
Boronic Acids as Prospective Inhibitors of Metallo-β-Lactamases: Efficient Chemical Reaction in the Enzymatic Active Site Revealed by Molecular Modeling


Abstract
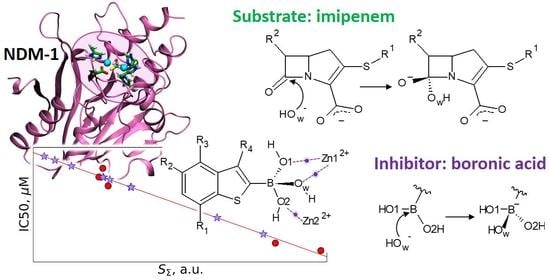
1. Introduction
2. Models and Methods
3. Results and Discussion
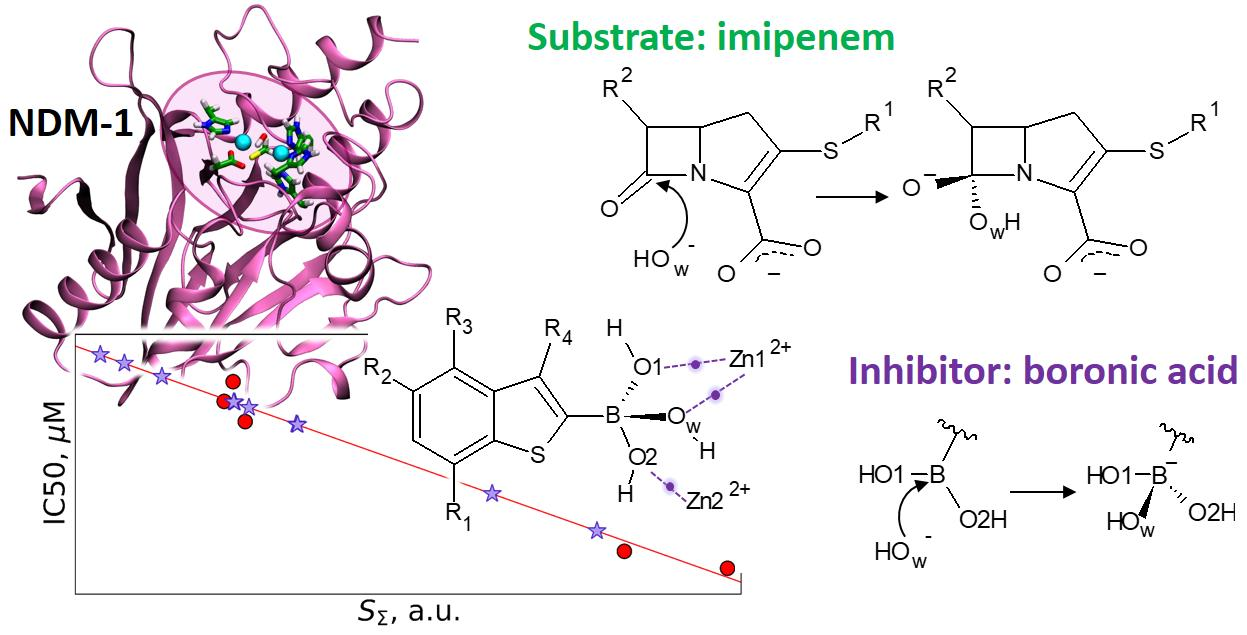
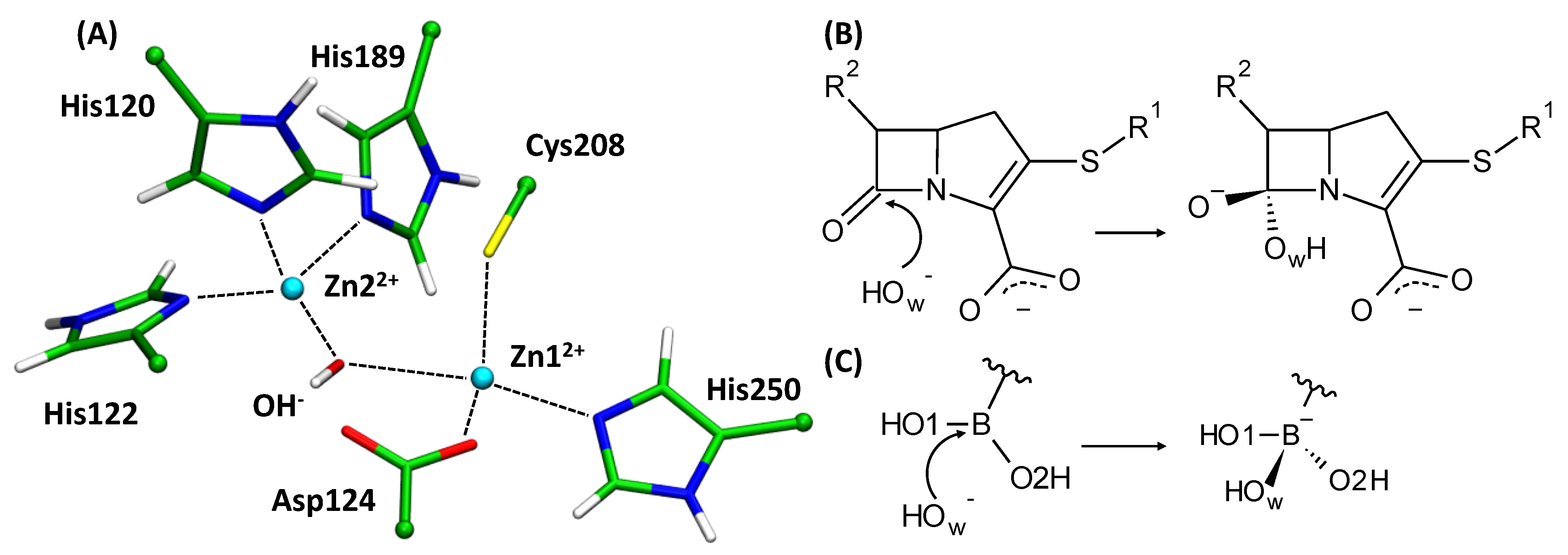
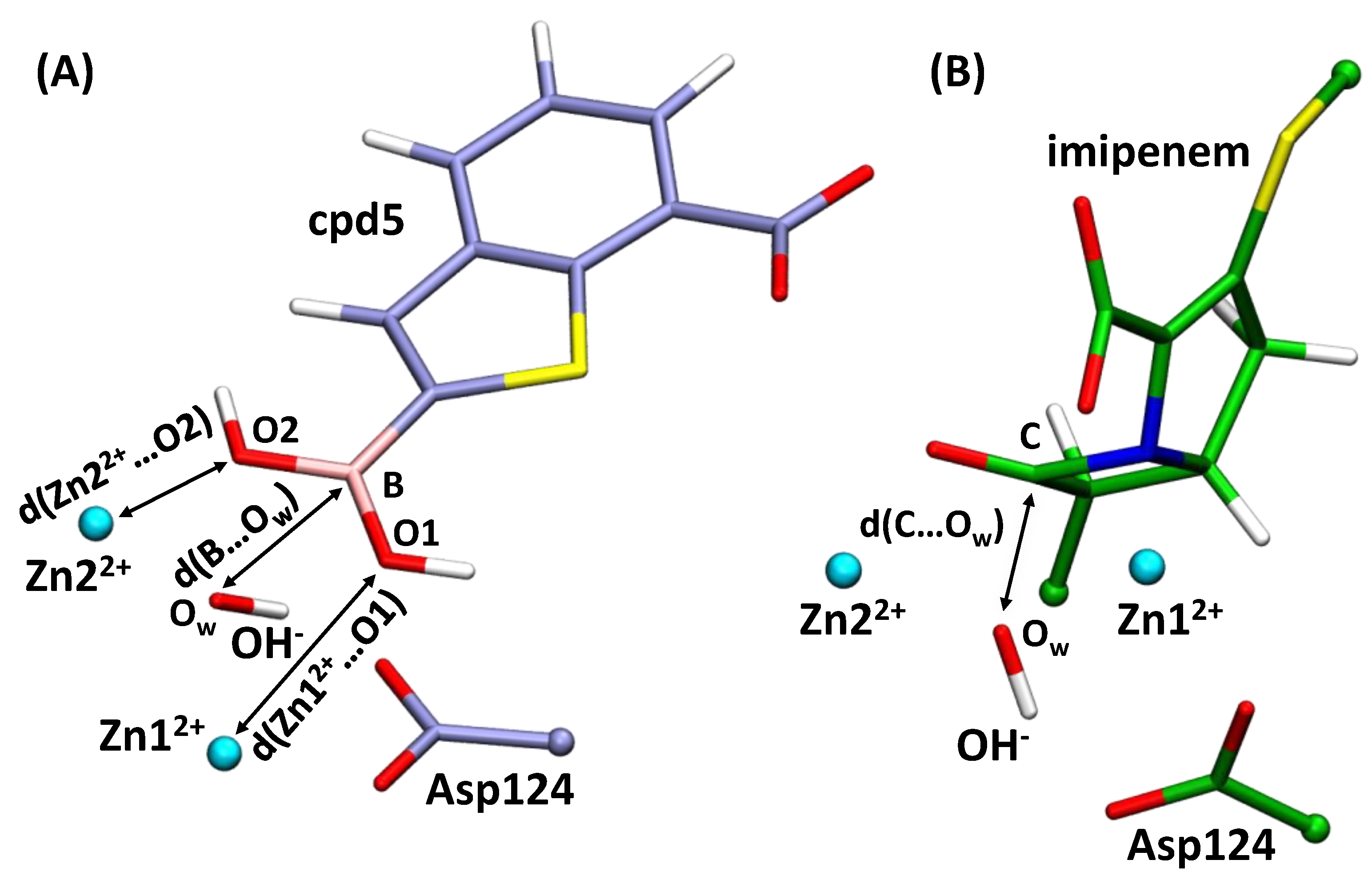
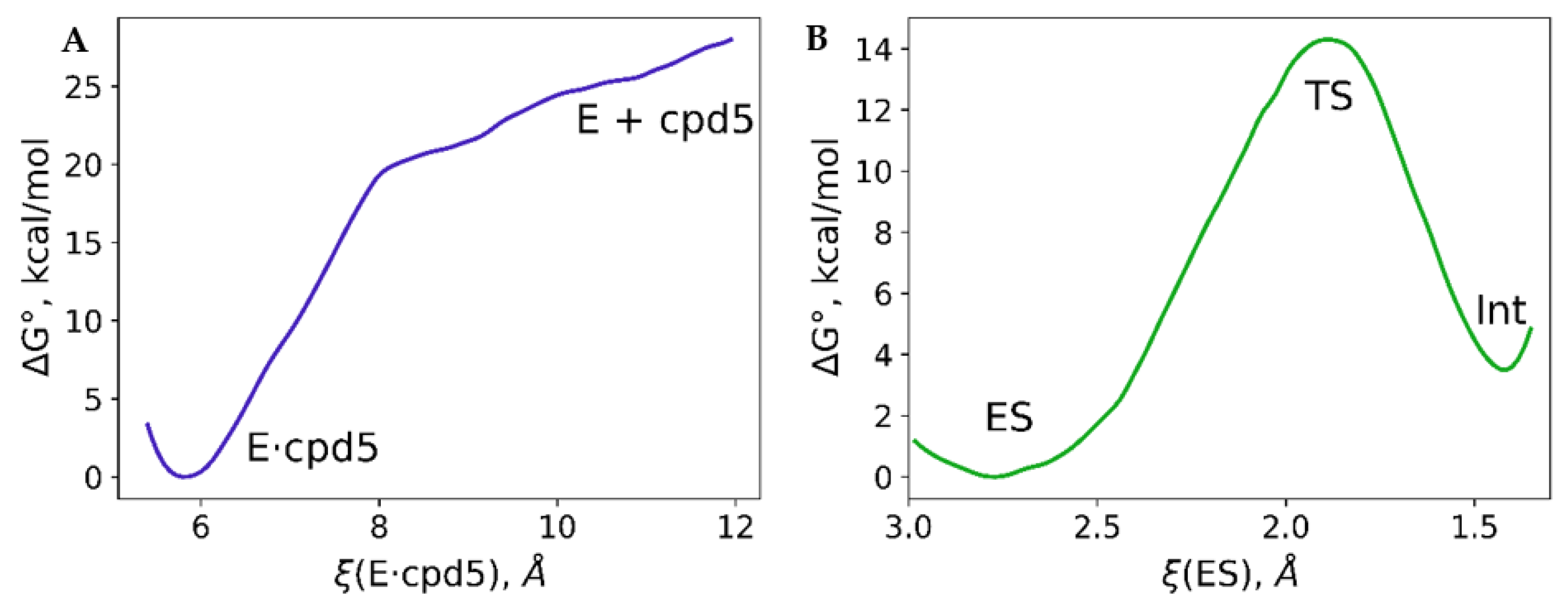
3.1. Imipenem and Boronic Acid Inhibitor Cpd5 in Water Solution and in the Active Site of NDM-1
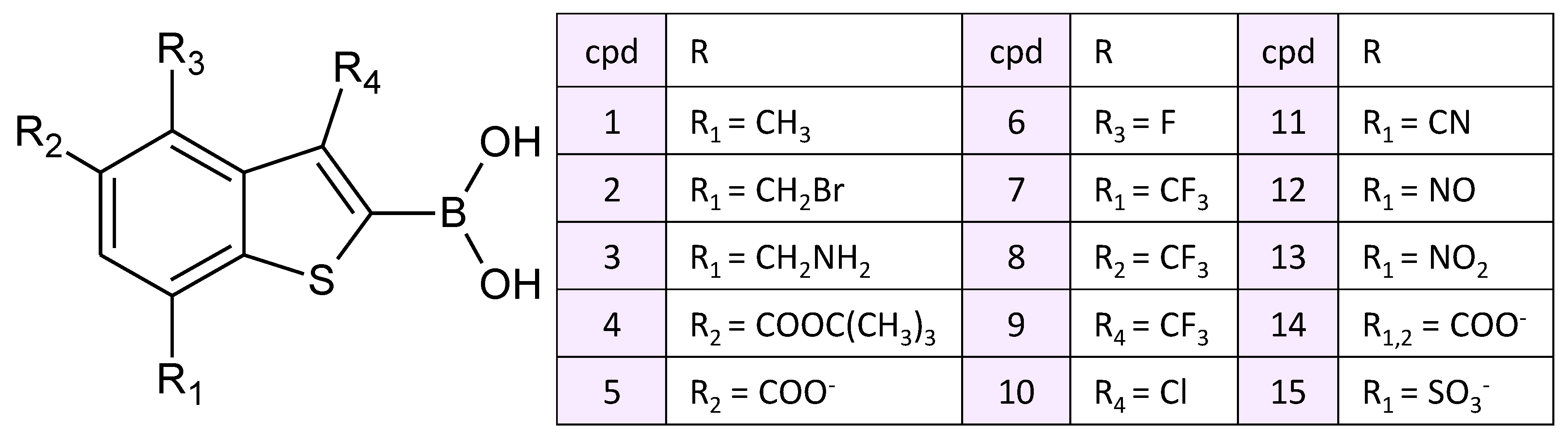
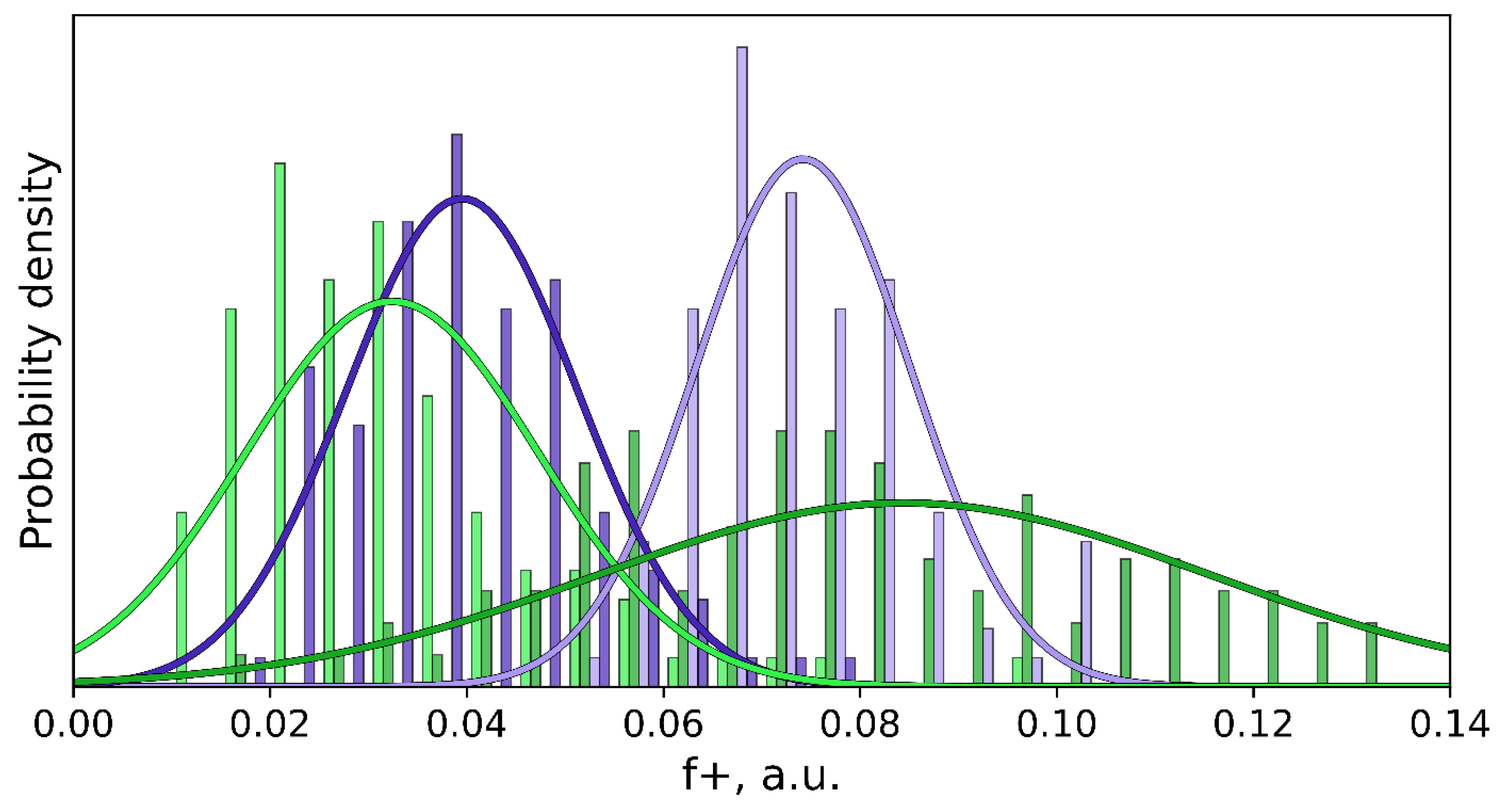
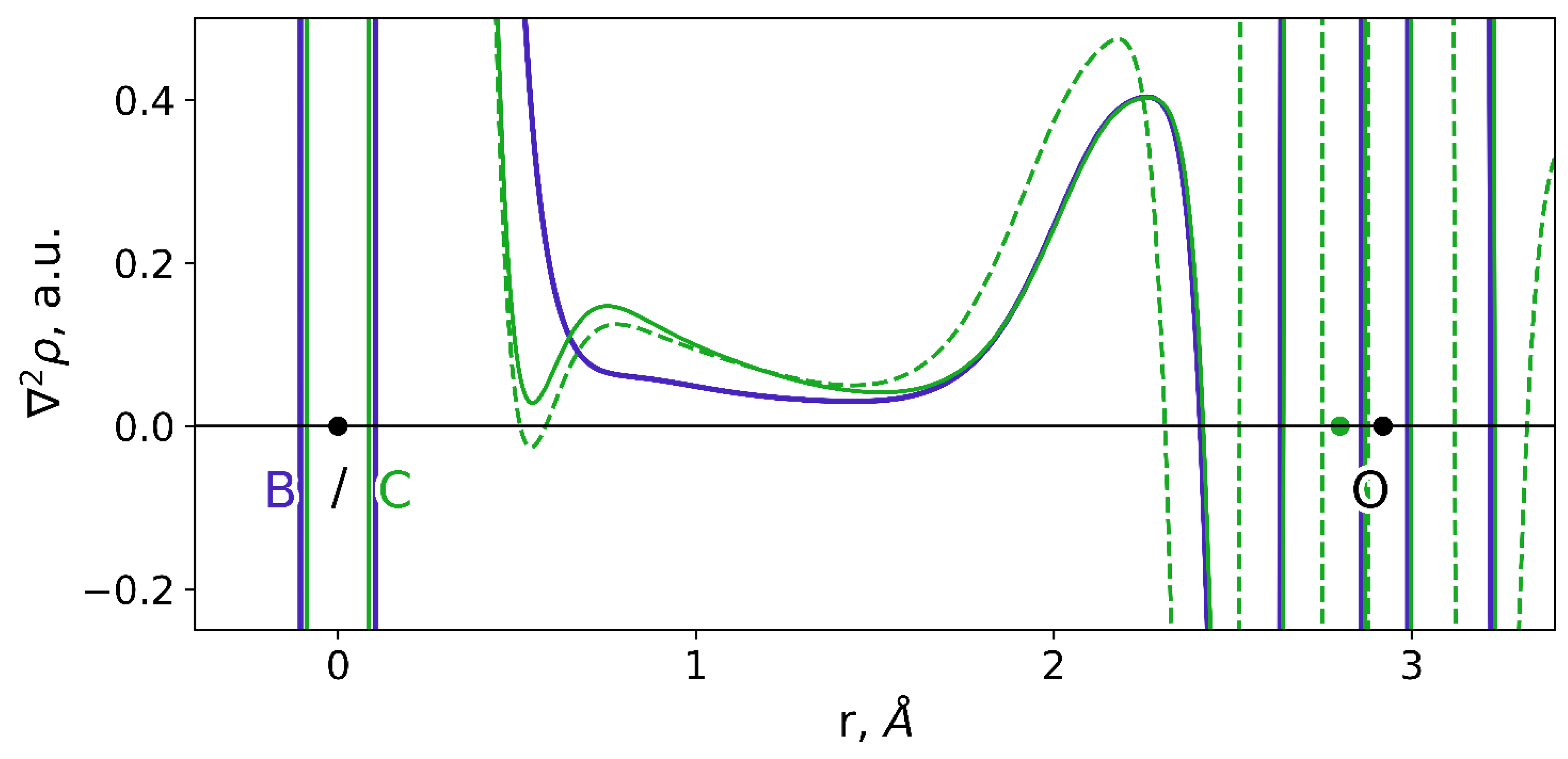
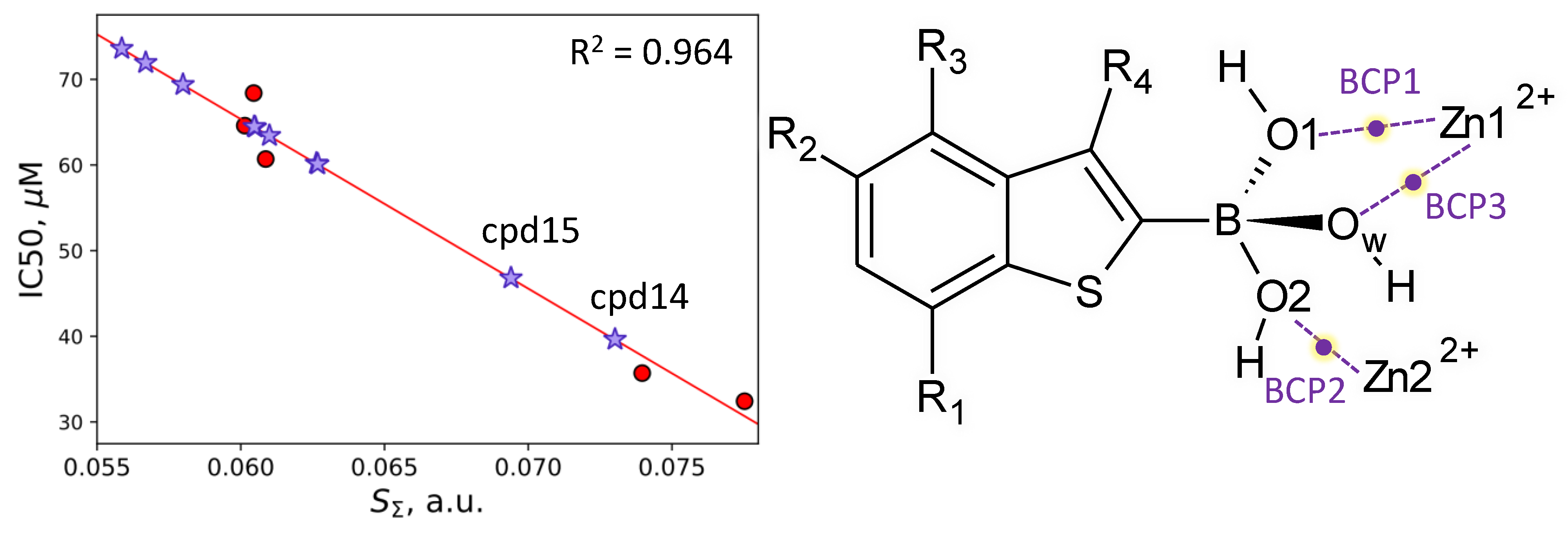
3.2. Interatomic Interactions Responsible for the Inhibition Potency
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Abraham, E.P.; Chain, E. An Enzyme from Bacteria able to Destroy Penicillin. Nature 1940, 146, 837. [Google Scholar] [CrossRef]
- Drawz, S.M.; Bonomo, R.A. Three Decades of β-Lactamase Inhibitors. Clin. Microbiol. Rev. 2010, 23, 160–201. [Google Scholar] [CrossRef]
- Lagadinou, M.; Onisor, M.O.; Rigas, A.; Musetescu, D.-V.; Gkentzi, D.; Assimakopoulos, S.F.; Panos, G.; Marangos, M. Antimicrobial Properties on Non-Antibiotic Drugs in the Era of Increased Bacterial Resistance. Antibiotics 2020, 9, 107. [Google Scholar] [CrossRef] [PubMed]
- Corrêa, R.C.G.; Heleno, S.A.; Alves, M.J.; Ferreira, I.C.F.R. Bacterial Resistance: Antibiotics of Last Generation used in Clinical Practice and the Arise of Natural Products as New Therapeutic Alternatives. Curr. Pharm. Des. 2020, 26, 815–837. [Google Scholar] [CrossRef]
- Munita, J.M.; Arias, C.A. Mechanisms of Antibiotic Resistance. Microbiol. Spectr. 2016, 4. [Google Scholar] [CrossRef] [PubMed]
- Hall, B.G.; Barlow, M. Revised Ambler classification of β-lactamases. J. Antimicrob. Chemother. 2005, 55, 1050–1051. [Google Scholar] [CrossRef] [PubMed]
- Levina, E.O.; Khrenova, M.G. Metallo-β-Lactamases: Influence of the Active Site Structure on the Mechanisms of Antibiotic Resistance and Inhibition. Biochemistry 2021, 86, S24–S37. [Google Scholar] [CrossRef]
- Mojica, M.F.; Bonomo, R.A.; Fast, W. B1-Metallo-β-Lactamases: Where Do We Stand? Curr. Drug Targets 2016, 17, 1029–1050. [Google Scholar] [CrossRef]
- Palzkill, T. Metallo-β-lactamase structure and function. Ann. N. Y. Acad. Sci. 2013, 1277, 91–104. [Google Scholar] [CrossRef]
