. Med. Chem. 1992, 35, 734–740. [Google Scholar] [CrossRef] [PubMed]
- McKenna, D.J.; Nazarali, A.J.; Himeno, A.; Saavedra, J.M. Chronic treatment with (+/−)DOI, a psychotomimetic 5-HT2 agonist, downregulates 5-HT2 receptors in rat brain. Neuropsychopharmacology 1989, 2, 81–87. [Google Scholar] [CrossRef]
- Shi, J.; Landry, M.; Carrasco, G.A.; Battaglia, G.; Muma, N.A. Sustained treatment with a 5-HT(2A) receptor agonist causes functional desensitization and reductions in agonist-labeled 5-HT(2A) receptors despite increases in receptor protein levels in rats. Neuropharmacology 2008, 55, 687–692. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, L.M.; Holm, N.B.; Leth-Petersen, S.; Kristensen, J.L.; Olsen, L.; Linnet, K. Characterization of the hepatic cytochrome P450 enzymes involved in the metabolism of 25I-NBOMe and 25I-NBOH. Drug Test. Anal. 2017, 9, 671–679. [Google Scholar] [CrossRef] [PubMed]
- Elmore, J.S.; Decker, A.M.; Sulima, A.; Rice, K.C.; Partilla, J.S.; Blough, B.E.; Baumann, M.H. Comparative neuropharmacology of N-(2-methoxybenzyl)-2,5-dimethoxyphenethylamine (NBOMe) hallucinogens and their 2C counterparts in male rats. Neuropharmacology 2018, 142, 240–250. [Google Scholar] [CrossRef]
- Wong, D.F.; Lever, J.R.; Hartig, P.R.; Dannals, R.F.; Villemagne, V.; Hoffman, B.J.; Wilson, A.A.; Ravert, H.T.; Links, J.M.; Scheffel, U.; et al. Localization of serotonin 5-HT2 receptors in living human brain by positron emission tomography using N1-([11C]-methyl)-2-Br-LSD. Synapse 1987, 1, 393–398. [Google Scholar] [CrossRef]
- Sargent, T., 3rd; Kalbhen, D.A.; Shulgin, A.T.; Stauffer, H.; Kusubov, N. A potential new brain-scanning agent: 4-77Br-2,5-dimethoxyphenylisoproplamine (4-Br-DPIA). J. Nucl. Med. 1975, 16, 243–245. [Google Scholar] [PubMed]
- Zea-Ponce, Y.; Kegeles, L.S.; Guo, N.; Raskin, L.; Bakthavachalam, V.; Laruelle, M. Pharmacokinetics and brain distribution in non human primate of R(-)[123I]DOI, A 5HT(2A/2C) serotonin agonist. Nucl. Med. Biol. 2002, 29, 575–583. [Google Scholar] [CrossRef]
- Ettrup, A.; Palner, M.; Gillings, N.; Santini, M.A.; Hansen, M.; Kornum, B.R.; Rasmussen, L.K.; Nagren, K.; Madsen, J.; Begtrup, M.; et al. Radiosynthesis and evaluation of 11C-CIMBI-5 as a 5-HT2A receptor agonist radioligand for PET. J. Nucl. Med. 2010, 51, 1763–1770. [Google Scholar] [CrossRef] [PubMed]
