
Is the Antidepressant Activity of Selective Serotonin Reuptake Inhibitors Mediated by Nicotinic Acetylcholine Receptors?


 and
and
Abstract
1. Introduction
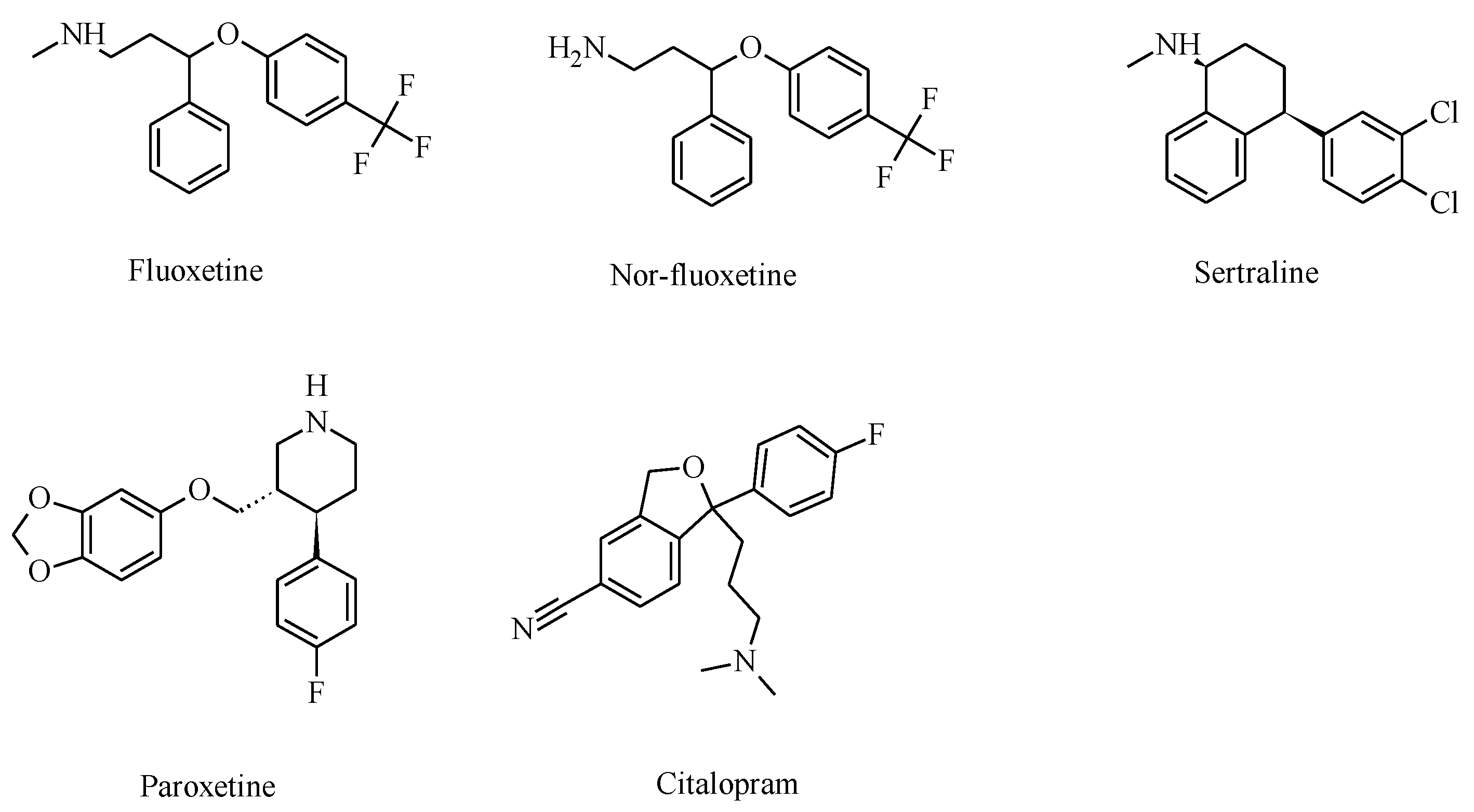
2. Selective Serotonin Reuptake Inhibitors (SSRIs)
3. Nicotinic Acetylcholine Receptor Functions Modulate Depression States
4. Relationship between Smoking and Depression
5. Preclinical Studies with Combinations of SSRIs and Nicotinic Ligands
6. SSRIs Inhibit nAChRs at Clinical Concentrations
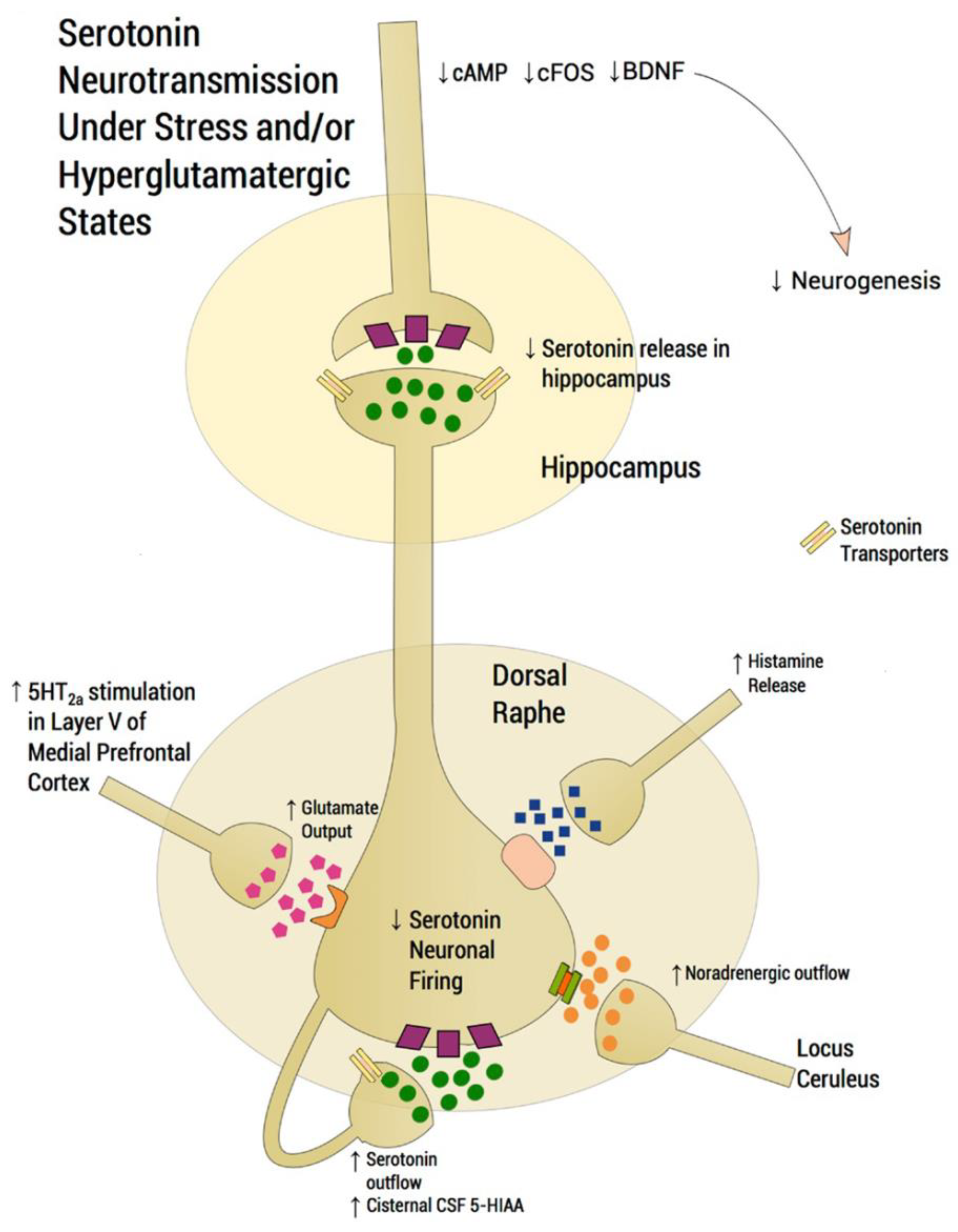
7. Neuronal Pathways Involved in the Antidepressant Activity of SSRIs
8. SSRIs Bind Distinct nAChR Subtypes with Different Affinities
9. SSRIs Interact with nAChRs in Different Conformational States
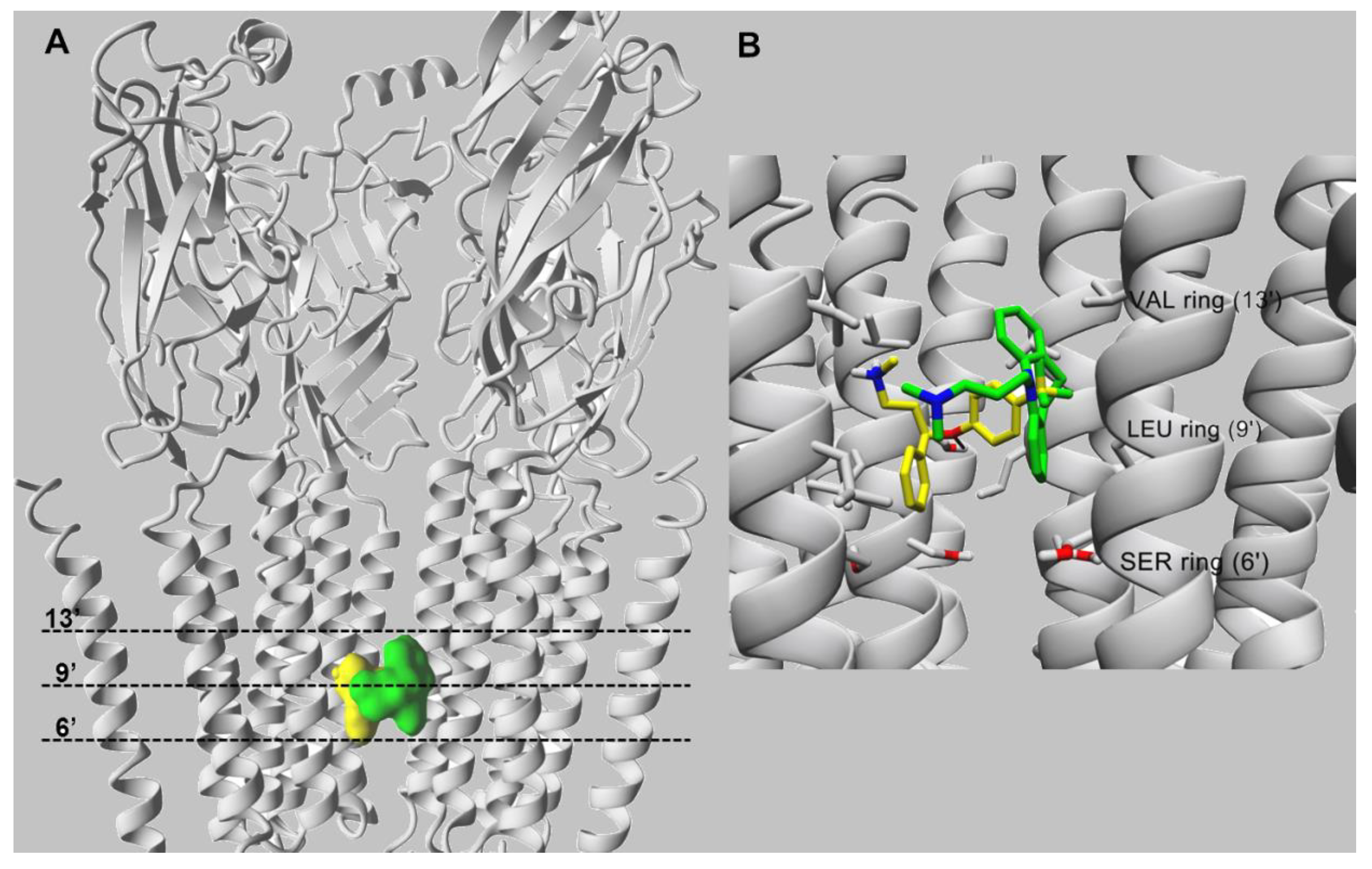
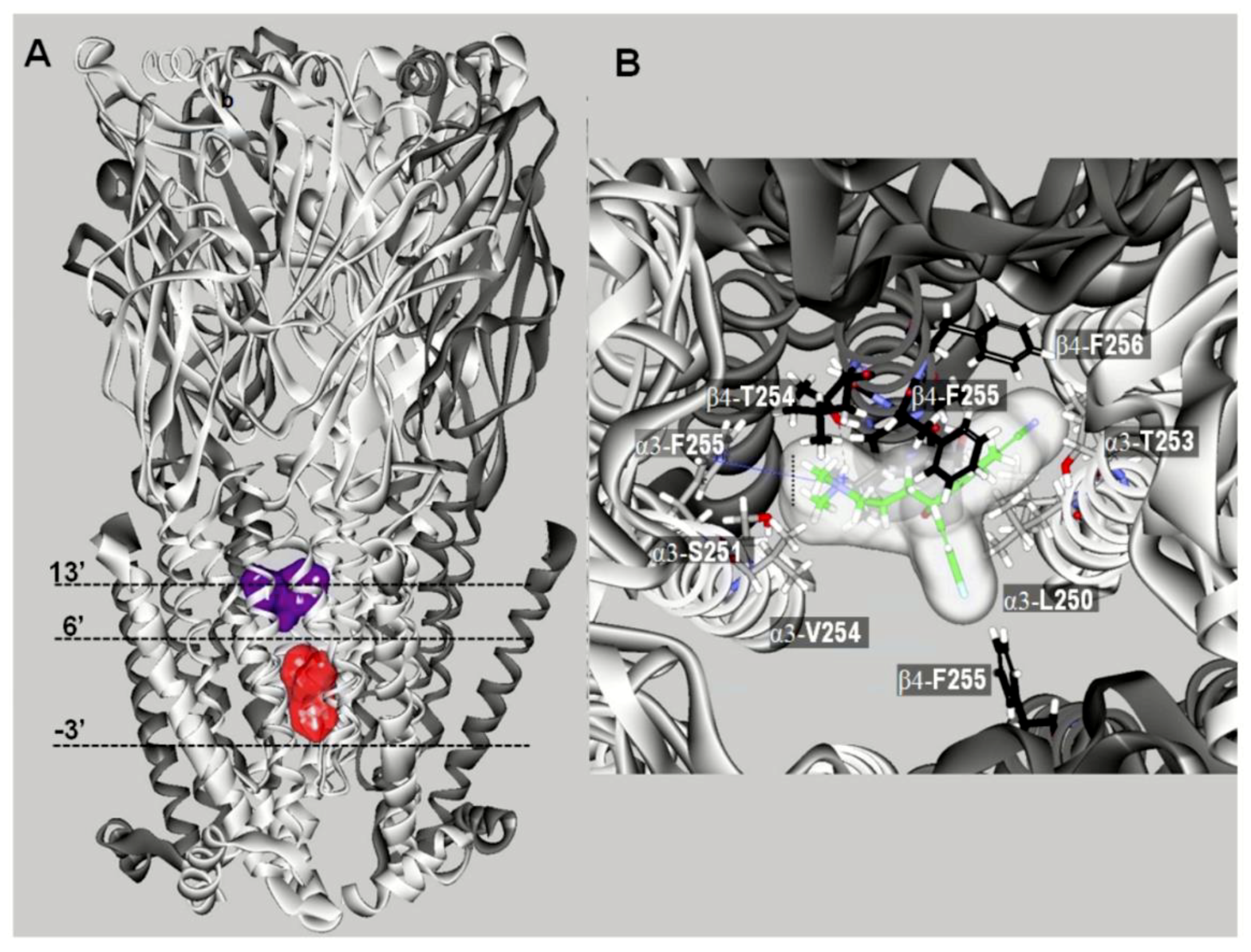
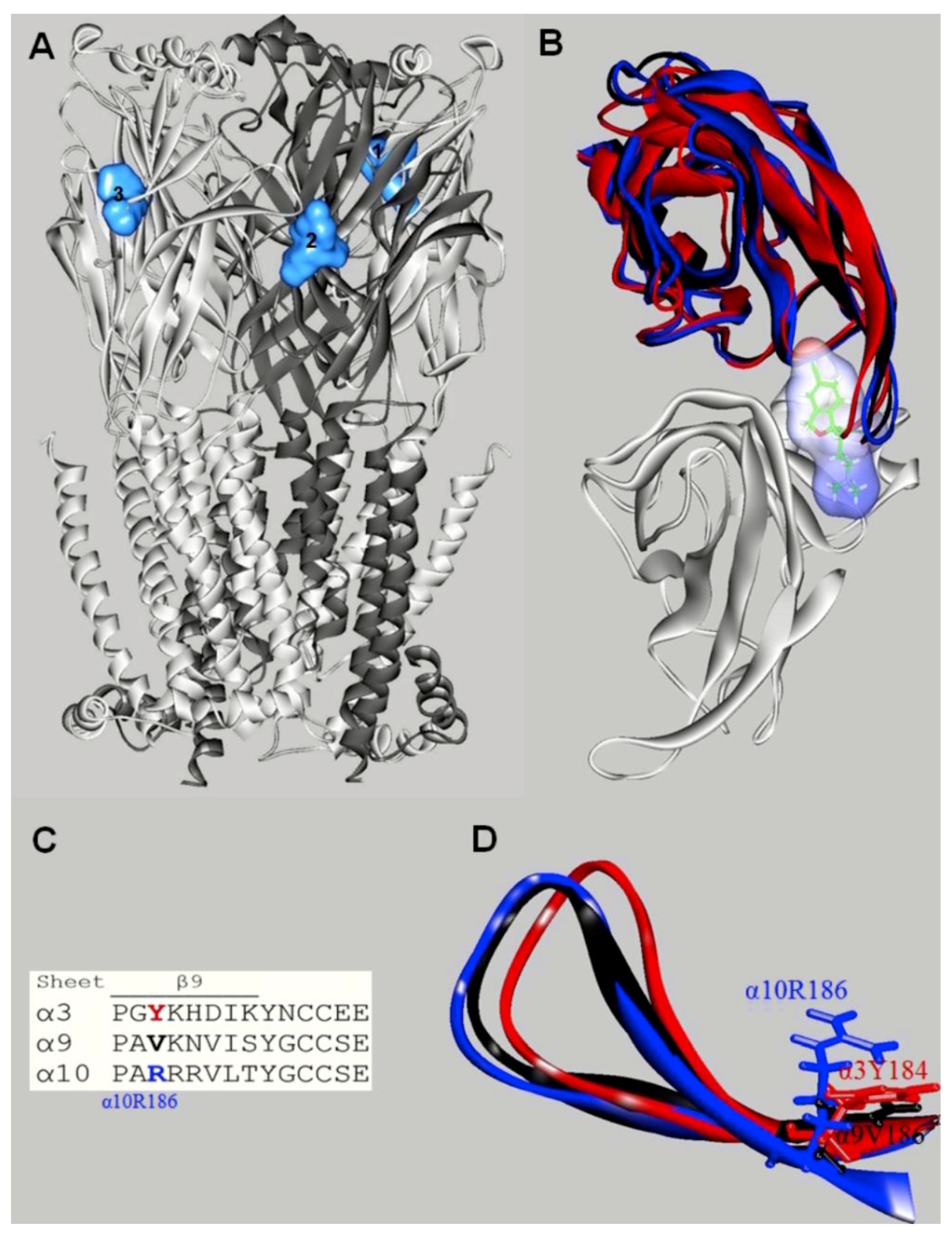
10. Characterization of SSRI Binding Sites for Different nAChR Subtypes
11. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- DSM-IV. Diagnostic and Statistical Manual of Mental Disorders IV-TR; American Psychiatric Press: Washington, DC, USA, 2020. [Google Scholar]
- Duman, R.S.; Aghajanian, G.K. Synaptic Dysfunction in Depression: Potential Therapeutic Targets. Science 2012, 338, 68–72. [Google Scholar] [CrossRef]
- Salerian, A.J.; Altar, C.A. The Prefrontal Cortex Influence over Subcortical and Limbic Regions Governs Antidepressant Response by N=H/(M+R). Psychiatry Res. 2012, 204, 1–12. [Google Scholar] [CrossRef]
- Zurkovsky, L.; Taylor, W.D.; Newhouse, P.A. Cognition as a Therapeutic Target in Late-Life Depression: Potential for Nicotinic Therapeutics. Biochem. Pharmacol. 2013, 86, 1133–1144. [Google Scholar] [CrossRef] [PubMed]
- Pandya, M.; Altinay, M.; Malone, D.A.; Anand, A. Where in the Brain Is Depression? Curr. Psychiatry Rep. 2012, 14, 634–642. [Google Scholar] [CrossRef]
- Drago, A.; Crisafulli, C.; Sidoti, A.; Serretti, A. The Molecular Interaction between the Glutamatergic, Noradrenergic, Dopaminergic and Serotoninergic Systems Informs a Detailed Genetic Perspective on Depressive Phenotypes. Prog. Neurobiol. 2011, 94, 418–460. [Google Scholar] [CrossRef]
- Pacher, P.; Kecskemeti, V. Trends in the Development of New Antidepressants. Is There a Light at the End of the Tunnel? Curr. Med. Chem. 2004, 11, 925–943. [Google Scholar] [CrossRef]
- Goldberg, J.S.; Bell, C.E.; Pollard, D.A. Revisiting the Monoamine Hypothesis of Depression: A New Perspective. Perspect. Med. Chem. 2014, 6, 1–8. [Google Scholar] [CrossRef]
- Ferguson, J.T. Treatment of Reserpine-Induced Depression with a New Analeptic: Phenidylate. Ann. N. Y. Acad. Sci. 1955, 61, 101–107. [Google Scholar] [CrossRef]
- Freis, E.D. Mental Depression in Hypertensive Patients Treated for Long Periods with Large Doses of Reserpine. N. Engl. J. Med. 1954, 251, 1006–1008. [Google Scholar] [CrossRef]
- Janowsky, D.S.; el-Yousef, M.K.; Davis, J.M.; Sekerke, H.J. A Cholinergic-Adrenergic Hypothesis of Mania and Depression. Lancet 1972, 2, 632–635. [Google Scholar] [CrossRef]
- Levin, E.D. Complex Relationships of Nicotinic Receptor Actions and Cognitive Functions. Biochem. Pharmacol. 2013, 86, 1145–1152. [Google Scholar] [CrossRef]
- Kenney, J.W.; Gould, T.J. Modulation of Hippocampus-Dependent Learning and Synaptic Plasticity by Nicotine. Mol. Neurobiol. 2008, 38, 101–121. [Google Scholar] [CrossRef]
- Mineur, Y.S.; Picciotto, M.R. Nicotine Receptors and Depression: Revisiting and Revising the Cholinergic Hypothesis. Trends Pharmacol. Sci. 2010, 31, 580–586. [Google Scholar] [CrossRef]
- Shytle, R.D.; Silver, A.A.; Lukas, R.J.; Newman, M.B.; Sheehan, D.V.; Sanberg, P.R. Nicotinic Acetylcholine Receptors as Targets for Antidepressants. Mol. Psychiatry 2002, 7, 525–535. [Google Scholar] [CrossRef]
- Steingard, R.J.; Yurgelun-Todd, D.A.; Hennen, J.; Moore, J.C.; Moore, C.M.; Vakili, K.; Young, A.D.; Katic, A.; Beardslee, W.R.; Renshaw, P.F. Increased Orbitofrontal Cortex Levels of Choline in Depressed Adolescents as Detected by in Vivo Proton Magnetic Resonance Spectroscopy. Biol. Psychiatry 2000, 48, 1053–1061. [Google Scholar] [CrossRef]
- Janowsky, D.S.; el-Yousef, M.K.; Davis, J.M. Acetylcholine and Depression. Psychosom. Med. 1974, 36, 248–257. [Google Scholar] [CrossRef]
