
FragNet, a Contrastive Learning-Based Transformer Model for Clustering, Interpreting, Visualizing, and Navigating Chemical Space



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
Molecular Similarity
Deep Learning for Molecular Similarity
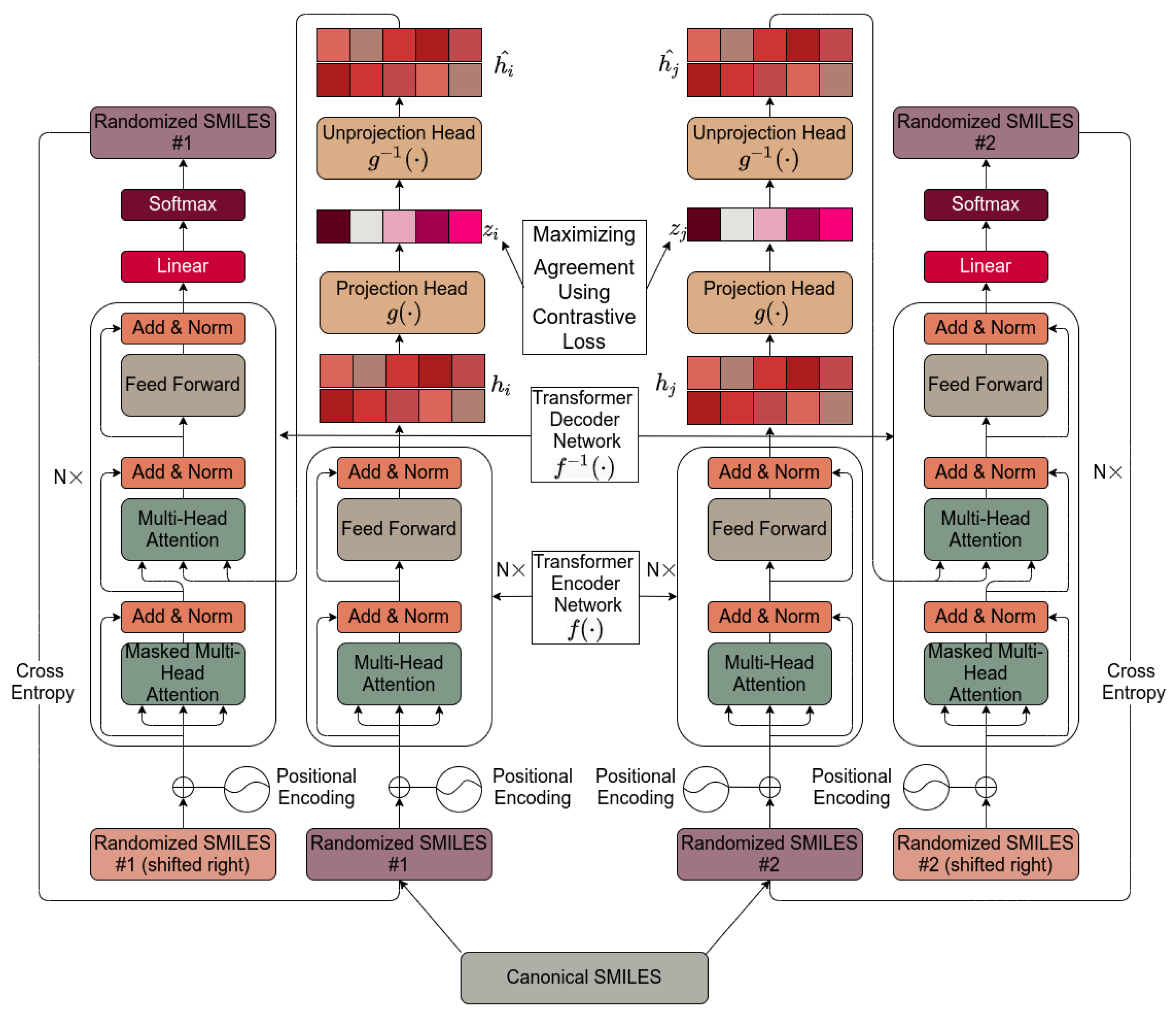
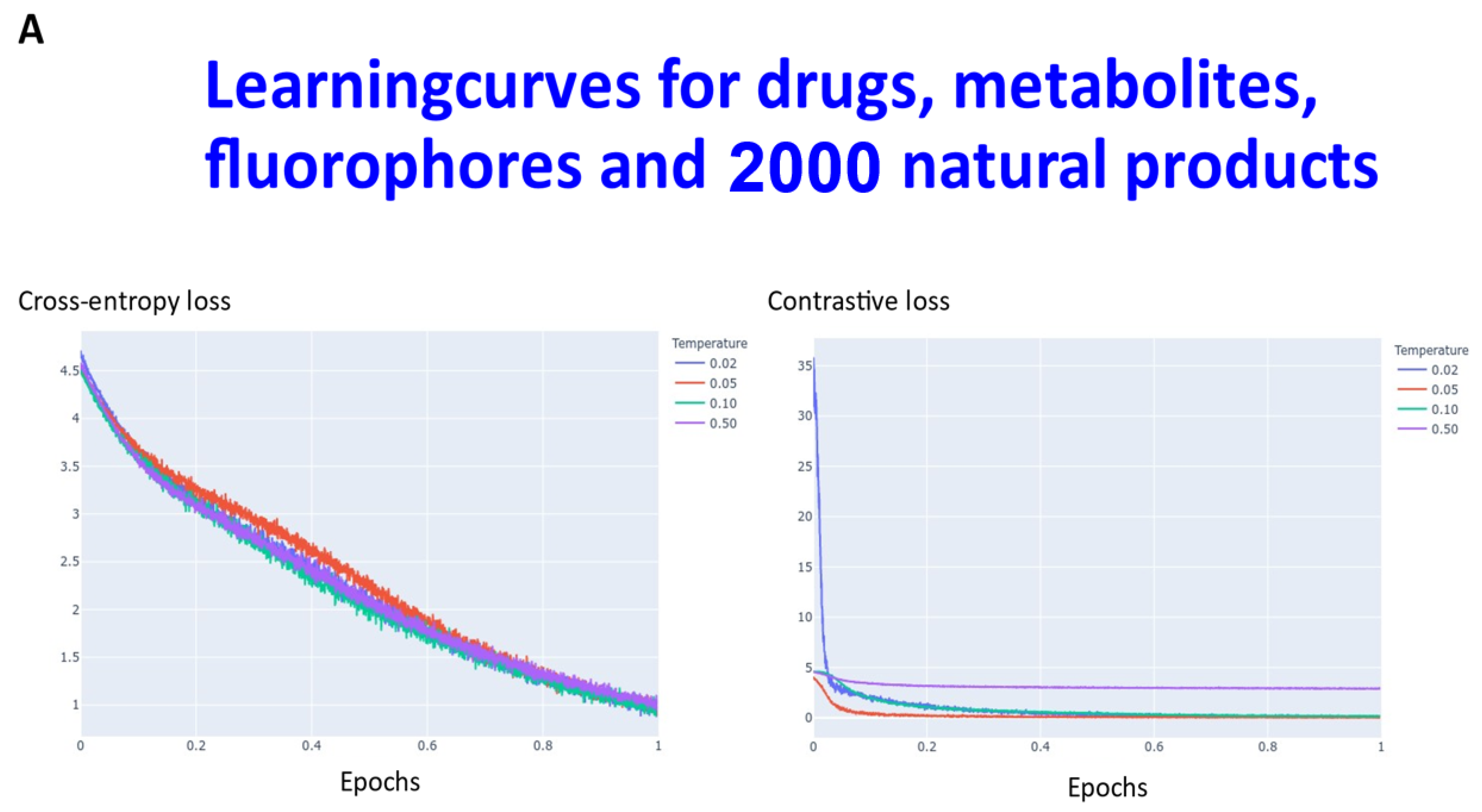
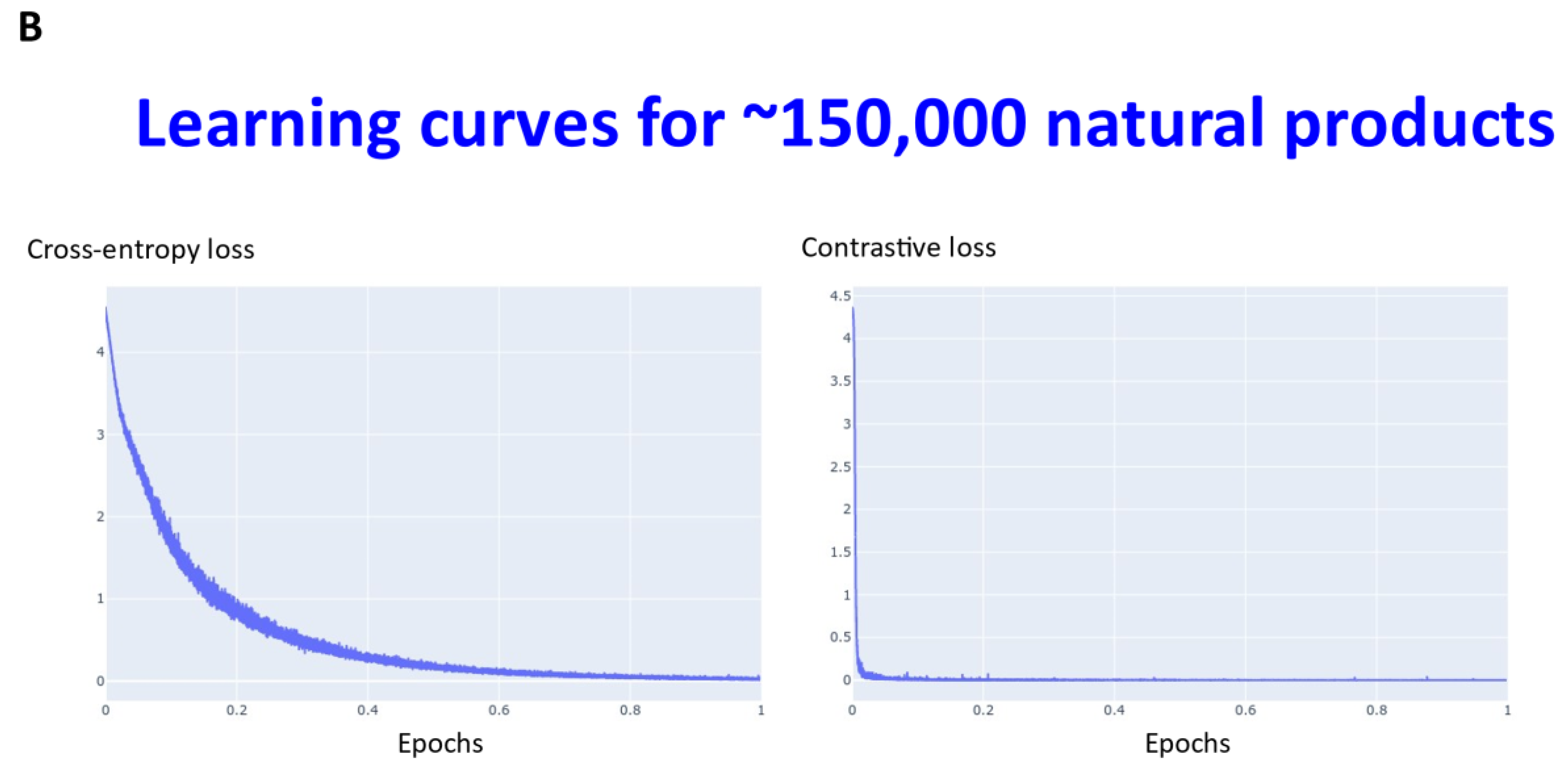
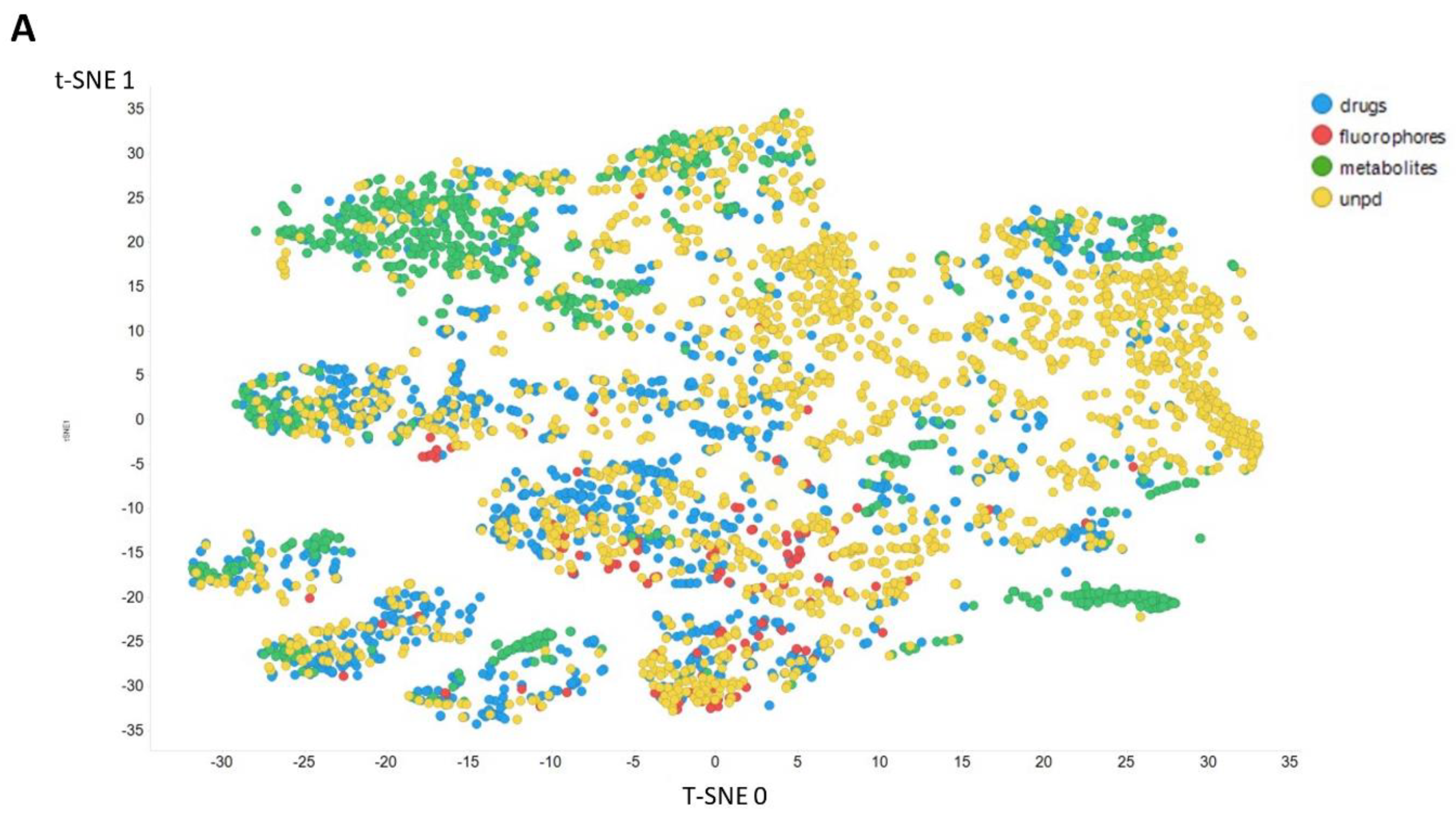
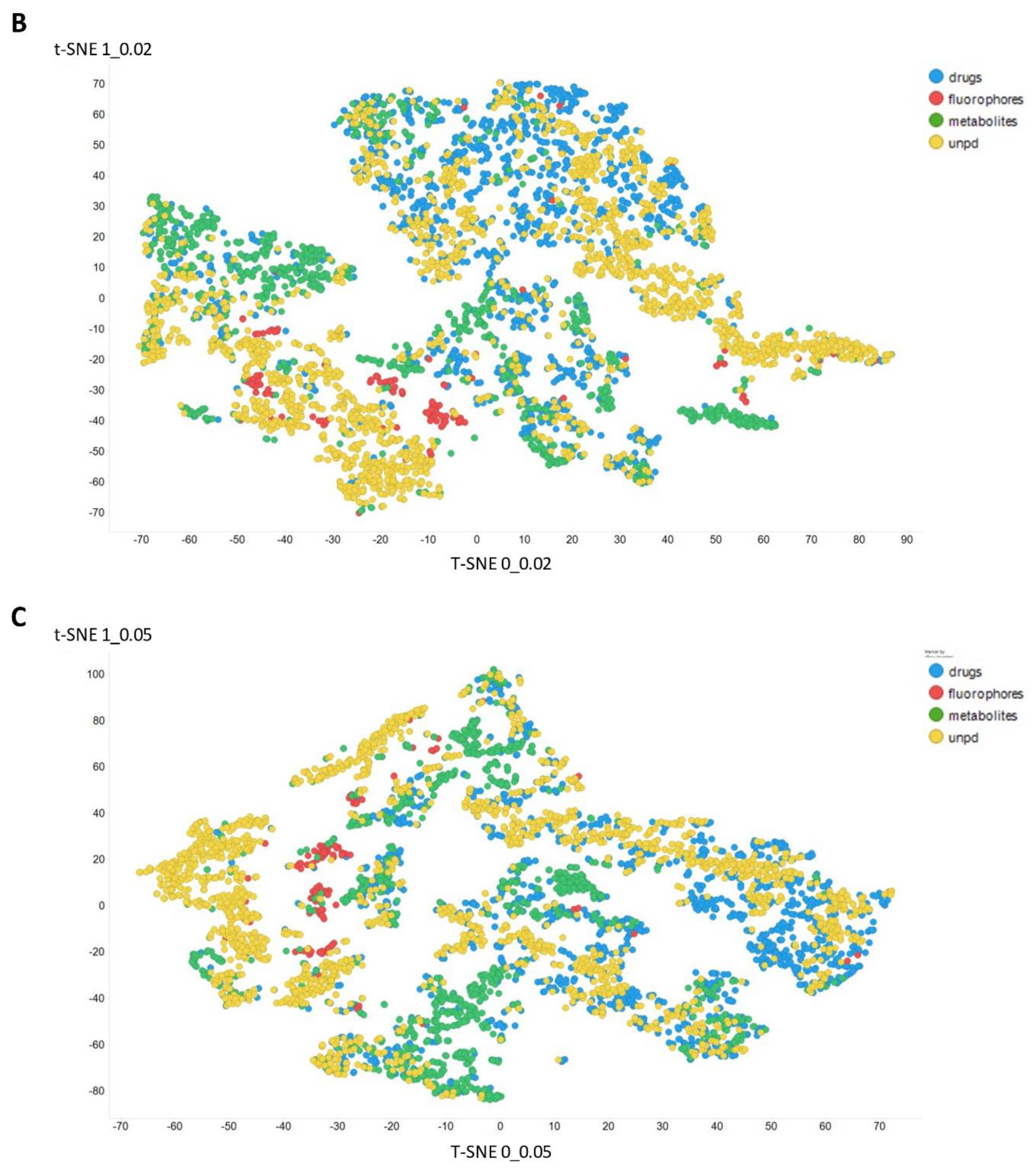
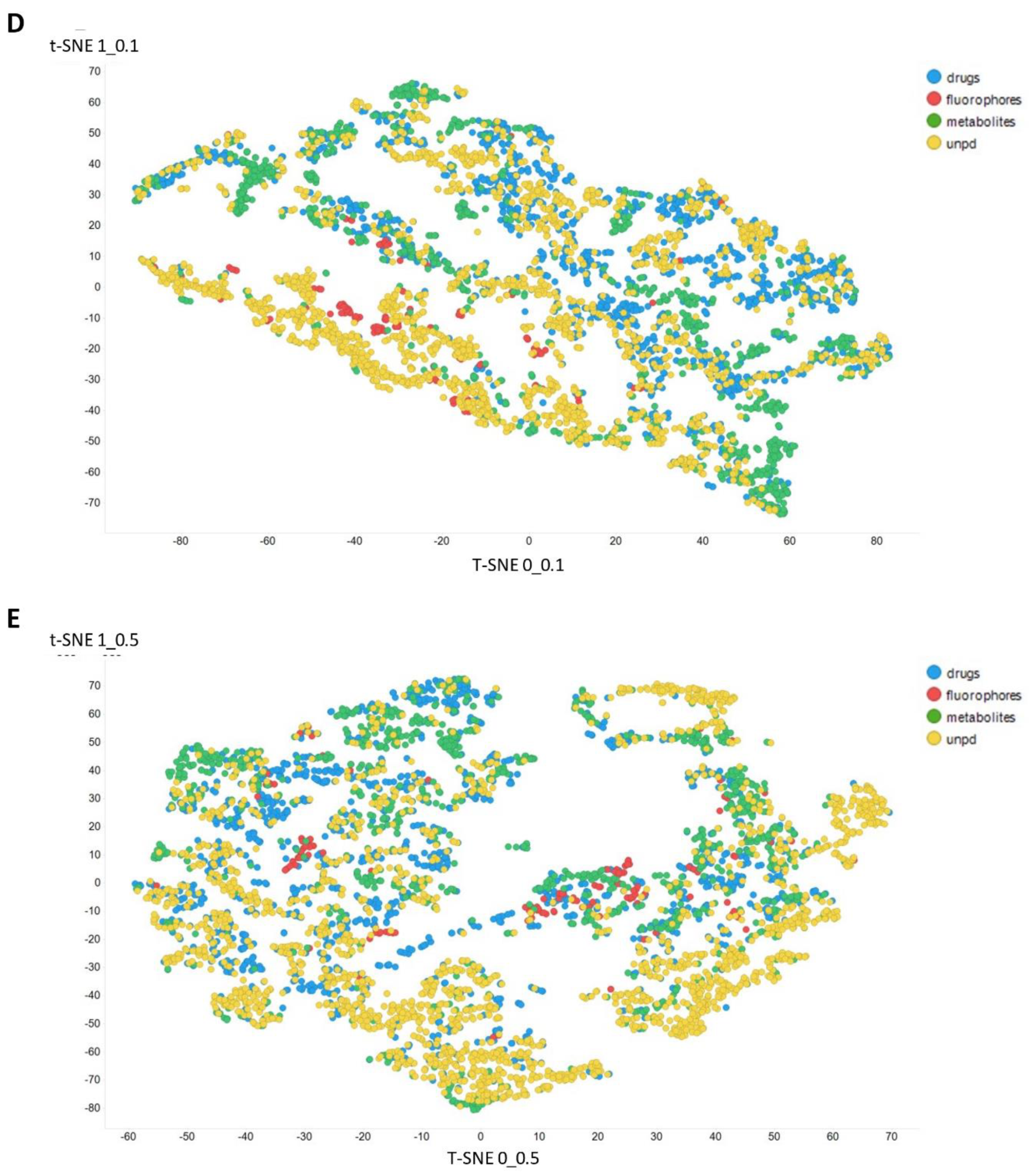
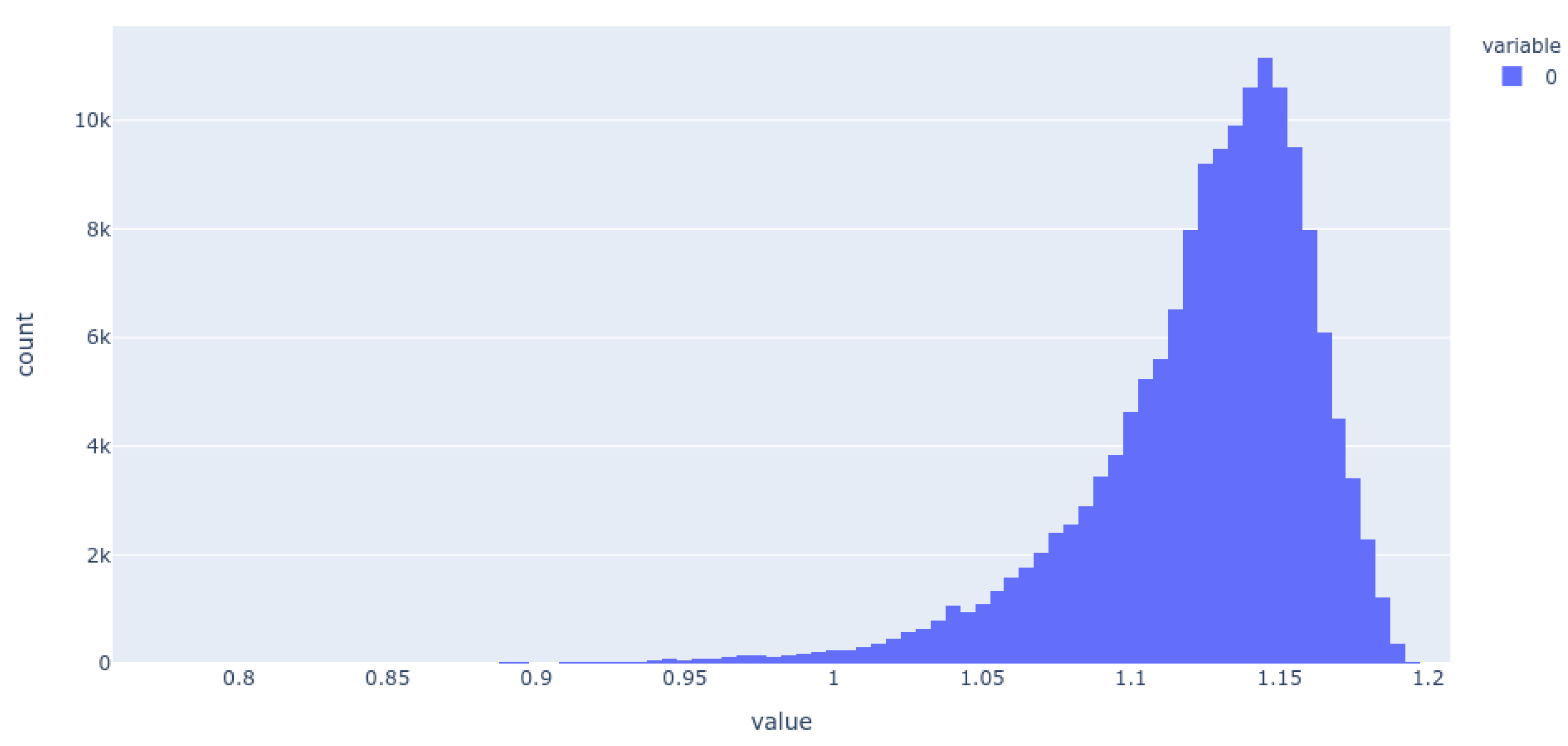
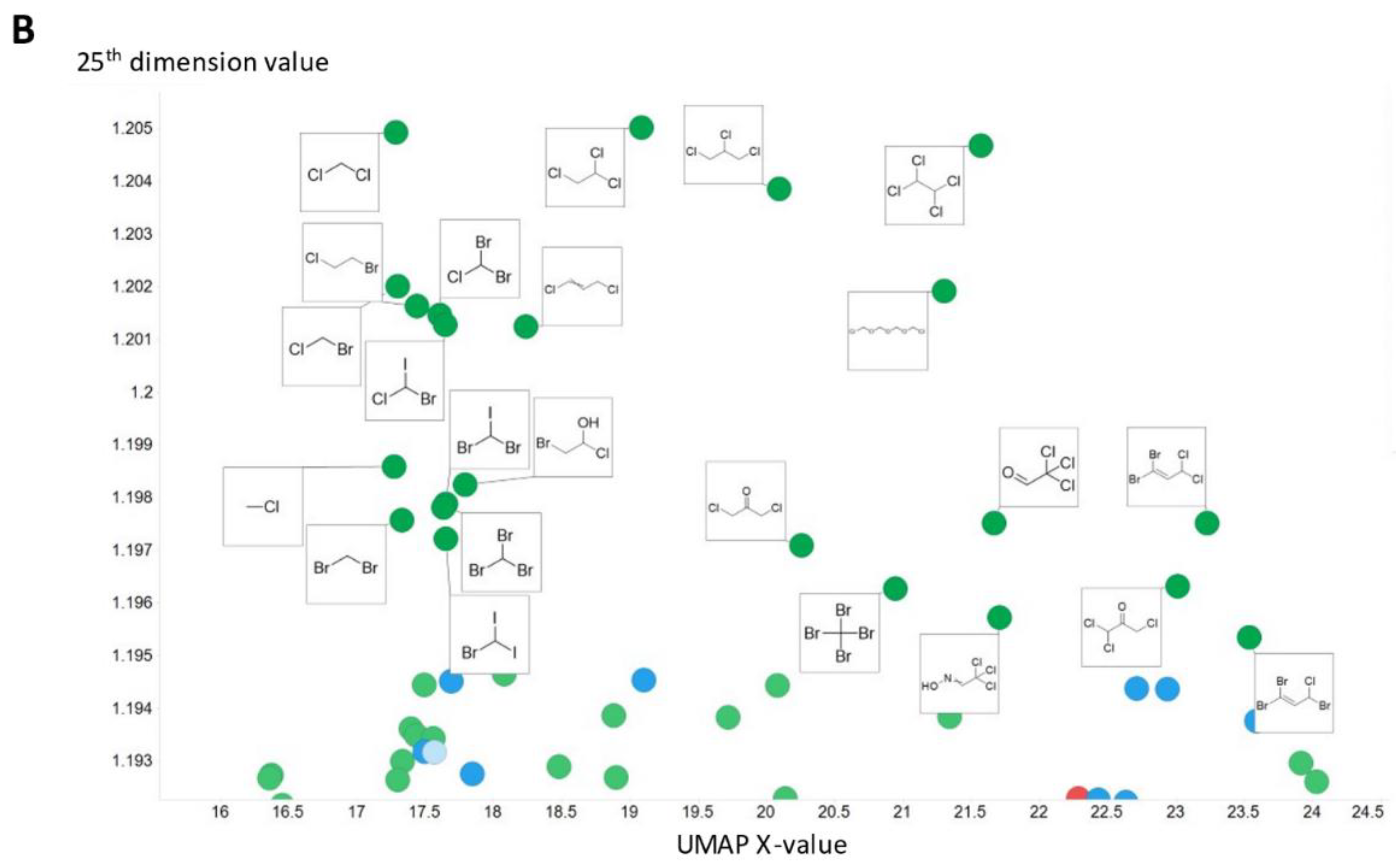
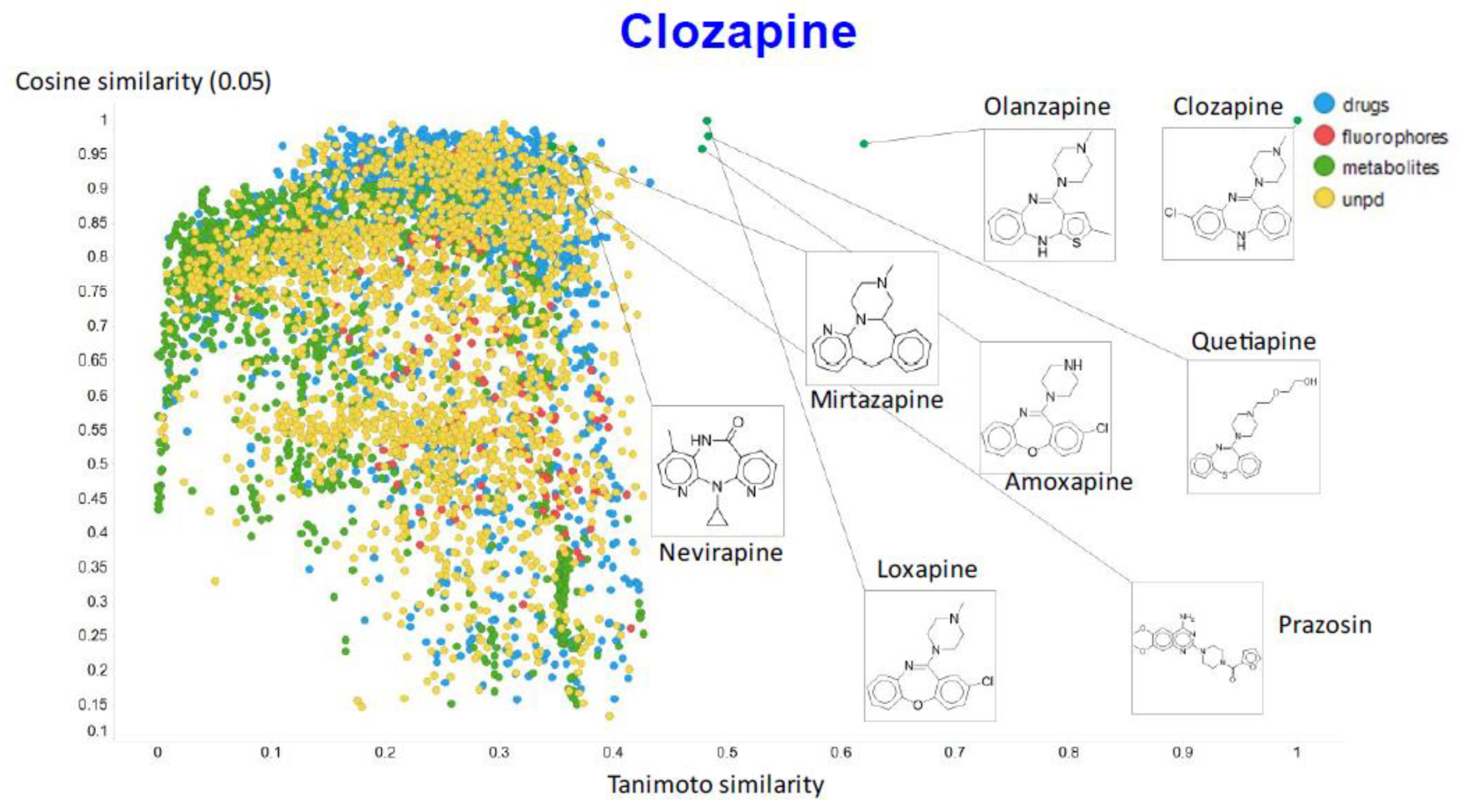
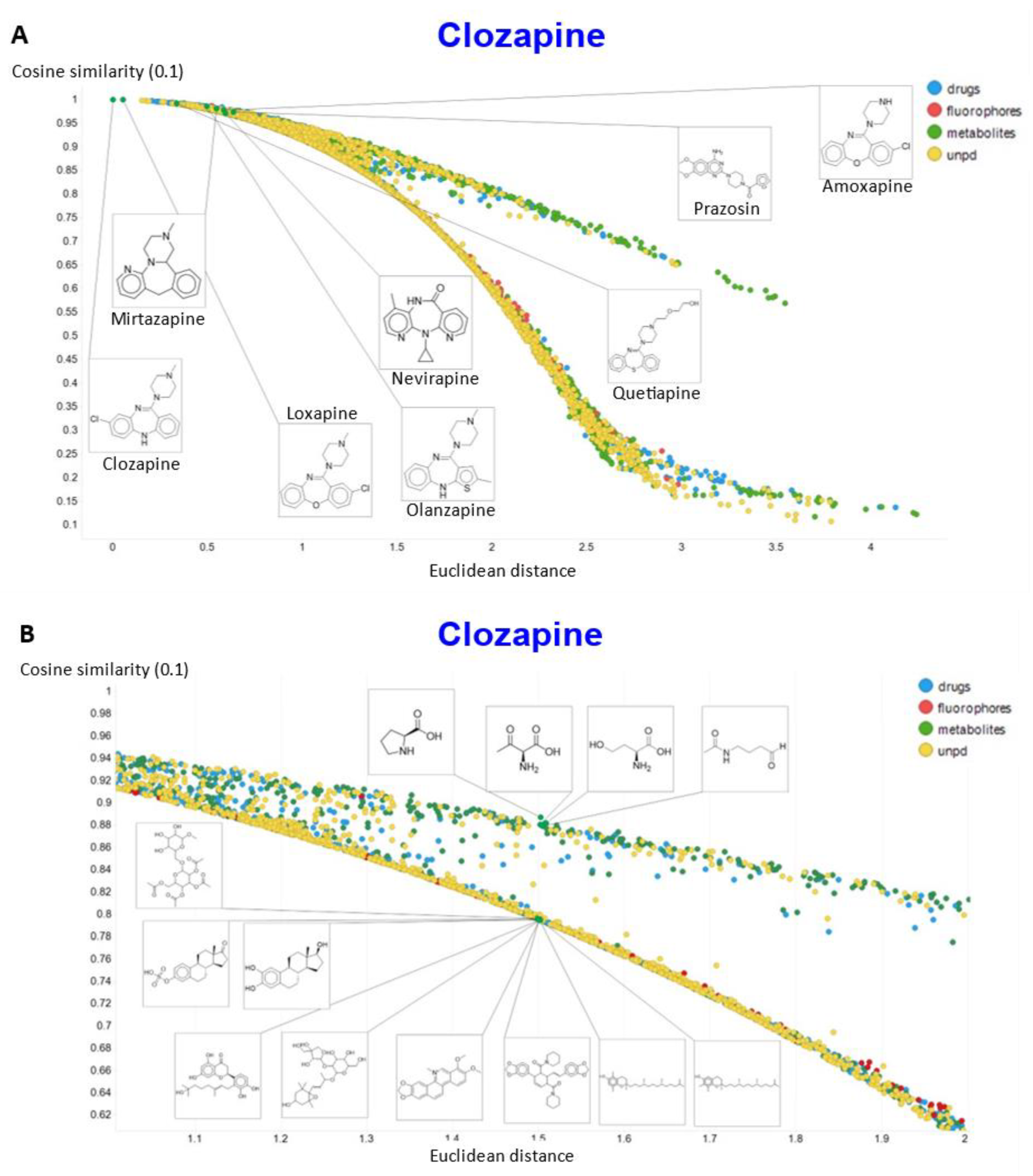
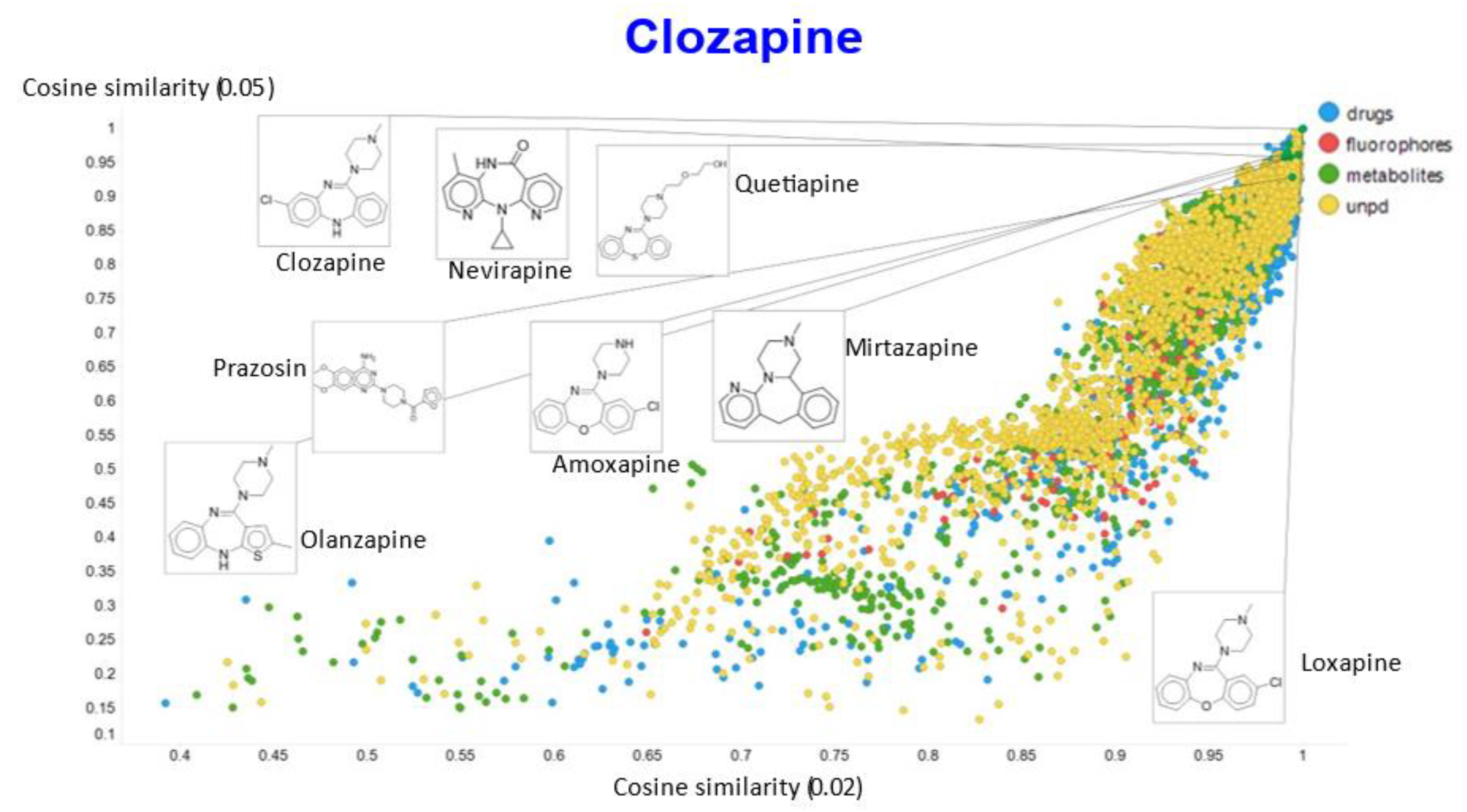
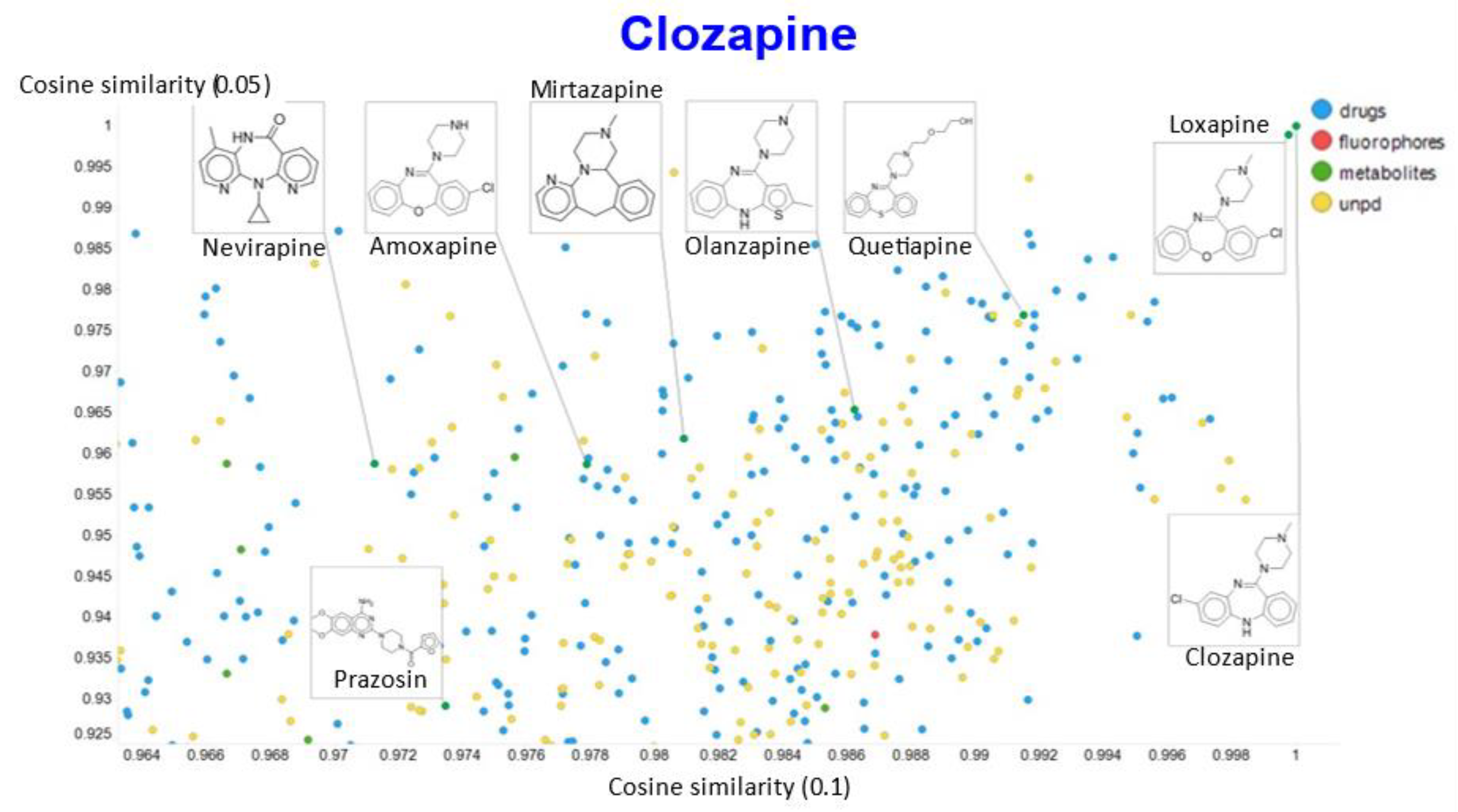
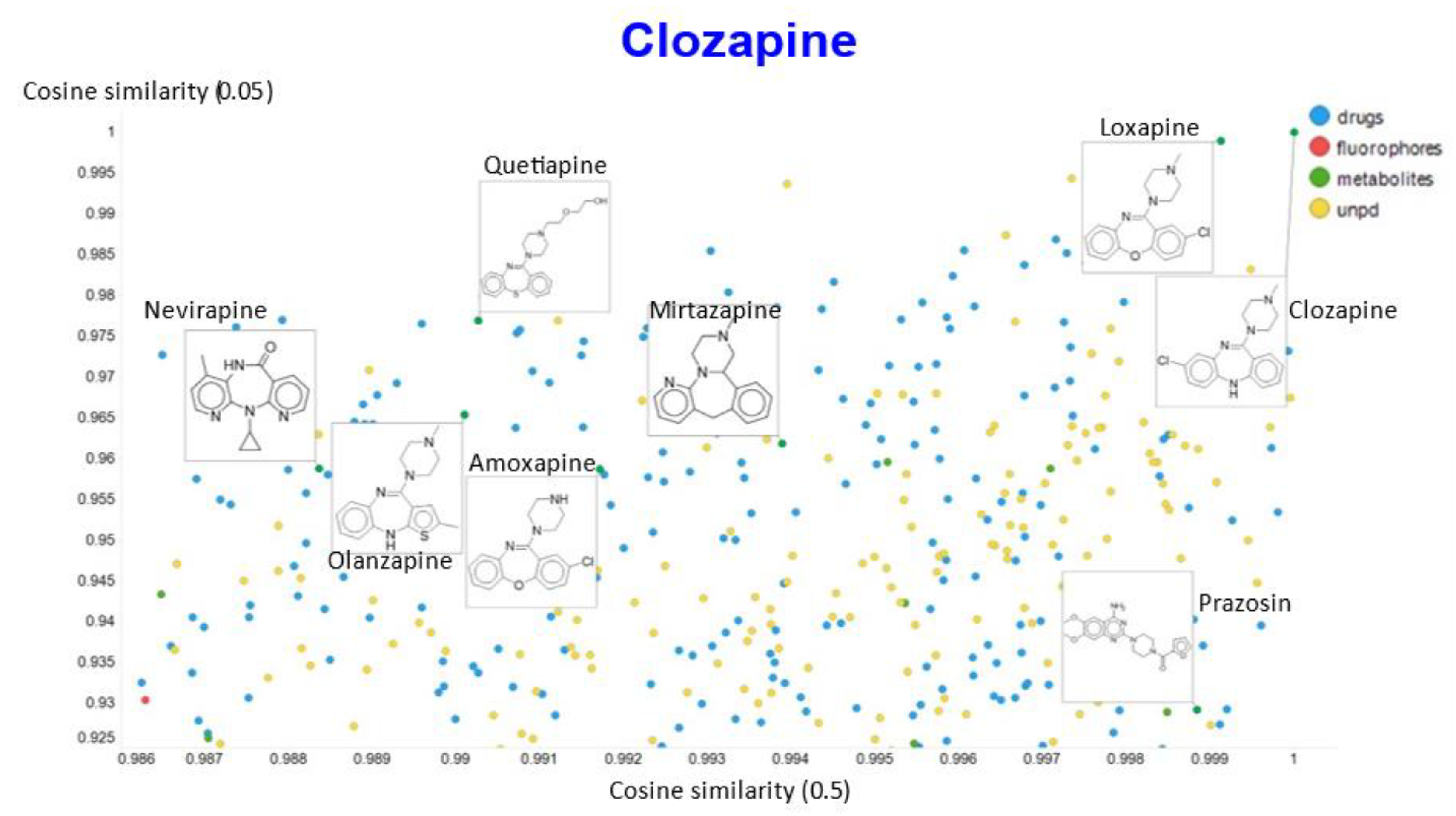
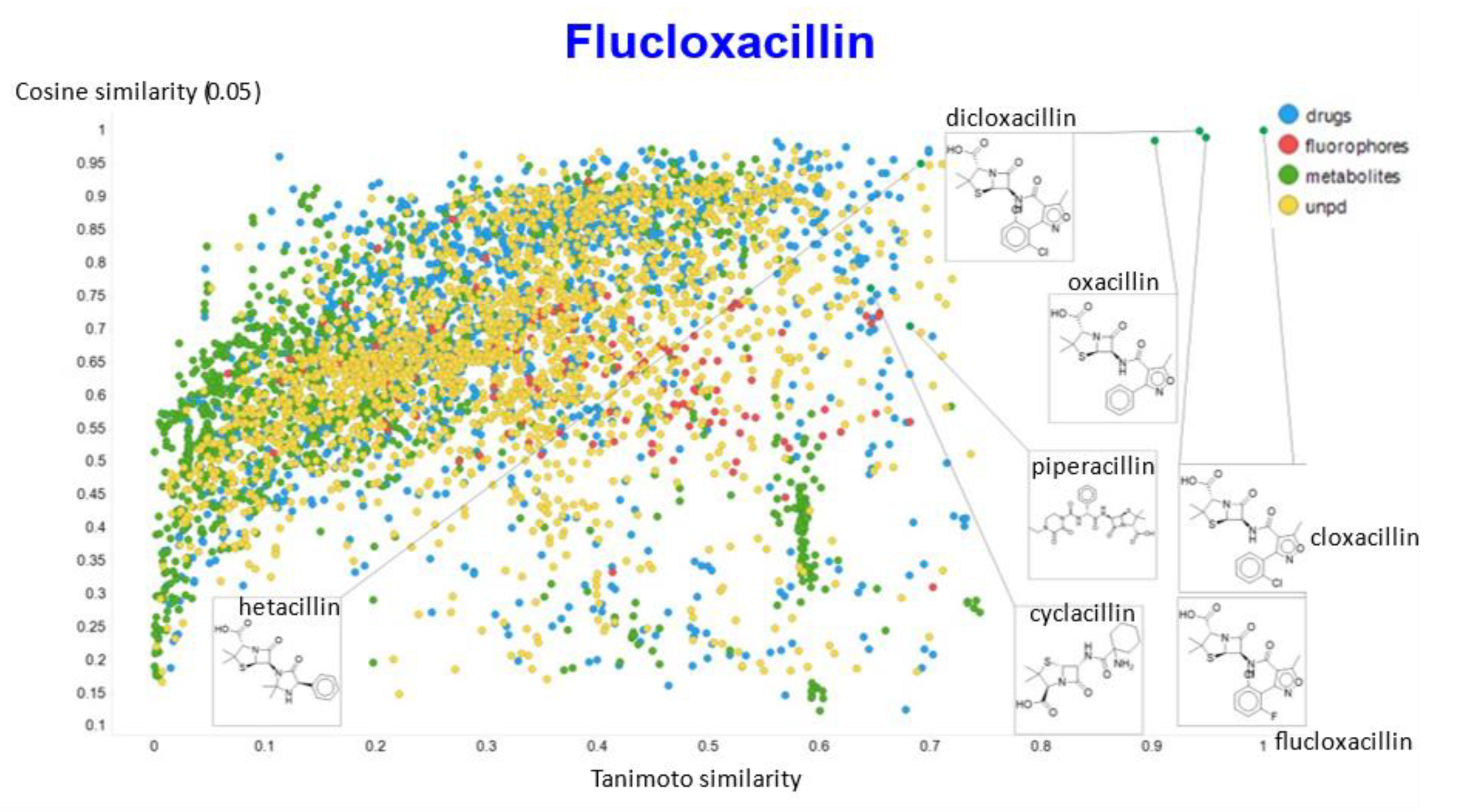
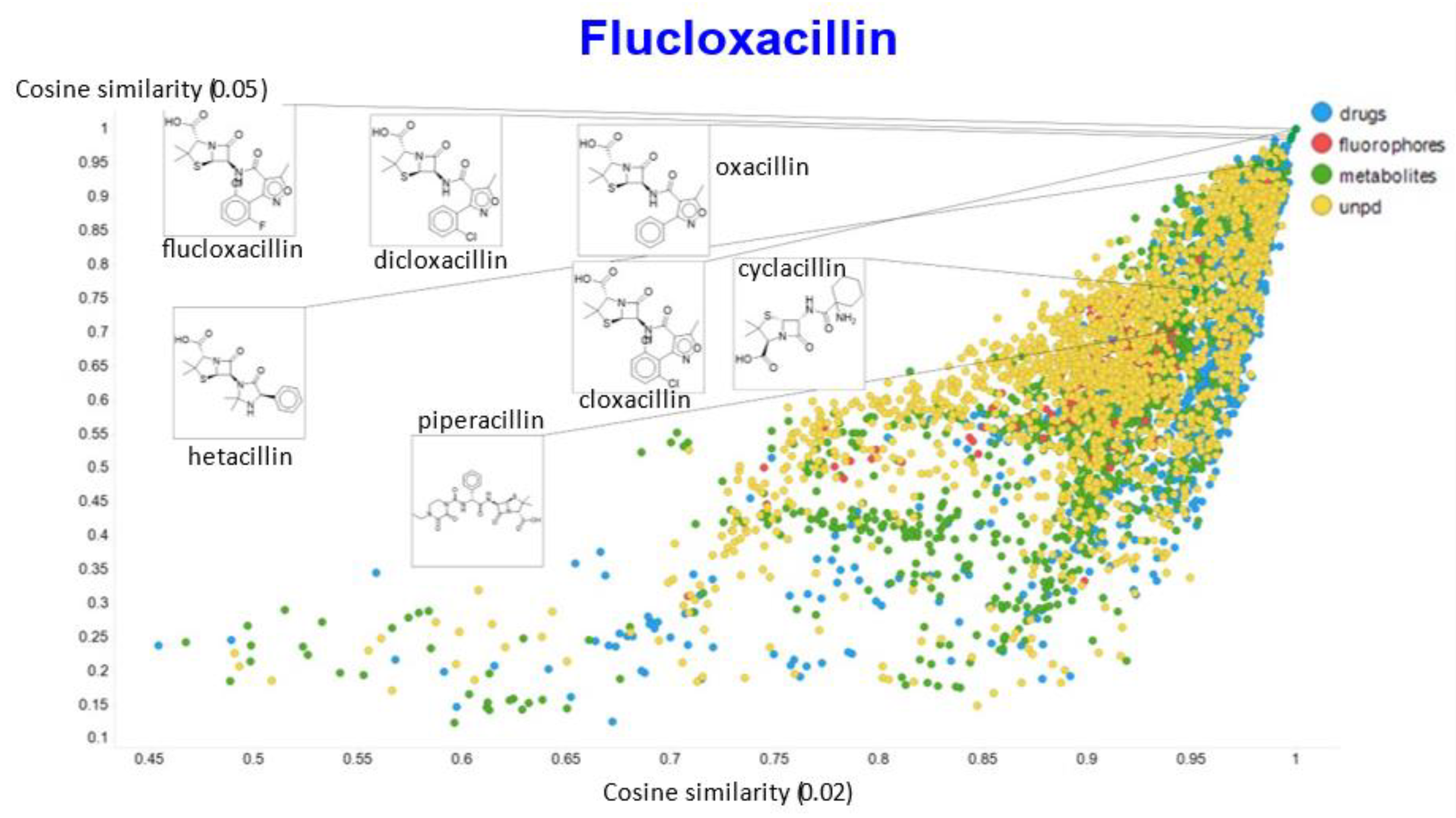
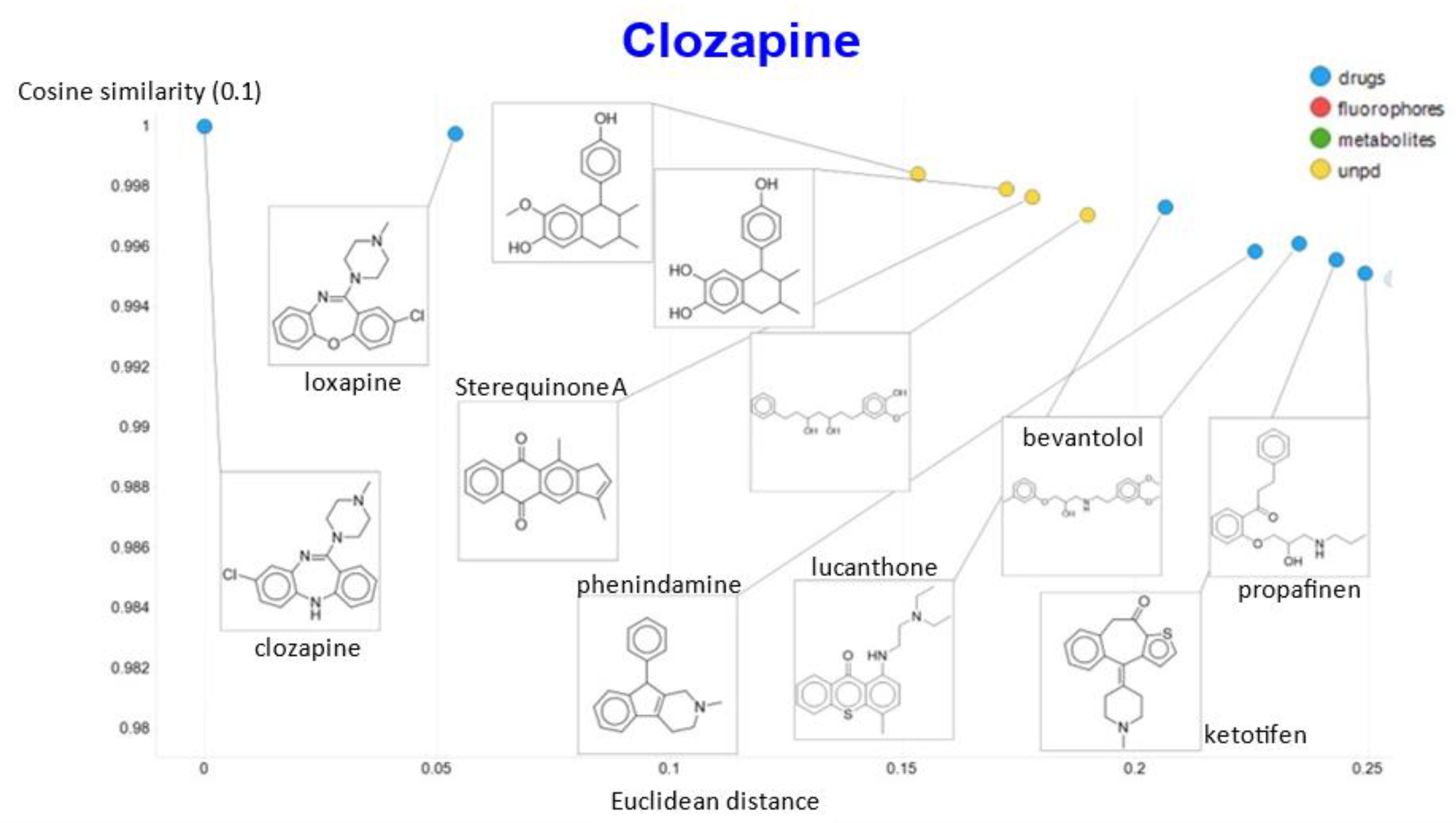
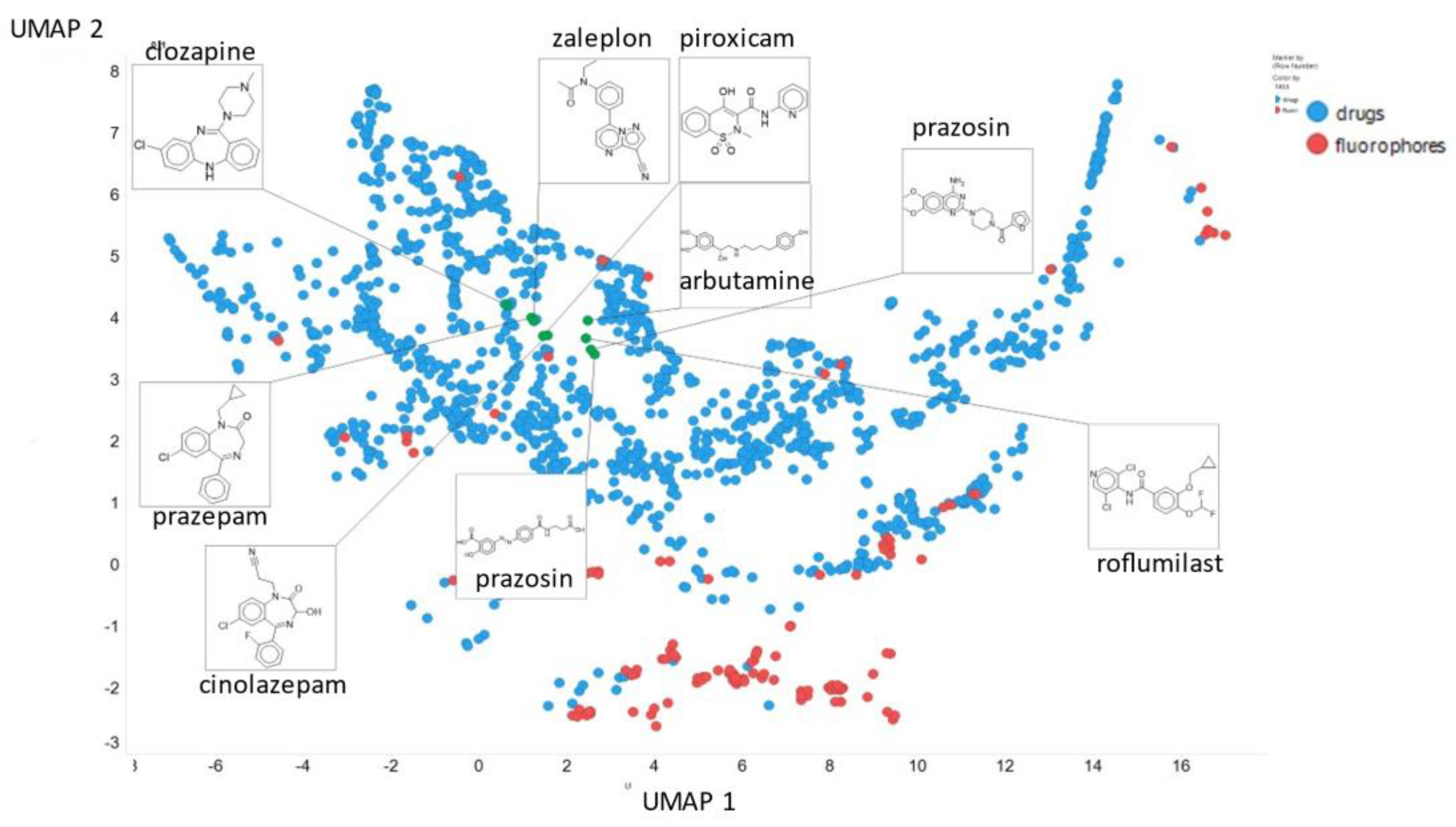
2. Results
3. Discussion
4. Materials and Methods
4.1. Molecular SMILES Augmentation
4.2. Base Encoder
4.3. Projection Head
4.4. Contrastive Loss
4.5. Unprojection Head
4.6. Base Decoder
4.7. Default Settings
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- LeCun, Y.; Bengio, Y.; Hinton, G. Deep learning. Nature 2015, 521, 436–444. [Google Scholar] [CrossRef]
- Schmidhuber, J. Deep learning in neural networks: An overview. Neural Netw. 2015, 61, 85–117. [Google Scholar] [CrossRef]
- Brown, T.B.; Mann, B.; Ryder, N.; Subbiah, M.; Kaplan, J.; Dhariwal, P.; Neelakantan, A.; Shyam, P.; Sastry, G.; Askell, A.; et al. Language models are few-shot learners. arXiv 2020, arXiv:2005.14165. [Google Scholar]
- Senior, A.W.; Evans, R.; Jumper, J.; Kirkpatrick, J.; Sifre, L.; Green, T.; Qin, C.; Zidek, A.; Nelson, A.W.R.; Bridgland, A.; et al. Improved protein structure prediction using potentials from deep learning. Nature 2020, 577, 706–710. [Google Scholar] [CrossRef] [PubMed]
- Samanta, S.; O’Hagan, S.; Swainston, N.; Roberts, T.J.; Kell, D.B. VAE-Sim: A novel molecular similarity measure based on a variational autoencoder. Molecules 2020, 25, 3446. [Google Scholar] [CrossRef]
- Kingma, D.; Welling, M. Auto-encoding variational Bayes. arXiv 2014, arXiv:1312.6114v1310. [Google Scholar]
- Kingma, D.P.; Welling, M. An introduction to variational autoencoders. arXiv 2019, arXiv:1906.02691v02691. [Google Scholar]
- Wei, R.; Mahmood, A. Recent advances in variational autoencoders with representation learning for biomedical informatics: A survey. IEEE Access 2021, 9, 4939–4956. [Google Scholar] [CrossRef]
- Wei, R.; Garcia, C.; El-Sayed, A.; Peterson, V.; Mahmood, A. Variations in variational autoencoders—A comparative evaluation. IEEE Access 2020, 8, 153651–153670. [Google Scholar] [CrossRef]
- Van Deursen, R.; Tetko, I.V.; Godin, G. Beyond chemical 1d knowledge using transformers. arXiv 2020, arXiv:2010.01027. [Google Scholar]
- Vaswani, A.; Shazeer, N.; Parmar, N.; Uszkoreit, J.; Jones, L.; Gomez, A.N.; Kaiser, L.; Polosukhin, I. Attention is all you need. arXiv 2017, arXiv:1706.03762. [Google Scholar]
- Chithrananda, S.; Grand, G.; Ramsundar, B. Chemberta: Large-scale self-supervised pretraining for molecular property prediction. arXiv 2020, arXiv:2010.09885. [Google Scholar]
- Sundararajan, M.; Taly, A.; Yan, Q. Axiomatic attribution for deep networks. arXiv 2017, arXiv:1703.01365. [Google Scholar]
- Simonyan, K.; Vedaldi, A.; Zisserman, A. Deep inside convolutional networks: Visualising image classification models and saliency maps. arXiv 2013, arXiv:1312.6034. [Google Scholar]
- Azodi, C.B.; Tang, J.; Shiu, S.H. Opening the black box: Interpretable machine learning for geneticists. Trends Genet. 2020, 36, 442–455. [Google Scholar] [CrossRef]
- Core, M.G.; Lane, H.C.; van Lent, M.; Gomboc, D.; Solomon, S.; Rosenberg, M. Building explainable artificial intelligence systems. AAAI 2006, 1766–1773. [Google Scholar] [CrossRef]
- Holzinger, A.; Biemann, C.; Pattichis, C.S.; Kell, D.B. What do we need to build explainable AI systems for the medical domain? arXiv 2017, arXiv:1712.09923v09921. [Google Scholar]
- Samek, W.; Montavon, G.; Vedaldi, A.; Hansen, L.K.; Müller, K.-R. Explainable AI: Interpreting, Explaining and Visualizing Deep Learning; Springer: Berlin, Germany, 2019. [Google Scholar]
- Singh, A.; Sengupta, S.; Lakshminarayanan, V. Explainable deep learning models in medical image analysis. arXiv 2020, arXiv:2005.13799. [Google Scholar]
- Tjoa, E.; Guan, C. A survey on explainable artificial intelligence (XAI): Towards medical XAI. arXiv 2019, arXiv:1907.07374. [Google Scholar] [CrossRef]