- Prabhakaran, J.; DeLorenzo, C.; Zanderigo, F.; Knudsen, G.M.; Gilling, N.; Pratap, M.; Jorgensen, M.J.; Daunais, J.; Kaplan, J.R.; Parsey, R.V.; et al. In vivo PET Imaging of [11C]CIMBI-5, a 5-HT2AR Agonist Radiotracer in Nonhuman Primates. J. Pharm. Pharm. Sci. 2019, 22, 352–364. [Google Scholar] [CrossRef]
- Ettrup, A.; Hansen, M.; Santini, M.A.; Paine, J.; Gillings, N.; Palner, M.; Lehel, S.; Herth, M.M.; Madsen, J.; Kristensen, J.; et al. Radiosynthesis and in vivo evaluation of a series of substituted 11C-phenethylamines as 5-HT (2A) agonist PET tracers. Eur. J. Nucl. Med. Mol. Imaging 2011, 38, 681–693. [Google Scholar] [CrossRef] [PubMed]
- Ettrup, A.; Svarer, C.; McMahon, B.; da Cunha-Bang, S.; Lehel, S.; Moller, K.; Dyssegaard, A.; Ganz, M.; Beliveau, V.; Jorgensen, L.M.; et al. Serotonin 2A receptor agonist binding in the human brain with [(11)C]Cimbi-36: Test-retest reproducibility and head-to-head comparison with the antagonist [(18)F]altanserin. NeuroImage 2016, 130, 167–174. [Google Scholar] [CrossRef] [PubMed]
- Leysen, J.E. Gaps and peculiarities in 5-HT2 receptor studies. Neuropsychopharmacology 1990, 3, 361–369. [Google Scholar]
- Finnema, S.J.; Stepanov, V.; Ettrup, A.; Nakao, R.; Amini, N.; Svedberg, M.; Lehmann, C.; Hansen, M.; Knudsen, G.M.; Halldin, C. Characterization of [(11)C]Cimbi-36 as an agonist PET radioligand for the 5-HT(2A) and 5-HT(2C) receptors in the nonhuman primate brain. NeuroImage 2014, 84, 342–353. [Google Scholar] [CrossRef]
- Johansen, A.; Holm, S.; Dall, B.; Keller, S.; Kristensen, J.L.; Knudsen, G.M.; Hansen, H.D. Human biodistribution and radiation dosimetry of the 5-HT2A receptor agonist Cimbi-36 labeled with carbon-11 in two positions. Ejnmmi Res 2019, 9, 71. [Google Scholar] [CrossRef] [PubMed]
- Cohen, I.; Vogel, W.H. Determination and physiological disposition of dimethyltryptamine and diethyltryptamine in rat brain, liver and plasma. Biochem. Pharmacol. 1972, 21, 1214–1216. [Google Scholar] [CrossRef]
- Vitale, A.A.; Pomilio, A.B.; Canellas, C.O.; Vitale, M.G.; Putz, E.M.; Ciprian-Ollivier, J. In vivo long-term kinetics of radiolabeled n,n-dimethyltryptamine and tryptamine. J. Nucl. Med. 2011, 52, 970–977. [Google Scholar] [CrossRef] [PubMed]
- Vollenweider, F.X.; Vontobel, P.; Hell, D.; Leenders, K.L. 5-HT modulation of dopamine release in basal ganglia in psilocybin-induced psychosis in man--a PET study with [11C]raclopride. Neuropsychopharmacology 1999, 20, 424–433. [Google Scholar] [CrossRef]