- Risch, S.C.; Cohen, R.M.; Janowsky, D.S.; Kalin, N.H.; Murphy, D.L. Mood and Behavioral Effects of Physostigmine on Humans Are Accompanied by Elevations in Plasma Beta-Endorphin and Cortisol. Science 1980, 209, 1545–1546. [Google Scholar] [CrossRef]
- Risch, S.C.; Cohen, R.M.; Janowsky, D.S.; Kalin, N.H.; Sitaram, N.; Gillin, J.C.; Murphy, D.L. Physostigmine Induction of Depressive Symptomatology in Normal Human Subjects. Psychiatry Res. 1981, 4, 89–94. [Google Scholar] [CrossRef]
- Mineur, Y.S.; Obayemi, A.; Wigestrand, M.B.; Fote, G.M.; Calarco, C.A.; Li, A.M.; Picciotto, M.R. Cholinergic Signaling in the Hippocampus Regulates Social Stress Resilience and Anxiety- and Depression-like Behavior. Proc. Natl. Acad. Sci. USA 2013, 110, 3573–3578. [Google Scholar] [CrossRef]
- Dailey, J.W.; Yan, Q.S.; Mishra, P.K.; Burger, R.L.; Jobe, P.C. Effects of Fluoxetine on Convulsions and on Brain Serotonin as Detected by Microdialysis in Genetically Epilepsy-Prone Rats. J. Pharmacol. Exp. Ther. 1992, 260, 533–540. [Google Scholar]
- Perry, K.W.; Fuller, R.W. Effect of Fluoxetine on Serotonin and Dopamine Concentration in Microdialysis Fluid from Rat Striatum. Life Sci. 1992, 50, 1683–1690. [Google Scholar] [CrossRef]
- Nagayasu, K.; Kitaichi, M.; Nishitani, N.; Asaoka, N.; Shirakawa, H.; Nakagawa, T.; Kaneko, S. Chronic Effects of Antidepressants on Serotonin Release in Rat Raphe Slice Cultures: High Potency of Milnacipran in the Augmentation of Serotonin Release. Int. J. Neuropsychopharmacol. 2013, 16, 2295–2306. [Google Scholar] [CrossRef]
- Elhwuegi, A.S. Central Monoamines and Their Role in Major Depression. Prog. Neuropsychopharmacol. Biol. Psychiatry 2004, 28, 435–451. [Google Scholar] [CrossRef]
- Fitzgerald, K.T.; Bronstein, A.C. Selective Serotonin Reuptake Inhibitor Exposure. Top. Companion Anim. Med. 2013, 28, 13–17. [Google Scholar] [CrossRef] [PubMed]
- García-Colunga, J.; Miledi, R. Blockage of Mouse Muscle Nicotinic Receptors by Serotonergic Compounds. Exp. Physiol. 1999, 84, 847–864. [Google Scholar] [CrossRef] [PubMed]
- García-Colunga, J.; Awad, J.N.; Miledi, R. Blockage of Muscle and Neuronal Nicotinic Acetylcholine Receptors by Fluoxetine (Prozac). Proc. Natl. Acad. Sci. USA 1997, 94, 2041–2044. [Google Scholar] [CrossRef]
- Gunasekara, N.S.; Noble, S.; Benfield, P. Paroxetine. An Update of Its Pharmacology and Therapeutic Use in Depression and a Review of Its Use in Other Disorders. Drugs 1998, 55, 85–120. [Google Scholar] [CrossRef] [PubMed]
- Wong, D.T.; Bymaster, F.P.; Engleman, E.A. Prozac (Fluoxetine, Lilly 110140), the First Selective Serotonin Uptake Inhibitor and an Antidepressant Drug: Twenty Years since Its First Publication. Life Sci. 1995, 57, 411–441. [Google Scholar] [CrossRef]
- Deflorio, C.; Catalano, M.; Fucile, S.; Limatola, C.; Grassi, F. Fluoxetine Prevents Acetylcholine-Induced Excitotoxicity Blocking Human Endplate Acetylcholine Receptor. Muscle Nerve 2014, 49, 90–97. [Google Scholar] [CrossRef]
- Staley, J.K.; Krishnan-Sarin, S.; Cosgrove, K.P.; Krantzler, E.; Frohlich, E.; Perry, E.; Dubin, J.A.; Estok, K.; Brenner, E.; Baldwin, R.M.; et al. Human Tobacco Smokers in Early Abstinence Have Higher Levels of β2* Nicotinic Acetylcholine Receptors than Nonsmokers. J. Neurosci. 2006, 26, 8707–8714. [Google Scholar] [CrossRef]
- Chaouch, A.; Müller, J.S.; Guergueltcheva, V.; Dusl, M.; Schara, U.; Rakocević-Stojanović, V.; Lindberg, C.; Scola, R.H.; Werneck, L.C.; Colomer, J.; et al. A Retrospective Clinical Study of the Treatment of Slow-Channel Congenital Myasthenic Syndrome. J. Neurol. 2012, 259, 474–481. [Google Scholar] [CrossRef]
- Bliziotes, M.; Eshleman, A.; Burt-Pichat, B.; Zhang, X.-W.; Hashimoto, J.; Wiren, K.; Chenu, C. Serotonin Transporter and Receptor Expression in Osteocytic MLO-Y4 Cells. Bone 2006, 39, 1313–1321. [Google Scholar] [CrossRef]
- Nencetti, S.; Demontis, G.C.; Mazzoni, M.R.; Betti, L.; Banti, I.; Rossello, A.; Lapucci, A. 3-[(Aryl)(4-Fluorobenzyloxy)Methyl]Piperidine Derivatives: High-Affinity Ligands for the Serotonin Transporter. J. Pharm. Pharmacol. 2007, 59, 1439–1445. [Google Scholar] [CrossRef]
- Wang, S.; Chen, Y.; Liu, X.; Xu, X.; Liu, X.; Liu, B.-F.; Zhang, G. Synthesis and Evaluation of Novel 2,3-Dihydrobenzo[b][1,4]Dioxin- and Indolealkylamine Derivatives as Potential Antidepressants. Arch. Pharm. 2014, 347, 32–41. [Google Scholar] [CrossRef]