- Arrieta, A.B.; Díaz-Rodríguez, N.; Del Ser, J.; Bennetot, A.; Tabik, S.; Barbado, A.; Garcia, S.; Gil-Lopez, S.; Molina, D.; Benjamins, R.; et al. Explainable artificial intelligence (XAI): Concepts, taxonomies, opportunities and challenges toward responsible AI. Inf. Fusion 2020, 58, 82–115. [Google Scholar] [CrossRef]
- Gunning, D.; Stefik, M.; Choi, J.; Miller, T.; Stumpf, S.; Yang, G.Z. XAI-explainable artificial intelligence. Sci. Robot. 2019, 4, eaay7120. [Google Scholar] [CrossRef]
- Parmar, G.; Li, D.; Lee, K.; Tu, Z. Dual contradistinctive generative autoencoder. arXiv 2020, arXiv:2011.10063. [Google Scholar]
- Peis, I.; Olmos, P.M.; Artés-Rodríguez, A. Unsupervised learning of global factors in deep generative models. arXiv 2020, arXiv:2012.08234. [Google Scholar]
- Klys, J.; Snell, J.; Zemel, R. Learning latent subspaces in variational autoencoders. arXiv 2018, arXiv:1812.06190. [Google Scholar]
- He, Z.; Kan, M.; Zhang, J.; Shan, S. PA-GAN: Progressive attention generative adversarial network for facial attribute editing. arXiv 2020, arXiv:2007.05892. [Google Scholar]
- Shen, X.; Liu, F.; Dong, H.; Lian, Q.; Chen, Z.; Zhang, T. Disentangled generative causal representation learning. arXiv 2020, arXiv:2010.02637. [Google Scholar]
- Esser, P.; Rombach, R.; Ommer, B. A note on data biases in generative models. arXiv 2020, arXiv:2012.02516. [Google Scholar]
- Kumar, A.; Sattigeri, P.; Balakrishnan, A. Variational inference of disentangled latent concepts from unlabeled observations. arXiv 2017, arXiv:1711.00848. [Google Scholar]
- Kim, H.; Mnih, A. Disentangling by factorising. arXiv 2018, arXiv:1802.05983. [Google Scholar]
- Locatello, F.; Bauer, S.; Lucic, M.; Rätsch, G.; Gelly, S.; Schölkopf, B.; Bachem, O. Challenging common assumptions in the unsupervised learning of disentangled representations. arXiv 2018, arXiv:1811.12359. [Google Scholar]
- Locatello, F.; Tschannen, M.; Bauer, S.; Rätsch, G.; Schölkopf, B.; Bachem, O. Disentangling factors of variation using few labels. arXiv 2019, arXiv:1905.01258v01251. [Google Scholar]
- Locatello, F.; Poole, B.; Rätsch, G.; Schölkopf, B.; Bachem, O.; Tschannen, M. Weakly-supervised disentanglement without compromises. arXiv 2020, arXiv:2002.02886. [Google Scholar]
- Oldfield, J.; Panagakis, Y.; Nicolaou, M.A. Adversarial learning of disentangled and generalizable representations of visual attributes. IEEE Trans. Neural Netw. Learn. Syst. 2021. [Google Scholar] [CrossRef] [PubMed]
- Pandey, A.; Schreurs, J.; Suykens, J.A.K. Generative restricted kernel machines: A framework for multi-view generation and disentangled feature learning. Neural Netw. 2021, 135, 177–191. [Google Scholar] [CrossRef] [PubMed]
- Hao, Z.; Lv, D.; Li, Z.; Cai, R.; Wen, W.; Xu, B. Semi-supervised disentangled framework for transferable named entity recognition. Neural Netw. 2021, 135, 127–138. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Yang, C.; Tang, X.; Zhou, B. Interfacegan: Interpreting the disentangled face representation learned by gans. IEEE Trans. Pattern Anal. Mach. Intell. 2020. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Tang, Y.; Zhu, Y.; Xiao, J.; Summers, R.M. A disentangled generative model for disease decomposition in chest x-rays via normal image synthesis. Med. Image Anal. 2021, 67, 101839. [Google Scholar] [CrossRef]
- Cootes, T.F.; Edwards, G.J.; Taylor, C.J. Active appearance models. IEEE Trans. Pattern Anal. Mach. Intell. 2001, 23, 681–685. [Google Scholar] [CrossRef]
- Cootes, T.F.; Taylor, C.J.; Cooper, D.H.; Graham, J. Active shape models—Their training and application. Comput. Vis. Image Underst. 1995, 61, 38–59. [Google Scholar] [CrossRef]
- Hill, A.; Cootes, T.F.; Taylor, C.J. Active shape models and the shape approximation problem. Image Vis. Comput. 1996, 14, 601–607. [Google Scholar] [CrossRef]
- Salam, H.; Seguier, R. A survey on face modeling: Building a bridge between face analysis and synthesis. Vis. Comput. 2018, 34, 289–319. [Google Scholar] [CrossRef]
- Bozkurt, A.; Esmaeili, B.; Brooks, D.H.; Dy, J.G.; van de Meent, J.-W. Evaluating combinatorial generalization in variational autoencoders. arXiv 2019, arXiv:1911.04594v04591. [Google Scholar]
- Alemi, A.A.; Poole, B.; Fischer, I.; Dillon, J.V.; Saurous, R.A.; Murphy, K. Fixing a broken ELBO. arXiv 2019, arXiv:1711.00464. [Google Scholar]
- Zhao, S.; Song, J.; Ermon, S. InfoVAE: Balancing learning and inference in variational autoencoders. arXiv 2017, arXiv:1706.02262v02263. [Google Scholar] [CrossRef]