- Sakashita, Y.; Abe, K.; Katagiri, N.; Kambe, T.; Saitoh, T.; Utsunomiya, I.; Horiguchi, Y.; Taguchi, K. Effect of psilocin on extracellular dopamine and serotonin levels in the mesoaccumbens and mesocortical pathway in awake rats. Biol. Pharm. Bull. 2015, 38, 134–138. [Google Scholar] [CrossRef] [PubMed]
- Minuzzi, L.; Nomikos, G.G.; Wade, M.R.; Jensen, S.B.; Olsen, A.K.; Cumming, P. Interaction between LSD and dopamine D2/3 binding sites in pig brain. Synapse 2005, 56, 198–204. [Google Scholar] [CrossRef] [PubMed]
- Dolder, P.C.; Schmid, Y.; Steuer, A.E.; Kraemer, T.; Rentsch, K.M.; Hammann, F.; Liechti, M.E. Pharmacokinetics and Pharmacodynamics of Lysergic Acid Diethylamide in Healthy Subjects. Clin. Pharmacokinet. 2017, 56, 1219–1230. [Google Scholar] [CrossRef] [PubMed]
- Jensen, S.B.; Olsen, A.K.; Pedersen, K.; Cumming, P. Effect of monoamine oxidase inhibition on amphetamine-evoked changes in dopamine receptor availability in the living pig: A dual tracer PET study with [11C]harmine and [11C]raclopride. Synapse 2006, 59, 427–434. [Google Scholar] [CrossRef]
- Pedersen, K.; Simonsen, M.; Ostergaard, S.D.; Munk, O.L.; Rosa-Neto, P.; Olsen, A.K.; Jensen, S.B.; Moller, A.; Cumming, P. Mapping the amphetamine-evoked changes in [11C]raclopride binding in living rat using small animal PET: Modulation by MAO-inhibition. NeuroImage 2007, 35, 38–46. [Google Scholar] [CrossRef] [PubMed]
- Leo, D.; Mus, L.; Espinoza, S.; Hoener, M.C.; Sotnikova, T.D.; Gainetdinov, R.R. Taar1-mediated modulation of presynaptic dopaminergic neurotransmission: Role of D2 dopamine autoreceptors. Neuropharmacology 2014, 81, 283–291. [Google Scholar] [CrossRef]
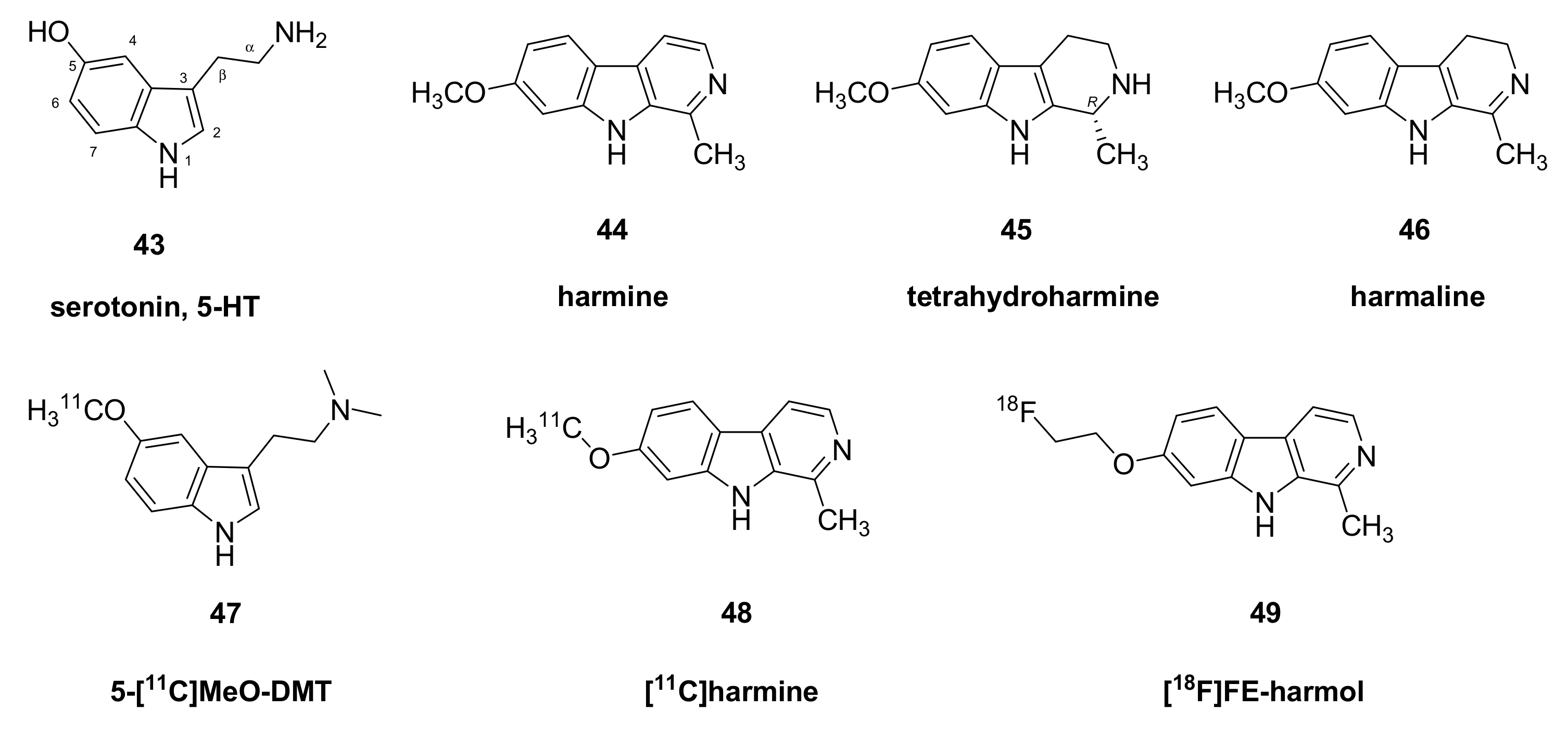
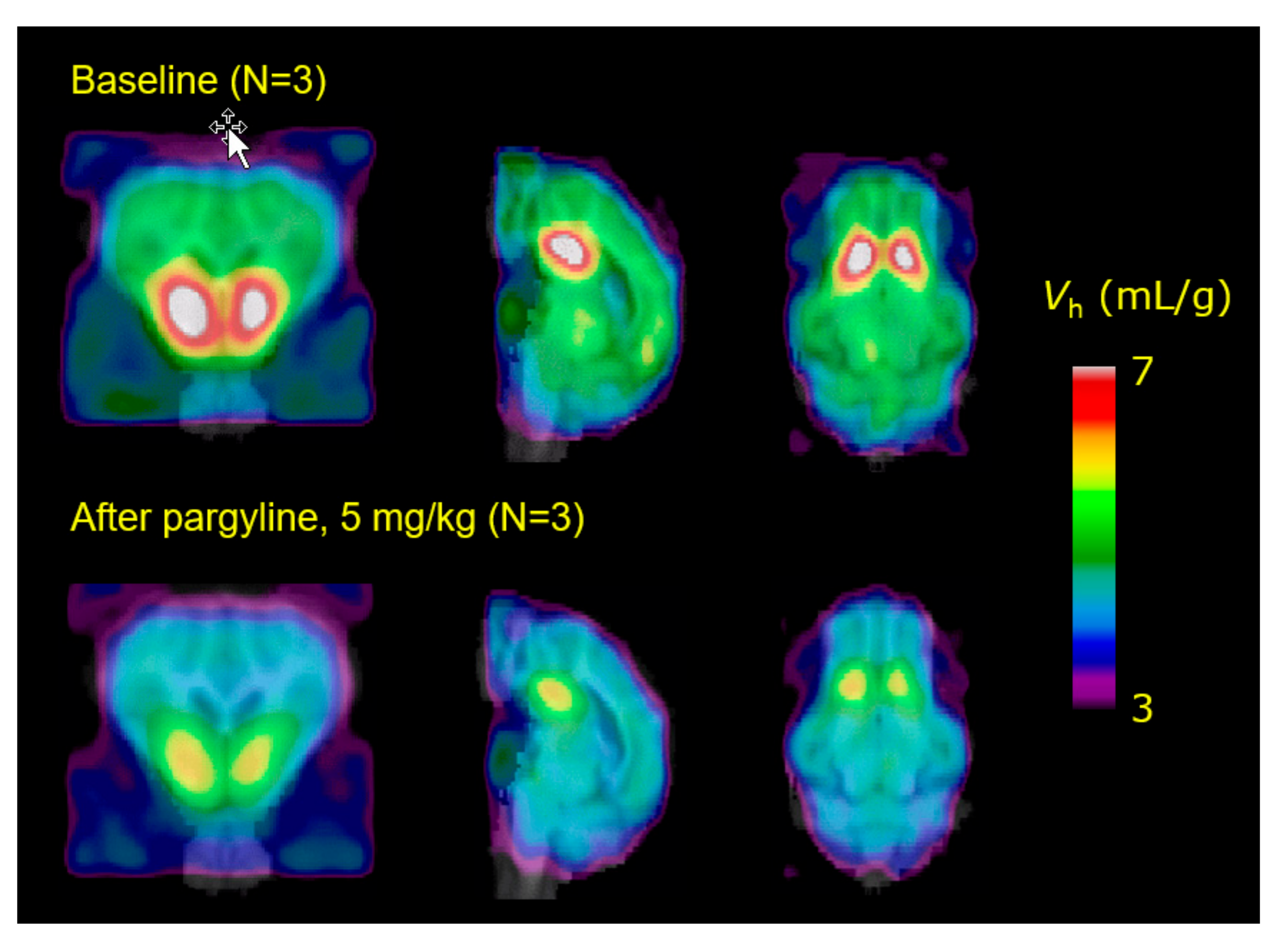
- Maschauer, S.; Haller, A.; Riss, P.J.; Kuwert, T.; Prante, O.; Cumming, P. Specific binding of [(18)F]fluoroethyl-harmol to monoamine oxidase A in rat brain cryostat sections, and compartmental analysis of binding in living brain. J. Neurochem. 2015, 135, 908–917. [Google Scholar] [CrossRef]
- Jorgensen, L.M.; Weikop, P.; Villadsen, J.; Visnapuu, T.; Ettrup, A.; Hansen, H.D.; Baandrup, A.O.; Andersen, F.L.; Bjarkam, C.R.; Thomsen, C.; et al. Cerebral 5-HT release correlates with [(11)C]Cimbi36 PET measures of 5-HT2A receptor occupancy in the pig brain. J. Cereb. Blood Flow Metab. 2017, 37, 425–434. [Google Scholar] [CrossRef]