- Linciano, P.; Cendron, L.; Gianquinto, E.; Spyrakis, F.; Tondi, D. Ten Years with New Delhi Metallo-β-lactamase-1 (NDM-1): From Structural Insights to Inhibitor Design. ACS Infect. Dis. 2019, 5, 9–34. [Google Scholar] [CrossRef]
- Kim, Y.; Cunningham, M.A.; Mire, J.; Tesar, C.; Sacchettini, J.; Joachimiak, A. NDM-1, the ultimate promiscuous enzyme: Substrate recognition and catalytic mechanism. FASEB J. 2013, 27, 1917–1927. [Google Scholar] [CrossRef] [PubMed]
- Feng, H.; Liu, X.; Wang, S.; Fleming, J.; Wang, D.-C.; Liu, W. The mechanism of NDM-1-catalyzed carbapenem hydrolysis is distinct from that of penicillin or cephalosporin hydrolysis. Nat. Commun. 2017, 8, 2242. [Google Scholar] [CrossRef] [PubMed]
- Rolain, J.M.; Parola, P.; Cornaglia, G. New Delhi metallo-beta-lactamase (NDM-1): Towards a new pandemia? Clin. Microbiol. Infect. 2010, 16, 1699–1701. [Google Scholar] [CrossRef] [PubMed]
- Isozumi, R.; Yoshimatsu, K.; Yamashiro, T.; Hasebe, F.; Nguyen, B.M.; Ngo, T.C.; Yasuda, S.P.; Koma, T.; Shimizu, K.; Arikawa, J. blaNDM-1–positive Klebsiella pneumoniae from Environment, Vietnam. Emerg. Infect. Dis. 2012, 18, 1383–1385. [Google Scholar] [CrossRef]
- Liang, Z.; Li, L.; Wang, Y.; Chen, L.; Kong, X.; Hong, Y.; Lan, L.; Zheng, M.; Guang-Yang, C.; Liu, H.; et al. Molecular Basis of NDM-1, a New Antibiotic Resistance Determinant. PLoS ONE 2011, 6, e23606. [Google Scholar] [CrossRef]
- Horie, R.; Hazbun, A.; Chen, K.; Cao, C.; Levine, M.; Horie, T. Shared evolutionary origin of vertebrate neural crest and cranial placodes. Nature 2018, 560, 228–232. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Sun, X.; Kong, F.; Xia, L.; Deng, X.; Wang, D.; Wang, J. Specific NDM-1 Inhibitor of Isoliquiritin Enhances the Activity of Meropenem against NDM-1-positive Enterobacteriaceae in vitro. Int. J. Environ. Res. Public Health 2020, 17, 2162. [Google Scholar] [CrossRef] [PubMed]
- Hecker, S.J.; Reddy, K.R.; Lomovskaya, O.; Griffith, D.C.; Rubio-Aparicio, D.; Nelson, K.; Tsivkovski, R.; Sun, D.; Sabet, M.; Tarazi, Z.; et al. Discovery of Cyclic Boronic Acid QPX7728, an Ultrabroad-Spectrum Inhibitor of Serine and Metallo-β-lactamases. J. Med. Chem. 2020, 63, 7491–7507. [Google Scholar] [CrossRef]
- Chen, C.; Liu, Y.; Zhang, Y.-J.; Ge, Y.; Lei, J.-E.; Yang, K.-W. The assemblage of covalent and metal binding dual functional scaffold for cross-class metallo-β-lactamases inhibition. Future Med. Chem. 2019, 11, 2381–2394. [Google Scholar] [CrossRef] [PubMed]
