3.1. Experimental
3.1.1. Chemistry
All chemicals and reagents were obtained from Acros Organic (Geel, Belgium), QRec (Penang, Malaysia, Asia), Sigma-Aldrich Chemical Co. or Merck (Darmstadt, Germany). Thin-layer chromatography (TLC) was performed on alumina plates pre-coated with silica gel (Merck 60 F254). The progress of reactions was determined by the appearance of product and disappearance of reactant spots under UV radiation (λmax = 254 nm, Muttenz, Switzerland), respectively. Melting points were determined on open capillary tubes and are uncorrected. All spectral data were obtained on the following instruments: infrared (IR) spectra were recorded on a Perkin-Elmer System FTIR-ATR spectrometer; 1D- and 2D-NMR spectra were recorded on a 500 FT-NMR Bruker Advance spectrometer (Bruker Bioscience, Billerica, MA, USA) in CD3COCD3, DMSO-d6, CDCl3 and tetramethylsilane (TMS) as internal standards. Chemical shifts are reported in part per million (δ-scale) and the coupling constants, J, are reported in Hertz (Hz). High resolution HRMS mass spectra were obtained from a Waters Xevo QTOF MS system.
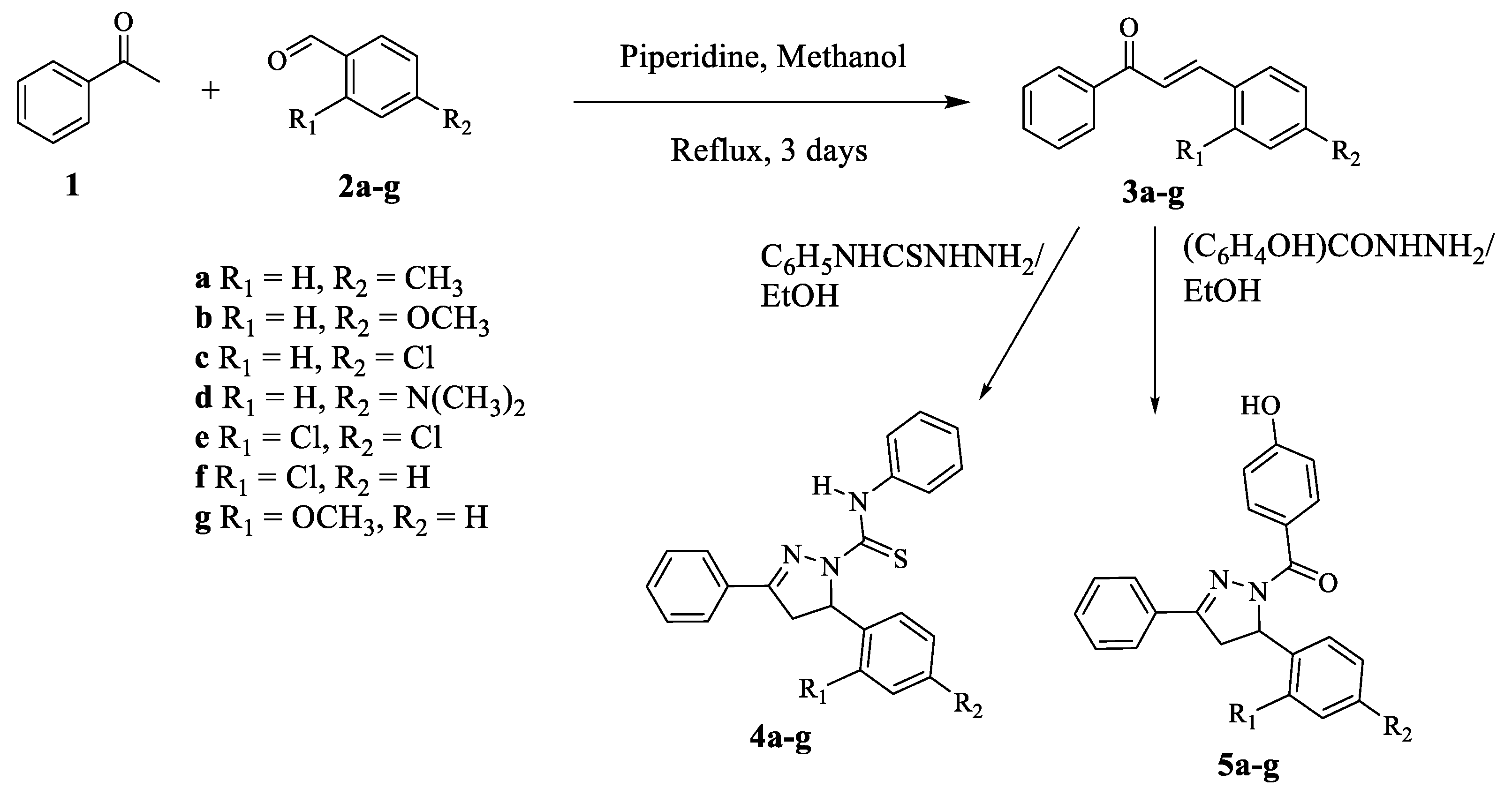
3.1.2. General Procedure for the Preparation of Chalcone Derivatives (3a–g) by the Claisen–Schmidt Condensation Reaction
Chalcone derivatives (
3a–
g) were synthesized following the method described in the literature. A mixture of acetophenone (
1) (0.01 mol) and substituted benzaldehyde (
2a–
g) (0.015 mol) in methanol (10 mL) was refluxed in the presence of few drops of piperidine for 72 h [
26,
27]. The solution was kept in an ice bath until a solid was obtained, and the chalcone compounds were filtered, washed with cold water, dried and recrystallized from ethanol. The results were compared with the literature [
28,
29,
30,
31].
(E)-3-(4-methylphenyl)-1-phenylprop-2-en-1-one (3a). Pale yellow crystalline solid; yield: 83%; m.p.: 94–96 °C; FTIR (ATR) νmax (cm−1): 3025 (Csp2–H), 2918 (Csp3–H), 1656 (C=O), 1595 (–CH=CH–), 1514 and 1449 (aromatic C=C); 1H-NMR (500MHz, CD3COCD3): δH 2.39 (3H, s, 4-CH3), 7.30 (2H, d, J = 8.0 Hz, H-3′ and H-5′), 7.58 (2H, t, J = 7.5 Hz, H-2″ and H-6″), 7.66 (1H, t, J = 7.5 Hz, H-4″), 7.75 (2H, t, J = 8.0 Hz, H-2′ and H-6′), 7.79 (1H, d, J = 15.5 Hz, H-2), 7.85 (1H, d, J = 15.5 Hz, H-3) and 8.16 (2H, d, J = 7.5 Hz, H-3″ and H-5′’); 13C-NMR (125 MHz; CD3COCD3): δC 20.6 (4-CH3), 121.0 (C-2), 128.4 (C-3″ and C-5″), 128.7 (C-2″ and C-6″), 128.7 (C-2′ and C-6′), 129.6 (C-3′ and C-5′), 132.4 (C-1′), 132.7 (C-4″), 138.4 (C-1″), 140.9 (C-4′), 144.1 (C-3) and 189.1 (1-C=O).