- Hasler, F.Q.B.B.; Treyer, V.; Schubiger, P.A.; Buck, A.; Vollenweider, F.X. Role of prefrontal serotonin-2A receptors in self experience during pilocybin-indiced altered states. Neuropsychobiology 2009, 59, 60. [Google Scholar] [CrossRef]
- Madsen, M.K.; Fisher, P.M.; Burmester, D.; Dyssegaard, A.; Stenbaek, D.S.; Kristiansen, S.; Johansen, S.S.; Lehel, S.; Linnet, K.; Svarer, C.; et al. Psychedelic effects of psilocybin correlate with serotonin 2A receptor occupancy and plasma psilocin levels. Neuropsychopharmacology 2019, 44, 1328–1334. [Google Scholar] [CrossRef]
- Stenbaek, D.S.; Kristiansen, S.; Burmester, D.; Madsen, M.K.; Frokjaer, V.G.; Knudsen, G.M.; Fisher, P.M. Trait Openness and serotonin 2A receptors in healthy volunteers: A positron emission tomography study. Hum. Brain Mapp. 2019, 40, 2117–2124. [Google Scholar] [CrossRef]
- Raval, N.R.; Johansen, A.; Donovan, L.L.; Ros, N.F.; Ozenne, B.; Hansen, H.D.; Knudsen, G.M. A Single Dose of Psilocybin Increases Synaptic Density and Decreases 5-HT2A Receptor Density in the Pig Brain. Int. J. Mol. Sci. 2021, 22. [Google Scholar] [CrossRef]
- de la Fuente Revenga, M.; Zhu, B.; Guevara, C.A.; Naler, L.B.; Saunders, J.B.; Zhou, Z.; Toneatti, R.; Sierra, S.; Wolstenholme, J.T.; Beardsley, P.M.; et al. Prolonged epigenetic and synaptic plasticity alterations following single exposre to a psychedelic in mice. bioRxiv 2021. [Google Scholar] [CrossRef]
- Shao, L.-X.; Liao, C.; Gregg, I.; Savalia, N.K.; Delagarza, K.; Kwan, A.C. Psilocybin iduces rapid and persistent growth of dendritic spines in frontal cortex in vivo. bioRxiv 2021. [Google Scholar] [CrossRef]
- Savalia, N.K.; Shao, L.X.; Kwan, A.C. A Dendrite-Focused Framework for Understanding the Actions of Ketamine and Psychedelics. Trends Neurosci. 2021, 44, 260–275. [Google Scholar] [CrossRef] [PubMed]
- Libanio Osorio Marta, R.F. Metabolism of lysergic acid diethylamide (LSD): An update. Drug Metab. Rev. 2019, 51, 378–387. [Google Scholar] [CrossRef] [PubMed]
- Kalberer, F.; Kreis, W.; Rutschmann, J. The fate of psilocin in the rat. Biochem. Pharmacol. 1962, 11, 261–269. [Google Scholar] [CrossRef]
- Kamata, T.; Katagi, M.; Kamata, H.T.; Miki, A.; Shima, N.; Zaitsu, K.; Nishikawa, M.; Tanaka, E.; Honda, K.; Tsuchihashi, H. Metabolism of the psychotomimetic tryptamine derivative 5-methoxy-N,N-diisopropyltryptamine in humans: Identification and quantification of its urinary metabolites. Drug Metab. Dispos. Biol. Fate Chem. 2006, 34, 281–287. [Google Scholar] [CrossRef] [PubMed]
