
Thrombin-Derived Peptides Potentiate the Activity of Gram-Positive-Specific Antibiotics against Gram-Negative Bacteria

 , ,
, ,
Abstract
1. Introduction
2. Results
2.1. Synergy with Thrombin-Derived C-terminal Peptides
2.2. Alanine Scan of Peptide 6
2.3. Exploring the Synergistic Range
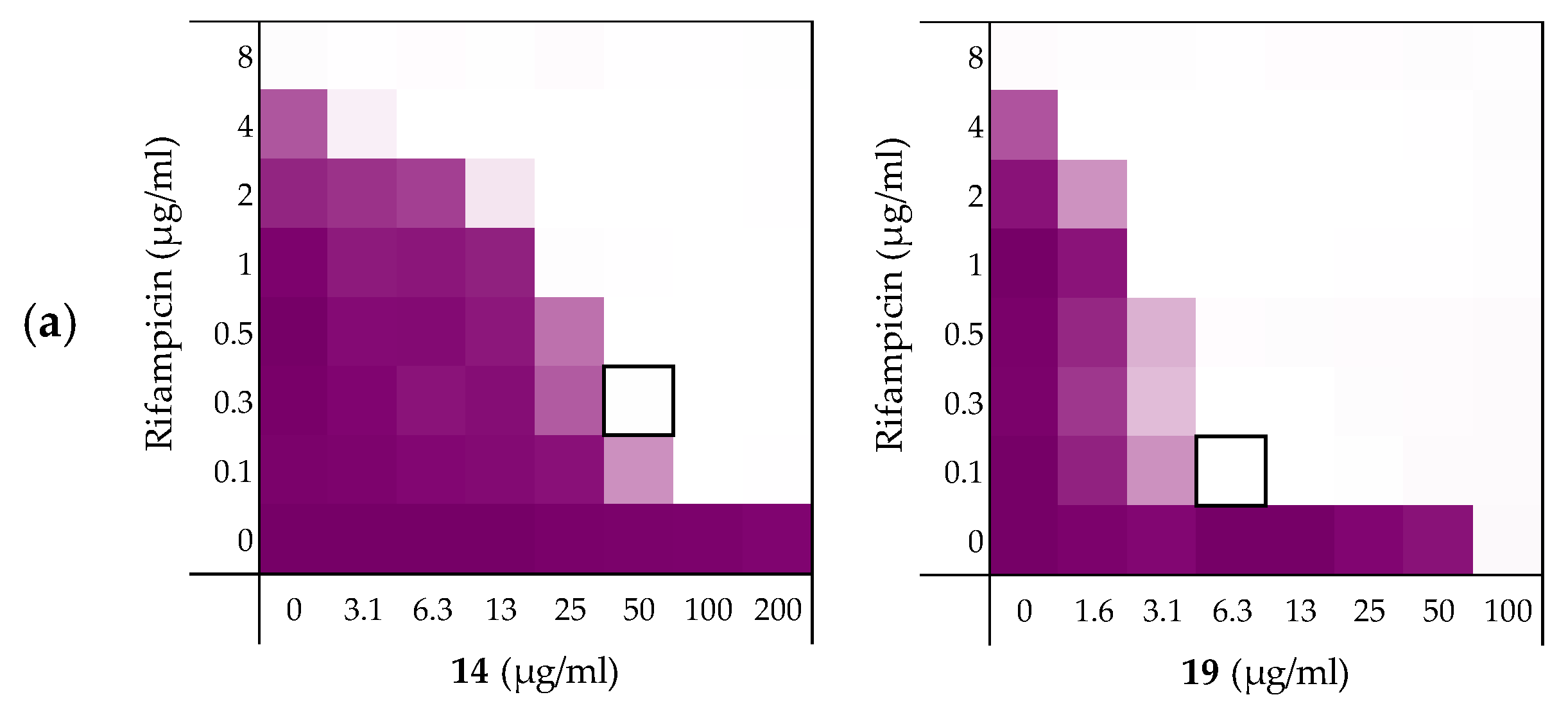
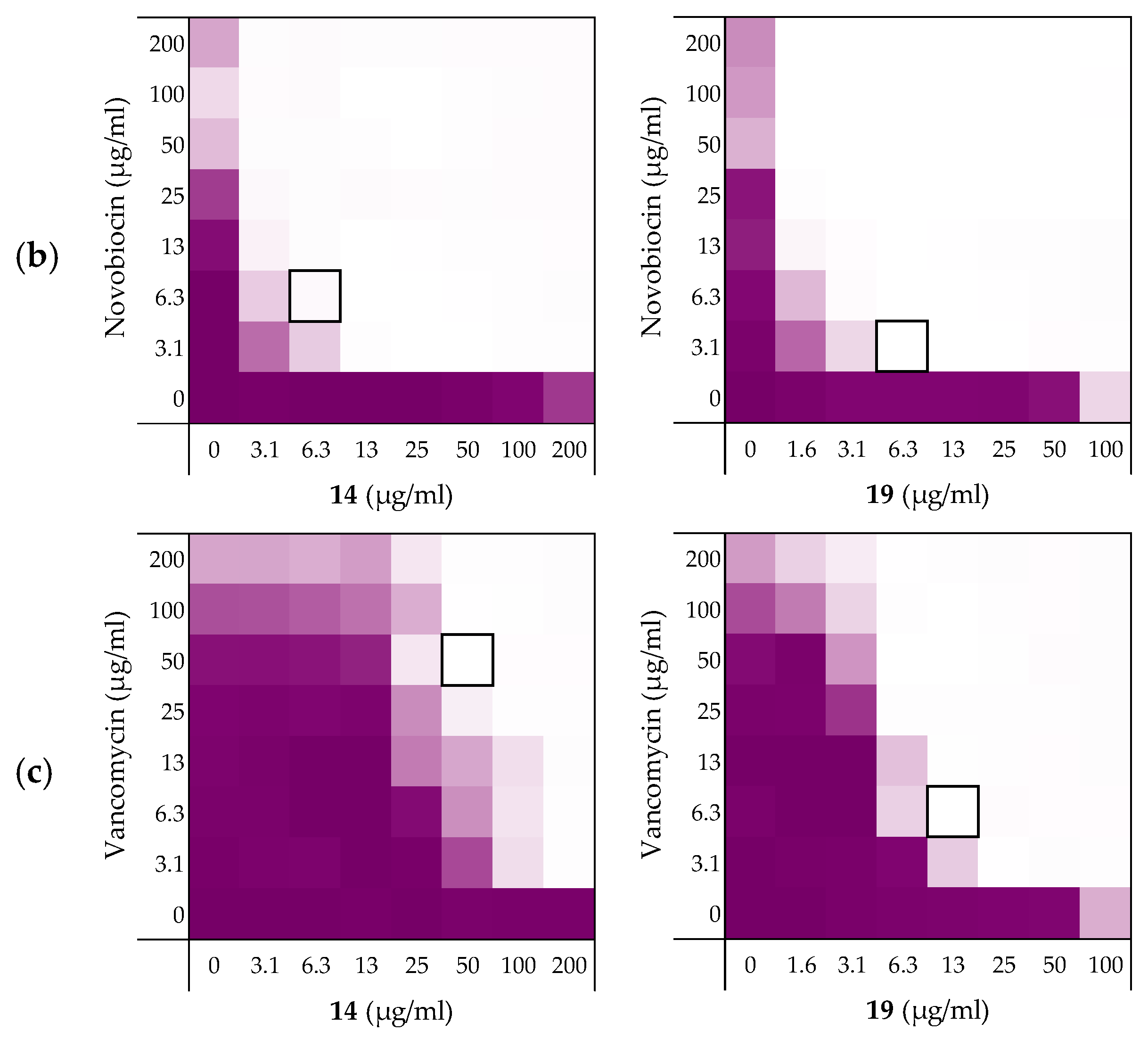
2.3.1. Synergy with Rifampicin, Novobiocin, and Vancomycin
2.3.2. Synergy towards Other E. coli Strains Including MCR-Positive Clinical Isolates
2.3.3. Synergy towards A. baumannii, K. pneumoniae, and P. aeruginosa
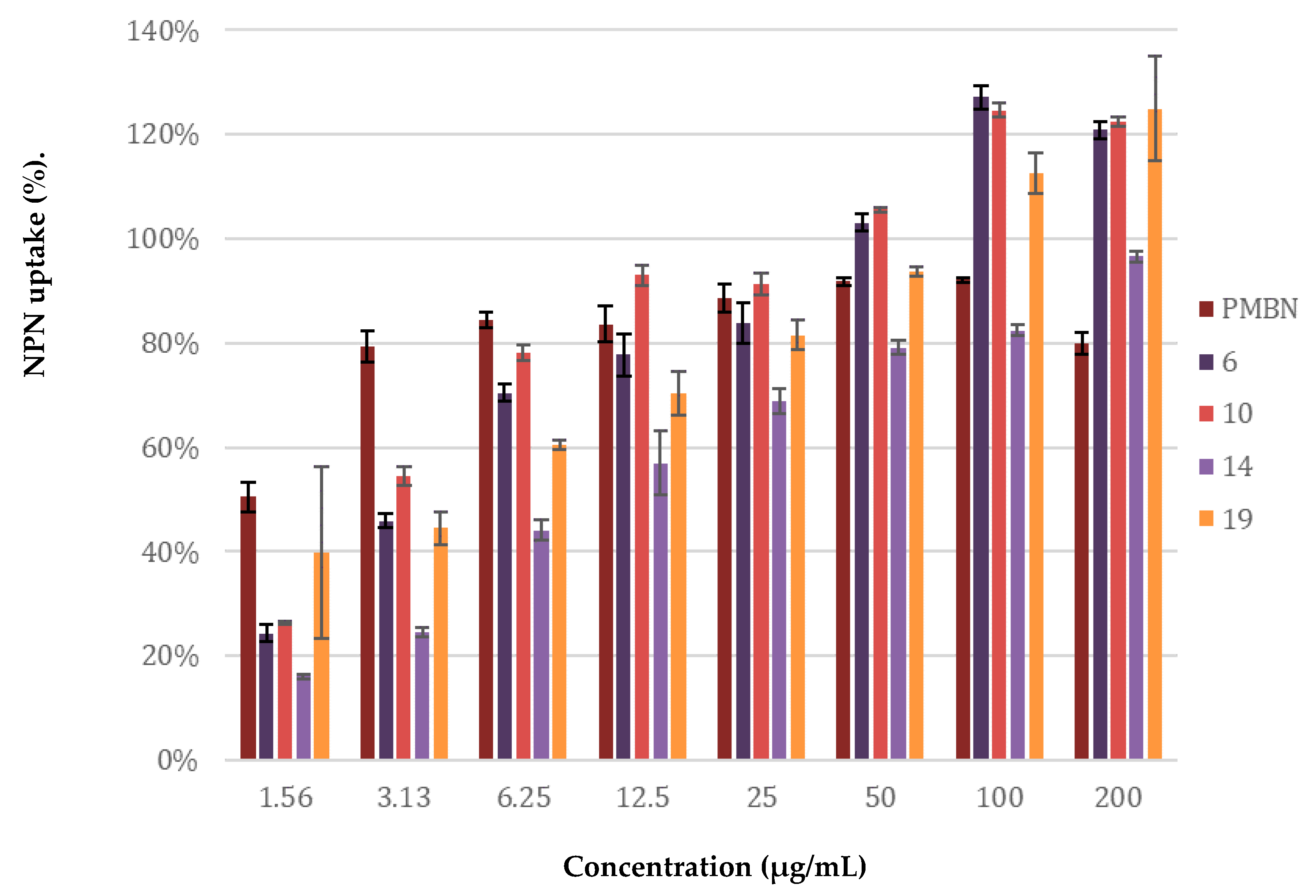
2.4. Mechanistic Studies
2.5. Stereochemical Studies
3. Discussion
4. Materials and Methods
4.1. Manual Peptide Synthesis for Carboxylic Acid C-terminus
4.2. Automated Peptide Synthesis for C-terminal Amides
4.3. Antimicrobial Assays
4.4. Minimal Inhibitory Concentration (MIC) Assay