- Lendvai, B.; Vizi, E.S. Nonsynaptic Chemical Transmission through Nicotinic Acetylcholine Receptors. Physiol. Rev. 2008, 88, 333–349. [Google Scholar] [CrossRef]
- Marchi, M.; Grilli, M. Presynaptic Nicotinic Receptors Modulating Neurotransmitter Release in the Central Nervous System: Functional Interactions with Other Coexisting Receptors. Prog. Neurobiol. 2010, 92, 105–111. [Google Scholar] [CrossRef] [PubMed]
- Changeux, J.-P. The Nicotinic Acetylcholine Receptor: The Founding Father of the Pentameric Ligand-Gated Ion Channel Superfamily. J. Biol. Chem. 2012, 287, 40207–40215. [Google Scholar] [CrossRef] [PubMed]
- Yakel, J.L. Cholinergic Receptors: Functional Role of Nicotinic ACh Receptors in Brain Circuits and Disease. Pflüg. Arch. Eur. J. Physiol. 2013, 465, 441–450. [Google Scholar] [CrossRef] [PubMed]
- Picciotto, M.R.; Mineur, Y.S. Molecules and Circuits Involved in Nicotine Addiction: The Many Faces of Smoking. Neuropharmacology 2014, 76 Pt B, 545–553. [Google Scholar] [CrossRef]
- Picciotto, M.R.; Brunzell, D.H.; Caldarone, B.J. Effect of Nicotine and Nicotinic Receptors on Anxiety and Depression. Neuroreport 2002, 13, 1097–1106. [Google Scholar] [CrossRef] [PubMed]
- Saricicek, A.; Esterlis, I.; Maloney, K.H.; Mineur, Y.S.; Ruf, B.M.; Muralidharan, A.; Chen, J.I.; Cosgrove, K.P.; Kerestes, R.; Ghose, S.; et al. Persistent β2*-Nicotinic Acetylcholinergic Receptor Dysfunction in Major Depressive Disorder. Am. J. Psychiatry 2012, 169, 851–859. [Google Scholar] [CrossRef]
- Hannestad, J.O.; Cosgrove, K.P.; DellaGioia, N.F.; Perkins, E.; Bois, F.; Bhagwagar, Z.; Seibyl, J.P.; McClure-Begley, T.D.; Picciotto, M.R.; Esterlis, I. Changes in the Cholinergic System between Bipolar Depression and Euthymia as Measured with [123I]5IA Single Photon Emission Computed Tomography. Biol. Psychiatry 2013, 74, 768–776. [Google Scholar] [CrossRef]
- Meyer, P.M.; Strecker, K.; Kendziorra, K.; Becker, G.; Hesse, S.; Woelpl, D.; Hensel, A.; Patt, M.; Sorger, D.; Wegner, F.; et al. Reduced α4β2*-Nicotinic Acetylcholine Receptor Binding and Its Relationship to Mild Cognitive and Depressive Symptoms in Parkinson Disease. Arch. Gen. Psychiatry 2009, 66, 866–877. [Google Scholar] [CrossRef]
- Andreasen, J.T.; Henningsen, K.; Bate, S.; Christiansen, S.; Wiborg, O. Nicotine Reverses Anhedonic-like Response and Cognitive Impairment in the Rat Chronic Mild Stress Model of Depression: Comparison with Sertraline. J. Psychopharmacol. Oxf. Engl. 2011, 25, 1134–1141. [Google Scholar] [CrossRef]
- Andreasen, J.T.; Redrobe, J.P.; Nielsen, E.Ø. Combined α7 Nicotinic Acetylcholine Receptor Agonism and Partial Serotonin Transporter Inhibition Produce Antidepressant-like Effects in the Mouse Forced Swim and Tail Suspension Tests: A Comparison of SSR180711 and PNU-282987. Pharmacol. Biochem. Behav. 2012, 100, 624–629. [Google Scholar] [CrossRef]
- Gentile, A.; Bianco, A.; Nordström, A.; Nordström, P. Use of Alcohol, Drugs, Inhalants, and Smoking Tobacco and the Long-Term Risk of Depression in Men: A Nationwide Swedish Cohort Study from 1969–2017. Drug Alcohol Depend. 2021, 221, 108553. [Google Scholar] [CrossRef]
- Glassman, A.H.; Helzer, J.E.; Covey, L.S.; Cottler, L.B.; Stetner, F.; Tipp, J.E.; Johnson, J. Smoking, Smoking Cessation, and Major Depression. JAMA 1990, 264, 1546–1549. [Google Scholar] [CrossRef]
- Kalman, D.; Morissette, S.B.; George, T.P. Co-Morbidity of Smoking in Patients with Psychiatric and Substance Use Disorders. Am. J. Addict. 2005, 14, 106–123. [Google Scholar] [CrossRef]
- Mendelsohn, C. Smoking and Depression—A Review. Aust. Fam. Physician 2012, 41, 304–307. [Google Scholar]
- Tsuang, M.T.; Francis, T.; Minor, K.; Thomas, A.; Stone, W.S. Genetics of Smoking and Depression. Hum. Genet. 2012, 131, 905–915. [Google Scholar] [CrossRef]
- Piirtola, M.; Kaprio, J.; Baker, T.B.; Piasecki, T.M.; Piper, M.E.; Korhonen, T. The Associations of Smoking Dependence Motives with Depression among Daily Smokers. Addict. Abingdon Engl. 2021. [Google Scholar] [CrossRef] [PubMed]
- Bacher, I.; Wu, B.; Shytle, D.R.; George, T.P. Mecamylamine—A Nicotinic Acetylcholine Receptor Antagonist with Potential for the Treatment of Neuropsychiatric Disorders. Expert Opin. Pharmacother. 2009, 10, 2709–2721. [Google Scholar] [CrossRef] [PubMed]