- Leiris, S.; Coelho, A.; Castandet, J.; Bayet, M.; Lozano, C.; Bougnon, J.; Bousquet, J.; Everett, M.; Lemonnier, M.; Sprynski, N.; et al. SAR Studies Leading to the Identification of a Novel Series of Metallo-β-lactamase Inhibitors for the Treatment of Carbapenem-Resistant Enterobacteriaceae Infections That Display Efficacy in an Animal Infection Model. ACS Infect. Dis. 2019, 5, 131–140. [Google Scholar] [CrossRef] [PubMed]
- Yuan, C.; Yan, J.; Song, C.; Yang, F.; Li, C.; Wang, C.; Su, H.; Chen, W.; Wang, L.; Wang, Z.; et al. Discovery of [1,2,4]Triazole Derivatives as New Metallo-β-Lactamase Inhibitors. Molecules 2019, 25, 56. [Google Scholar] [CrossRef]
- Zhang, Y.; Yan, Y.; Liang, L.; Feng, J.; Wang, X.; Li, L.; Yang, K. Halogen-Substituted Triazolethioacetamides as a Potent Skeleton for the Development of Metallo-β-Lactamase Inhibitors. Molecules 2019, 24, 1174. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Lu, X.; Wang, Y.; Hu, X.; Fu, H.; Gao, L.; You, X.; Tang, S.; Song, D. Synthesis and antibacterial evaluation against resistant Gram-negative bacteria of monobactams bearing various substituents on oxime residue. Bioorg. Chem. 2020, 94, 103487. [Google Scholar] [CrossRef]
- Maryam, L.; Khalid, S.; Ali, A.; Khan, A.U. Synergistic effect of doripenem in combination with cefoxitin and tetracycline in inhibiting NDM-1 producing bacteria. Future Microbiol. 2019, 14, 671–689. [Google Scholar] [CrossRef]
- Carosso, S.; Miller, M.J. Syntheses and studies of new forms of N-sulfonyloxy β-lactams as potential antibacterial agents and β-lactamase inhibitors. Bioorg. Med. Chem. 2015, 23, 6138–6147. [Google Scholar] [CrossRef] [PubMed]
- Santucci, M.; Spyrakis, F.; Cross, S.; Quotadamo, A.; Farina, D.; Tondi, D.; De Luca, F.; Docquier, J.-D.; Prieto, A.I.; Ibacache, C.; et al. Computational and biological profile of boronic acids for the detection of bacterial serine- and metallo-β-lactamases. Sci. Rep. 2017, 7, 17716. [Google Scholar] [CrossRef]
- Groundwater, P.W.; Xu, S.; Lai, F.; Váradi, L.; Tan, J.; Perry, J.D.; Hibbs, D.E. New Delhi metallo-β-lactamase-1: Structure, inhibitors and detection of producers. Future Med. Chem. 2016, 8, 993–1012. [Google Scholar] [CrossRef]
- Tooke, C.L.; Hinchliffe, P.; Bragginton, E.C.; Colenso, C.K.; Hirvonen, V.H.A.; Takebayashi, Y.; Spencer, J. β-Lactamases and β-Lactamase Inhibitors in the 21st Century. J. Mol. Biol. 2019, 431, 3472–3500. [Google Scholar] [CrossRef] [PubMed]