- Brown, R.T.; Nicholas, C.R.; Cozzi, N.V.; Gassman, M.C.; Cooper, K.M.; Muller, D.; Thomas, C.D.; Hetzel, S.J.; Henriquez, K.M.; Ribaudo, A.S.; et al. Pharmacokinetics of Escalating Doses of Oral Psilocybin in Healthy Adults. Clin. Pharmacokinet. 2017, 56, 1543–1554. [Google Scholar] [CrossRef]
- Leth-Petersen, S.; Gabel-Jensen, C.; Gillings, N.; Lehel, S.; Hansen, H.D.; Knudsen, G.M.; Kristensen, J.L. Metabolic Fate of Hallucinogenic NBOMes. Chem. Res. Toxicol. 2016, 29, 96–100. [Google Scholar] [CrossRef] [PubMed]
- Johansen, A.; Hansen, H.D.; Svarer, C.; Lehel, S.; Leth-Petersen, S.; Kristensen, J.L.; Gillings, N.; Knudsen, G.M. The importance of small polar radiometabolites in molecular neuroimaging: A PET study with [(11)C]Cimbi-36 labeled in two positions. J. Cereb. Blood Flow Metab. 2018, 38, 659–668. [Google Scholar] [CrossRef]
- Breusova, K.; Ernstsen, K.G.; Palner, M.; Linnet, K.; Kristensen, J.L.; Kretschmann, A.C. A quantitative method for the selective 5-HT2A agonist 25CN-NBOH in rat plasma and brain. J. Pharm. Biomed. Anal. 2021, 199, 114016. [Google Scholar] [CrossRef]
- Christian, S.T.; Harrison, R.; Quayle, E.; Pagel, J.; Monti, J. The in vitro identification of dimethyltryptamine (DMT) in mammalian brain and its characterization as a possible endogenous neuroregulatory agent. Biochem. Med. 1977, 18, 164–183. [Google Scholar] [CrossRef]
- Axelrod, J. Enzymatic formation of psychotomimetic metabolites from normally occurring compounds. Science 1961, 134, 343. [Google Scholar] [CrossRef]
- Dean, J.G. Indolethylamine-N-methyltransferase Polymorphisms: Genetic and Biochemical Approaches for Study of Endogenous N,N,-dimethyltryptamine. Front. Neurosci. 2018, 12, 232. [Google Scholar] [CrossRef]
- Barker, S.A. N, N-Dimethyltryptamine (DMT), an Endogenous Hallucinogen: Past, Present, and Future Research to Determine Its Role and Function. Front. Neurosci. 2018, 12, 536. [Google Scholar] [CrossRef] [PubMed]
- Mavlyutov, T.A.; Baker, E.M.; Losenegger, T.M.; Kim, J.R.; Torres, B.; Epstein, M.L.; Ruoho, A.E. The Sigma-1 Receptor-A Therapeutic Target for the Treatment of ALS? Adv. Exp. Med. Biol. 2017, 964, 255–265. [Google Scholar] [CrossRef]