- George, T.P.; Sacco, K.A.; Vessicchio, J.C.; Weinberger, A.H.; Shytle, R.D. Nicotinic Antagonist Augmentation of Selective Serotonin Reuptake Inhibitor-Refractory Major Depressive Disorder: A Preliminary Study. J. Clin. Psychopharmacol. 2008, 28, 340–344. [Google Scholar] [CrossRef] [PubMed]
- Weinberger, A.H.; McKee, S.A.; Picciotto, M.R.; Mazure, C.M. Examining Antidepressant Drug Response by Smoking Status: Why Is It Important and How Often Is It Done? J. Psychopharmacol. Oxf. Engl. 2011, 25, 1269–1276. [Google Scholar] [CrossRef]
- Belujon, P.; Grace, A.A. Dopamine System Dysregulation in Major Depressive Disorders. Int. J. Neuropsychopharmacol. 2017, 20, 1036–1046. [Google Scholar] [CrossRef]
- Ortells, M.O.; Barrantes, G.E. Tobacco Addiction: A Biochemical Model of Nicotine Dependence. Med. Hypotheses 2010, 74, 884–894. [Google Scholar] [CrossRef] [PubMed]
- Fenster, C.P.; Hicks, J.H.; Beckman, M.L.; Covernton, P.J.; Quick, M.W.; Lester, R.A. Desensitization of Nicotinic Receptors in the Central Nervous System. Ann. N. Y. Acad. Sci. 1999, 868, 620–623. [Google Scholar] [CrossRef]
- Giniatullin, R.; Nistri, A.; Yakel, J.L. Desensitization of Nicotinic ACh Receptors: Shaping Cholinergic Signaling. Trends Neurosci. 2005, 28, 371–378. [Google Scholar] [CrossRef]
- Cuevas-Olguin, R.; Esquivel-Rendon, E.; Vargas-Mireles, J.; Barajas-Lόpez, C.; Salgado-Delgado, R.; Saderi, N.; Arias, H.R.; Atzori, M.; Miranda-Morales, M. Nicotine Smoking Concentrations Modulate GABAergic Synaptic Transmission in Murine Medial Prefrontal Cortex by Activation of α7* and β2* Nicotinic Receptors. Eur. J. Neurosci. 2020, 51, 781–792. [Google Scholar] [CrossRef]
- Wanaverbecq, N.; Semyanov, A.; Pavlov, I.; Walker, M.C.; Kullmann, D.M. Cholinergic Axons Modulate GABAergic Signaling among Hippocampal Interneurons via Postsynaptic α7 Nicotinic Receptors. J. Neurosci. 2007, 27, 5683–5693. [Google Scholar] [CrossRef]
- Hughes, J.R.; Stead, L.F.; Hartmann-Boyce, J.; Cahill, K.; Lancaster, T. Antidepressants for Smoking Cessation. Cochrane Database Syst. Rev. 2014, CD000031. [Google Scholar] [CrossRef] [PubMed]
- Brown, R.A.; Abrantes, A.M.; Strong, D.R.; Niaura, R.; Kahler, C.W.; Miller, I.W.; Price, L.H. Efficacy of Sequential Use of Fluoxetine for Smoking Cessation in Elevated Depressive Symptom Smokers. Nicotine Tob. Res. 2014, 16, 197–207. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, E.B.; Bettiker, R.L.; Bogdanov, M.; Wurtman, R.J. Effects of Systemic Nicotine on Serotonin Release in Rat Brain. Brain Res. 1993, 621, 311–318. [Google Scholar] [CrossRef]
- Redrobe, J.P.; Nielsen, E.Ø.; Christensen, J.K.; Peters, D.; Timmermann, D.B.; Olsen, G.M. α7 Nicotinic Acetylcholine Receptor Activation Ameliorates Scopolamine-Induced Behavioural Changes in a Modified Continuous Y-Maze Task in Mice. Eur. J. Pharmacol. 2009, 602, 58–65. [Google Scholar] [CrossRef]
- Andreasen, J.T.; Nielsen, E.Ø.; Christensen, J.K.; Olsen, G.M.; Peters, D.; Mirza, N.R.; Redrobe, J.P. Subtype-Selective Nicotinic Acetylcholine Receptor Agonists Enhance the Responsiveness to Citalopram and Reboxetine in the Mouse Forced Swim Test. J. Psychopharmacol. Oxf. Engl. 2011, 25, 1347–1356. [Google Scholar] [CrossRef] [PubMed]
- Popik, P.; Kozela, E.; Krawczyk, M. Nicotine and Nicotinic Receptor Antagonists Potentiate the Antidepressant-like Effects of Imipramine and Citalopram. Br. J. Pharmacol. 2003, 139, 1196–1202. [Google Scholar] [CrossRef]
- Arias, H.R.; Jin, X.-T.; Gallino, S.; Peng, C.; Feuerbach, D.; García-Colunga, J.; Elgoyhen, A.B.; Drenan, R.M.; Ortells, M.O. Selectivity of (±)-Citalopram at Nicotinic Acetylcholine Receptors and Different Inhibitory Mechanisms between Habenular α3β4* and α9α10 Subtypes. Neurochem. Int. 2019, 131, 104552. [Google Scholar] [CrossRef]
- Chen, M.-F.; Huang, Y.-C.; Long, C.; Yang, H.-I.; Lee, H.-C.; Chen, P.-Y.; Hoffer, B.J.; Lee, T.J.-F. Bimodal Effects of Fluoxetine on Cerebral Nitrergic Neurogenic Vasodilation in Porcine Large Cerebral Arteries. Neuropharmacology 2012, 62, 1651–1658. [Google Scholar] [CrossRef]
- Arias, H.R.; Feuerbach, D.; Targowska-Duda, K.M.; Russell, M.; Jozwiak, K. Interaction of Selective Serotonin Reuptake Inhibitors with Neuronal Nicotinic Acetylcholine Receptors. Biochemistry 2010, 49, 5734–5742. [Google Scholar] [CrossRef]