4.5. Checkerboard Assays
4.6. Hemolysis Assay
4.7. LPS Competition Assay
4.8. Membrane Permeability Assay Using N-phenylnaphthalen-1-amine (NPN)
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Freire-Moran, L.; Aronsson, B.; Manz, C.; Gyssens, I.C.; So, A.D.; Monnet, D.L.; Cars, O. Critical shortage of new antibiotics in development against multidrug-resistant bacteria—Time to react is now. Drug Resist. Updat. 2011, 14, 118–124. [Google Scholar] [CrossRef]
- Amann, S.; Neef, K.; Kohl, S. Antimicrobial resistance (AMR). Eur. J. Hosp. Pharm. 2019, 26, 175–177. [Google Scholar] [CrossRef]
- Willyard, C. The drug-resistant bacteria that pose the greatest health threats. Nat. Cell Biol. 2017, 543, 15. [Google Scholar] [CrossRef]
- Nikaido, H. Molecular Basis of Bacterial Outer Membrane Permeability Revisited. Microbiol. Mol. Biol. Rev. 2003, 67, 593–656. [Google Scholar] [CrossRef] [PubMed]
- Silhavy, T.J.; Kahne, D.; Walker, S. The Bacterial Cell Envelope. Cold Spring Harb. Perspect. Biol. 2010, 2, a000414. [Google Scholar] [CrossRef] [PubMed]
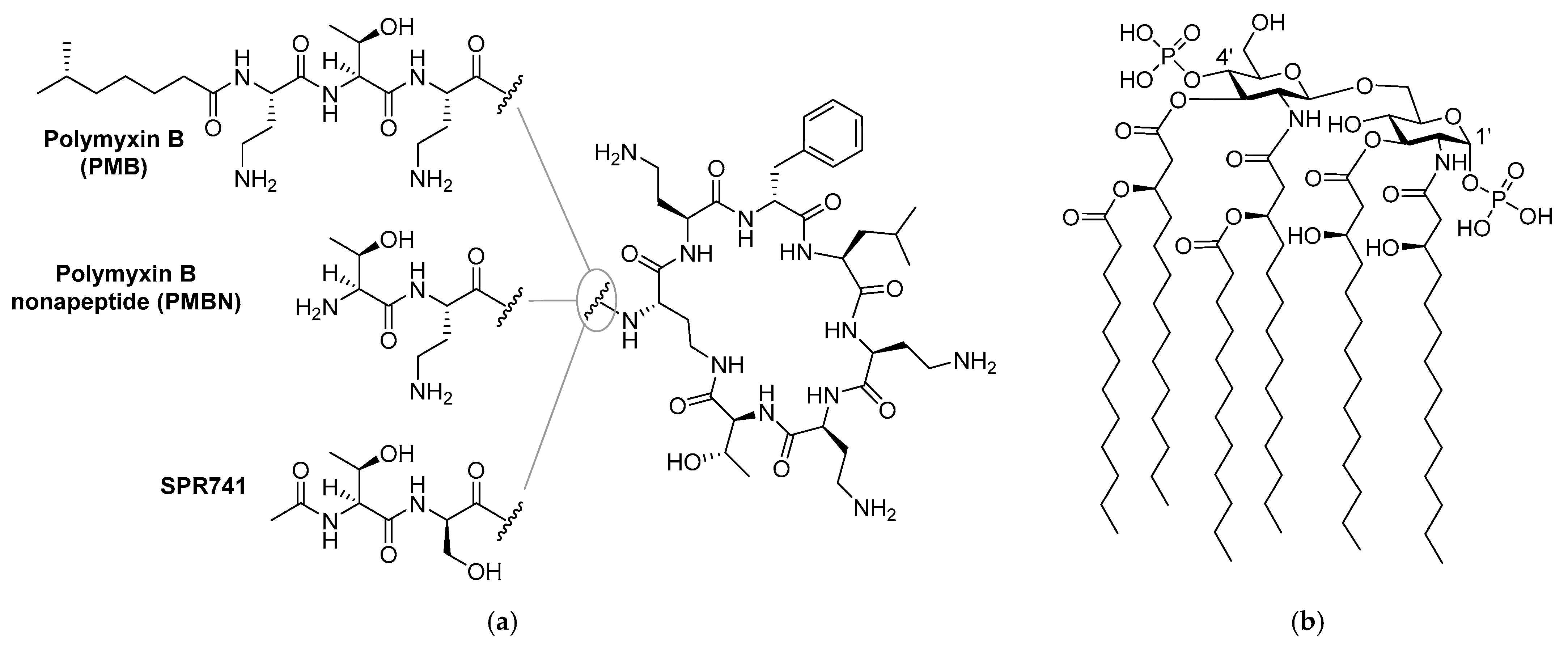
- Vaara, M.; Vaara, T. Sensitization of Gram-negative bacteria to antibiotics and complement by a nontoxic oligopeptide. Nat. Cell Biol. 1983, 303, 526–528. [Google Scholar] [CrossRef] [PubMed]
- Vaara, M. Agents that increase the permeability of the outer membrane. Microbiol. Rev. 1992, 56, 395–411. [Google Scholar] [CrossRef] [PubMed]
- Rojas, E.R.; Billings, G.; Odermatt, P.D.; Auer, G.K.; Zhu, L.; Miguel, A.; Chang, F.; Weibel, D.B.; Theriot, J.A.; Huang, K.C. The outer membrane is an essential load-bearing element in Gram-negative bacteria. Nat. Cell Biol. 2018, 559, 617–621. [Google Scholar] [CrossRef] [PubMed]