- Leibfried, F.; Dutordoir, V.; John, S.T.; Durrande, N. A tutorial on sparse Gaussian processes and variational inference. arXiv 2020, arXiv:2012.13962. [Google Scholar]
- Rezende, D.J.; Viola, F. Taming VAEs. arXiv 2018, arXiv:1810.00597v00591. [Google Scholar]
- Dai, B.; Wipf, D. Diagnosing and enhancing VAE models. arXiv 2019, arXiv:1903.05789v05782. [Google Scholar]
- Li, Y.; Yu, S.; Principe, J.C.; Li, X.; Wu, D. PRI-VAE: Principle-of-relevant-information variational autoencoders. arXiv 2020, arXiv:2007.06503. [Google Scholar]
- Higgins, I.; Matthey, L.; Pal, A.; Burgess, C.; Glorot, X.; Botvinick, M.; Mohamed, S.; Lerchner, A. β-VAE: Learning basic visual concepts with a constrained variational framework. In Proceedings of the ICLR 2017, Toulon, France, 24–26 April 2017. [Google Scholar]
- Burgess, C.P.; Higgins, I.; Pal, A.; Matthey, L.; Watters, N.; Desjardins, G.; Lerchner, A. Understanding disentangling in β-VAE. arXiv 2018, arXiv:1804.03599. [Google Scholar]
- Havtorn, J.D.; Frellsen, J.; Hauberg, S.; Maaløe, L. Hierarchical vaes know what they don’t know. arXiv 2021, arXiv:2102.08248. [Google Scholar]
- Kumar, A.; Poole, B. On implicit regularization in β-VAEs. arXiv 2021, arXiv:2002.00041. [Google Scholar]
- Yang, T.; Ren, X.; Wang, Y.; Zeng, W.; Zheng, N.; Ren, P. GroupifyVAE: From group-based definition to VAE-based unsupervised representation disentanglement. arXiv 2021, arXiv:2102.10303. [Google Scholar]
- Gatopoulos, I.; Tomczak, J.M. Self-supervised variational auto-encoders. arXiv 2020, arXiv:2010.02014. [Google Scholar]
- Rong, Y.; Bian, Y.; Xu, T.; Xie, W.; Wei, Y.; Huang, W.; Huang, J. Self-supervised graph transformer on large-scale molecular data. arXiv 2020, arXiv:2007.02835. [Google Scholar]
- Saeed, A.; Grangier, D.; Zeghidour, N. Contrastive learning of general-purpose audio representations. arXiv 2020, arXiv:2010.10915. [Google Scholar]
- Aneja, J.; Schwing, A.; Kautz, J.; Vahdat, A. NCP-VAE: Variational autoencoders with noise contrastive priors. arXiv 2020, arXiv:2010.02917. [Google Scholar]
- Artelt, A.; Hammer, B. Efficient computation of contrastive explanations. arXiv 2020, arXiv:2010.02647. [Google Scholar]
- Ciga, O.; Martel, A.L.; Xu, T. Self supervised contrastive learning for digital histopathology. arXiv 2020, arXiv:2011.13971. [Google Scholar]
- Chen, T.; Kornblith, S.; Norouzi, M.; Hinton, G. A simple framework for contrastive learning of visual representations. arXiv 2020, arXiv:2002.05709. [Google Scholar]
- Jaiswal, A.; Babu, A.R.; Zadeh, M.Z.; Banerjee, D.; Makedon, F. A survey on contrastive self-supervised learning. arXiv 2020, arXiv:2011.00362. [Google Scholar]
- Purushwalkam, S.; Gupta, A. Demystifying contrastive self-supervised learning: Invariances, augmentations and dataset biases. arXiv 2020, arXiv:2007.13916. [Google Scholar]
- Van den Oord, A.; Li, Y.; Vinyals, O. Representation learning with contrastive predictive coding. arXiv 2018, arXiv:1807.03748v03742. [Google Scholar]
- Verma, V.; Luong, M.-T.; Kawaguchi, K.; Pham, H.; Le, Q.V. Towards domain-agnostic contrastive learning. arXiv 2020, arXiv:2011.04419. [Google Scholar]
- Le-Khac, P.H.; Healy, G.; Smeaton, A.F. Contrastive representation learning: A framework and review. arXiv 2020, arXiv:2010.05113. [Google Scholar] [CrossRef]
- Wang, Q.; Meng, F.; Breckon, T.P. Data augmentation with norm-VAE for unsupervised domain adaptation. arXiv 2020, arXiv:2012.00848. [Google Scholar]
- Li, H.; Zhang, X.; Sun, R.; Xiong, H.; Tian, Q. Center-wise local image mixture for contrastive representation learning. arXiv 2020, arXiv:2011.02697. [Google Scholar]
- You, Y.; Chen, T.; Sui, Y.; Chen, T.; Wang, Z.; Shen, Y. Graph contrastive learning with augmentations. arXiv 2020, arXiv:2010.13902. [Google Scholar]
- Willett, P. Similarity-based data mining in files of two-dimensional chemical structures using fingerprint measures of molecular resemblance. Wires Data Min. Knowl. 2011, 1, 241–251. [Google Scholar] [CrossRef]
- Stumpfe, D.; Bajorath, J. Similarity searching. Wires Comput. Mol. Sci. 2011, 1, 260–282. [Google Scholar] [CrossRef]
- Maggiora, G.; Vogt, M.; Stumpfe, D.; Bajorath, J. Molecular similarity in medicinal chemistry. J. Med. Chem. 2014, 57, 3186–3204. [Google Scholar] [CrossRef]
- Irwin, J.J.; Shoichet, B.K. ZINC--a free database of commercially available compounds for virtual screening. J. Chem. Inf. Model. 2005, 45, 177–182. [Google Scholar] [CrossRef]
