
Structure, Z′ = 2 Crystal Packing Features of 3-(2-Chlorobenzylidene)-5-(p-tolyl)furan-2(3H)-one

 ,
,  ,
,
Abstract
1. Introduction
2. Results and Discussion
2.1. Solving Structure Attempts
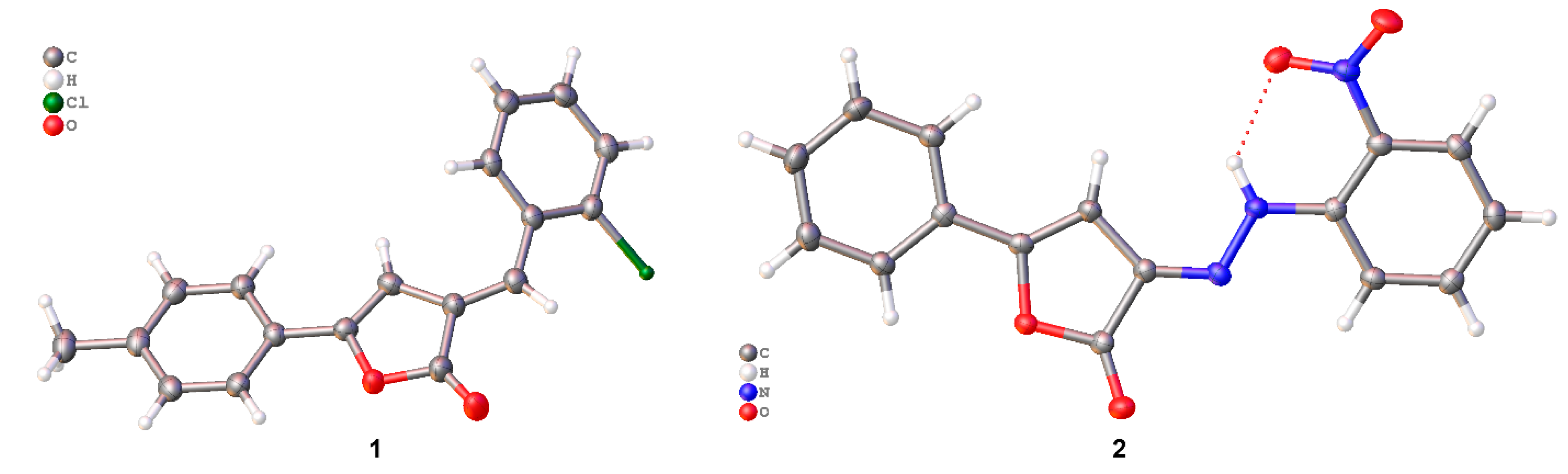
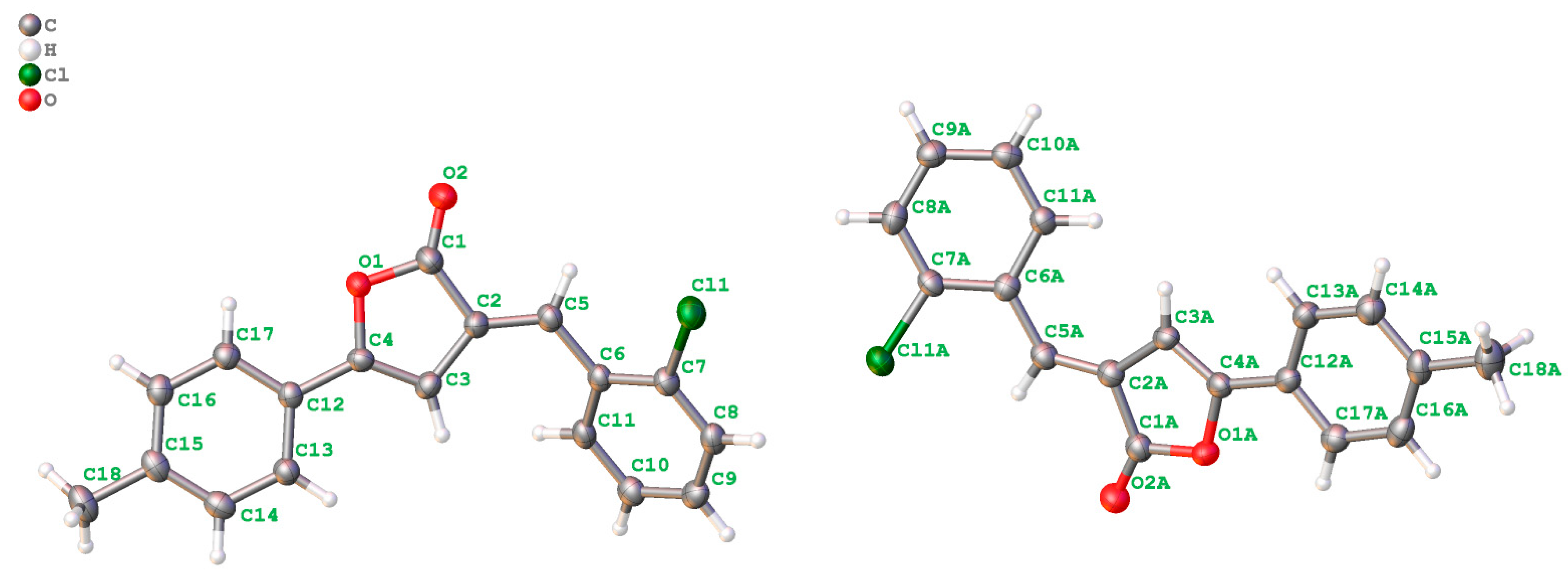
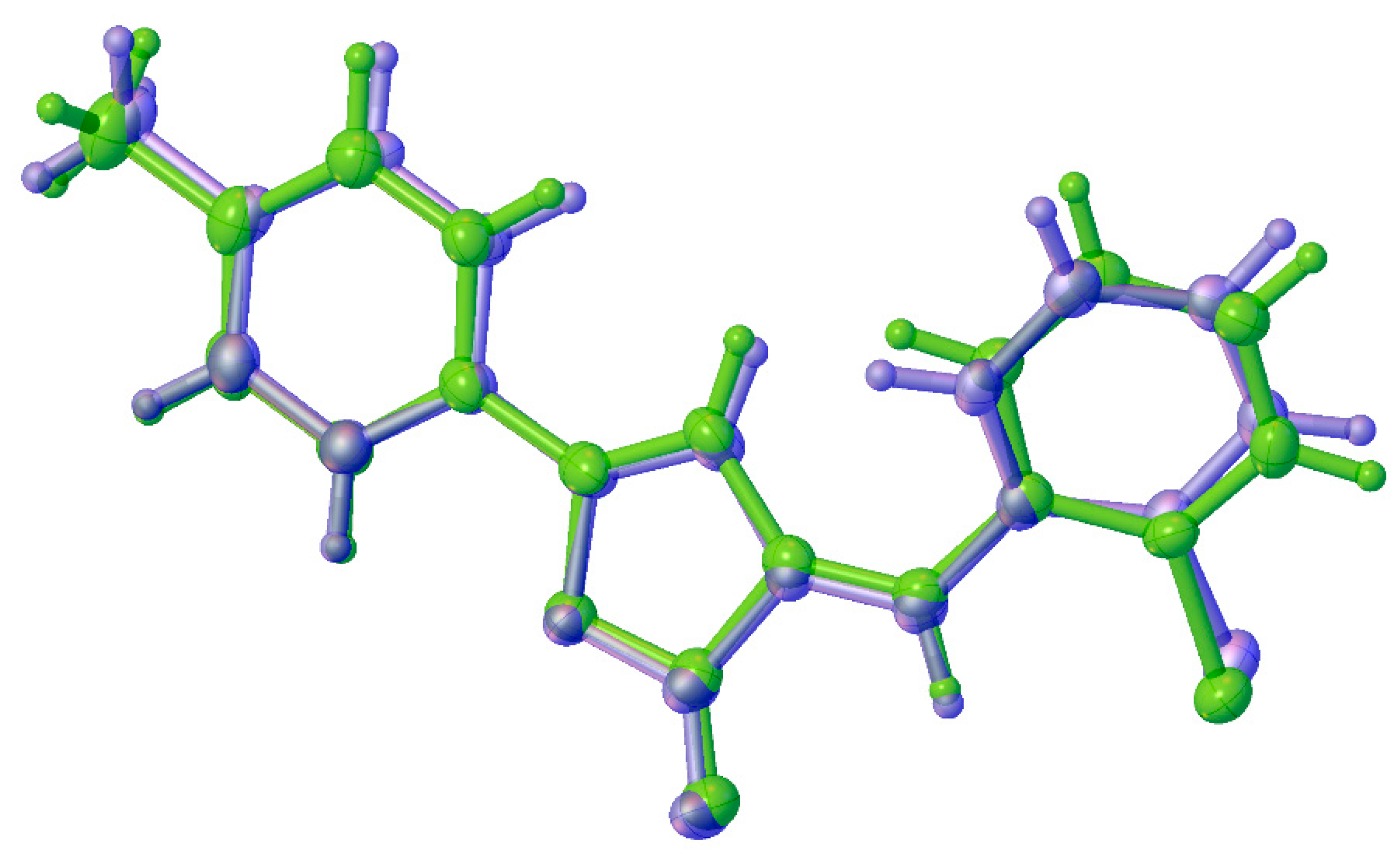
2.2. Crystal Structure Analysis
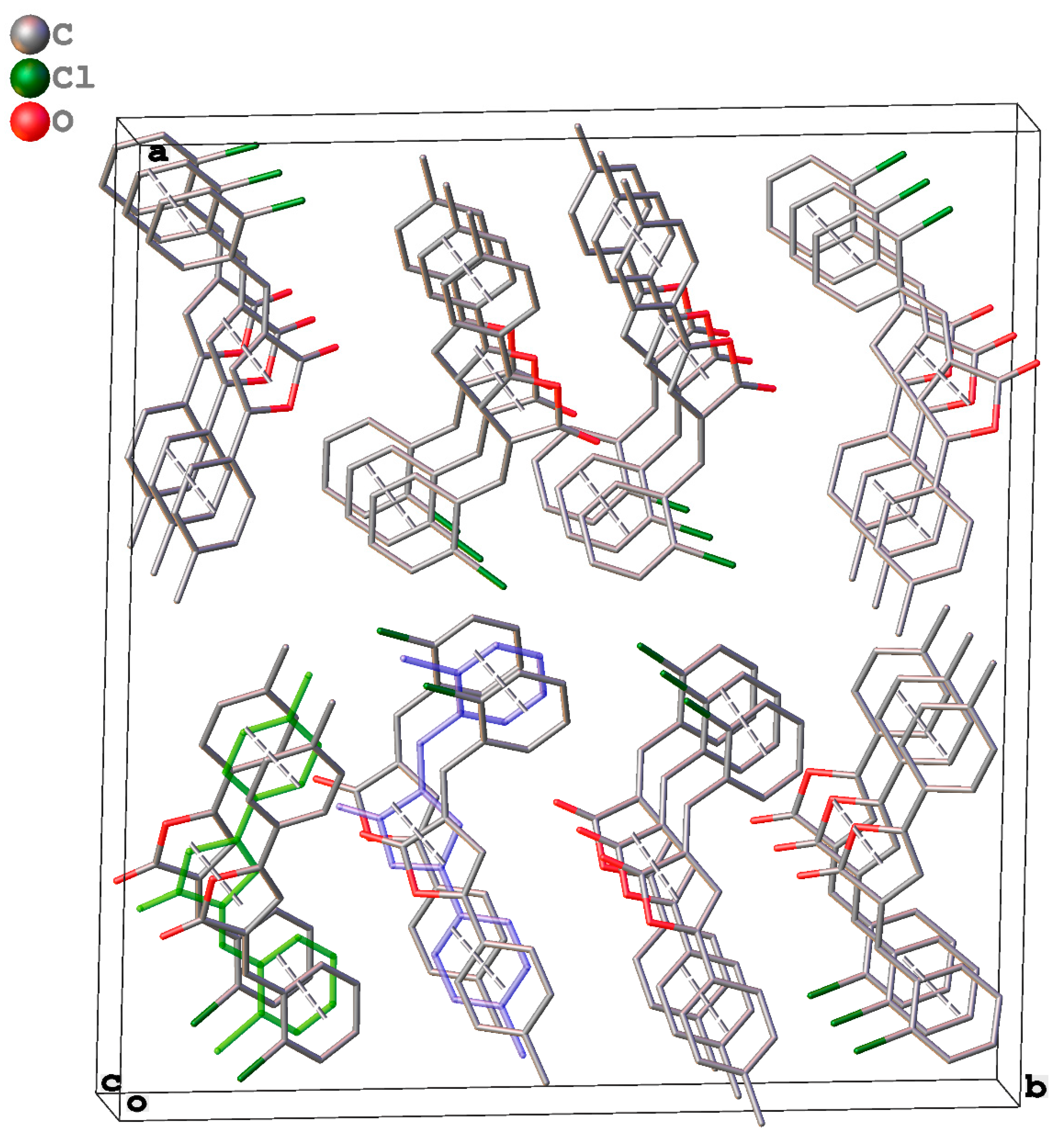
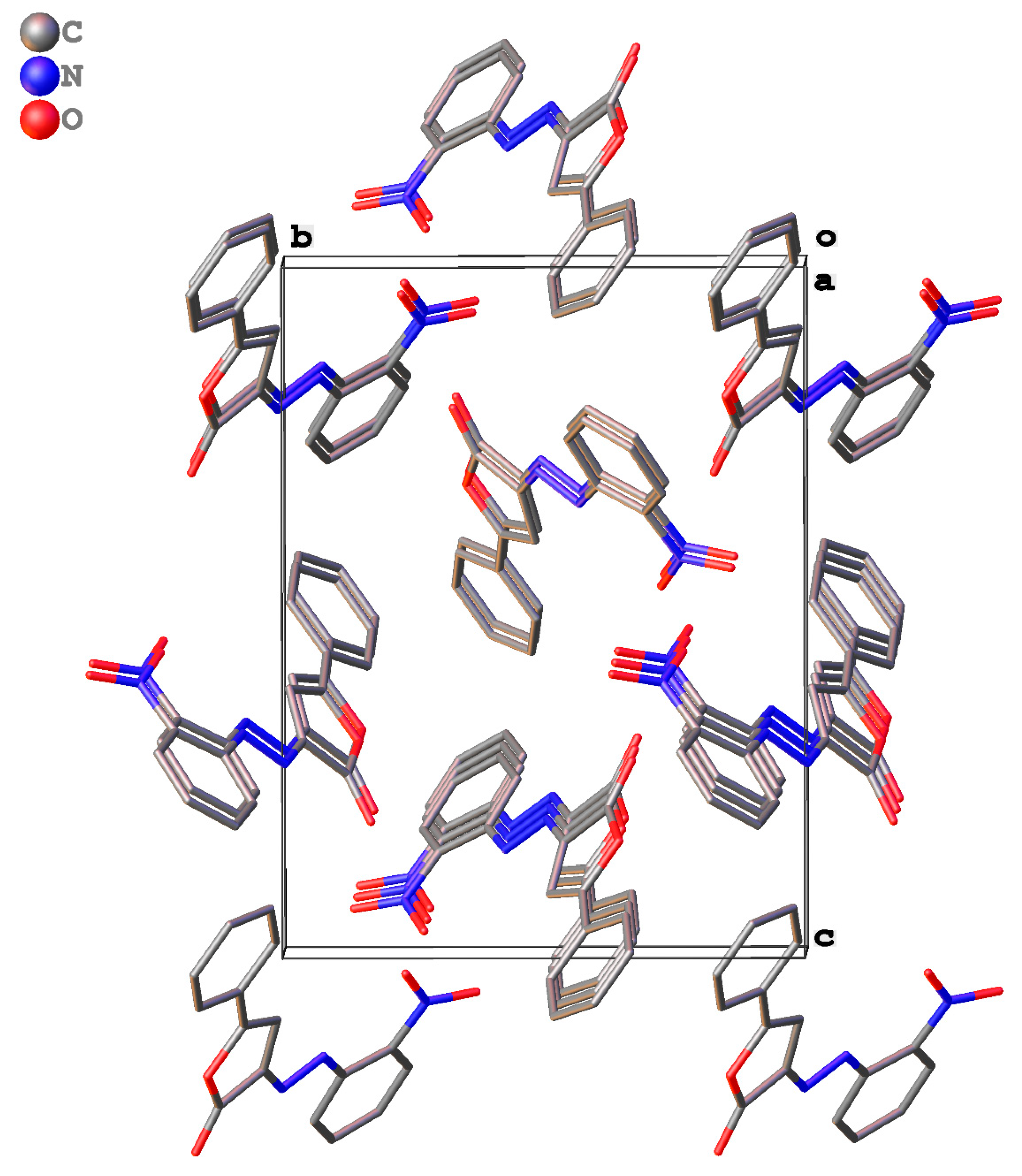
2.3. Packing and π–π Stacking Interactions
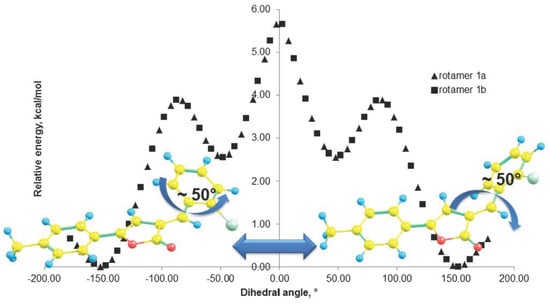
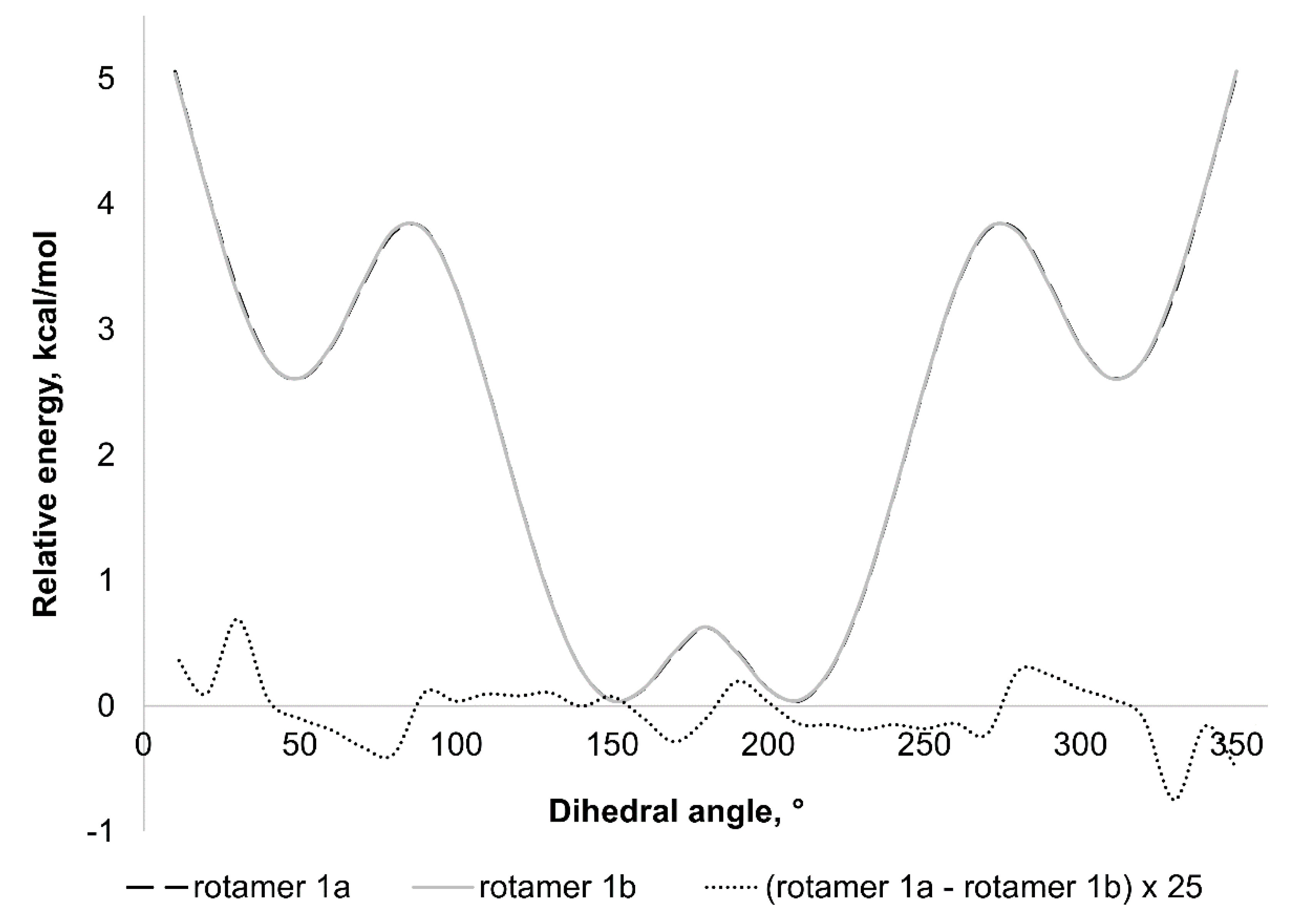
2.4. Theoretical Investigation of the Rotational Barrier
3. Material and Methods
3.1. Physical Measurements
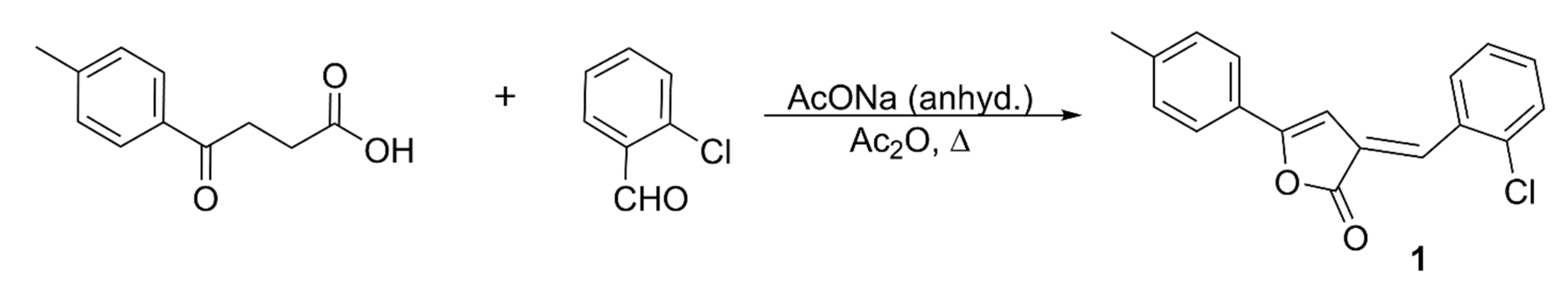
3.2. Synthesis, Characterization, and Crystallization
3.3. Crystal Structure Determinations and Refinement
3.4. DFT Calculations
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Behr, L.C.; Fusco, R.; Jarboe, C.H. Pyrazoles and Reduced and Condensed Pyrazoles. Chemistry of Heterocyclic Compounds; Wiley: New York, NY, USA, 1967; Volume 22, p. 888. [Google Scholar]
- Karci, F. Synthesis of disazo dyes derived from heterocyclic components. Color. Technol. 2005, 121, 275–280. [Google Scholar] [CrossRef]
- Ledenyova, I.V.; Didenko, V.V.; Shikhaliev, K.S. The chemistry of pyrazole-3(5)-diazonium salts. Chem. Heterocycl. Compd. 2014, 50, 1318–1349. [Google Scholar] [CrossRef]
- Maiorova, O.A.; Babkina, N.V.; Egorova, A.Y. Studies of stereochemistry of 3-(arylhydrazono)furan-2(3H)-ones, synthesis of 4-(arylhydrazono)pyridazin-3(1H)-ones. Chem. Heterocycl. Compd. 2015, 51, 514–517. [Google Scholar] [CrossRef]
- Maiorova, O.A.; Grinev, V.S.; Yegorova, A.Y. Crystal structure of 3-(2-(2-nitrophenyl)hydrazono)-5-phenyl-3H-furan-2-one. J. Struct. Chem. 2015, 56, 803–805. [Google Scholar] [CrossRef]
- Maksimov, E.A.; Mayorova, O.A.; Yegorova, A.Y. Acid- and base-catalyzed modifications of 3-[aryl(hetaryl)hydrazinylidene]-3H-furan-2-ones. Russ. J. Org. Chem. 2015, 51, 1305–1307. [Google Scholar] [CrossRef]