- Kimura, Y.; Matsunaga, H.; Vaara, M. Polymyxin B octapeptide and polymyxin B heptapeptide are potent outer membrane permeability-increasing agents. J. Antibiot. 1992, 45, 742–749. [Google Scholar] [CrossRef]
- Lam, C.; Hildebrandt, J.; Schütze, E.; Wenzel, A.F. Membrane-disorganizing property of polymyxin B nonapeptide. J. Antimicrob. Chemother. 1986, 18, 9–15. [Google Scholar] [CrossRef]
- Viljanen, P.; Vaara, M. Susceptibility of gram-negative bacteria to polymyxin B nonapeptide. Antimicrob. Agents Chemother. 1984, 25, 701–705. [Google Scholar] [CrossRef]
- Viljanen, P.; Koski, P.; Vaara, M. Effect of small cationic leukocyte peptides (defensins) on the permeability barrier of the outer membrane. Infect. Immun. 1988, 56, 2324–2329. [Google Scholar] [CrossRef]
- Tyrrell, J.M.; Aboklaish, A.F.; Walsh, T.R.; Vaara, T.; Vaara, M. The polymyxin derivative NAB739 is synergistic with several antibiotics against polymyxin-resistant strains of Escherichia coli, Klebsiella pneumoniae and Acinetobacter baumannii. Peptides 2019, 112, 149–153. [Google Scholar] [CrossRef]
- Vaara, M. Polymyxin Derivatives that Sensitize Gram-Negative Bacteria to Other Antibiotics. Molecules 2019, 24, 249. [Google Scholar] [CrossRef]
- FDA Approves New Antibacterial Drug to Treat Complicated Urinary Tract Infections as Part of Ongoing Efforts to Address Antimicrobial Resistance|FDA. Available online: https://www.fda.gov/news-events/press-announcements/fda-approves-new-antibacterial-drug-treat-complicated-urinary-tract-infections-part-ongoing-efforts (accessed on 23 March 2020).
- Spero Therapeutics Highlights SPR741 Phase 1 and Preclinical Data at the 28th European Congress of Clinical Microbiology and Infectious Diseases|Spero Therapeutics, Inc. Available online: https://investors.sperotherapeutics.com/news-releases/news-release-details/spero-therapeutics-highlights-spr741-phase-1-and-preclinical (accessed on 23 March 2020).