(E)-3-(4-methoxyphenyl)-1-phenylprop-2-en-1-one (3b). Yellow crystalline solid; yield: 74%; m.p.: 73–75 °C; FTIR (ATR) νmax cm−1: 3018 (Csp2–H), 2842 (Csp3–H), 1656 (C=O), 1575 (–CH=CH–), 1510, 1445 (aromatic C=C), 1253 (C–O); 1H-NMR (500MHz; CD3COCD3): δH 3.88 (3H, s, 4-OCH3), 7.03 (2H, d, J = 8.5 Hz, H-3′ and H-5′), 7.57 (2H, t, J = 7.5 Hz, H-2″ and H-6″), 7.65 (1H, t, J = 7.5 Hz, H-4″), 7.75 (1H, d, J = 15.5 Hz, H-2), 7.80 (1H, d, J = 15.5 Hz, H-3), 7.83 (2H, d, J = 7.5 Hz, H-2′ and H-6′), and 8.15 (2H, d, J = 7.5 Hz, H-3″ and H-5″); 13C-NMR (125 MHz; CD3COCD3); δC 54.9 (4-OCH3), 114.4 (C-3′ and C-5′), 119.6 (C-2), 127.7 (C-1′), 128.3 (C-3′’ and C-5″), 128.6 (C-2′’ and C-6″), 130.5 (C-2′ and C-6′), 132.6 (C-4″), 138.6 (C-1″), 143.9 (C-3), 161.9 (C-4′) and 189.0 (1-C=O).
(E)-3-(4-chlorophenyl)-1-phenylprop-2-en-1-one (3c). Pale yellow crystalline solid; yield: 82%; m.p.: 113–115 °C; FTIR (ATR) νmax cm−1: 3059 (Csp2–H), 2931 (Csp3–H), 1653 (C=O), 1591 (–C=C–), 1563, 1489 (aromatic C=C), 1221 (C–O), 823 (C–Cl); 1H-NMR (500 MHz, CD3COCD3): δH 7.52 (2H, d, J = 8.5 Hz, H-3′ and H-5′), 7.58 (2H, t, J = 8.0 Hz, H-2″ and H-6″), 7.68 (1H, t, J = 7.5 Hz, H-4″), 7.79 (1H, d, J = 16.0 Hz, H-2), 7.90 (2H, d, J = 8.5 Hz, H-2′ and H-6′), 7.93 (1H, d, J = 16.0 Hz, H-3), and 8.17 (2H, d, J = 7.5 Hz, H-3″ and H-5″); 13C-NMR (125 MHz, CD3COCD3); δC 122.8 (C-2), 128.5 (C-3″ and C-5″), 128.7 (C-2″ and C-6″), 129.1 (C-2′ and C-‘6), 130.2 (C-3′ and C-5′), 132.9 (C-4″), 134.0 (C-1′), 135.7 (C-4′), 138.1 (C-1″), 142.4 (C-3) and 188.9 (1-C =O).
(E)-3-(4-dimethylaminophenyl)-1-phenylprop-2-en-1-one (3d). Orange crystalline solid; yield: 57%; m.p.: 113–115 °C; FTIR (ATR) νmax cm−1: 3053.1 (Csp2–H), 2909.8 (Csp3–H), 1646 (C=O), 1611 (–C=C–), 1511, 1457 (C=C aromatic), 1322 (C–N); 1H-NMR (500 MHz; CD3COCD3): δH 3.07 (6H, s, 4-CH3 and 5-CH3), 6.80 (2H, d, J =9.0 Hz, H-3′ and H-5′), 7.55 (2H, t, J =7.5 Hz, H-2″ and H-6″), 7.61 (1H, d, J = 7.5 Hz, H-4″), 7.62 (1H, d, J = 15.5 Hz, H-2), 7.69 (2H, d, J = 9.0 Hz, H-2′ and H-6′), 7.77 (1H, d, J = 15.5 Hz, H-3) and 8.12 (2H, d, J = 7.5 Hz, H-3″ and H-5″); 13C-NMR (125 MHz; CD3COCD3); δC 39.3 (4-CH3 and 5-CH3), 111.8 (C-3′ and C-5′), 116.3 (C-2), 122.6 (C-1′), 128.1 (C-3″ and C-5″), 128.5 (C-2″ and C-6′’), 130.5 (C-2′ and C-6′), 132.2 (C-4″), 139.1 (C-1″), 145.1 (C-3), 152.3 (C-4′) and 188.7 (1-C=O).
(E)-3-(2,4-dichlorophenyl)-1-phenylprop-2-en-1-one (3e). Yellow solid; yield: 52%; m.p.: 69–73 °C; FTIR (ATR) νmax cm−1: 3064 (Csp2–H), 2931 (Csp3–H), 1652 (C=O), 1607 (–C=C–), 1514, 1475.1 (C=C aromatic); 1H-NMR (500 MHz, CD3COCD3): δH 7.49 (1H, dd, J = 2.0 Hz and 8.5 Hz, H-5′), 7.59 (2H, t, J = 8.0 Hz, H-2″ and H-6′’), 7.64 (1H, d, J = 2.0 Hz, H-3′), 7.69 (1H, d, J = 7.5 Hz, H-4″), 7.96 (1H, d, J = 16.0 Hz, H-2), 8.12 (1H, d, J = 15.5 Hz, H-3) and 8.18 (3H, t, J = 7.5 Hz, H-6′, H-3″ and H-5″); 13C-NMR (125 MHz, CD3COCD3); δC 125.2 (C-2), 127.9 (C-5′), 128.6 (C-3′’ and C-5″), 128.8 (C-2″ and C-6″), 129.4 (C-3′), 129.6 (C-6′), 132.0 (C-4′), 133.2 (C-4″), 135.6 (C-1′), 136.0 (C-2′), 137.6 (C-3), 137.8 (C-1′’) and 188.7 (1-C=O).
(E)-3-(2-chlorophenyl)-1-phenylprop-2-en-1-one (3f). Pale yellow crystalline solid; yield: 57%; m.p.: 45–48 °C; FTIR (ATR) νmax cm−1: 3060 (Csp2–H), 2931 (Csp3–H), 1662 (C=O), 1603 (–C=C–), 1514, 1463 (C=C aromatic), 748 (C–Cl); 1H-NMR (500 MHz, CD3COCD3): δH 7.47 (2H, m, H-4′ and H-5′), 7.56 (1H, m, H-3′), 7.60 (2H, d, J = 7.5 Hz, H-2″ and H-6″), 7.69 (1H, d, J = 7.5 Hz, H-4″), 7.92 (1H, d, J = 15.5 Hz, H-2), 8.14 (1H, d, J = 7.5 Hz, H-6′), 8.19 (2H, d, J = 8.5 Hz, H-3″ and H-5″) and 8.20 (1H, d, J = 15.5 Hz, H-3); 13C-NMR (125 MHz, CD3COCD3); δC 124.7 (C-2), 127.6 (C-4′), 128.2 (C-6′), 128.5 (C-3″ and C-5″), 128.7 (C-2″ and C-6″), 130.1 (C-5′), 131.6 (C-3′), 133.0 (C-1′), 133.1 (C-4″), 134.9 (C-2′), 137.9 (C-1″), 139.0 (C-3) and 188.9 (1-C=O).