- Ertl, P.; Schuffenhauer, A. Estimation of synthetic accessibility score of drug-like molecules based on molecular complexity and fragment contributions. J. Cheminform. 2009, 1, 8. [Google Scholar] [CrossRef]
- Patel, H.; Ihlenfeldt, W.D.; Judson, P.N.; Moroz, Y.S.; Pevzner, Y.; Peach, M.L.; Delannee, V.; Tarasova, N.I.; Nicklaus, M.C. Savi, in silico generation of billions of easily synthesizable compounds through expert-system type rules. Sci. Data 2020, 7, 384. [Google Scholar] [CrossRef]
- Bickerton, G.R.; Paolini, G.V.; Besnard, J.; Muresan, S.; Hopkins, A.L. Quantifying the chemical beauty of drugs. Nat. Chem. 2012, 4, 90–98. [Google Scholar] [CrossRef]
- Cernak, T.; Dykstra, K.D.; Tyagarajan, S.; Vachal, P.; Krska, S.W. The medicinal chemist’s toolbox for late stage functionalization of drug-like molecules. Chem. Soc. Rev. 2016, 45, 546–576. [Google Scholar] [CrossRef]
- Lovrić, M.; Molero, J.M.; Kern, R. PySpark and RDKit: Moving towards big data in cheminformatics. Mol. Inform. 2019, 38, e1800082. [Google Scholar] [CrossRef]
- Clyde, A.; Ramanathan, A.; Stevens, R. Scaffold embeddings: Learning the structure spanned by chemical fragments, scaffolds and compounds. arXiv 2021, arXiv:2103.06867. [Google Scholar]
- Arús-Pous, J.; Awale, M.; Probst, D.; Reymond, J.L. Exploring chemical space with machine learning. Chem. Int. J. Chem. 2019, 73, 1018–1023. [Google Scholar] [CrossRef]
- Awale, M.; Probst, D.; Reymond, J.L. WebMolCS: A web-based interface for visualizing molecules in three-dimensional chemical spaces. J. Chem. Inf. Model. 2017, 57, 643–649. [Google Scholar] [CrossRef]
- Baldi, P.; Muller, K.R.; Schneider, G. Charting chemical space: Challenges and opportunities for artificial intelligence and machine learning. Mol. Inform. 2011, 30, 751–752. [Google Scholar] [CrossRef]
- Chen, Y.; Garcia de Lomana, M.; Friedrich, N.O.; Kirchmair, J. Characterization of the chemical space of known and readily obtainable natural products. J. Chem. Inf. Model. 2018, 58, 1518–1532. [Google Scholar] [CrossRef] [PubMed]
- Drew, K.L.M.; Baiman, H.; Khwaounjoo, P.; Yu, B.; Reynisson, J. Size estimation of chemical space: How big is it? J. Pharm. Pharmacol. 2012, 64, 490–495. [Google Scholar] [CrossRef] [PubMed]
- Ertl, P. Visualization of chemical space for medicinal chemists. J. Cheminform. 2014, 6, O4. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Medina, M.; Prieto-Martinez, F.D.; Naveja, J.J.; Mendez-Lucio, O.; El-Elimat, T.; Pearce, C.J.; Oberlies, N.H.; Figueroa, M.; Medina-Franco, J.L. Chemoinformatic expedition of the chemical space of fungal products. Future Med. Chem. 2016, 8, 1399–1412. [Google Scholar] [CrossRef]
- Klimenko, K.; Marcou, G.; Horvath, D.; Varnek, A. Chemical space mapping and structure-activity analysis of the chembl antiviral compound set. J. Chem. Inf. Model. 2016, 56, 1438–1454. [Google Scholar] [CrossRef]
- Lin, A.; Horvath, D.; Afonina, V.; Marcou, G.; Reymond, J.L.; Varnek, A. Mapping of the available chemical space versus the chemical universe of lead-like compounds. ChemMedChem 2018, 13, 540–554. [Google Scholar] [CrossRef]
- Lucas, X.; Gruning, B.A.; Bleher, S.; Günther, S. The purchasable chemical space: A detailed picture. J. Chem. Inf. Model. 2015, 55, 915–924. [Google Scholar] [CrossRef]
- Nigam, A.; Friederich, P.; Krenn, M.; Aspuru-Guzik, A. Augmenting genetic algorithms with deep neural networks for exploring the chemical space. arXiv 2019, arXiv:1909.11655. [Google Scholar]
- O’Hagan, S.; Kell, D.B. Generation of a small library of natural products designed to cover chemical space inexpensively. Pharm. Front. 2019, 1, e190005. [Google Scholar]
- Polishchuk, P.G.; Madzhidov, T.I.; Varnek, A. Estimation of the size of drug-like chemical space based on GDB-17 data. J. Comput. Aided Mol. Des. 2013, 27, 675–679. [Google Scholar] [CrossRef]
- Reymond, J.L. The chemical space project. Acc. Chem. Res. 2015, 48, 722–730. [Google Scholar] [CrossRef] [PubMed]
- Rosén, J.; Gottfries, J.; Muresan, S.; Backlund, A.; Oprea, T.I. Novel chemical space exploration via natural products. J. Med. Chem. 2009, 52, 1953–1962. [Google Scholar] [CrossRef] [PubMed]
- Thakkar, A.; Selmi, N.; Reymond, J.L.; Engkvist, O.; Bjerrum, E. ‘Ring breaker’: Neural network driven synthesis prediction of the ring system chemical space. J. Med. Chem. 2020, 63, 8791–8808. [Google Scholar] [CrossRef] [PubMed]
- Thiede, L.A.; Krenn, M.; Nigam, A.; Aspuru-Guzik, A. Curiosity in exploring chemical space: Intrinsic rewards for deep molecular reinforcement learning. arXiv 2020, arXiv:2012.11293. [Google Scholar]