- Vaara, M.; Siikanen, O.; Apajalahti, J.; Fox, J.; Frimodt-Møller, N.; He, H.; Poudyal, A.; Li, J.; Nation, R.L.; Vaara, T. A Novel Polymyxin Derivative That Lacks the Fatty Acid Tail and Carries Only Three Positive Charges Has Strong Synergism with Agents Excluded by the Intact Outer Membrane. Antimicrob. Agents Chemother. 2010, 54, 3341–3346. [Google Scholar] [CrossRef] [PubMed]
- Kubesch, P.; Boggs, J.; Luciano, L.; Maass, G.; Tuemmler, B. Interaction of polymyxin B nonapeptide with anionic phospholipids. Biochemistry 1987, 26, 2139–2149. [Google Scholar] [CrossRef]
- Vaara, M.; Viljanen, P. Binding of polymyxin B nonapeptide to gram-negative bacteria. Antimicrob. Agents Chemother. 1985, 27, 548–554. [Google Scholar] [CrossRef] [PubMed]
- Vaara, M. Polymyxin B nonapeptide complexes with lipopolysaccharide. FEMS Microbiol. Lett. 1983, 18, 117–121. [Google Scholar] [CrossRef]
- A Peterson, A.; E Hancock, R.; McGroarty, E.J. Binding of polycationic antibiotics and polyamines to lipopolysaccharides of Pseudomonas aeruginosa. J. Bacteriol. 1985, 164, 1256–1261. [Google Scholar] [CrossRef]
- Cavaillon, J.-M.; Adib-Conquy, M. Bench-to-bedside review: Endotoxin tolerance as a model of leukocyte reprogramming in sepsis. Crit. Care 2006, 10, 233. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Fenton, M.J.; Golenbock, D.T. LPS-binding proteins and receptors. J. Leukoc. Biol. 1998, 64, 25–32. [Google Scholar] [CrossRef] [PubMed]
- Rosenfeld, Y.; Shai, Y. Lipopolysaccharide (Endotoxin)-host defense antibacterial peptides interactions: Role in bacterial resistance and prevention of sepsis. Biochim. Biophys. Acta (BBA) Biomembr. 2006, 1758, 1513–1522. [Google Scholar] [CrossRef]
- Brandenburg, K.; Heinbockel, L.; Correa, W.; Lohner, K. Peptides with dual mode of action: Killing bacteria and preventing endotoxin-induced sepsis. Biochim. Biophys. Acta (BBA) Biomembr. 2016, 1858, 971–979. [Google Scholar] [CrossRef]
- Saravanan, R.; A Holdbrook, D.; Petrlova, J.; Singh, S.; A Berglund, N.; Choong, Y.K.; Kjellström, S.; Bond, P.J.; Malmsten, M.; Schmidtchen, A. Structural basis for endotoxin neutralisation and anti-inflammatory activity of thrombin-derived C-terminal peptides. Nat. Commun. 2018, 9, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Dong, N.; Li, X.R.; Xu, X.Y.; Lv, Y.F.; Li, Z.Y.; Shan, A.S.; Wang, J.L. Correction to: Characterization of bactericidal efficiency, cell selectivity, and mechanism of short interspecific hybrid peptides. Amino Acids 2018, 50, 967. [Google Scholar] [CrossRef]
- Jerala, R.; Porro, M. Endotoxin neutralizing peptides. Curr. Top. Med. Chem. 2004, 4, 1173–1184. [Google Scholar] [CrossRef]
- Kaconis, Y.; Kowalski, I.; Howe, J.; Brauser, A.; Richter, W.; Razquin-Olazarán, I.; Iñigo-Pestaña, M.; Garidel, P.; Rössle, M.; De Tejada, G.M.; et al. Biophysical Mechanisms of Endotoxin Neutralization by Cationic Amphiphilic Peptides. Biophys. J. 2011, 100, 2652–2661. [Google Scholar] [CrossRef]