- López-Valdés, H.E.; García-Colunga, J. Antagonism of Nicotinic Acetylcholine Receptors by Inhibitors of Monoamine Uptake. Mol. Psychiatry 2001, 6, 511–519. [Google Scholar] [CrossRef][Green Version]
- García-Colunga, J.; Vázquez-Gómez, E.; Miledi, R. Combined Actions of Zinc and Fluoxetine on Nicotinic Acetylcholine Receptors. Pharm. J. 2004, 4, 388–393. [Google Scholar] [CrossRef]
- Fryer, J.D.; Lukas, R.J. Antidepressants Noncompetitively Inhibit Nicotinic Acetylcholine Receptor Function. J. Neurochem. 1999, 72, 1117–1124. [Google Scholar] [CrossRef] [PubMed]
- Maggi, L.; Palma, E.; Miledi, R.; Eusebi, F. Effects of Fluoxetine on Wild and Mutant Neuronal α7 Nicotinic Receptors. Mol. Psychiatry 1998, 3, 350–355. [Google Scholar] [CrossRef] [PubMed]
- Feuerbach, D.; Lingenhöhl, K.; Dobbins, P.; Mosbacher, J.; Corbett, N.; Nozulak, J.; Hoyer, D. Coupling of Human Nicotinic Acetylcholine Receptors α7 to Calcium Channels in GH3 Cells. Neuropharmacology 2005, 48, 215–227. [Google Scholar] [CrossRef]
- Duarte, Y.; Rojas, M.; Canan, J.; Pérez, E.G.; González-Nilo, F.; García-Colunga, J. Different Classes of Antidepressants Inhibit the Rat α7 Nicotinic Acetylcholine Receptor by Interacting within the Ion Channel: A Functional and Structural Study. Molecules 2021, 26, 998. [Google Scholar] [CrossRef]
- Arias, H.R.; Feuerbach, D.; Bhumireddy, P.; Ortells, M.O. Inhibitory Mechanisms and Binding Site Location for Serotonin Selective Reuptake Inhibitors on Nicotinic Acetylcholine Receptors. Int. J. Biochem. Cell Biol. 2010, 42, 712–724. [Google Scholar] [CrossRef] [PubMed]
- Hennings, E.C.; Kiss, J.P.; De Oliveira, K.; Toth, P.T.; Vizi, E.S. Nicotinic Acetylcholine Receptor Antagonistic Activity of Monoamine Uptake Blockers in Rat Hippocampal Slices. J. Neurochem. 1999, 73, 1043–1050. [Google Scholar] [CrossRef]
- Arias, H.R.; Vázquez-Gómez, E.; Hernández-Abrego, A.; Gallino, S.; Feuerbach, D.; Ortells, M.O.; Elgoyhen, A.B.; García-Colunga, J. Tricyclic Antidepressants Inhibit Hippocampal α7* and α9α10 Nicotinic Acetylcholine Receptors by Different Mechanisms. Int. J. Biochem. Cell Biol. 2018, 100, 1–10. [Google Scholar] [CrossRef]
- Peng, H.; Ferris, R.L.; Matthews, T.; Hiel, H.; Lopez-Albaitero, A.; Lustig, L.R. Characterization of the Human Nicotinic Acetylcholine Receptor Subunit alpha (α) 9 (CHRNA9) and alpha (α) 10 (CHRNA10) in Lymphocytes. Life Sci. 2004, 76, 263–280. [Google Scholar] [CrossRef]
- Caiaffo, V.; Oliveira, B.D.R.; de Sá, F.B.; Evêncio Neto, J. Anti-inflammatory, Antiapoptotic, and Antioxidant Activity of Fluoxetine. Pharmacol. Res. Perspect. 2016, 4. [Google Scholar] [CrossRef]
- Goodnick, P.J. Pharmacokinetics of Second Generation Antidepressants: Fluoxetine. Psychopharmacol. Bull. 1991, 27, 503–512. [Google Scholar] [PubMed]
- Henry, M.E.; Schmidt, M.E.; Hennen, J.; Villafuerte, R.A.; Butman, M.L.; Tran, P.; Kerner, L.T.; Cohen, B.; Renshaw, P.F. A Comparison of Brain and Serum Pharmacokinetics of R-Fluoxetine and Racemic Fluoxetine: A 19-F MRS Study. Neuropsychopharmacology 2005, 30, 1576–1583. [Google Scholar] [CrossRef]
- Karson, C.N.; Newton, J.E.; Livingston, R.; Jolly, J.B.; Cooper, T.B.; Sprigg, J.; Komoroski, R.A. Human Brain Fluoxetine Concentrations. J. Neuropsychiatry Clin. Neurosci. 1993, 5, 322–329. [Google Scholar] [PubMed]
- Renshaw, P.F.; Guimaraes, A.R.; Fava, M.; Rosenbaum, J.F.; Pearlman, J.D.; Flood, J.G.; Puopolo, P.R.; Clancy, K.; Gonzalez, R.G. Accumulation of Fluoxetine and Norfluoxetine in Human Brain during Therapeutic Administration. Am. J. Psychiatry 1992, 149, 1592–1594. [Google Scholar]
- Bolo, N.R.; Hodé, Y.; Nédélec, J.F.; Lainé, E.; Wagner, G.; Macher, J.P. Brain Pharmacokinetics and Tissue Distribution in Vivo of Fluvoxamine and Fluoxetine by Fluorine Magnetic Resonance Spectroscopy. Neuropsychopharmacology 2000, 23, 428–438. [Google Scholar] [CrossRef]
- Nedahl, M.; Johansen, S.S.; Linnet, K. Reference Brain/Blood Concentrations of Citalopram, Duloxetine, Mirtazapine and Sertraline. J. Anal. Toxicol. 2018, 42, 149–156. [Google Scholar] [CrossRef] [PubMed]
- Weber, M.L.; Hofland, C.M.; Shaffer, C.L.; Flik, G.; Cremers, T.; Hurst, R.S.; Rollema, H. Therapeutic Doses of Antidepressants Are Projected Not to Inhibit Human α4β2 Nicotinic Acetylcholine Receptors. Neuropharmacology 2013, 72, 88–95. [Google Scholar] [CrossRef]