- Krajnc, A.; Lang, P.A.; Panduwawala, T.D.; Brem, J.; Schofield, C.J. Will morphing boron-based inhibitors beat the β-lactamases? Curr. Opin. Chem. Biol. 2019, 50, 101–110. [Google Scholar] [CrossRef] [PubMed]
- Song, S.; Gao, P.; Sun, L.; Kang, D.; Kongsted, J.; Poongavanam, V.; Zhan, P.; Liu, X. Recent developments in the medicinal chemistry of single boron atom-containing compounds. Acta Pharm. Sin. B 2021, in press. [Google Scholar] [CrossRef]
- Fernandes, G.F.S.; Denny, W.A.; Dos Santos, J.L. Boron in drug design: Recent advances in the development of new therapeutic agents. Eur. J. Med. Chem. 2019, 179, 791–804. [Google Scholar] [CrossRef] [PubMed]
- Kahlert, J.; Austin, C.J.D.; Kassiou, M.; Rendina, L.M. The Fifth Element in Drug Design: Boron in Medicinal Chemistry. Aust. J. Chem. 2013, 66, 1118. [Google Scholar] [CrossRef]
- Richardson, P.G.; Hideshima, T.; Anderson, K.C. Bortezomib (PS-341): A Novel, First-in-Class Proteasome Inhibitor for the Treatment of Multiple Myeloma and Other Cancers. Cancer Control 2003, 10, 361–369. [Google Scholar] [CrossRef]
- Cahill, S.T.; Cain, R.; Wang, D.Y.; Lohans, C.T.; Wareham, D.W.; Oswin, H.P.; Mohammed, J.; Spencer, J.; Fishwick, C.W.G.; McDonough, M.A.; et al. Cyclic Boronates Inhibit All Classes of β-Lactamases. Antimicrob. Agents Chemother. 2017, 61, e02260-16. [Google Scholar] [CrossRef] [PubMed]
- Cendron, L.; Quotadamo, A.; Maso, L.; Bellio, P.; Montanari, M.; Celenza, G.; Venturelli, A.; Costi, M.P.; Tondi, D. X-ray Crystallography Deciphers the Activity of Broad-Spectrum Boronic Acid β-Lactamase Inhibitors. ACS Med. Chem. Lett. 2019, 10, 650–655. [Google Scholar] [CrossRef] [PubMed]
- Thomas, P.W.; Cammarata, M.; Brodbelt, J.S.; Fast, W. Covalent Inhibition of New Delhi Metallo-β-Lactamase-1 (NDM-1) by Cefaclor. ChemBioChem 2014, 15, 2541–2548. [Google Scholar] [CrossRef]
- Lang, P.A.; Parkova, A.; Leissing, T.M.; Calvopiña, K.; Cain, R.; Krajnc, A.; Panduwawala, T.D.; Philippe, J.; Fishwick, C.W.G.; Trapencieris, P.; et al. Bicyclic Boronates as Potent Inhibitors of AmpC, the Class C β-Lactamase from Escherichia coli. Biomolecules 2020, 10, 899. [Google Scholar] [CrossRef]
- Bader, R.F.W. Atoms in Molecules-A Quantum Theory; Oxford University Press: Oxford, UK, 1990. [Google Scholar]
- Bader, R.F.W. A quantum theory of molecular structure and its applications. Chem. Rev. 1991, 91, 893–928. [Google Scholar] [CrossRef]