(E)-3-(2-methoxyphenyl)-1-phenylprop-2-en-1-one (3g). Yellow crystalline solid; yield: 62%; m.p.: 51–53 °C; FTIR (ATR) νmax cm−1: 3019 (Csp2–H), 2948 (Csp3–H), 1659 (C=O), 1594 (–C=C–), 1513, 1439 (C=C aromatic), 1247 (C–O); 1H-NMR (500 MHz; CD3COCD3): δH 3.98 (3H, s, 4-OCH3), 7.05 (1H, t, J = 7.5 Hz, H-5′), 7.13 (1H, d, J = 8.5 Hz, H-3′), 7.64 (1H, m, H-4′), 7.58 (2H, t, J = 7.5 Hz, H-2″ and H-6″), 7.66 (1H, d, J = 7.5 Hz, H-4″), 7.88 (1H, d, J = 15.5 Hz, H-2), 7.90 (1H, t, J = 7.5 Hz, H-6′), 8.14 (2H, d, J = 8.5 Hz, H-3″ and H-5″) and 8.17 (1H, d, J = 15.5 Hz, H-3); 13C-NMR (125 MHz; CD3COCD3); δC 55.2 (4-OCH3), 111.5 (C-3′), 120.7 (C-5′), 122.1 (C-2), 123.6 (C-1′), 128.4 (C-3″ and C-5″), 128.6 (C-6′), 128.6 (C-2″ and C-6″), 132.0 (C-4′), 132.7 (C-4″), 138.5 (C-1″), 139.0 (C-3), 158.8 (C-2′) and 189.4 (1-C=O).
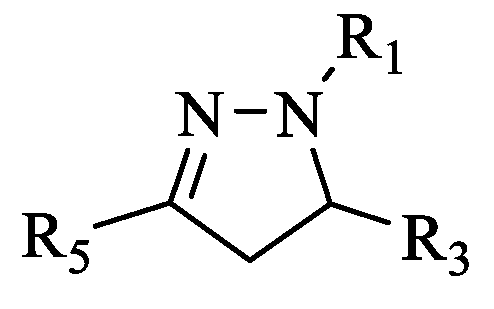
3.1.3. General Procedure for the Preparation of the First Series of 3,5-disubstituted-4,5-dihydro-N-phenyl-1H-pyrazole-1-carbothioamide (4a–g)
The carbothioamide compounds (
4a–g) were synthesized according to a previously reported method with slight modifications. The cyclo-condensation of chalcone derivatives (
3a–g) (1 mmol) with 4-phenyl-3-thiosemicarbazide (1.5 mmol) was carried out in ethanol (6 mL) in the presence of NaOH (3 mmol) for 3 to 8 h [
32]. The reaction progress was observed on a TLC plate. After the reaction was over, the reaction mixture was left at room temperature overnight. A solid was formed after crushed ice was added. The solid was filtered, washed with cold water, dried and recrystallized from ethanol. Please refer the
Supplementary Data for full spectra.
5-(4-methylphenyl)-N,3-diphenyl-4,5-dihydro-1H-pyrazole-1-carbothioamide (4a). Cream-colored solid; yield: 26%; m.p.: 133–136 °C; FTIR (ATR) νmax cm−1: 3291 (N–H), 3028 (Csp2–H), 2919 (Csp3–H), 1594 (C=N), 1520, 1449 (aromatic C=C), 1399 (C=S); 1H-NMR (500 MHz, CD3COCD3): δH 2.29 (3H, s, 14-CH3), 3.26 (1H, dd, J = 3.5 Hz and 18 Hz, 4α-CH), 4.05 (1H, dd, J = 11.5 Hz and 18 Hz, 4β-CH), 6.14 (1H, dd, J = 3.5 Hz and 11.5 Hz, 5-CH), 7.16 (5H, m, H-2′, H-3′, H-5′, H-6′, H-11), 7.33 (2H, t, J = 7.5 Hz, H-10 and H-12), 7.49 (3H, m, H-3″, H-4″ and H-5″), 7.72 (2H, m, H-9 and H-13), 7.98 (2H, dd, J = 2.0 Hz and 7.5 Hz, H-2″ and H-6″), 9.93 (1H, s, 7-NH); 13C NMR (125 MHz, CD3COCD3): δC 20.2 (14-CH3), 42.3 (4-CH2), 63.3 (5-CH), 124.3 (C-9 and C-13), 124.6 (C-11), 125.5 (C-2′ and C-6′), 127.2 (C-2″ and C-6″), 128.0 (C-10 and C-12), 128.7 (C-3″ and C-5″), 129.1 (C-3′ and C-5′), 130.7 (C-4″), 131.3 (C-1″), 136.5 (C-4′), 139.8 (C-8), 140.1 (C-1′), 155.3 (3-C=N), 174.2 (6-C=S); HRMS: 394.1156 [M + Na]+ {calcd. 394.1354 for C23H21N3SNa}.
5-(4-methoxyphenyl)-N,3-diphenyl-4,5-dihydro-1H-pyrazole-1-carbothioamide (4b). Pale yellow crystals; yield: 39%; m.p.: 173–175 °C; FTIR (ATR) νmax cm−1: 3334 (N–H), 3058 (Csp2–H), 2842 (Csp3–H), 1592 (C=N), 1510, 1445 (aromatic C=C), 1396 (C=S), 1244 (C–O); 1H-NMR (500 MHz, CD3COCD3): δH 3.29 (1H, dd, J = 3.5 Hz and 18.0 Hz, 4α-CH), 3.78 (3H, s, 14-OCH3), 4.05 (1H, dd, J = 11.5 Hz and 18.0 Hz, 4β-CH), 6.13 (1H, dd, J = 3.5 Hz and 11.5 Hz, 5-CH), 6.89 (2H, d, J = 9.0 Hz, H-3′ and H-5′), 7.15 (1H, t, J = 7.5 Hz, H-11), 7.22 (2H, d, J = 8.5 Hz, H-2′ and H-6′), 7.33 (2H, t, J = 7.5 Hz, H-10 and H-12), 7.50 (3H, m, H-3″, H-4″ and H-5″), 7.73 (2H, d, J = 7.5 Hz, H-9 and H-13), 7.99 (2H, m, H-2″ and H-6″), 9.92 (1H, s, 7-NH); 13C NMR (125 MHz, CD3COCD3): δC 42.3 (4-CH2), 54.6 (14-OCH3), 63.1 (5-CH), 113.8 (C-3′ and C-5′), 124.3 (C-9 and C-13), 124.6 (C-11), 126.9 (C-2′ and C-6′), 127.2 (C-2″ and C-6″), 128.0 (C-10 and C-12), 128.7 (C-3″ and C-5″), 130.7 (C-4″), 131.4 (C-1″), 135.1 (C-1′), 139.8 (C-8), 155.3 (3-C=N), 158.9 (C-4′), 174.2 (6-C=S); HRMS: 410.1098 [M + Na]+ {calcd. 410.1303 for C23H21N3OSNa}.
5-(4-chlorophenyl)-N,3-diphenyl-4,5-dihydro-1H-pyrazole-1-carbothioamide (4c). Yellow solid; yield: 34%; m.p.: 150–153 °C; FTIR (ATR) νmax cm−1: 3306 (N–H), 3034 (Csp2–H), 1594 (C=N), 1538, 1489 (aromatic C=C), 1380 (C=S); 1H-NMR (500 MHz, CD3COCD3): δH 3.33 (1H, dd, J = 3.5 Hz and 18.0 Hz, 4α-CH), 4.11 (1H, dd, J = 11.5 Hz and 18.0 Hz, 4β-CH), 6.18 (1H, dd, J = 3.5 Hz and 11.5 Hz, 5-CH), 7.16 (1H, t, J = 7.5 Hz, H-11), 7.35 (6H, m, H-2′, H-3′, H-5′, H-6′, H-10 and H-12), 7.50 (3H, m, H-3″, H-4″ and H-5″), 7.72 (2H, t, J = 7.5 Hz, H-9 and H-13), 7.99 (2H, d, J = 7.0 Hz, H-2″ and H-6″), 9.96 (1H, s, 7-NH); 13C-NMR (125 MHz, CD3COCD3): δC 42.1 (4-CH2), 63.0 (5-CH), 124.4 (C-9 and C-13), 124.7 (C-11), 127.2 (C-2′ and C-6′), 127.6 (C-2″ and C-6″), 128.1 (C-10 and C-12), 128.6 (C-3′ and C-5′), 128.7 (C-3″ and C-5″), 130.7 (C-4″), 131.2 (C-1″), 132.2 (C-4′), 139.7 (C-8), 142.0 (C-1′), 155.2 (3-C=N), 174.3 (6-C=S); HRMS: 392.0952 [M + H]+ {calcd. 392.0988 for (C22H19ClN3S}.