- Coley, C.W. Defining and exploring chemical spaces. Trends Chem. 2021, 3, 133–145. [Google Scholar] [CrossRef]
- Bender, A.; Glen, R.C. Molecular similarity: A key technique in molecular informatics. Org. Biomol. Chem. 2004, 2, 3204–3218. [Google Scholar] [CrossRef]
- O’Hagan, S.; Kell, D.B. Consensus rank orderings of molecular fingerprints illustrate the ‘most genuine’ similarities between marketed drugs and small endogenous human metabolites, but highlight exogenous natural products as the most important ‘natural’ drug transporter substrates. ADMET DMPK 2017, 5, 85–125. [Google Scholar]
- Sterling, T.; Irwin, J.J. ZINC 15—Ligand discovery for everyone. J. Chem. Inf. Model. 2015, 55, 2324–2337. [Google Scholar] [CrossRef]
- Raffel, C.; Shazeer, N.; Roberts, A.; Lee, K.; Narang, S.; Matena, M.; Zhou, Y.; Li, W.; Liu, P.J. Exploring the limits of transfer learning with a unified text-to-text transformer. arXiv 2019, arXiv:1910.10683. [Google Scholar]
- Rives, A.; Goyal, S.; Meier, J.; Guo, D.; Ott, M.; Zitnick, C.L.; Ma, J.; Fergus, R. Biological structure and function emerge from scaling unsupervised learning to 250 million protein sequences. bioRxiv 2019, 622803. [Google Scholar] [CrossRef]
- So, D.R.; Liang, C.; Le, Q.V. The evolved transformer. arXiv 2019, arXiv:1901.11117. [Google Scholar]
- Grechishnikova, D. Transformer neural network for protein specific de novo drug generation as machine translation problem. bioRxiv 2020. [Google Scholar] [CrossRef]
- Choromanski, K.; Likhosherstov, V.; Dohan, D.; Song, X.; Gane, A.; Sarlos, T.; Hawkins, P.; Davis, J.; Mohiuddin, A.; Kaiser, L.; et al. Rethinking attention with Performers. arXiv 2020, arXiv:2009.14794. [Google Scholar]
- Yun, C.; Bhojanapalli, S.; Rawat, A.S.; Reddi, S.J.; Kumar, S. Are transformers universal approximators of sequence-to-sequence functions? arXiv 2019, arXiv:1912.10077. [Google Scholar]
- Dosovitskiy, A.; Beyer, L.; Kolesnikov, A.; Weissenborn, D.; Zhai, X.; Unterthiner, T.; Dehghani, M.; Minderer, M.; Heigold, G.; Gelly, S.; et al. An image is worth 16 × 16 words: Transformers for image recognition at scale. arXiv 2020, arXiv:2010.11929. [Google Scholar]
- Fedus, W.; Zoph, B.; Shazeer, N. Switch transformers: Scaling to trillion parameter models with simple and efficient sparsity. arXiv 2021, arXiv:2101.03961. [Google Scholar]
- Lu, K.; Grover, A.; Abbeel, P.; Mordatch, I. Pretrained transformers as universal computation engines. arXiv 2021, arXiv:2103.05247. [Google Scholar]
- Goyal, P.; Caron, M.; Lefaudeux, B.; Xu, M.; Wang, P.; Pai, V.; Singh, M.; Liptchinsky, V.; Misra, I.; Joulin, A.; et al. Self-supervised pretraining of visual features in the wild. arXiv 2021, arXiv:2103.01988v01981. [Google Scholar]
- Wang, Y.; Wang, J.; Cao, Z.; Farimani, A.B. MolCLR: Molecular contrastive learning of representations via graph neural networks. arXiv 2021, arXiv:2102.10056. [Google Scholar]
- Chen, T.; Kornblith, S.; Swersky, K.; Norouzi, M.; Hinton, G. Big self-supervised models are strong semi-supervised learners. arXiv 2020, arXiv:2006.10029. [Google Scholar]
- O’Hagan, S.; Kell, D.B. Structural similarities between some common fluorophores used in biology, marketed drugs, endogenous metabolites, and natural products. Mar. Drugs 2020, 18, 582. [Google Scholar] [CrossRef] [PubMed]
- Ji, Z.; Zou, X.; Huang, T.; Wu, S. Unsupervised few-shot feature learning via self-supervised training. Front. Comput. Neurosci. 2020, 14, 83. [Google Scholar] [CrossRef]
- Wang, Y.; Yao, Q.; Kwok, J.; Ni, L.M. Generalizing from a few examples: A survey on few-shot learning. arXiv 2019, arXiv:1904.05046. [Google Scholar]
- Ma, J.; Fong, S.H.; Luo, Y.; Bakkenist, C.J.; Shen, J.P.; Mourragui, S.; Wessels, L.F.A.; Hafner, M.; Sharan, R.; Peng, J.; et al. Few-shot learning creates predictive models of drug response that translate from high-throughput screens to individual patients. Nat. Cancer 2021, 2, 233–244. [Google Scholar] [CrossRef]
- Li, F.-F.; Fergus, R.; Perona, P. One-shot learning of object categories. IEEE Trans. Pattern Anal. Mach. Intell. 2006, 28, 594–611. [Google Scholar]
- Rezende, D.J.; Mohamed, S.; Danihelka, I.; Gregor, K.; Wierstra, D. One-shot generalization in deep generative models. arXiv 2016, arXiv:1603.05106v05101. [Google Scholar]
- Altae-Tran, H.; Ramsundar, B.; Pappu, A.S.; Pande, V. Low data drug discovery with one-shot learning. ACS Cent. Sci. 2017, 3, 283–293. [Google Scholar] [CrossRef] [PubMed]