- Dean, J.G.; Liu, T.; Huff, S.; Sheler, B.; Barker, S.A.; Strassman, R.J.; Wang, M.M.; Borjigin, J. Biosynthesis and Extracellular Concentrations of N,N-dimethyltryptamine (DMT) in Mammalian Brain. Sci. Rep. 2019, 9, 9333. [Google Scholar] [CrossRef]
- Nichols, D.E. N,N-dimethyltryptamine and the pineal gland: Separating fact from myth. J. Psychopharmacol. 2018, 32, 30–36. [Google Scholar] [CrossRef]
- Shen, H.W.; Jiang, X.L.; Winter, J.C.; Yu, A.M. Psychedelic 5-methoxy-N,N-dimethyltryptamine: Metabolism, pharmacokinetics, drug interactions, and pharmacological actions. Curr. Drug Metab. 2010, 11, 659–666. [Google Scholar] [CrossRef]
- Agurell, S.; Holmstedt, B.; Lindgren, J.E. Metabolism of 5-methoxy-N,-N dimethyltryptamine- 14 C in the rat. Biochem. Pharmacol. 1969, 18, 2771–2781. [Google Scholar] [CrossRef]
- Keiser, M.J.; Setola, V.; Irwin, J.J.; Laggner, C.; Abbas, A.I.; Hufeisen, S.J.; Jensen, N.H.; Kuijer, M.B.; Matos, R.C.; Tran, T.B.; et al. Predicting new molecular targets for known drugs. Nature 2009, 462, 175–181. [Google Scholar] [CrossRef]
- Samoylenko, V.; Rahman, M.M.; Tekwani, B.L.; Tripathi, L.M.; Wang, Y.H.; Khan, S.I.; Khan, I.A.; Miller, L.S.; Joshi, V.C.; Muhammad, I. Banisteriopsis caapi, a unique combination of MAO inhibitory and antioxidative constituents for the activities relevant to neurodegenerative disorders and Parkinson’s disease. J. Ethnopharmacol. 2010, 127, 357–367. [Google Scholar] [CrossRef]
- Palhano-Fontes, F.; Barreto, D.; Onias, H.; Andrade, K.C.; Novaes, M.M.; Pessoa, J.A.; Mota-Rolim, S.A.; Osorio, F.L.; Sanches, R.; Dos Santos, R.G.; et al. Rapid antidepressant effects of the psychedelic ayahuasca in treatment-resistant depression: A randomized placebo-controlled trial. Psychol. Med. 2019, 49, 655–663. [Google Scholar] [CrossRef] [PubMed]
- Berlowitz, I.; Walt, H.; Ghasarian, C.; Mendive, F.; Martin-Soelch, C. Short-Term Treatment Effects of a Substance Use Disorder Therapy Involving Traditional Amazonian Medicine. J. Psychoact. Drugs 2019, 51, 323–334. [Google Scholar] [CrossRef]
- Jiang, X.L.; Shen, H.W.; Yu, A.M. Modification of 5-methoxy-N,N-dimethyltryptamine-induced hyperactivity by monoamine oxidase A inhibitor harmaline in mice and the underlying serotonergic mechanisms. Pharmacol. Rep. PR 2016, 68, 608–615. [Google Scholar] [CrossRef] [PubMed]