- Levina, E.O.; Khrenova, M.G.; Astakhov, A.A.; Tsirelson, V.G. Revealing electronic features governing hydrolysis of cephalosporins in the active site of the L1 metallo-β-lactamase. RSC Adv. 2020, 10, 8664–8676. [Google Scholar] [CrossRef]
- Khrenova, M.G.; Krivitskaya, A.V.; Tsirelson, V.G. The QM/MM-QTAIM approach reveals the nature of the different reactivity of cephalosporins in the active site of L1 metallo-β-lactamase. New J. Chem. 2019, 43, 7329–7338. [Google Scholar] [CrossRef]
- Khrenova, M.G.; Nemukhin, A.V.; Tsirelson, V.G. Discrimination of enzyme–substrate complexes by reactivity using the electron density analysis: Peptide bond hydrolysis by the matrix metalloproteinase-2. Mendeleev Commun. 2020, 30, 583–585. [Google Scholar] [CrossRef]
- Khrenova, M.G.; Tsirelson, V.G.; Nemukhin, A.V. Dynamical properties of enzyme–substrate complexes disclose substrate specificity of the SARS-CoV-2 main protease as characterized by the electron density descriptors. Phys. Chem. Chem. Phys. 2020, 22, 19069–19079. [Google Scholar] [CrossRef]
- Khrenova, M.G.; Tsirelson, V.G. The N···H hydrogen bond strength in the transition state at the limiting step determines the reactivity of cephalosporins in the active site of L1 metallo-β-lactamase. Mendeleev Commun. 2019, 29, 492–494. [Google Scholar] [CrossRef]
- Kim, S.; Chen, J.; Cheng, T.; Gindulyte, A.; He, J.; He, S.; Li, Q.; Shoemaker, B.A.; Thiessen, P.A.; Yu, B.; et al. PubChem in 2021: New data content and improved web interfaces. Nucleic Acids Res. 2021, 49, D1388–D1395. [Google Scholar] [CrossRef] [PubMed]
- Word, J.M.; Lovell, S.C.; Richardson, J.S.; Richardson, D.C. Asparagine and glutamine: Using hydrogen atom contacts in the choice of side-chain amide orientation. J. Mol. Biol. 1999, 285, 1735–1747. [Google Scholar] [CrossRef] [PubMed]
- Best, R.B.; Zhu, X.; Shim, J.; Lopes, P.E.M.; Mittal, J.; Feig, M.; MacKerell, A.D. Optimization of the Additive CHARMM All-Atom Protein Force Field Targeting Improved Sampling of the Backbone ϕ, ψ and Side-Chain χ1 and χ2 Dihedral Angles. J. Chem. Theory Comput. 2012, 8, 3257–3273. [Google Scholar] [CrossRef] [PubMed]
- Denning, E.J.; Priyakumar, U.D.; Nilsson, L.; Mackerell, A.D. Impact of 2′-hydroxyl sampling on the conformational properties of RNA: Update of the CHARMM all-atom additive force field for RNA. J. Comput. Chem. 2011, 32, 1929–1943. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Vanommeslaeghe, K.; Hatcher, E.; Acharya, C.; Kundu, S.; Zhong, S.; Shim, J.; Darian, E.; Guvench, O.; Lopes, P.; Vorobyov, I.; et al. CHARMM general force field (CGenFF): A force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields. J. Comput. Chem. 2010, 31, 671–690. [Google Scholar] [CrossRef] [PubMed]