- De Tejada, G.M.; Heinbockel, L.; Ferrer-Espada, R.; Heine, H.; Alexander, C.; Bárcena-Varela, S.; Goldmann, T.; Correa, W.; Wiesmüller, K.-H.; Gisch, N.; et al. Lipoproteins/peptides are sepsis-inducing toxins from bacteria that can be neutralized by synthetic anti-endotoxin peptides. Sci. Rep. 2015, 5, 14292. [Google Scholar] [CrossRef]
- Stokes, J.M.; MacNair, C.R.; Ilyas, B.; French, S.; Côté, J.-P.; Bouwman, C.; Farha, M.A.; Sieron, A.O.; Whitfield, C.; Coombes, B.K.; et al. Pentamidine sensitizes Gram-negative pathogens to antibiotics and overcomes acquired colistin resistance. Nat. Microbiol. 2017, 2, 1–8. [Google Scholar] [CrossRef]
- E Hancock, R.; Wong, P.G. Compounds which increase the permeability of the Pseudomonas aeruginosa outer membrane. Antimicrob. Agents Chemother. 1984, 26, 48–52. [Google Scholar] [CrossRef]
- Li, Q.; Cebrián, R.; Montalbán-López, M.; Ren, H.; Wu, W.; Kuipers, O.P. Outer-membrane-acting peptides and lipid II-targeting antibiotics cooperatively kill Gram-negative pathogens. Commun. Biol. 2021, 4, 1–11. [Google Scholar] [CrossRef]
- Papareddy, P.; Rydengård, V.; Pasupuleti, M.; Walse, B.; Mörgelin, M.; Chalupka, A.; Malmsten, M.; Schmidtchen, A. Proteolysis of Human Thrombin Generates Novel Host Defense Peptides. PLoS Pathog. 2010, 6, e1000857. [Google Scholar] [CrossRef]
- Kasetty, G.; Papareddy, P.; Kalle, M.; Rydengård, V.; Mörgelin, M.; Albiger, B.; Malmsten, M.; Schmidtchen, A. Structure-Activity Studies and Therapeutic Potential of Host Defense Peptides of Human Thrombin. Antimicrob. Agents Chemother. 2011, 55, 2880–2890. [Google Scholar] [CrossRef] [PubMed]
- Holdbrook, D.A.; Singh, S.; Choong, Y.K.; Petrlova, J.; Malmsten, M.; Bond, P.J.; Verma, N.K.; Schmidtchen, A.; Saravanan, R. Influence of pH on the activity of thrombin-derived antimicrobial peptides. Biochim. Biophys. Acta (BBA) Biomembr. 2018, 1860, 2374–2384. [Google Scholar] [CrossRef]
- Odds, F.C. Synergy, antagonism, and what the chequerboard puts between them. J. Antimicrob. Chemother. 2003, 52, 1. [Google Scholar] [CrossRef] [PubMed]
- Washington, J.A.; Wilson, W.R. Erythromycin: A Microbial and Clinical Perspective after 30 Years of Clinical Use (First of Two Parts). Mayo Clin. Proc. 1985, 60, 189–203. [Google Scholar] [CrossRef]
- Washington, J.A.; Wilson, W.R. Erythromycin: A Microbial and Clinical Perspective after 30 Years of Clinical Use (Second of Two Parts). Mayo Clin. Proc. 1985, 60, 271–278. [Google Scholar] [CrossRef]
- Farr, B.; Mandell, G.L. Rifampin. Med. Clin. N. Am. 1982, 66, 157–168. [Google Scholar] [CrossRef]
- Furevi, A.; Ståhle, J.; Muheim, C.; Gkotzis, S.; Udekwu, K.I.; Daley, D.O.; Widmalm, G. Structural analysis of the O-antigen polysaccharide from Escherichia coli O188. Carbohydr. Res. 2020, 498, 108051. [Google Scholar] [CrossRef] [PubMed]
- Uchida, K.; Mizushima, S. A Simple Method for Isolation of Lipopolysaccharides fromPseudomonas aeruginosaand Some Other Bacterial Strains. Agric. Biol. Chem. 1987, 51, 3107–3114. [Google Scholar] [CrossRef][Green Version]