- Halberstadt, A.L. Behavioral and pharmacokinetic interactions between monoamine oxidase inhibitors and the hallucinogen 5-methoxy-N,N-dimethyltryptamine. Pharmacol. Biochem. Behav. 2016, 143, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Grome, J.J.; Harper, A.M. Local cerebral glucose utilisation following indoleamine- and piperazine-containing 5-hydroxytryptamine agonists. J. Neurochem. 1986, 46, 117–124. [Google Scholar] [CrossRef] [PubMed]
- Kelly, P.A.; Davis, C.J.; Goodwin, G.M. Differential patterns of local cerebral glucose utilization in response to 5-hydroxytryptamine agonists. Neuroscience 1988, 25, 907–915. [Google Scholar] [CrossRef]
- Tyls, F.; Palenicek, T.; Kaderabek, L.; Lipski, M.; Kubesova, A.; Horacek, J. Sex differences and serotonergic mechanisms in the behavioural effects of psilocin. Behav. Pharmacol. 2016, 27, 309–320. [Google Scholar] [CrossRef]
- Vollenweider, F.X.; Leenders, K.L.; Scharfetter, C.; Maguire, P.; Stadelmann, O.; Angst, J. Positron emission tomography and fluorodeoxyglucose studies of metabolic hyperfrontality and psychopathology in the psilocybin model of psychosis. Neuropsychopharmacology 1997, 16, 357–372. [Google Scholar] [CrossRef]
- Gouzoulis-Mayfrank, E.; Schreckenberger, M.; Sabri, O.; Arning, C.; Thelen, B.; Spitzer, M.; Kovar, K.A.; Hermle, L.; Bull, U.; Sass, H. Neurometabolic effects of psilocybin, 3,4-methylenedioxyethylamphetamine (MDE) and d-methamphetamine in healthy volunteers. A double-blind, placebo-controlled PET study with [18F]FDG. Neuropsychopharmacology 1999, 20, 565–581. [Google Scholar] [CrossRef]
- Carhart-Harris, R.L.; Muthukumaraswamy, S.; Roseman, L.; Kaelen, M.; Droog, W.; Murphy, K.; Tagliazucchi, E.; Schenberg, E.E.; Nest, T.; Orban, C.; et al. Neural correlates of the LSD experience revealed by multimodal neuroimaging. Proc. Natl. Acad. Sci. USA 2016, 113, 4853–4858. [Google Scholar] [CrossRef]
- Hermle, L.; Gouzoulis-Mayfrank, E.; Spitzer, M. Blood flow and cerebral laterality in the mescaline model of psychosis. Pharmacopsychiatry 1998, 31 (Suppl. 2), 85–91. [Google Scholar] [CrossRef]
- Riba, J.; Romero, S.; Grasa, E.; Mena, E.; Carrio, I.; Barbanoj, M.J. Increased frontal and paralimbic activation following ayahuasca, the pan-Amazonian inebriant. Psychopharmacology 2006, 186, 93–98. [Google Scholar] [CrossRef]