- Phillips, J.C.; Braun, R.; Wang, W.; Gumbart, J.; Tajkhorshid, E.; Villa, E.; Chipot, C.; Skeel, R.D.; Kalé, L.; Schulten, K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005, 26, 1781–1802. [Google Scholar] [CrossRef]
- Adamo, C.; Barone, V. Toward reliable density functional methods without adjustable parameters: The PBE0 model. J. Chem. Phys. 1999, 110, 6158. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef]
- Hehre, W.J.; Ditchfield, R.; Pople, J.A. Self—Consistent Molecular Orbital Methods. XII. Further Extensions of Gaussian—Type Basis Sets for Use in Molecular Orbital Studies of Organic Molecules. J. Chem. Phys. 1972, 56, 2257. [Google Scholar] [CrossRef]
- Hariharan, P.C.; Pople, J.A. The influence of polarization functions on molecular orbital hydrogenation energies. Theor. Chim. Acta 1973, 28, 213–222. [Google Scholar] [CrossRef]
- Valiev, M.; Bylaska, E.J.; Govind, N.; Kowalski, K.; Straatsma, T.P.; Van Dam, H.J.J.; Wang, D.; Nieplocha, J.; Apra, E.; Windus, T.L.; et al. NWChem: A comprehensive and scalable open-source solution for large scale molecular simulations. Comput. Phys. Commun. 2010, 181, 1477–1489. [Google Scholar] [CrossRef]
- Seritan, S.; Bannwarth, C.; Fales, B.S.; Hohenstein, E.G.; Isborn, C.M.; Kokkila-Schumacher, S.I.L.; Li, X.; Liu, F.; Luehr, N.; Snyder, J.W.; et al. TeraChem: A graphical processing unit-accelerated electronic structure package for large-scale ab initio molecular dynamics. WIREs Comput. Mol. Sci. 2020. [Google Scholar] [CrossRef]
- Melo, M.C.R.; Bernardi, R.C.; Rudack, T.; Scheurer, M.; Riplinger, C.; Phillips, J.C.; Maia, J.D.C.; Rocha, G.B.; Ribeiro, J.V.; Stone, J.E.; et al. NAMD goes quantum: An integrative suite for hybrid simulations. Nat. Methods 2018, 15, 351–354. [Google Scholar] [CrossRef]
- Khrenova, M.G.; Kulakova, A.M.; Nemukhin, A.V. Light-Induced Change of Arginine Conformation Modulates the Rate of Adenosine Triphosphate to Cyclic Adenosine Monophosphate Conversion in the Optogenetic System Containing Photoactivated Adenylyl Cyclase. J. Chem. Inf. Model. 2021, 61, 1215–1225. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Lu, T.; Chen, F. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef]