- Mayorova, O.A.; Yegorova, A.Y. 13C and1H NMR study of azo coupling products from diazonium salts and furan-2-(3H)-ones. Magn. Reson. Chem. 2015, 53, 853–856. [Google Scholar] [CrossRef]
- Mokhonova, I.D.; Maksimov, E.A.; Ledenyona, I.V.; Yegorova, A.Y.; Shikhaliev, K.S. Reactions of 3H-furan-2-ones and 2H-chromen-2-ones with pyrazole-3(5)-diazonium salts. Heterocycl. Commun. 2018, 24, 183–185. Available online: https://www.degruyter.com/view/journals/hc/24/4/article-p183.xml (accessed on 19 March 2021). [CrossRef]
- Butler, R.N. Diazotization of heterocyclic primary amines. Chem. Rev. 1975, 75, 241–257. [Google Scholar] [CrossRef]
- Elnagdi, M.H.; Abdel-Galil, F.M.; Riad, B.Y.; Elgemeie, G.E.H. Recent Developments in Chemistry of 3(5)-Aminopyrazoles. Heterocycles 1983, 20, 2437–2470. Available online: https://www.heterocycles.jp/newlibrary/downloads/PDF/10478/20/12 (accessed on 19 March 2021). [CrossRef]
- Makino, K.; Kim, H.S.; Kurasawa, Y. Synthesis of pyrazoles and condensed pyrazoles. J. Heterocycl. Chem. 1999, 36, 321–332. [Google Scholar] [CrossRef]
- Ledenyova, I.V.; Gracheva, A.A.; Shikhaliev, K.S. ChemInform Abstract: Reactions of Pyrazole-3(5)-diazonium Salts with 4-Hydroxy-2H-chromen-2-one and Isochroman-1,3-dione. Chem. Heterocycl. Compd. 2015, 51, 734–737. [Google Scholar] [CrossRef]
- Deeb, A.; Kotb, M. Pyridazine Derivatives and Related Compounds Part 10. Reactions of 3-Diazopyrazolo[3,4-c]pyridazine with Reactive Methylene Compounds and Other Groups. Heterocycles 2004, 63, 1143. Available online: https://www.heterocycles.jp/newlibrary/downloads/PDF/01198/63/5 (accessed on 19 March 2021). [CrossRef]
- Elmaati, T.M.A.; El-Taweel, F.M. New trends in the chemistry of 5-aminopyrazoles. J. Heterocycl. Chem. 2004, 41, 109–134. [Google Scholar] [CrossRef]