- Sanches, R.F.; de Lima Osorio, F.; Dos Santos, R.G.; Macedo, L.R.; Maia-de-Oliveira, J.P.; Wichert-Ana, L.; de Araujo, D.B.; Riba, J.; Crippa, J.A.; Hallak, J.E. Antidepressant Effects of a Single Dose of Ayahuasca in Patients With Recurrent Depression: A SPECT Study. J. Clin. Psychopharmacol. 2016, 36, 77–81. [Google Scholar] [CrossRef]
- Barsuglia, J.P.; Polanco, M.; Palmer, R.; Malcolm, B.J.; Kelmendi, B.; Calvey, T. A case report SPECT study and theoretical rationale for the sequential administration of ibogaine and 5-MeO-DMT in the treatment of alcohol use disorder. Prog. Brain Res. 2018, 242, 121–158. [Google Scholar] [CrossRef] [PubMed]
- Lewis, C.R.; Preller, K.H.; Kraehenmann, R.; Michels, L.; Staempfli, P.; Vollenweider, F.X. Two dose investigation of the 5-HT-agonist psilocybin on relative and global cerebral blood flow. NeuroImage 2017, 159, 70–78. [Google Scholar] [CrossRef] [PubMed]
- Carhart-Harris, R.L.; Erritzoe, D.; Williams, T.; Stone, J.M.; Reed, L.J.; Colasanti, A.; Tyacke, R.J.; Leech, R.; Malizia, A.L.; Murphy, K.; et al. Neural correlates of the psychedelic state as determined by fMRI studies with psilocybin. Proc. Natl. Acad. Sci. USA 2012, 109, 2138–2143. [Google Scholar] [CrossRef] [PubMed]
- McBride, M.C. Bufotenine: Toward an understanding of possible psychoactive mechanisms. J. Psychoact. Drugs 2000, 32, 321–331. [Google Scholar] [CrossRef] [PubMed]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cumming, P.; Scheidegger, M.; Dornbierer, D.; Palner, M.; Quednow, B.B.; Martin-Soelch, C. Molecular and Functional Imaging Studies of Psychedelic Drug Action in Animals and Humans. Molecules 2021, 26, 2451. https://doi.org/10.3390/molecules26092451
Cumming P, Scheidegger M, Dornbierer D, Palner M, Quednow BB, Martin-Soelch C. Molecular and Functional Imaging Studies of Psychedelic Drug Action in Animals and Humans. Molecules. 2021; 26(9):2451. https://doi.org/10.3390/molecules26092451
Chicago/Turabian StyleCumming, Paul, Milan Scheidegger, Dario Dornbierer, Mikael Palner, Boris B. Quednow, and Chantal Martin-Soelch. 2021. "Molecular and Functional Imaging Studies of Psychedelic Drug Action in Animals and Humans" Molecules 26, no. 9: 2451. https://doi.org/10.3390/molecules26092451
APA StyleCumming, P., Scheidegger, M., Dornbierer, D., Palner, M., Quednow, B. B., & Martin-Soelch, C. (2021). Molecular and Functional Imaging Studies of Psychedelic Drug Action in Animals and Humans. Molecules, 26(9), 2451. https://doi.org/10.3390/molecules26092451