5-(4-dimethylaminohenyl)-N,3-diphenyl-4,5-dihydro-1H-pyrazole-1-carbothioamide (4d). Orange oil; yield: 12%; FTIR (ATR) νmax cm−1: 3338 (N–H), 3031 (Csp2–H), 2921 (Csp3–H), 1595 (C=N), 1517, 1446 (aromatic C=C), 1395 (C=S), 1320 (C–N); 1H-NMR (500 MHz, CD3COCD3): δH 2.90(6H, s, 14-CH3 and 15-CH3), 3.09 (1H, dd, J = 3.5 Hz and 18.0 Hz, 4α-CH), 3.99 (1H, dd, J = 11.5 Hz and 18.0 Hz, 4β-CH), 6.08 (1H, dd, J = 3.5 Hz and 11.0 Hz, 5-CH), 6.69 (2H, d, J = 9.0 Hz, H-3′ and H-5′), 7.13 (3H, m, H-2′, H-6′ and H-11), 7.33 (2H, m, H-10 and H-12), 7.50 (3H, m, H-3″, H-4″ and H-5″), 7.74 (2H, m, H-9 and H-13), 7.98 (2H, m, H-2″ and H-6″), 9.88 (1H, s, 7-NH); 13C-NMR (125 MHz, CD3COCD3): δC 39.8 (14-CH3 and 15-CH3), 42.3 (4-CH2), 63.2 (5-CH), 112.4 (C-3′ and C-5′), 124.2 (C-9 and C-13), 124.5 (C-11), 126.6 (C-2′ and C-6′), 127.2 (C-2″ and C-6″), 128.0 (C-10 and C-12), 128.7 (C-3″ and C-5″), 130.6 (C-4″), 130.6 (C-1′), 131.5 (C-1″), 139.9 (C-8), 150.0 (C-4′), 155.3 (3-C=N), 174.1 (6-C=S); HRMS: 401.1848 [M + H]+ {calcd. 401.1800 for C24H25N4S}.
5-(2,4-dichlorophenyl)-N,3-diphenyl-4,5-dihydro-1H-pyrazole-1-carbothioamide (4e). Yellowish powder; yield: 15%; m.p.: 175–177 °C; FTIR (ATR) νmax cm−1: 3345 (N–H), 3056 (Csp2–H), 2925 (Csp3–H), 1589 (C=N), 1510, 1447 (aromatic C=C), 1398 (C=S); 1H-NMR (500 MHz, CD3COCD3): δH 3.14 (1H, dd, J = 4.0 Hz and 18.0 Hz, 4α-CH), 4.03 (1H, dd, J = 12.0 Hz and 18.0 Hz, 4β-CH), 6.25 (1H, dd, J = 4.5 Hz and 12.0 Hz, 5-CH), 7.04 (2H, m, H-10 and H-12), 7.21 (3H, m, H-5′, H-6′ and H-11), 7.36 (3H, m, H-3″, H-4″ and H-5″), 7.40 (1H, s, H-3′), 7.59 (2H, dd, J = 1.0 Hz and 8.5 Hz, H-9 and H-13), 7.84 (2H, dd, J = 1.5 Hz and 8.0 Hz, H-2″ and H-6″), 9.88 (1H, s, 7-NH); 13C-NMR (125 MHz, CD3COCD3): δC 40.7 (4-CH2), 61.1 (5-CH), 124.4 (C-10 and C-12), 124.9 (C-11), 127.3 (C-2″ and C-6″), 127.6 (C-5′ and C-6′), 128.1 (C-9 and C-13), 128.7 (C-3″ and C-5″), 129.2 (C-3′), 130.8 (C-4″), 131.1 (C-1″), 132.0 (C-2′), 132.9 (C-4′), 139.0 (C-8), 139.6 (C-1′), 155.4 (3-C=N), 174.3 (6-C=S); HRMS: 426.0573 [M + H]+ {calcd. 426.0599 for (C22H18Cl2N3S}.
5-(2-chlorophenyl)-N,3-diphenyl-4,5-dihydro-1H-pyrazole-1-carbothioamide (
4f). Yellow solid; yield: 34%; m.p.: 59–62 °C ([
33]: 58–60 °C); FTIR (ATR) ν
max cm
−1: 3310 (N–H), 3051 (C
sp2–H), 2936 (C
sp3–H), 1588 (C=N), 1516, 1447 (aromatic C=C), 1400 (C=S);
1H-NMR (500 MHz, CDCl
3): δ
H 3.19 (1H, dd,
J = 4.0 Hz and 18.0 Hz, 4α-C
H), 3.99 (1H, dd,
J = 11.5 Hz and 18.0 Hz, 4β-C
H), 6.50 (1H, dd,
J = 3.5 Hz and 12.0 Hz, 5-C
H), 7.20 (5H, m, H-3′, H-4′, H-5′, H-6′ and H-11), 7.40 (2H, t,
J = 7.5 Hz, H-10 and H-12), 7.46 (3H, m, H-3″, H-4″ and H-5″), 7.79 (2H, d,
J = 7.5 Hz, H-9 and H-13), 7.83 (2H, m, H-2″ and H-6″), 9.37 (1H, s, 7-N
H);
13C-NMR (125 MHz, CDCl
3): δ
C 41.5 (4-
CH
2), 61.3 (5-
CH), 124.2 (C-9 and C-13), 125.5 (C-11), 126.9 (C-2″ and C-6″), 127.2 (C-5′), 128.7 (C-10 and C-12), 128.7 (C-6′), 128.9 (C-3″ and C-5″), 129.4 (C-3′), 130.0 (C-4′), 130.6 (C-4″), 131.1 (C-2′), 131.3 (C-1″), 138.7 (C-1′), 138.8 (C-8), 155.4 (3-C=N), 174.1 (6-
C=S); HRMS: 414.0771 [M + Na]
+ {calcd. 414.0808 for C
22H
18ClN
3SNa}.
5-(2-methoxyphenyl)-N,3-diphenyl-4,5-dihydro-1H-pyrazole-1-carbothioamide (4g). Yellow solid; yield: 22%; m.p.: 193–196 °C; FTIR (ATR) νmax cm−1: 3359 (N–H), 3055 (Csp2–H), 2920 (Csp3–H), 1572 (C=N), 1520, 1445 (aromatic C=C), 1379 (C=S); 1H-NMR (500 MHz, CD3COCD3): δH 3.27 (1H, dd, J = 3.5 Hz and 18.0 Hz, 4α-CH), 3.82 (3H, s, 14-OCH3), 4.06 (1H, dd, J = 11.5 Hz and 18.0 Hz, 4β-CH), 6.15 (1H, dd, J = 3.5 Hz and 11.5 Hz, 5-CH), 7.16 (5H, q, J = 8.0 Hz and 17.5 Hz, H-3′, H-4′, H-5′, H-6′ and H-11), 7.33 (2H, t, J = 7.5 Hz, H-10 and H-12), 7.50 (3H, m, H-3″, H-4″ and H-5″), 7.73 (2H, d, J = 7.5 Hz, H-9 and H-13), 7.98 (2H, dd, J = 1.5 Hz and 8.0 Hz, H-2″ and H-6″), 9.93 (1H, s, 7-NH); 13C-NMR (125 MHz, CD3COCD3): δC 42.3 (4-CH2), 55.8 (14-OCH3), 63.3 (5-CH), 124.3 (C-9 and C-13), 124.6 (C-11), 125.6 (C-5′ and C-6′), 127.2 (C-2″ and C-6″), 128.0 (C-10 and C-12), 128.7 (C-3″ and C-5″), 129.1 (C-3′ and C-4′),130.7 (C-4″), 131.3 (C-1″), 136.5 (C-2′), 139.8 (C-8), 140.1 (C-1′), 155.3 (3-C=N), 174.2 (6-C=S); HRMS: 410.1319 [M + Na]+ {calcd. 410.1303 for C23H21N3OSNa}.