- Baskin, I.I. Is one-shot learning a viable option in drug discovery? Expert Opin. Drug Discov. 2019, 14, 601–603. [Google Scholar] [CrossRef]
- He, X.; Zhao, K.Y.; Chu, X.W. AutoML: A survey of the state-of-the-art. Knowl. Based Syst. 2021, 212, 106622. [Google Scholar] [CrossRef]
- Chochlakis, G.; Georgiou, E.; Potamianos, A. End-to-end generative zero-shot learning via few-shot learning. arXiv 2021, arXiv:2102.04379. [Google Scholar]
- Majumder, O.; Ravichandran, A.; Maji, S.; Polito, M.; Bhotika, R.; Soatto, S. Revisiting contrastive learning for few-shot classification. arXiv 2021, arXiv:2101.11058. [Google Scholar]
- Dasari, S.; Gupta, A. Transformers for one-shot visual imitation. arXiv 2020, arXiv:2011.05970. [Google Scholar]
- Logeswaran, L.; Lee, A.; Ott, M.; Lee, H.; Ranzato, M.A.; Szlam, A. Few-shot sequence learning with transformers. arXiv 2020, arXiv:2012.09543. [Google Scholar]
- Belkin, M.; Hsu, D.; Ma, S.; Mandal, S. Reconciling modern machine-learning practice and the classical bias-variance trade-off. Proc. Natl. Acad. Sci. USA 2019, 116, 15849–15854. [Google Scholar] [CrossRef] [PubMed]
- Van der Maaten, L.; Hinton, G. Visualizing data using t-sne. J. Mach. Learn. Res. 2008, 9, 2579–2605. [Google Scholar]
- Van der Maaten, L. Learning a parametric embedding by preserving local structure. Proc. AISTATS 2009, 384–391. [Google Scholar]
- McInnes, L.; Healy, J.; Melville, J. UMAP: Uniform manifold approximation and projection for dimension reduction. arXiv 2018, arXiv:1802.03426v03422. [Google Scholar]
- McInnes, L.; Healy, J.; Saul, N.; Großberger, L. UMAP: Uniform manifold approximation and projection. J. Open Source Softw. 2018. [Google Scholar] [CrossRef]
- Dickens, D.; Rädisch, S.; Chiduza, G.N.; Giannoudis, A.; Cross, M.J.; Malik, H.; Schaeffeler, E.; Sison-Young, R.L.; Wilkinson, E.L.; Goldring, C.E.; et al. Cellular uptake of the atypical antipsychotic clozapine is a carrier-mediated process. Mol. Pharm. 2018, 15, 3557–3572. [Google Scholar] [CrossRef]
- Horvath, D.; Jeandenans, C. Neighborhood behavior of in silico structural spaces with respect to in vitro activity spaces-a novel understanding of the molecular similarity principle in the context of multiple receptor binding profiles. J. Chem. Inf. Comput. Sci. 2003, 43, 680–690. [Google Scholar] [CrossRef]
- Bender, A.; Jenkins, J.L.; Li, Q.L.; Adams, S.E.; Cannon, E.O.; Glen, R.C. Molecular similarity: Advances in methods, applications and validations in virtual screening and qsar. Annu. Rep. Comput. Chem. 2006, 2, 141–168. [Google Scholar] [PubMed]
- Horvath, D.; Koch, C.; Schneider, G.; Marcou, G.; Varnek, A. Local neighborhood behavior in a combinatorial library context. J. Comput. Aid. Mol. Des. 2011, 25, 237–252. [Google Scholar] [CrossRef] [PubMed]
- Gasteiger, J. Handbook of Chemoinformatics: From Data to Knowledge; Wiley/VCH: Weinheim, Germany, 2003. [Google Scholar]
- Bajorath, J. Chemoinformatics: Concepts, Methods and Tools for Drug Discovery; Humana Press: Totowa, NJ, USA, 2004. [Google Scholar]
- Sutherland, J.J.; Raymond, J.W.; Stevens, J.L.; Baker, T.K.; Watson, D.E. Relating molecular properties and in vitro assay results to in vivo drug disposition and toxicity outcomes. J. Med. Chem. 2012, 55, 6455–6466. [Google Scholar] [CrossRef] [PubMed]
- Capecchi, A.; Probst, D.; Reymond, J.L. One molecular fingerprint to rule them all: Drugs, biomolecules, and the metabolome. J. Cheminform. 2020, 12, 43. [Google Scholar] [CrossRef] [PubMed]
- Muegge, I.; Mukherjee, P. An overview of molecular fingerprint similarity search in virtual screening. Expert Opin. Drug Discov. 2016, 11, 137–148. [Google Scholar] [CrossRef] [PubMed]
- Nisius, B.; Bajorath, J. Rendering conventional molecular fingerprints for virtual screening independent of molecular complexity and size effects. ChemMedChem 2010, 5, 859–868. [Google Scholar] [CrossRef] [PubMed]
- Riniker, S.; Landrum, G.A. Similarity maps—A visualization strategy for molecular fingerprints and machine-learning methods. J. Cheminform. 2013, 5, 43. [Google Scholar] [CrossRef] [PubMed]
- Vogt, I.; Stumpfe, D.; Ahmed, H.E.; Bajorath, J. Methods for computer-aided chemical biology. Part 2: Evaluation of compound selectivity using 2d molecular fingerprints. Chem. Biol. Drug Des. 2007, 70, 195–205. [Google Scholar] [CrossRef]
- O’Hagan, S.; Swainston, N.; Handl, J.; Kell, D.B. A ‘rule of 0.5′ for the metabolite-likeness of approved pharmaceutical drugs. Metabolomics 2015, 11, 323–339. [Google Scholar] [CrossRef]