- Bakbardina, O.V.; Nurmagambetova, R.T.; Gazalieva, M.A.; Fazylov, S.D.; Temreshev, I.I. Synthesis and fungicidal activity of pseudo-thiohydantoins, their 5-arylidene derivatives, and 5-arylidene-3-β-aminothiazolid-2,4-one hydrochlorides. Pharm. Chem. J. 2006, 40, 537–539. [Google Scholar] [CrossRef]
- Zhang, S.; Cheng, K.; Wang, X.; Yin, H. Selection, synthesis, and anti-inflammatory evaluation of the arylidene malonate derivatives as TLR4 signaling inhibitors. Bioorg. Med. Chem. 2012, 20, 6073–6079. [Google Scholar] [CrossRef] [PubMed]
- Zoto, C.A.; Ucak-Astarlioglu, M.G.; Connors, R.E. Photochemistry of oxygenated and deoxygenated solutions of the photosensitizer (2E,5E)-2,5-bis(4-dimethylaminobenzylidene)-cyclopentanone, a ketocyanine dye. J. Mol. Struct. 2016, 1105, 396–402. [Google Scholar] [CrossRef]
- Anis’Kova, T.V.; Yegorova, A.Y.; Chadina, V.V. Reactions of 5-aryl-3-arylmethylene substituted 3H-furan-2-ones and 3H-pyrrol-2-ones with acetoacetic ester. Mendeleev Commun. 2008, 18, 167–168. [Google Scholar] [CrossRef]
- Egorova, A.Y.; Chadina, V.V. Arylmethylene derivatives of 5-R-3H-furan-2-ones and N-aryl-5-R-3H-pyrrol-2-ones in reaction with acetylacetone. Chem. Heterocycl. Compd. 2007, 43, 1233–1238. [Google Scholar] [CrossRef]
- Timofeeva, Z.Y.; Yegorova, A.Y. Michael condensation of 3-arylidene-3H-pyrrol-2-ones and 3-arylidene-3H-furan-2-ones with cyclohexanone. Chem. Heterocycl. Compd. 2007, 43, 690–694. [Google Scholar] [CrossRef]
- Egorova, A.Y. Synthesis of arylidene derivatives of N-unsubstituted pyrrolin-2-ones. Russ. Chem. Bull. 2002, 51, 183–184. [Google Scholar] [CrossRef]
- Anis’Kova, T.V.; Chadina, V.V.; Yegorova, A.Y. Reaction of 3-Arylmethylidene-3H-furan-2-ones with 3-Amino-1,2,4-triazole as a Convenient Technique to Synthesize Condensed Diazepinones. Synth. Commun. 2011, 41, 2315–2322. [Google Scholar] [CrossRef]
- Velezheva, V.S.; Dvorkin, V.V.; Suvorov, N.N. Synthesis of 2-amino-3-aryl-4-quinolones and 2-(aminoarylmethylidene)-3-indolinones from 2-arylidene-3-indolinones. Chem. Heterocycl. Compd. 1985, 21, 234. [Google Scholar] [CrossRef]