3.1.4. General Procedure for the Preparation of (3,5-disubstituted-4,5-dihydro-1H-pyrazol-yl) (4-hydroxyphenyl)methanone (5a–g)
The new methanone compounds (
5a–g) were synthesized by the cyclo-condensation of chalcone (
3a–g) (1 mmol) with 4-hydroxybenzhydrazide (1.5 mmol) in ethanol (6 mL) in the presence of NaOH (3 mmol) for 3 to 8 h. The reaction progress was observed on a TLC plate. After the reaction was over, the reaction mixture was left at room temperature overnight. The reaction mixture was neutralized by adding 1%
v/v of HCl and monitored by pH paper until the precipitate formed. A solid was formed after crushed ice was added. The solid was filtered, washed with cold water and dried. The products were purified by CC using silica gel with the eluent
n-hexane:ethyl acetate to give the compounds
5a–g. Please refer the
Supplementary Data for full spectra.
N-(4-hydroxyphenyl)(5-(4-methylphenyl)-3-phenyl-4,5-dihydro-1H-pyrazol-1-yl)methanone (5a). Pale yellow powder; yield: 29%; m.p.: 224–228 °C; FTIR (ATR) νmax cm−1: 3209 (O–H), 3055 (Csp2–H), 2923 (Csp3–H), 1740 (C=O), 1588 (C=N), 1511, 1439 (aromatic C=C), 1230 (C–O); 1H-NMR (500 MHz, DMSO-d6): δH 2.27 (3H, s, 14-CH3), 3.12 (1H, dd, J = 5.5 Hz and 18.0 Hz, 4α-CH), 3.87 (1H, dd, J = 12.0 Hz and 18.0 Hz, 4β-CH), 5.71 (1H, dd, J = 5.0 Hz and 11.5 Hz, 5-CH), 6.85 (2H, d, J = 9.0 Hz, H-8 and H-12), 7.16 (4H, m, H-2′, H-3′, H-5′ and H-6′), 7.47 (3H, m, H-3″, H-4″ and H-5″), 7.75 (2H, m, H-2″ and H-6″), 7.86 (2H, d, J = 8.5 Hz, H-9 and H-11), 10.04 (1H, s, 13-OH); 13C-NMR (125 MHz, DMSO-d6): δC 21.1 (14-CH3), 41.6 (4-CH2), 61.0 (5-CH), 115.0 (C-8 and C-12), 125.3 (C-7), 126.0 (C-2″ and C-6″), 127.1 (C-2′ and C-6′), 129.3 (C-3′ and C-5′), 129.7 (C-3″ and C-5″), 130.7 (C-4″), 131.7 (C-1″), 132.4 (C-9 and C-11), 136.8 (C-4′), 140.2 (C-1′), 155.9 (3-C=N), 160.4 (10-C-OH), 165.2 (6-C=O); HRMS: 357.1591 [M + H]+ {calcd. 357.1603 for C23H21N2O2}.
N-(4-hydroxyphenyl)(5-(4-methoxyphenyl)-3-phenyl-4,5-dihydro-1H-pyrazol-1-yl)methanone (5b). Pale yellow powder; yield: 30%; m.p.: 228–232 °C; FTIR (ATR) νmax cm−1: 3211.5 (O–H), 3066 (Csp2–H), 2929 (Csp3–H), 1741 (C=O), 1605 (C=N), 1513, 1437 (aromatic C=C), 1236 (C–O); 1H-NMR (500 MHz, DMSO-d6): δH 3.14 (1H, dd, J = 5.0 Hz and 18.0 Hz, 4α-CH), 3.72 (3H, s, 14-OCH3), 3.86 (1H, dd, J = 12.0 Hz and 18.0 Hz, 4β-CH), 5.70 (1H, dd, J = 5.0 Hz and 11.5 Hz, 5-CH), 6.84 (2H, d, J = 9.0 Hz, H-8 and H-12), 6.90 (2H, d, J = 8.5 Hz, H-2′ and H-6′), 7.21 (2H, d, J = 8.5 Hz, H-3′ and H-5′), 7.47 (3H, m, H-3″, H-4″ and H-5″), 7.76 (2H, m, H-2″ and H-6″), 7.85 (2H, d, J = 9.0 Hz, H-9 and H-11), 10.03 (1H, s, 13-OH); 13C-NMR (125 MHz, DMSO-d6): δC 41.6 (4-CH2), 55.6 (14-OCH3), 60.7 (5-CH), 114.5 (C-2′ and C-6′), 114.9 (C-8 and C-12), 125.4 (C-7), 127.1 (C-2″ and C-6″), 127.4 (C-3′ and C-5′), 129.3 (C-3″ and C-5″), 130.7 (C-4″), 131.8 (C-1″), 132.4 (C-9 and C-11), 135.2 (C-1′), 155.0 (3-C=N), 158.9 (C-4′), 160.4 (10-C-OH), 165.2 (6-C=O); HRMS: 373.1583 [M + H]+ {calcd. 373.1552 for C23H21N2O3}.
N-(4-hydroxyphenyl)(5-(4-chlorolphenyl)-3-phenyl-4,5-dihydro-1H-pyrazol-1-yl)methanone (5c). Yellowish needles; yield: 16%; m.p.: 263–266 °C; FTIR (ATR) νmax cm−1: 3255 (O–H), 3070 (Csp2–H), 2926 (Csp3–H), 1738 (C=O), 1572 (C=N), 1516, 1438 (aromatic C=C), 1238 (C–O); 1H-NMR (δ/ppm, 500 MHz, DMSO-d6): δH 3.16 (1H, dd, J = 5.5 Hz and 18.0 Hz, 4α-CH), 3.89 (1H, dd, J = 12.0 Hz and 18.0 Hz, 4β-CH), 5.76 (1H, dd, J = 5.0 Hz and 11.5 Hz, 5-CH), 6.85 (2H, d, J = 9.0 Hz, H-8 and H-12), 7.32 (2H, d, J = 8.5 Hz, H-2′ and H-6′), 7.41 (2H, d, J = 8.5 Hz, H-3′ and H-5′), 7.47 (3H, m, H-3″, H-4″ and H-5″), 7.75 (2H, m, H-2″ and H-6″), 7.87 (2H, d, J = 8.5 Hz, H-9 and H-11), 10.07 (1H, s, 13-OH); 13C-NMR (δ/ppm, 125 MHz, DMSO-d6): δC 41.4 (4-CH2), 60.7 (5-CH), 114.9 (C-8 and C-12), 125.1 (C-7), 127.2 (C-2″ and C-6″), 128.1 (C-2′ and C-6′), 129.1 (C-3′ and C-5′), 129.3 (C-3″ and C-5″), 130.8 (C-4″), 131.6 (C-1″), 132.2 (C-4′), 132.5 (C-9 and C-11), 142.1 (C-1′), 155.1 (3-C=N), 160.5 (10-C-OH), 165.3 (6-C=O); HRMS: 377.1030 [M + H]+ {calcd. 376.0979 for C22H17ClN2O2}.