- Bartashevich, E.V.; Troitskaya, E.A.; Tsirelson, V.G. The N⋯I halogen bond in substituted pyridines as viewed by the source function and delocalization indices. Chem. Phys. Lett. 2014, 601, 144–148. [Google Scholar] [CrossRef]
- Gatti, C.; Cargnoni, F.; Bertini, L. Chemical information from the source function. J. Comput. Chem. 2003, 24, 422–436. [Google Scholar] [CrossRef] [PubMed]
- Bader, R.F.W.; Gatti, C. A Green’s function for the density. Chem. Phys. Lett. 1998, 287, 233–238. [Google Scholar] [CrossRef]
- Hirshfeld, F.L. Bonded-atom fragments for describing molecular charge densities. Theor. Chim. Acta 1977, 44, 129–138. [Google Scholar] [CrossRef]
- Liu, S.; Rong, C.; Lu, T. Information Conservation Principle Determines Electrophilicity, Nucleophilicity, and Regioselectivity. J. Phys. Chem. A 2014, 118, 3698–3704. [Google Scholar] [CrossRef]
- Roy, R.K. Stockholders Charge Partitioning Technique. A Reliable Electron Population Analysis Scheme to Predict Intramolecular Reactivity Sequence. J. Phys. Chem. A 2003, 107, 10428–10434. [Google Scholar] [CrossRef]
- Oláh, J.; Van Alsenoy, C.; Sannigrahi, A.B. Condensed Fukui Functions Derived from Stockholder Charges: Assessment of Their Performance as Local Reactivity Descriptors. J. Phys. Chem. A 2002, 106, 3885–3890. [Google Scholar] [CrossRef]
- Khrenova, M.G.; Kulakova, A.M.; Nemukhin, A.V. Proof of concept for poor inhibitor binding and efficient formation of covalent adducts of KRAS G12C and ARS compounds. Org. Biomol. Chem. 2020, 18, 3069–3081. [Google Scholar] [CrossRef]
- Khrenova, M.G.; Nemukhin, A.V. Modeling the Transient Kinetics of the L1 Metallo-β-Lactamase. J. Phys. Chem. B 2018, 122, 1378–1386. [Google Scholar] [CrossRef]
- Vasilevskaya, T.; Khrenova, M.G.; Nemukhin, A.V.; Thiel, W. Mechanism of proteolysis in matrix metalloproteinase-2 revealed by QM/MM modeling. J. Comput. Chem. 2015, 36, 1621–1630. [Google Scholar] [CrossRef]
- Carroll, M.T.; Cheeseman, J.R.; Osman, R.; Weinstein, H. Nucleophilic addition to activated double bonds: Predictions of reactivity from the Laplacian of the charge density. J. Phys. Chem. 1989, 93, 5120–5123. [Google Scholar] [CrossRef]
- Jin, J.-L.; Li, H.-B.; Lu, T.; Duan, Y.-A.; Geng, Y.; Wu, Y.; Su, Z.-M. Density functional studies on photophysical properties and chemical reactivities of the triarylboranes: Effect of the constraint of planarity. J. Mol. Model. 2013, 19, 3437–3446. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Descriptor | Descriptor Range | Slope | Intercept, μM | R2 | Error, μM | RSS, μM−2 |