- Krysin, M.Y.; Anokhina, I.K.; Zyk, N.V.; Zalukaev, L.P. Diphthalolylpyrkolidines from 2-arylidene-1,3-indandiones and phenyl azide. Chem. Heterocycl. Compd. 1989, 25, 226. [Google Scholar] [CrossRef]
- Adam, W.; Nemes, C.; Patonay, T.; Halasz, J.; Jambor, Z.; Levai, A.; Toth, G. Stereoselective epoxidation of 2-arylidene-1-indanones and 2-arylidene-1-benzosuberones. Monatshefte Chem. 1996, 127, 683–690. [Google Scholar] [CrossRef]
- Bortnikov, S.V.; Efremova, I.E.; Berestovitskaya, V.M. Halogenation of 2-Arylidene-3-methyl-4-nitro-3-thiolene-1,1-dioxides. Chem. Heterocycl. Compd. 2001, 37, 775–776. [Google Scholar] [CrossRef]
- Cheney, B.V. Structure-activity relationships for drugs binding to the agonist and antagonist states of the primary morphine receptor. J. Med. Chem. 1988, 31, 521–531. [Google Scholar] [CrossRef]
- Bradbury, S.P.; Ankley, G.T.; Mekenyan, O.G. Quantitative structure-activity relationships for polychlorinated hydroxybiphenyl estrogen receptor binding affinity: An Assessment of conformer flexibility. Environ. Toxicol. Chem. 1996, 15, 1945–1954. [Google Scholar] [CrossRef]
- Dahl, S.G. Molecular Models and Structure—Activity Relationships. Antipsychotics 1996, 120, 29–41. [Google Scholar] [CrossRef]
- Steed, K.M.; Steed, J.W. Packing Problems: High Z′ Crystal Structures and Their Relationship to Cocrystals, Inclusion Compounds, and Polymorphism. Chem. Rev. 2015, 115, 2895–2933. [Google Scholar] [CrossRef] [PubMed]
- Ruck, M. Zeitschrift für Kristallographie—Crystalline. Materials 2000, 215, 148–156. [Google Scholar] [CrossRef]
- Nelyubina, Y.V.; Barzilovich, P.Y.; Antipin, M.Y.; Aldoshin, S.M.; Lyssenko, K.A. Cation-π and Lone Pair-π Interactions Combined in One: The First Experimental Evidence of (H3O-lp)+⋅⋅⋅π-System Binding in a Crystal. ChemPhysChem 2011, 12, 2895–2898. [Google Scholar] [CrossRef] [PubMed]