- O’Hagan, S.; Kell, D.B. Understanding the foundations of the structural similarities between marketed drugs and endogenous human metabolites. Front. Pharm. 2015, 6, 105. [Google Scholar] [CrossRef]
- O’Hagan, S.; Kell, D.B. The apparent permeabilities of Caco-2 cells to marketed drugs: Magnitude, and independence from both biophysical properties and endogenite similarities. Peer J. 2015, 3, e1405. [Google Scholar] [CrossRef]
- O’Hagan, S.; Kell, D.B. MetMaxStruct: A Tversky-similarity-based strategy for analysing the (sub)structural similarities of drugs and endogenous metabolites. Front. Pharm. 2016, 7, 266. [Google Scholar] [CrossRef]
- O’Hagan, S.; Kell, D.B. Analysis of drug-endogenous human metabolite similarities in terms of their maximum common substructures. J. Cheminform. 2017, 9, 18. [Google Scholar] [CrossRef] [PubMed]
- O’Hagan, S.; Kell, D.B. Analysing and navigating natural products space for generating small, diverse, but representative chemical libraries. Biotechnol. J. 2018, 13, 1700503. [Google Scholar] [CrossRef] [PubMed]
- Gawehn, E.; Hiss, J.A.; Schneider, G. Deep learning in drug discovery. Mol. Inform. 2016, 35, 3–14. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Bombarelli, R.; Wei, J.N.; Duvenaud, D.; Hernández-Lobato, J.M.; Sánchez-Lengeling, B.; Sheberla, D.; Aguilera-Iparraguirre, J.; Hirzel, T.D.; Adams, R.P.; Aspuru-Guzik, A. Automatic chemical design using a data-driven continuous representation of molecules. ACS Cent. Sci. 2018, 4, 268–276. [Google Scholar] [CrossRef]
- Sanchez-Lengeling, B.; Aspuru-Guzik, A. Inverse molecular design using machine learning: Generative models for matter engineering. Science 2018, 361, 360–365. [Google Scholar] [CrossRef]
- Arús-Pous, J.; Probst, D.; Reymond, J.L. Deep learning invades drug design and synthesis. Chimia 2018, 72, 70–71. [Google Scholar] [CrossRef]
- Yang, K.; Swanson, K.; Jin, W.; Coley, C.; Eiden, P.; Gao, H.; Guzman-Perez, A.; Hopper, T.; Kelley, B.; Mathea, M.; et al. Analyzing learned molecular representations for property prediction. J. Chem. Inf. Model. 2019, 59, 3370–3388. [Google Scholar] [CrossRef]
- Zhavoronkov, A.; Ivanenkov, Y.A.; Aliper, A.; Veselov, M.S.; Aladinskiy, V.A.; Aladinskaya, A.V.; Terentiev, V.A.; Polykovskiy, D.A.; Kuznetsov, M.D.; Asadulaev, A.; et al. Deep learning enables rapid identification of potent DDR1 kinase inhibitors. Nat. Biotechnol. 2019, 37, 1038–1040. [Google Scholar] [CrossRef]
- Khemchandani, Y.; O’Hagan, S.; Samanta, S.; Swainston, N.; Roberts, T.J.; Bollegala, D.; Kell, D.B. DeepGraphMolGen, a multiobjective, computational strategy for generating molecules with desirable properties: A graph convolution and reinforcement learning approach. J. Cheminform. 2020, 12, 53. [Google Scholar] [CrossRef] [PubMed]
- Shen, C.; Krenn, M.; Eppel, S.; Aspuru-Guzik, A. Deep molecular dreaming: Inverse machine learning for de-novo molecular design and interpretability with surjective representations. arXiv 2020, arXiv:2012.09712. [Google Scholar]
- Moret, M.; Friedrich, L.; Grisoni, F.; Merk, D.; Schneider, G. Generative molecular design in low data regimes. Nat. Mach. Intell. 2020, 2, 171–180. [Google Scholar] [CrossRef]
- Kell, D.B.; Samanta, S.; Swainston, N. Deep learning and generative methods in cheminformatics and chemical biology: Navigating small molecule space intelligently. Biochem. J. 2020, 477, 4559–4580. [Google Scholar] [CrossRef]
- Walters, W.P.; Barzilay, R. Applications of deep learning in molecule generation and molecular property prediction. Acc. Chem. Res. 2021, 54, 263–270. [Google Scholar] [CrossRef]
- Zaheer, M.; Guruganesh, G.; Dubey, A.; Ainslie, J.; Alberti, C.; Ontanon, S.; Pham, P.; Ravula, A.; Wang, Q.; Yang, L.; et al. Big bird: Transformers for longer sequences. arXiv 2020, arXiv:2007.14062. [Google Scholar]
- Hutson, M. The language machines. Nature 2021, 591, 22–25. [Google Scholar] [CrossRef]
- Topal, M.O.; Bas, A.; van Heerden, I. Exploring transformers in natural language generation: GPT, BERT, and XLNET. arXiv 2021, arXiv:2102.08036. [Google Scholar]
- Zandie, R.; Mahoor, M.H. Topical language generation using transformers. arXiv 2021, arXiv:2103.06434. [Google Scholar]
- Weininger, D. Smiles, a chemical language and information system.1. Introduction to methodology and encoding rules. J. Chem. Inf. Comput. Sci. 1988, 28, 31–36. [Google Scholar] [CrossRef]
- Tetko, I.V.; Karpov, P.; Van Deursen, R.; Godin, G. State-of-the-art augmented NLP transformer models for direct and single-step retrosynthesis. Nat. Commun. 2020, 11, 5575. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.; Lee, Y.O. Predicting chemical properties using self-attention multi-task learning based on SMILES representation. arXiv 2020, arXiv:2010.11272. [Google Scholar]