- Yu, L.F.; Brek Eaton, J.; Zhang, H.K.; Sabath, E.; Hanania, T.; Li, G.N.; van Breemen, R.B.; Whiteaker, P.; Liu, Q.; Wu, J.; et al. The Potent and Selective α4β2*/α6*-Nicotinic Acetylcholine Receptor Partial Agonist 2-[5-[5-((S)Azetidin-2-Ylmethoxy)-3-Pyridinyl]-3-Isoxazolyl]Ethanol Demonstrates Antidepressive-like Behavior in Animal Models and a Favorable ADME-Tox Profile. Pharmacol. Res. Perspect. 2014, 2, e00026. [Google Scholar] [CrossRef] [PubMed]
- Dulawa, S.C.; Janowsky, D.S. Cholinergic Regulation of Mood: From Basic and Clinical Studies to Emerging Therapeutics. Mol. Psychiatry 2019, 24, 694–709. [Google Scholar] [CrossRef]
- Coplan, J.D.; Gopinath, S.; Abdallah, C.G.; Berry, B.R. A Neurobiological Hypothesis of Treatment-Resistant Depression-Mechanisms for Selective Serotonin Reuptake Inhibitor Non-Efficacy. Front. Behav. Neurosci. 2014, 8, 189. [Google Scholar] [CrossRef]
- Van Dyke, A.M.; Francis, T.C.; Chen, H.; Bailey, A.M.; Thompson, S.M. Chronic Fluoxetine Treatment in Vivo Enhances Excitatory Synaptic Transmission in the Hippocampus. Neuropharmacology 2019, 150, 38–45. [Google Scholar] [CrossRef]
- Luscher, B.; Shen, Q.; Sahir, N. The GABAergic Deficit Hypothesis of Major Depressive Disorder. Mol. Psychiatry 2011, 16, 383–406. [Google Scholar] [CrossRef] [PubMed]
- Komlósi, G.; Molnár, G.; Rózsa, M.; Oláh, S.; Barzó, P.; Tamás, G. Fluoxetine (Prozac) and Serotonin Act on Excitatory Synaptic Transmission to Suppress Single Layer 2/3 Pyramidal Neuron-Triggered Cell Assemblies in the Human Prefrontal Cortex. J. Neurosci. 2012, 32, 16369–16378. [Google Scholar] [CrossRef]
- Chang, B.; Daniele, C.A.; Gallagher, K.; Madonia, M.; Mitchum, R.D.; Barrett, L.; Vezina, P.; McGehee, D.S. Nicotinic Excitation of Serotonergic Projections from Dorsal Raphe to the Nucleus Accumbens. J. Neurophysiol. 2011, 106, 801–808. [Google Scholar] [CrossRef] [PubMed]
- Galindo-Charles, L.; Hernandez-Lopez, S.; Galarraga, E.; Tapia, D.; Bargas, J.; Garduño, J.; Frías-Dominguez, C.; Drucker-Colin, R.; Mihailescu, S. Serotoninergic Dorsal Raphe Neurons Possess Functional Postsynaptic Nicotinic Acetylcholine Receptors. Synapse 2008, 62, 601–615. [Google Scholar] [CrossRef]
- Garduño, J.; Galindo-Charles, L.; Jiménez-Rodríguez, J.; Galarraga, E.; Tapia, D.; Mihailescu, S.; Hernandez-Lopez, S. Presynaptic α4β2 Nicotinic Acetylcholine Receptors Increase Glutamate Release and Serotonin Neuron Excitability in the Dorsal Raphe Nucleus. J. Neurosci. 2012, 32, 15148–15157. [Google Scholar] [CrossRef] [PubMed]
- Tucci, S.A.; Genn, R.F.; File, S.E. Methyllycaconitine (MLA) Blocks the Nicotine Evoked Anxiogenic Effect and 5-HT Release in the Dorsal Hippocampus: Possible Role of α7 Receptors. Neuropharmacology 2003, 44, 367–373. [Google Scholar] [CrossRef]
- Méndez, P.; Pazienti, A.; Szabó, G.; Bacci, A. Direct Alteration of a Specific Inhibitory Circuit of the Hippocampus by Antidepressants. J. Neurosci. 2012, 32, 16616–16628. [Google Scholar] [CrossRef]
- Griguoli, M.; Cherubini, E. Regulation of Hippocampal Inhibitory Circuits by Nicotinic Acetylcholine Receptors. J. Physiol. 2012, 590, 655–666. [Google Scholar] [CrossRef]
- Chamberland, S.; Topolnik, L. Inhibitory Control of Hippocampal Inhibitory Neurons. Front. Neurosci. 2012, 6, 165. [Google Scholar] [CrossRef]
- Zhang, G.-F.; Zhang, G.-F.; Zhang, G.-F. The Lateral Habenula: Role in Chronic Pain and Depression. Transl. Perioper. Pain Med. 2020, 7, 271–278. [Google Scholar]
- Castillo-Rolón, D.; Ramírez-Sánchez, E.; Arenas-López, G.; Garduño, J.; Hernández-González, O.; Mihailescu, S.; Hernández-López, S. Nicotine Increases Spontaneous Glutamate Release in the Rostromedial Tegmental Nucleus. Front. Neurosci. 2020, 14, 604583. [Google Scholar] [CrossRef]
- Elmer, G.I.; Palacorolla, H.; Mayo, C.L.; Brown, P.L.; Jhou, T.C.; Brady, D.; Shepard, P.D. The Rostromedial Tegmental Nucleus Modulates the Development of Stress-Induced Helpless Behavior. Behav. Brain Res. 2019, 359, 950–957. [Google Scholar] [CrossRef]
- Arias, H.R.; Targowska-Duda, K.M.; Feuerbach, D.; Jozwiak, K. Coronaridine Congeners Inhibit Human α3β4 Nicotinic Acetylcholine Receptors by Interacting with Luminal and Non-Luminal Sites. Int. J. Biochem. Cell Biol. 2015, 65, 81–90. [Google Scholar] [CrossRef]