- Nelyubina, Y.V.; Antipin, M.Y.; Cherepanov, I.A.; Lyssenko, K.A. Pseudosymmetry as viewed using charge density analysis. CrystEngComm 2010, 12, 77–81. [Google Scholar] [CrossRef]
- Nelyubina, Y.; Dalinger, I.L.; Lyssenko, K.A. Pseudosymmetry in Trinitropyrazole: The Cost of Error in Space-Group Determination. Angew. Chem. Int. Ed. 2011, 50, 2892–2894. [Google Scholar] [CrossRef]
- Rowland, R.S.; Taylor, R. Intermolecular Nonbonded Contact Distances in Organic Crystal Structures: Comparison with Distances Expected from van der Waals Radii. J. Phys. Chem. 1996, 100, 7384–7391. [Google Scholar] [CrossRef]
- Wilson, C.C. Single Crystal Neutron Diffraction from Molecular Materials: Series on Neutron Techniques and Applications; World Scientific Publishing Co. Pte Ltd.: Singapore, 2000; Volume 2. [Google Scholar]
- Brancatelli, G.; Bruno, G.; Nicolò, F.; Cordaro, M.; Grassi, G.; Risitano, F.; Scala, A. X-ray crystallography and computational studies of a variety of pyrrole derivatives obtained from mesoionic oxazoles and selected chromenones. J. Mol. Struct. 2011, 990, 132–139. [Google Scholar] [CrossRef]
- Liu, J.; Brooks, N.R. Stereoselective Stobbe Condensation of Ethyl Methyl Diphenylmethylenesuccinate with Aromatic Aldehydes. Org. Lett. 2002, 4, 3521–3524. [Google Scholar] [CrossRef]
- Belguedj, R.; Bouacida, S.; Merazig, H.; Belfaitah, A.; Chibani, A.; Bouraiou, A. Synthesis and crystal structures of three novel benzimidazole/benzoindolizine hybrids. Z. Naturforschung B 2016, 71, 231–239. [Google Scholar] [CrossRef]
- Geiger, D.; Geiger, H.C.; Deck, J.M. Structure determination of three furan-substituted benzimidazoles and calculation of π–π and C—H...π interaction energies. Acta Crystallogr. C 2014, 70, 1125–1132. [Google Scholar] [CrossRef] [PubMed]
- Avasthi, K.; Farooq, S.M.; Raghunandan, R.; Maulik, P.R. Studies on arene interactions in flexible pyrazolo[3,4-d]pyrimidine core based symmetrical ‘propylene/Leonard linker’ models: X-ray crystallographic evidence for disappearance of intramolecular stacking due to presence of chloro- or cyano-groups in place of methylsulfanyl or alkoxy substituents. J. Mol. Struct. 2009, 927, 27–36. [Google Scholar] [CrossRef]