|---|---|---|---|---|---|---|
| d(Zn12+…O1), Å | 2.32–2.51 | 184 ± 41 | −397 ± 100 | 0.827 | 195–203 | 150 |
| ρ(rBCP1), a.u. | 0.025–0.038 | −2667 ± 618 | 131 ± 19 | 0.815 | 34–42 | 160 |
| ∇2ρ(rBCP1), a.u. | 0.11–0.17 | −644 ± 159 | 137 ± 21 | 0.793 | 39–48 | 179 |
| S(rBCP1, O1), a.u. | 0.0018–0.0081 | −5489 ± 1238 | 74 ± 6 | 0.823 | 8–16 | 153 |
| d(Zn22+…O2), Å | 1.95–2.03 | 415 ± 51 | −778 ± 102 | 0.942 | 202–206 | 50 |
| ρ(rBCP2), a.u. | 0.072–0.094 | −1643 ± 259 | 183 ± 21 | 0.908 | 39–45 | 80 |
| ∇2ρ(rBCP2), a.u. | 0.28–0.34 | −602 ± 102 | 234 ± 31 | 0.894 | 60–66 | 92 |
| S(rBCP2, O2), a.u. | 0.026–0.036 | −3413 ± 432 | 152 ± 13 | 0.939 | 24–28 | 53 |
| d(Zn12+…Ow), Å | 1.94–1.96 | 1094 ± 640 | −2088 ± 1251 | 0.325 | 2490–2507 | 583 |
| ρ(rBCP3), a.u. | 0.088–0.094 | −3348 ± 3930 | 354 ± 354 | – | 700–723 | 927 |
| ∇2ρ(rBCP3), a.u. | 0.33–0.36 | −1207 ± 569 | 458 ± 191 | 0.467 | 378–393 | 461 |
| S(rBCP3, Ow), a.u. | 0.033–0.036 | −8964 ± 5351 | 352 ± 179 | 0.311 | 353–370 | 595 |
| ρΣ, a.u. | 0.19–0.22 | −1017 ± 114 | 255 ± 23 | 0.952 | 44–48 | 42 |
| ∇2ρΣ, a.u. | 0.72–0.84 | −292 ± 26 | 277 ± 20 | 0.968 | 39–43 | 28 |
| SΣ, a.u. | 0.060–0.078 | −1979 ± 192 | 184 ± 13 | 0.964 | 24–28 | 31 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Krivitskaya, A.V.; Khrenova, M.G. Boronic Acids as Prospective Inhibitors of Metallo-β-Lactamases: Efficient Chemical Reaction in the Enzymatic Active Site Revealed by Molecular Modeling. Molecules 2021, 26, 2026. https://doi.org/10.3390/molecules26072026
Krivitskaya AV, Khrenova MG. Boronic Acids as Prospective Inhibitors of Metallo-β-Lactamases: Efficient Chemical Reaction in the Enzymatic Active Site Revealed by Molecular Modeling. Molecules. 2021; 26(7):2026. https://doi.org/10.3390/molecules26072026
Chicago/Turabian StyleKrivitskaya, Alexandra V., and Maria G. Khrenova. 2021. "Boronic Acids as Prospective Inhibitors of Metallo-β-Lactamases: Efficient Chemical Reaction in the Enzymatic Active Site Revealed by Molecular Modeling" Molecules 26, no. 7: 2026. https://doi.org/10.3390/molecules26072026
APA StyleKrivitskaya, A. V., & Khrenova, M. G. (2021). Boronic Acids as Prospective Inhibitors of Metallo-β-Lactamases: Efficient Chemical Reaction in the Enzymatic Active Site Revealed by Molecular Modeling. Molecules, 26(7), 2026. https://doi.org/10.3390/molecules26072026