N-(4-hydroxyphenyl)(5-(4-dimethylaminophenyl)-3-phenyl-4,5-dihydro-1H-pyrazol-1-yl) methanone (5d). Brownish solid; yield: 9%; m.p.: 236–240 °C; FTIR (ATR) νmax cm−1: 3241.4 (O–H), 3065.5 (Csp2–H), 2929 (Csp3–H), 1720 (C=O), 1604 (C=N), 1511, 1423 (aromatic C=C), 1339 (C–N), 1226 (C–O). 1H-NMR (500 MHz, CDCl3): δH 2.77 (6H, s, 14-CH3 and 15-CH3), 3.09 (1H, dd, J = 5.0 Hz and 17.5 Hz, 4α-CH), 3.62 (1H, dd, J = 11.5 Hz and 18.0 Hz, 4β-CH), 5.65 (1H, dd, J = 5.0 Hz and 11.5 Hz, 5-CH), 6.64 (2H, d, J = 8.5 Hz, H-8 and H-12), 6.71 (2H, d, J = 9.0 Hz, H-2′ and H-6′), 7.12 (2H, d, J = 8.5 Hz, H-3′ and H-5′), 7.32 (3H, m, H-3″, H-4″ and H-5″), 7.63 (2H, m, H-2″ and H-6″), 7.85 (2H, d, J = 7.5 Hz, H-9 and H-11); 13C NMR (125 MHz, CDCl3): δC 41.1 (14-CH3 and 15-CH3), 41.4 (4-CH2), 61.1 (5-CH), 113.6 (C-3′ and C-5′), 114.7 (C-8 and C-12), 125.4 (C-7), 126.8 (C-2′ and C-6′), 126.9 (C-2″ and C-6″), 128.7 (C-3″ and C-5″), 130.4 (C-4″), 130.6 (C-1′), 131.5 (C-1″), 132.5 (C-9 and C-11), 149.5(C-4′), 155.1 (3-C=N), 159.4 (10-C-OH), 166.4 (6-C=O); HRMS: 386.1891 [M + H]+ {calcd. 386.1869 for C24H24N3O2}.
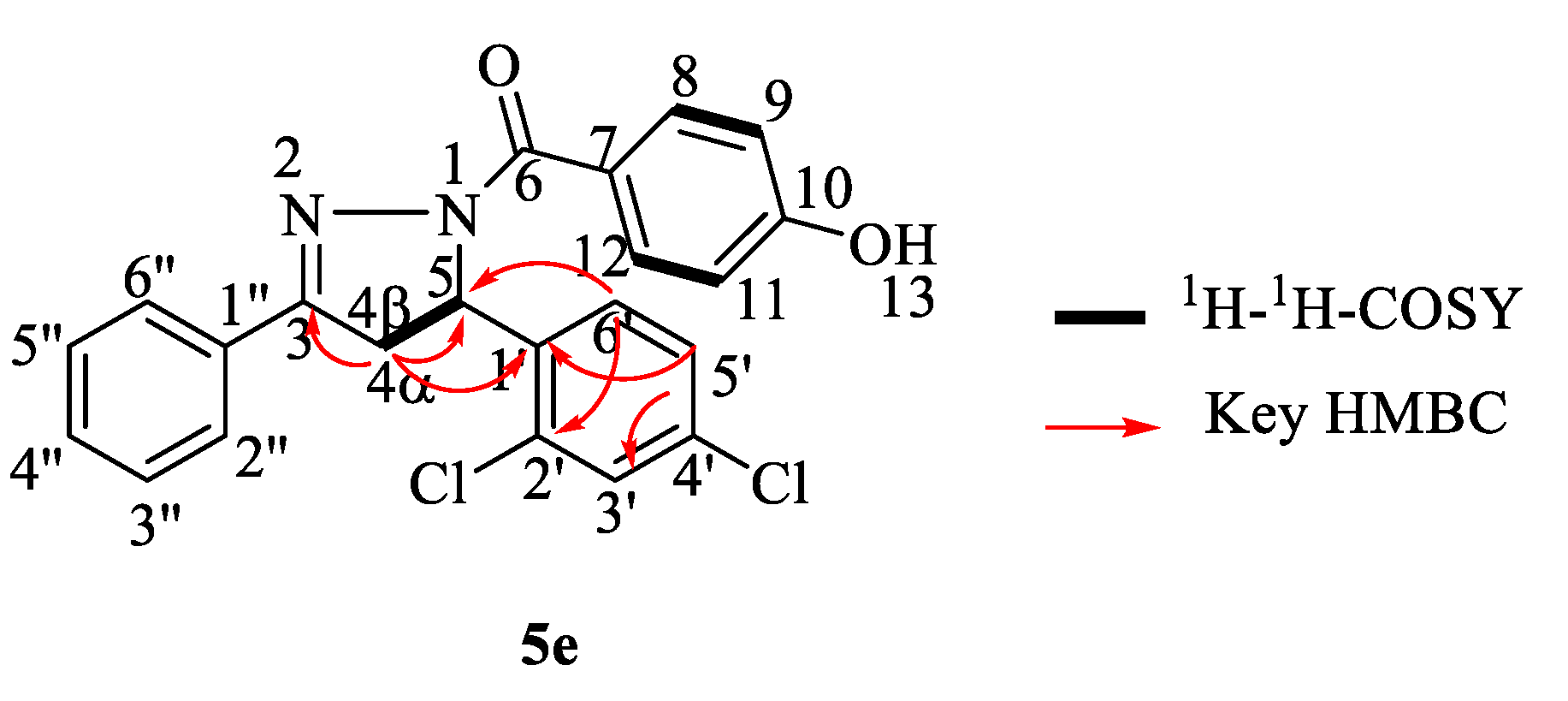
N-(4-hydroxyphenyl)(5-(2,4-dichlorolphenyl)-3-phenyl-4,5-dihydro-1H-pyrazol-1-yl)methanone (5e). Pale yellow powder; yield: 17%; m.p.: 237–241 °C; FTIR (ATR) νmax cm−1: 3137 (O–H), 3066 (Csp2–H), 2926 (Csp3–H), 1737 (C=O), 1618 (C=N), 1573, 1427 (aromatic C=C), 1244 (C–O); 1H-NMR (500 MHz, DMSO-d6): δH 3.13 (1H, dd, J = 6.0 Hz and 18.0 Hz, 4α-CH), 3.96 (1H, dd, J = 12.0 Hz and 18.0 Hz, 4β-CH), 5.95 (1H, dd, J = 5.5 Hz and 12.0 Hz, 5-CH), 6.87 (2H, d, J = 8.5 Hz, H-8 and H-12), 7.22 (1H, d, J = 8.0 Hz, H-6′), 7.40 (1H, dd, J = 2.5 Hz and 8.5 Hz, H-5′), 7.47 (3H, m, H-3″, H-4″ and H-5″), 7.69 (1H, d, J = 2.5 Hz, H-3′), 7.75 (2H, m, H-2″ and H-6″), 7.91 (2H, d, J = 8.5 Hz, H-9 and H-11), 10.11 (1H, s, 13-OH); 13C NMR (125 MHz, DMSO-d6): δC 40.1 (4-CH2), 58.8 (5-CH), 114.9 (C-8 and C-12), 124.7 (C-7), 127.2 (C-2″ and C-6″), 128.4 (C-5′ and C-6′), 129.3 (C-3″ and C-5″), 129.6 (C-3′), 130.9 (C-4″), 131.5 (C-1″), 132.4 (C-2′), 132.6 (C-9 and C-11), 133.0 (C-4′), 138.9 (C-1′), 155.3 (3-C=N), 160.7 (10-C-OH), 165.2 (6-C=O); HRMS: 411.0694 [M + H]+ {calcd. 411.0667 for C22H17Cl2N2O2}.