- Pflüger, P.M.; Glorius, F. Molecular machine learning: The future of synthetic chemistry? Angew. Chem. Int. Ed. Engl. 2020. [Google Scholar] [CrossRef] [PubMed]
- Shin, B.; Park, S.; Bak, J.; Ho, J.C. Controlled molecule generator for optimizing multiple chemical properties. arXiv 2020, arXiv:2010.13908. [Google Scholar]
- Liu, X.; Zhang, F.; Hou, Z.; Mian, L.; Wang, Z.; Zhang, J.; Tang, J. Self-supervised learning: Generative or contrastive. arXiv 2020, arXiv:2006.08218v08214. [Google Scholar]
- Wanyan, T.; Honarvar, H.; Jaladanki, S.K.; Zang, C.; Naik, N.; Somani, S.; Freitas, J.K.D.; Paranjpe, I.; Vaid, A.; Miotto, R.; et al. Contrastive learning improves critical event prediction in COVID-19 patients. arXiv 2021, arXiv:2101.04013. [Google Scholar]
- Kostas, D.; Aroca-Ouellette, S.; Rudzicz, F. Bendr: Using transformers and a contrastive self-supervised learning task to learn from massive amounts of EEG data. arXiv 2021, arXiv:2101.12037. [Google Scholar]
- Everitt, B.S. Cluster Analysis; Edward Arnold: London, UK, 1993. [Google Scholar]
- Botvinick, M.; Barrett, D.G.T.; Battaglia, P.; de Freitas, N.; Kumaran, D.; Leibo, J.Z.; Lillicrap, T.; Modayil, J.; Mohamed, S.; Rabinowitz, N.C.; et al. Building machines that learn and think for themselves. Behav. Brain Sci. 2017, 40, e255. [Google Scholar] [CrossRef]
- Hassabis, D.; Kumaran, D.; Summerfield, C.; Botvinick, M. Neuroscience-inspired artificial intelligence. Neuron 2017, 95, 245–258. [Google Scholar] [CrossRef]
- Shevlin, H.; Vold, K.; Crosby, M.; Halina, M. The limits of machine intelligence despite progress in machine intelligence, artificial general intelligence is still a major challenge. EMBO Rep. 2019, 20. [Google Scholar] [CrossRef]
- Pei, J.; Deng, L.; Song, S.; Zhao, M.; Zhang, Y.; Wu, S.; Wang, G.; Zou, Z.; Wu, Z.; He, W.; et al. Towards artificial general intelligence with hybrid Tianjic chip architecture. Nature 2019, 572, 106–111. [Google Scholar] [CrossRef]
- Stanley, K.O.; Clune, J.; Lehman, J.; Miikkulainen, R. Designing neural networks through neuroevolution. Nat. Mach. Intell. 2019, 1, 24–35. [Google Scholar] [CrossRef]
- Zhang, Y.; Qu, P.; Ji, Y.; Zhang, W.; Gao, G.; Wang, G.; Song, S.; Li, G.; Chen, W.; Zheng, W.; et al. A system hierarchy for brain-inspired computing. Nature 2020, 586, 378–384. [Google Scholar] [CrossRef]
- Nadji-Tehrani, M.; Eslami, A. A brain-inspired framework for evolutionary artificial general intelligence. IEEE Trans. Neural Netw. Learn. Syst. 2020, 31, 5257–5271. [Google Scholar] [CrossRef]
- Bjerrum, E.J. SMILES enumeration as data augmentation for neural network modeling of molecules. arXiv 2017, arXiv:1703.07076. [Google Scholar]
- Sohn, K. Improved deep metric learning with multi-class n-pair loss objective. NIPS 2016, 30, 1857–1865. [Google Scholar]
- Wu, Z.; Xiong, Y.; Yu, S.; Lin, D. Unsupervised feature learning via non-parametric instance-level discrimination. arXiv 2018, arXiv:1805.01978. [Google Scholar]
- Kingma, D.P.; Ba, J.L. Adam: A method for stochastic optimization. arXiv 2015, arXiv:1412.6980v1418. [Google Scholar]
- Srivastava, N.; Hinton, G.; Krizhevsky, A.; Sutskever, I.; Salakhutdinov, R. Dropout: A simple way to prevent neural networks from overfitting. J. Mach. Learn. Res. 2014, 15, 1929–1958. [Google Scholar]

























Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shrivastava, A.D.; Kell, D.B. FragNet, a Contrastive Learning-Based Transformer Model for Clustering, Interpreting, Visualizing, and Navigating Chemical Space. Molecules 2021, 26, 2065. https://doi.org/10.3390/molecules26072065
Shrivastava AD, Kell DB. FragNet, a Contrastive Learning-Based Transformer Model for Clustering, Interpreting, Visualizing, and Navigating Chemical Space. Molecules. 2021; 26(7):2065. https://doi.org/10.3390/molecules26072065
Chicago/Turabian StyleShrivastava, Aditya Divyakant, and Douglas B. Kell. 2021. "FragNet, a Contrastive Learning-Based Transformer Model for Clustering, Interpreting, Visualizing, and Navigating Chemical Space" Molecules 26, no. 7: 2065. https://doi.org/10.3390/molecules26072065
APA StyleShrivastava, A. D., & Kell, D. B. (2021). FragNet, a Contrastive Learning-Based Transformer Model for Clustering, Interpreting, Visualizing, and Navigating Chemical Space. Molecules, 26(7), 2065. https://doi.org/10.3390/molecules26072065