- Arias, H.R.; Jin, X.; Feuerbach, D.; Drenan, R.M. Selectivity of Coronaridine Congeners at Nicotinic Acetylcholine Receptors and Inhibitory Activity on Mouse Medial Habenula. Int. J. Biochem. Cell Biol. 2017, 92, 202–209. [Google Scholar] [CrossRef]
- Rodrı Guez, P.; Urbanavicius, J.; Prieto, J.P.; Fabius, S.; Reyes, A.L.; Havel, V.; Sames, D.; Scorza, C.; Carrera, I. A Single Administration of the Atypical Psychedelic Ibogaine or Its Metabolite Noribogaine Induces an Antidepressant-Like Effect in Rats. ACS Chem. Neurosci. 2020, 11, 1661–1672. [Google Scholar] [CrossRef]
- Arias, H.R.; Fedorov, N.B.; Benson, L.C.; Lippiello, P.M.; Gatto, G.J.; Feuerbach, D.; Ortells, M.O. Functional and Structural Interaction of (-)-Reboxetine with the Human α4β2 Nicotinic Acetylcholine Receptor. J. Pharmacol. Exp. Ther. 2013, 344, 113–123. [Google Scholar] [CrossRef]
- Arias, H.R.; Ortells, M.O.; Feuerbach, D. (-)-Reboxetine Inhibits Muscle Nicotinic Acetylcholine Receptors by Interacting with Luminal and Non-Luminal Sites. Neurochem. Int. 2013, 63, 423–431. [Google Scholar] [CrossRef]
- Gumilar, F.; Arias, H.R.; Spitzmaul, G.; Bouzat, C. Molecular Mechanisms of Inhibition of Nicotinic Acetylcholine Receptors by Tricyclic Antidepressants. Neuropharmacology 2003, 45, 964–976. [Google Scholar] [CrossRef]
- Arias, H.R.; Bhumireddy, P.; Bouzat, C. Molecular Mechanisms and Binding Site Locations for Noncompetitive Antagonists of Nicotinic Acetylcholine Receptors. Int. J. Biochem. Cell Biol. 2006, 38, 1254–1276. [Google Scholar] [CrossRef]
- Calimet, N.; Simoes, M.; Changeux, J.-P.; Karplus, M.; Taly, A.; Cecchini, M. A Gating Mechanism of Pentameric Ligand-Gated Ion Channels. Proc. Natl. Acad. Sci. USA 2013, 110, E3987–E3996. [Google Scholar] [CrossRef]
- Arias, H.R.; Feuerbach, D.; Targowska-Duda, K.M.; Aggarwal, S.; Lapinsky, D.J.; Jozwiak, K. Structural and Functional Interaction of (±)-2-(N-Tert-Butylamino)-3′-Iodo-4′-Azidopropiophenone, a Photoreactive Bupropion Derivative, with Nicotinic Acetylcholine Receptors. Neurochem. Int. 2012, 61, 1433–1441. [Google Scholar] [CrossRef]
- Arias, H.R.; Gumilar, F.; Rosenberg, A.; Targowska-Duda, K.M.; Feuerbach, D.; Jozwiak, K.; Moaddel, R.; Wainer, I.W.; Bouzat, C. Interaction of Bupropion with Muscle-Type Nicotinic Acetylcholine Receptors in Different Conformational States. Biochemistry 2009, 48, 4506–4518. [Google Scholar] [CrossRef]
- Morales-Perez, C.L.; Noviello, C.M.; Hibbs, R.E. X-Ray Structure of the Human α4β2 Nicotinic Receptor. Nature 2016, 538, 411–415. [Google Scholar] [CrossRef] [PubMed]
- Unwin, N. Refined Structure of the Nicotinic Acetylcholine Receptor at 4Å Resolution. J. Mol. Biol. 2005, 346, 967–989. [Google Scholar] [CrossRef]
- Arias, H.R.; Targowska-Duda, K.M.; Feuerbach, D.; Sullivan, C.J.; Maciejewski, R.; Jozwiak, K. Different Interaction between Tricyclic Antidepressants and Mecamylamine with the Human α3β4 Nicotinic Acetylcholine Receptor Ion Channel. Neurochem. Int. 2010, 56, 642–649. [Google Scholar] [CrossRef] [PubMed]
- Arias, H.R.; Rosenberg, A.; Targowska-Duda, K.M.; Feuerbach, D.; Jozwiak, K.; Moaddel, R.; Wainer, I.W. Tricyclic Antidepressants and Mecamylamine Bind to Different Sites in the Human α4β2 Nicotinic Receptor Ion Channel. Int. J. Biochem. Cell Biol. 2010, 42, 1007–1018. [Google Scholar] [CrossRef]
- Targowska-Duda, K.M.; Arias, H.R.; Jozwiak, K. Application of in Silico Methods to Support Experimental Data: Interactions of Antidepressants with Nicotinic Acetylcholine Receptors. Open Conf. Proc. J. 2013, 4, 11–22. [Google Scholar]
- Harms, M.J.; Schlessman, J.L.; Chimenti, M.S.; Sue, G.R.; Damjanović, A.; García-Moreno, B. A Buried Lysine That Titrates with a Normal pKa: Role of Conformational Flexibility at the Protein-Water Interface as a Determinant of pKa Values. Protein Sci. Publ. Protein Soc. 2008, 17, 833–845. [Google Scholar] [CrossRef] [PubMed]
- Harms, M.J.; Schlessman, J.L.; Sue, G.R.; García-Moreno, E.B. Arginine Residues at Internal Positions in a Protein Are Always Charged. Proc. Natl. Acad. Sci. USA 2011, 108, 18954–18959. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| nAChR Subtype | SSRI | Method | IC50 (μM) | References |
|---|---|---|---|---|