- Ivanov, D.M.; Kirina, Y.V.; Novikov, A.S.; Starova, G.L.; Kukushkin, V.Y. Efficient π-stacking with benzene provides 2D assembly of trans-[PtCl2(p-CF3C6H4CN)2]. J. Mol. Struct. 2016, 1104, 19–23. [Google Scholar] [CrossRef]
- Sedavkina, V.A.; Morozova, N.A.; Egorova, A.Y.; Ostroumov, I.G. Synthesis of 5-alkyl-3H-thiolen-2-ones and 5-alkyl-3H-furan-2-ones and condensation reactions at the heterocyclic methylene group. Chem. Heterocycl. Compd. 1987, 23, 377–380. [Google Scholar] [CrossRef]
- Bruker. SADABS; Version 2008/1; Bruker AXS Inc.: Madison, WI, USA, 2008. [Google Scholar]
- Bruker. APEX II; Version 20-1; Bruker AXS Inc.: Madison, WI, USA, 2007. [Google Scholar]
- Bruker. SAINT; Version 723A; Bruker AXS Inc.: Madison, WI, USA, 2001. [Google Scholar]
- Bruker. XPREP; Version 2005/2; Bruker AXS Inc.: Madison, WI, USA, 2005. [Google Scholar]
- Sheldrick, G.M. SHELXT– Integrated space-group and crystal-structure determination. Acta Cryst. 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Macrae, C.F.; Bruno, I.J.; Chisholm, J.A.; Edgington, P.R.; McCabe, P.; Pidcock, E.; Rodriguez-Monge, L.; Taylor, R.J.; Van De Streek, J.; Wood, P.A. Mercury CSD 2.0– new features for the visualization and investigation of crystal structures. J. Appl. Crystallogr. 2008, 41, 466–470. [Google Scholar] [CrossRef]
- Becke, A.D. Perspective on “Density functional thermochemistry. III. The role of exact exchange”. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 1988, 38, 3098–3100. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ring | Interplanar Distance, Å | Intercentroid Distance, Å | Ring Offset, Å | Angle θ, ° |
|---|---|---|---|---|
| 1a | ||||
| o-Cl-Ar | 3.470(3) | 3.8037(2) | 1.559(7) | 24.19(5) |