N-(4-hydroxyphenyl)(5-(2-chlorolphenyl)-3-phenyl-4,5-dihydro-1H-pyrazol-1-yl)methanone (5f). Pale yellow powder; yield: 15%; m.p.: 216–219 °C; FTIR (ATR) νmax cm−1: 3125 (O–H), 3066 (Csp2–H), 2928 (Csp3–H), 1720 (C=O), 1605 (C=N), 1514, 1431 (aromatic C=C), 1240 (C–O); 1H-NMR (500 MHz, DMSO-d6): δH 3.05 (1H, dd, J = 5.5 Hz and 18.0 Hz, 4α-CH), 3.96 (1H, dd, J = 12.0 Hz and 18.0 Hz, 4β-CH), 5.96 (1H, dd, J = 5.5 Hz and 12.0 Hz, 5-CH), 6.86 (2H, d, J = 9.0 Hz, H-8 and H-12), 7.18 (1H, m, H-5′), 7.30 (2H, m, H-3′ and H-6′), 7.44 (3H, m, H-3″, H-4″ and H-5″), 7.50 (1H, m, H-4′), 7.72 (2H, m, H-2″ and H-6″), 7.89 (2H, d, J = 8.5 Hz, H-9 and H-11), 10.27 (1H, s, 13-OH); 13C NMR (125 MHz, DMSO-d6): δC 40.4 (4-CH2), 59.1 (5-CH), 115.0 (C-8 and C-12), 124.9 (C-7), 127.1 (C-2″, C-6″ and C-5′), 128.2 (C-6′), 129.3 (C-3″ and C-5″), 129.5 (C-3′), 130.3 (C-4′), 130.9 (C-4″), 131.4 (C-1″ and C-2′), 132.5 (C-9 and C-11), 139.6 (C-1′), 155.3 (3-C=N), 160.5 (10-C-OH), 165.4 (6-C=O); HRMS: 377.1011 [M + H]+ {calcd. 377.1057 for C22H18ClN2O2}.
N-(4-hydroxyphenyl)(5-(2-methoxyphenyl)-3-phenyl-4,5-dihydro-1H-pyrazol-1-yl)methanone (5g). Pale yellow powder; yield: 22%; m.p.: 220–223 °C; FTIR (ATR) νmax cm−1: 3198 (O–H), 3029 (Csp2–H), 2925 (Csp3–H), 1738 (C=O), 1603 (C=N), 1513, 1429 (aromatic C=C), 1245 (C–O); 1H-NMR (500 MHz, DMSO-d6): δH 3.00 (1H, dd, J = 5.0 Hz and 18.0 Hz, 4α-CH), 3.84 (1H, dd, J = 10.0 Hz and 19.0 Hz, 4β-CH), 3.85 (3H, s, 14-OCH3), 5.89 (1H, dd, J = 4.5 Hz and 12.0 Hz, 5-CH), 6.87 (3H, m, H-5′, H-8 and H-12), 7.05 (2H, m, H-3′ and H-6′), 7.26 (1H, m, H-4′), 7.45 (3H, m, H-3″, H-4″ and H-5″), 7.74 (2H, m, H-2″ and H-6″), 7.89 (2H, d, J = 9.0 Hz, H-9 and H-11), 10.09 (1H, s, 13-OH); 13C NMR (125 MHz, DMSO-d6): δC 40.1 (4-CH2), 56.1 (14-OCH3), 56.9 (5-CH), 111.9 (C-6′), 114.9 (C-8 and C-12), 120.9 (C-5′), 125.4 (C-7), 125.9 (C-3′), 127.1 (C-2″ and C-6″), 128.9 (C-4′), 129.3 (C-3″ and C-5″), 131.0 (C-1′), 130.7 (C-4″), 131.8 (C-1″), 132.4 (C-9 and C-11), 155.5 (3-C=N), 156.4 (C-2′), 160.4 (10-C-OH), 165.1 (6-C=O); HRMS: 373.1599 [M + H]+ {calcd. 373.1552 for C23H21N2O3}.
3.1.5. Screening of Anti-Tuberculosis Activity against M. tuberculosis H37Ra
The tetrazolium microplate assay (TEMA) method was performed to evaluate the anti-mycobacterial activity of the derivatives as described by Caviedes et al. with minor modifications [
34]. The assay was performed in 96-well plates in duplicate and at least three times independently. The derivatives were dissolved in DMSO and serially diluted to the desired concentration in complete Middlebrook 7H9 media enriched with albumin dextrose catalase supplement to reduce the DMSO concentration below 1%. DMSO at this concentration did not inhibit the growth of
M. tuberculosis H37Ra (unreported data). Briefly, 200 μL of sterile distilled water was added to the outer wells of the microplate and 100 µL of Middlebrook 7H9-ADC media into the wells in columns C to G, rows 2 to 11. Then, 100 μL of working solution containing the compounds was added into the wells in columns B and C, rows 2 to 11, in duplicate. A two-fold serial dilution of the compounds was made by transferring 100 μL from the wells in column C to column D, the content was mixed well, and the dilutions were continued until column G, where 100 μL of the excess medium from the wells in column G was discarded. Log phase
M. tuberculosis H37Ra at a density of (~1.5 × 107 CFU/mL) was added and incubated at 37 °C with 5% CO
2 for 5 days. On day 5, 50 μL of tetrazolium reagent mixture was added to all wells, and the plates were re-incubated for 24 h. The results were read visually the following day. The MIC is defined as the lowest drug concentration that prevented the color change from yellow to purple. A small volume of the culture from the 96-well plate was transferred into Middlebrook 7H10 agar media and the plates were incubated at 37 °C with 5% CO
2 for 28 days. The MBC is defined as the lowest concentration of compound that did not show any bacterial colony growth.