| hα4β2 | Fluoxetine | Voltage-clamp | 1.83 | [69] |
| Ca2+ influx | 4.4 ± 0.6 | [70] | ||
| Paroxetine | Ca2+ influx | 8.6 ± 2.3 | [70] | |
| Citalopram | Ca2+ influx | 19.0 ± 4.2 | [68] | |
| rα2β4 | Fluoxetine | Voltage-clamp | 0.37 b | [71] |
| 0.45 ± 0.03 c | [27] | |||
| Nor-fluoxetine | Voltage-clamp | 0.13 b | [71] | |
| Zimelidine | Voltage-clamp | 0.67 b | [71] | |
| hα3β4 | Fluoxetine | Ca2+ influx | 2.0 ± 0.4 | [70] |
| Paroxetine | Ca2+ influx | 2.6 ± 0.3 | [70] | |
| Citalopram | Ca2+ influx | 5.1 ± 1.3 | [68] | |
| rα3β4 | Fluoxetine | Voltage-clamp | 0.64 ± 0.03 c | [27] |
| rα4β4 | Fluoxetine | Voltage-clamp (+100–200 µM Zn2+) | 0.3 ± 0.045 d 0.17 ± 0.018 d | [72] |
| rα3β4α5 | Fluoxetine | 86Rb+ efflux | 2.5 | [73] |
| Paroxetine | 86Rb+ efflux | 4.9 | [73] | |
| Sertraline | 86Rb+ efflux | 3.1 | [73] | |
| hα7 | Fluoxetine | Voltage-clamp | 1.6 | [74] |
| Ca2+ influx | 4.7 ± 0.83 | [75] | ||
| Ca2+ influx | 4.9 ± 1.0 | [70] | ||
| Voltage-clamp | 5.27 | [69] | ||
| Paroxetine | Ca2+ influx | 8.6 ± 2.0 | [70] | |
| Ca2+ influx | 9.1 ± 0.6 | [75] | ||
| Sertraline | Ca2+ influx | 10.0 ± 0.81 | [75] | |
| Citalopram | Ca 2+ influx | 18.8 ± 1.1 | [68] | |
| Hippocampal rα7 * | Nor-fluoxetine | Voltage-clamp | 0.82 ± 0.04 a | [76] |
| Fluoxetine | Voltage-clamp | 0.66 ± 0.06 a | [76] | |
| Escitalopram | Voltage-clamp | 28.9 ± 5.1 | [76] | |
| hα3β2 | Fluoxetine | Voltage-clamp | 4.14 | [69] |
| rα9α10 | Citalopram | Voltage-clamp | 7.5 ± 0.9 | [68] |
| mα1β1γδ | Fluoxetine | 86Rb+ efflux | 2.1 | [73] |
| Voltage-clamp (0 mV) | 2.2 | [26] | ||
| Voltage-clamp (+100–200 µM Zn2+) | 0.45± 0.04 d 0.19 ± 0.02 d | [72] | ||
| Voltage-clamp | 0.22 b | [71] | ||
| Voltage-clamp | 0.3 e | [27] | ||
| hα1β1γδ | Fluoxetine | Ca2+ influx: 5 min pre-incubation 240 min pre-incubation 1440 min pre-incubation | 1.8 ± 0.6 0.22 ± 0.01 0.17 ± 0.08 | [77] |
| hα1β1εδ | Fluoxetine | Ca2+ influx | 0.85 b 0.55 b | [30] |
| mα1β1γδ | Nor-fluoxetine | Voltage-clamp | 0.07 b | [71] |
| hα1β1γδ | Paroxetine | Ca2+ influx: 5 min pre-incubation | 4.8 ± 0.6 | [77] |
| mα1β1γδ | Paroxetine | 86Rb+ efflux | 5.6 | [73] |
| Sertraline | 86Rb+ efflux | 3.5 | [73] | |
| Habenular mα3β4 * | Citalopram | Patch-clamp | 7.6 ± 1.0 | [68] |
| Hippocampal nAChRs | Citalopram | Nicotine-evoked NA release | 0.93 | [78] |
| Fluoxetine | Nicotine-evoked NA release | 0.57 | [78] | |
| nAChRs from superior cervical ganglion | Fluoxetine | Ca2+ influx | 2.0 | [69] |
| Radioligand | nAChR Subtype | SSRI | Ki (μM) a | Reference | |
|---|---|---|---|---|---|
| Resting State | Desensitized State | ||||
| [3H]Imipramine | hα4β2 | Fluoxetine | 3.2 ± 0.4 | 1.0 ± 0.1 | [70] |
| Paroxetine | 16.1 ± 1.3 | 6.7 ± 0.9 | [70] | ||
| Citalopram | ND | 4.1 ± 0.3 | [68] | ||
| hα3β4 | Citalopram | ND | 1.8 ± 0.1 | [68] | |
| Fluoxetine | 14.9 ± 1.4 | 4.8 ± 0.5 | [70] | ||
| Paroxetine | 24.0 ± 1.8 | 6.9 ± 0.6 | [70] | ||
| hα7 | Fluoxetine | 16.9 ± 1.4 | 11.0 ± 1.0 | [70] | |
| Paroxetine | 8.9 ± 0.8 | 8.7 ± 0.6 | [70] | ||
| [3H]TCP | Torpedo α1β1γδ | Fluoxetine | 1.9 ± 0.2 | 0.96 ± 0.04 | [77] |
| Paroxetine | 34 ± 2 | 2.5 ± 0.1 | [77] | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Arias, H.R.; Targowska-Duda, K.M.; García-Colunga, J.; Ortells, M.O. Is the Antidepressant Activity of Selective Serotonin Reuptake Inhibitors Mediated by Nicotinic Acetylcholine Receptors? Molecules 2021, 26, 2149. https://doi.org/10.3390/molecules26082149
Arias HR, Targowska-Duda KM, García-Colunga J, Ortells MO. Is the Antidepressant Activity of Selective Serotonin Reuptake Inhibitors Mediated by Nicotinic Acetylcholine Receptors? Molecules. 2021; 26(8):2149. https://doi.org/10.3390/molecules26082149
Chicago/Turabian StyleArias, Hugo R., Katarzyna M. Targowska-Duda, Jesús García-Colunga, and Marcelo O. Ortells. 2021. "Is the Antidepressant Activity of Selective Serotonin Reuptake Inhibitors Mediated by Nicotinic Acetylcholine Receptors?" Molecules 26, no. 8: 2149. https://doi.org/10.3390/molecules26082149
APA StyleArias, H. R., Targowska-Duda, K. M., García-Colunga, J., & Ortells, M. O. (2021). Is the Antidepressant Activity of Selective Serotonin Reuptake Inhibitors Mediated by Nicotinic Acetylcholine Receptors? Molecules, 26(8), 2149. https://doi.org/10.3390/molecules26082149