| Fur-2(3H)-one | 3.478(4) | 3.8037(2) | 1.540(8) | 23.89(3) |
| p-Tol | 3.380(3) | 3.8037(2) | 1.745(7) | 27.31(4) |
| 1b | ||||
| o-Cl-Ar | 3.519(3) | 3.8037(2) | 1.444(7) | 22.33(8) |
| Fur-2(3H)-one | 3.453(4) | 3.8037(2) | 1.596(8) | 24.81(8) |
| p-Tol | 3.464(3) | 3.8037(2) | 1.572(7) | 24.41(9) |
| Extremum | Contact | C(5)H–Cl(1) | C(3)H–C(11)H | C(5)H–C(11)H | C(3)H–Cl(1) | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| Angle | 1a | 1b | 1a | 1b | 1a | 1b | 1a | 1b | ||
| Global min | −152.303 | −147.749 | 2.690 | 2.722 | 2.242 | 2.307 | 3.711 | 3.692 | 5.657 | 5.583 |
| Local max | −82.303 | −87.749 | 3.647 | 3.535 | 4.095 | 3.899 | 3.014 | 3.121 | 3.776 | 3.995 |
| Local min | −52.303 | −47.749 | 4.065 | 4.105 | 4.852 | 4.932 | 2.536 | 2.483 | 2.943 | 2.881 |
| Global max * | −2.303 | 2.2511 | 4.409 | 4.409 | 5.551 | 5.551 | 2.070 | 2.070 | 2.502 | 2.502 |
| Local min | 47.697 | 52.251 | 4.105 | 4.065 | 4.932 | 4.853 | 2.483 | 2.535 | 2.880 | 2.942 |
| Local max | 87.697 | 82.2509 | 3.536 | 3.648 | 3.900 | 4.097 | 3.120 | 3.013 | 3.994 | 3.774 |
| Global min | 147.697 | 152.251 | 2.723 | 2.690 | 2.308 | 2.243 | 3.692 | 3.711 | 5.581 | 5.655 |
| Local max ** | 177.697 | −177.749 | 2.543 | 2.543 | 1.968 | 1.968 | 3.807 | 3.807 | 6.009 | 6.010 |
| Geometrical Parameter | Experimental | Calculated | ||
|---|---|---|---|---|
| 1a | 1b | 1a | 1b | |
| C(2)–C(5) | 1.354(4) | 1.351(5) | 1.356 | 1.356 |
| C(2)–C(3) | 1.437(5) | 1.447(5) | 1.442 | 1.442 |
| C(5)–C(6) | 1.461(5) | 1.452(4) | 1.457 | 1.457 |
| O(2)···C(5)H | 2.714(4) | 2.628(4) | 2.629 | 2.629 |
| C(5)H···Cl(1) | 2.785(3) | 2.651(3) | 2.690 | 2.690 |
| C(3)H···C(11)H | 2.303(5) | 2.151(5) | 2.242 | 2.243 |
| C(2)–C(5)–C(6) | 124.4(3) | 128.4(3) | 127.858 | 127.843 |
| C(2)–C(5)–C(6)–C(7) | −144.5(3) | 157.6(4) | −152.303 | 152.251 |
| Crystal data | |
| Chemical formula | C18H13ClO2 |
| Mr | 296.73 |
| Crystal system, space group | Orthorhombic, Pna21 |
| Temperature (K) | 100 |