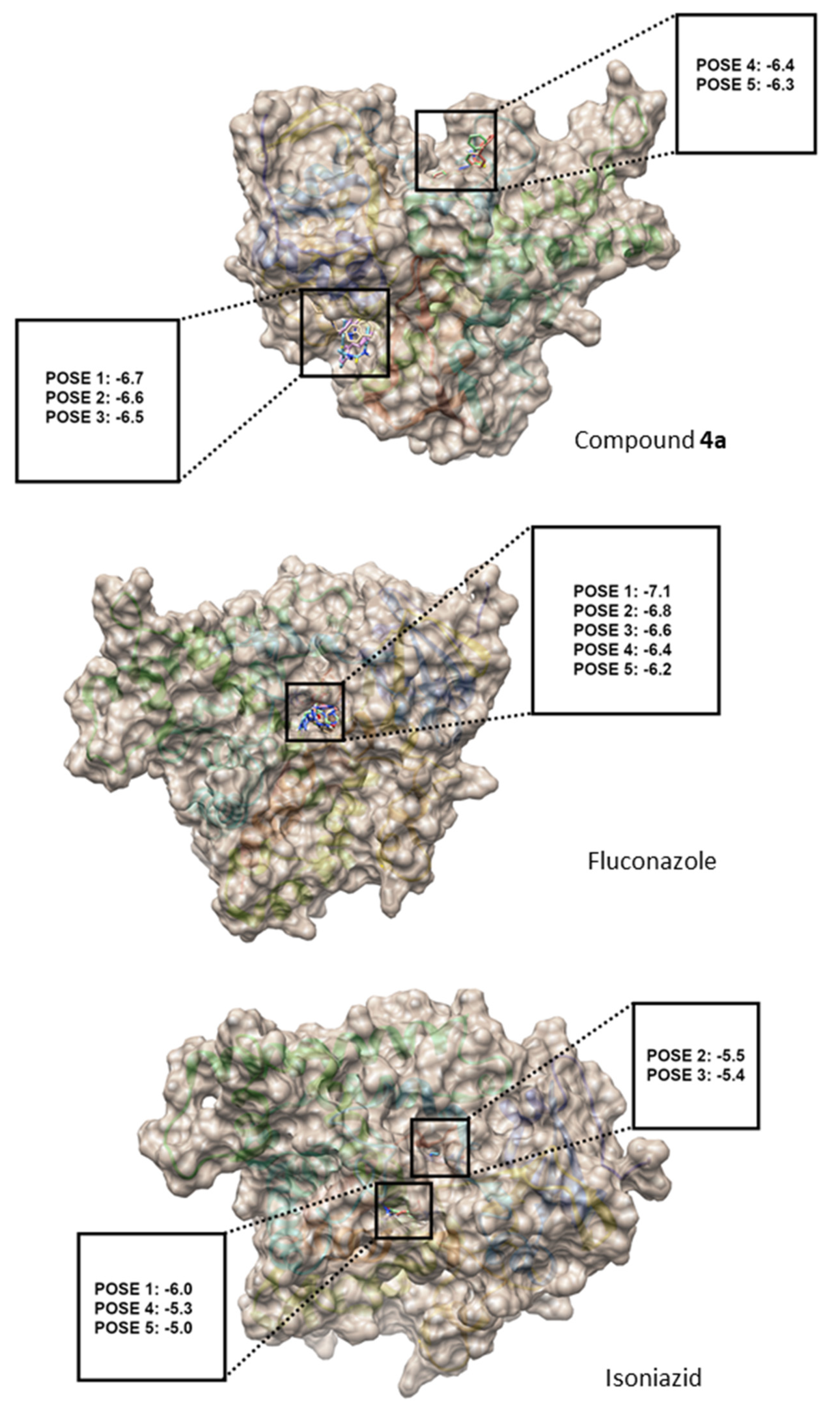
3.1.6. Molecular Docking
The three-dimensional coordinates of the reference structure, cytochrome P45014 alpha-sterol demethylase (CYP51) complexed with fluconazole (PDB ID: 1EA1), and the structure of the experimental control, isoniazid (PDB ID: 2VCF), were fetched by ID from the Protein Data Bank (PDB) database in UCSF Chimera version 1.14 [
35]. Before docking, the crystal PDBs were processed using the Dock Prep tool, starting with the removal of water molecules and unrelated hetam (i.e., refer to any ions molecules and any atoms that not belong to the protein) followed by separation of the receptor and inhibitor from the complexes into individual structures and finally the minimization of individual structures by steepest descent steps [
36]. The compound
4a was built using ChemDraw and subsequently converted from cdx format to PDB.
To recognize the binding sites in CYP51, the grid size was set to −18, −2.6 and 63 along the X-, Y- and Z-axes, respectively, with a 0.375 Å grid spacing. The grid center along the X-, Y- and Z-axes was set to 76, 72, and 83 Å. The AutoDock Vina tool was then used to calculate possible bindings and energies [
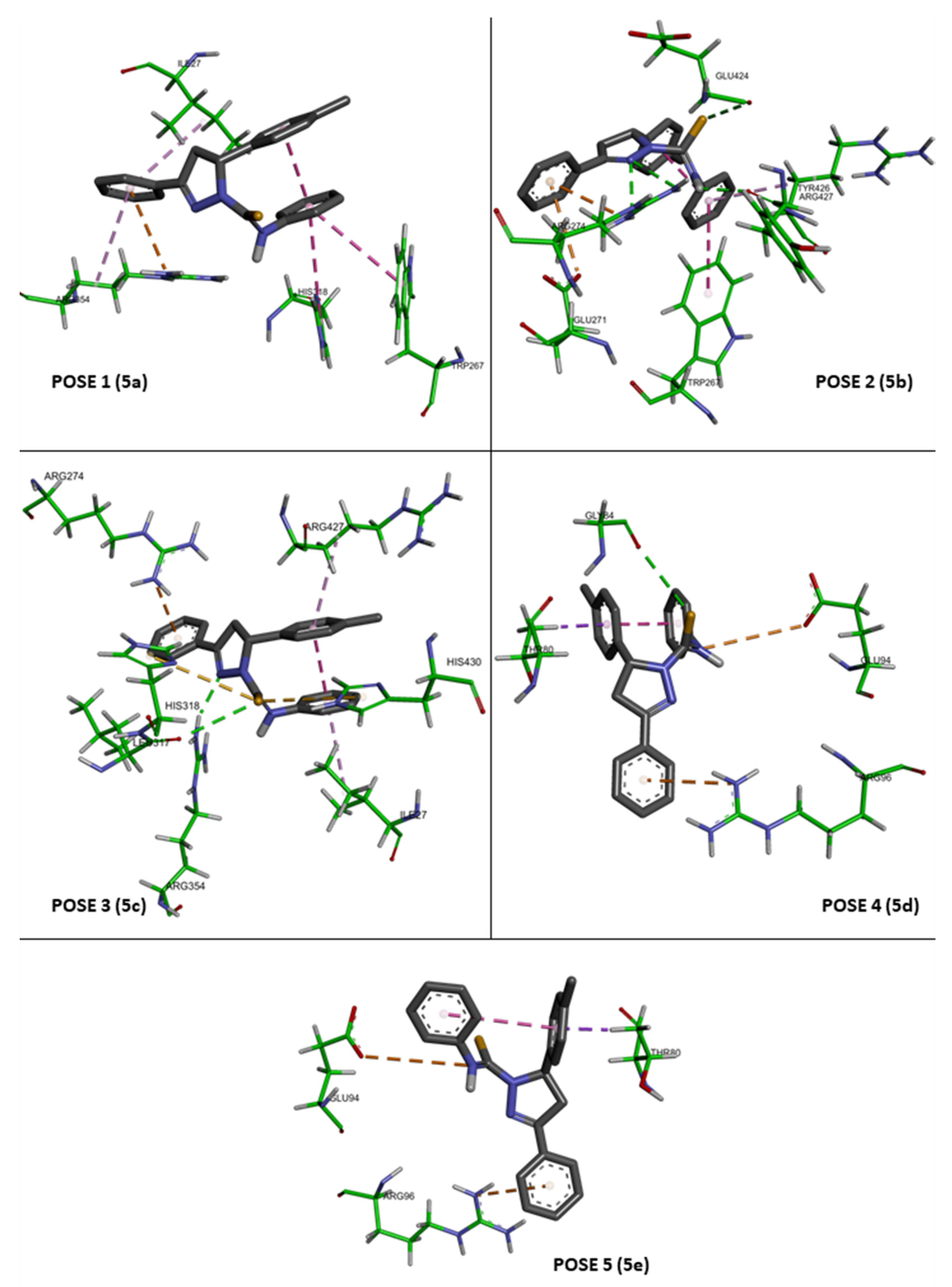
37]. All poses were combined using UCSF Chimera version 1.14 and further labelled by the GNU Image Manipulation program. The interaction modes between CYP51 and compound
4 for each pose were further analyzed using Biovia Discovery Studio Visualizer Client 2020 (Dassault Systèmes BIOVIA, Discovery Studio Modeling Environment, Release 2017, San Diego: Dassault Systèmes, 2016).










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}