| a, b, c (Å) | 28.2038(14), 25.8791(14), 3.8037(2) |
| V (Å3) | 2776.3(3) |
| Z | 8 |
| Z′ | 2 |
| Radiation type | CuKα |
| µ (mm−1) | 2.44 |
| Crystal size (mm) | 0.30 × 0.05 × 0.05 |
| Data collection | |
| Diffractometer | Bruker APEX II CCD (charge-coupled device) area detector’ diffractometer |
| Absorption correction | Multi-scan SADABS |
| No. of measured, independent, and observed [I > 2σ(I)] reflections | 26,357, 4925, 4084 |
| Rint | 0.083 |
| (sin θ/λ)max (Å−1) | 0.618 |
| Refinement | |
| R[F2 > 2σ(F2)], wR(F2), S | 0.051, 0.119, 1.00 |
| No. of reflections | 4925 |
| No. of parameters | 382 |
| No. of restraints | 1 |
| H-atom treatment | H-atom parameters constrained |
| Δρmax, Δρmin (e Å−3) | 0.24, −0.42 |
| Absolute structure | Refined as an inversion twin |
| Absolute structure parameter | 0.36(2) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Grinev, V.S.; Mayorova, O.A.; Anis’kova, T.V.; Tikhomolova, A.S.; Yegorova, A.Y. Structure, Z′ = 2 Crystal Packing Features of 3-(2-Chlorobenzylidene)-5-(p-tolyl)furan-2(3H)-one. Molecules 2021, 26, 2137. https://doi.org/10.3390/molecules26082137
Grinev VS, Mayorova OA, Anis’kova TV, Tikhomolova AS, Yegorova AY. Structure, Z′ = 2 Crystal Packing Features of 3-(2-Chlorobenzylidene)-5-(p-tolyl)furan-2(3H)-one. Molecules. 2021; 26(8):2137. https://doi.org/10.3390/molecules26082137
Chicago/Turabian StyleGrinev, Vyacheslav S., Oksana A. Mayorova, Tatyana V. Anis’kova, Alexandra S. Tikhomolova, and Alevtina Yu. Yegorova. 2021. "Structure, Z′ = 2 Crystal Packing Features of 3-(2-Chlorobenzylidene)-5-(p-tolyl)furan-2(3H)-one" Molecules 26, no. 8: 2137. https://doi.org/10.3390/molecules26082137
APA StyleGrinev, V. S., Mayorova, O. A., Anis’kova, T. V., Tikhomolova, A. S., & Yegorova, A. Y. (2021). Structure, Z′ = 2 Crystal Packing Features of 3-(2-Chlorobenzylidene)-5-(p-tolyl)furan-2(3H)-one. Molecules, 26(8), 2137. https://doi.org/10.3390/molecules26082137