- Ebbensgaard, A.; Mordhorst, H.; Aarestrup, F.M.; Hansen, E.B. The Role of Outer Membrane Proteins and Lipopolysaccharides for the Sensitivity of Escherichia coli to Antimicrobial Peptides. Front. Microbiol. 2018, 9, 2153. [Google Scholar] [CrossRef]
- Wang, Z.; Wang, J.; Ren, G.; Molinaro, A.; Wang, X. Influence of Core Oligosaccharide of Lipopolysaccharide to Outer Membrane Behavior of Escherichia coli. Mar. Drugs 2015, 13, 3325–3339. [Google Scholar] [CrossRef]
- Liu, Y.-Y.; Wang, Y.; Walsh, T.R.; Yi, L.-X.; Zhang, R.; Spencer, J.; Doi, Y.; Tian, G.; Dong, B.; Huang, X.; et al. Emergence of plasmid-mediated colistin resistance mechanism MCR-1 in animals and human beings in China: A microbiological and molecular biological study. Lancet Infect. Dis. 2016, 16, 161–168. [Google Scholar] [CrossRef]
- Nang, S.C.; Li, J.; Velkov, T. The rise and spread of mcr plasmid-mediated polymyxin resistance. Crit. Rev. Microbiol. 2019, 45, 131–161. [Google Scholar] [CrossRef]
- Helander, I.M.; Mattila-Sandholm, T. Fluorometric assessment of Gram-negative bacterial permeabilization. J. Appl. Microbiol. 2000, 88, 213–219. [Google Scholar] [CrossRef]
- Macnair, C.R.; Stokes, J.M.; Carfrae, L.A.; Fiebig-Comyn, A.A.; Coombes, B.K.; Mulvey, M.R.; Brown, E.D. Overcoming mcr-1 mediated colistin resistance with colistin in combination with other antibiotics. Nat. Commun. 2018, 9, 1–8. [Google Scholar] [CrossRef]
- Van Regenmortel, M.H.; Muller, S. D-peptides as immunogens and diagnostic reagents. Curr. Opin. Biotechnol. 1998, 9, 377–382. [Google Scholar] [CrossRef]
- Singh, S.; Kalle, M.; Papareddy, P.; Schmidtchen, A.; Malmsten, M. Lipopolysaccharide Interactions of C-Terminal Peptides from Human Thrombin. Biomacromolecules 2013, 14, 1482–1492. [Google Scholar] [CrossRef]
- Oliva, R.; Battista, F.; Cozzolino, S.; Notomista, E.; Winter, R.; Del Vecchio, P.; Petraccone, L. Encapsulating properties of sulfobutylether-β-cyclodextrin toward a thrombin-derived antimicrobial peptide. J. Therm. Anal. Calorim. 2019, 138, 3249–3256. [Google Scholar] [CrossRef]
- Burdukiewicz, M.; Sidorczuk, K.; Rafacz, D.; Pietluch, F.; Chilimoniuk, J.; Rödiger, S.; Gagat, P. Proteomic Screening for Prediction and Design of Antimicrobial Peptides with AmpGram. Int. J. Mol. Sci. 2020, 21, 4310. [Google Scholar] [CrossRef]
- Mura, M.; Wang, J.; Zhou, Y.; Pinna, M.; Zvelindovsky, A.V.; Dennison, S.R.; Phoenix, D.A. The effect of amidation on the behaviour of antimicrobial peptides. Eur. Biophys. J. 2016, 45, 195–207. [Google Scholar] [CrossRef]
- Tsubery, H.; Ofek, I.; Cohen, S.; Fridkin, M. The functional association of polymyxin B with bacterial lipopolysaccharide is stereospecific: Studies on polymyxin B nonapeptide. Biochemistry 2000, 39, 11837–11844. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.; Mozsolits, H.; Hammer, J.; Zmuda, E.; Zhu, F.; Zhang, Y.; Aguilar, M.I.; Blazyk, J. Influence of Tryptophan on Lipid Binding of Linear Amphipathic Cationic Antimicrobial Peptides. Biochemistry 2003, 42, 9395–9405. [Google Scholar] [CrossRef] [PubMed]
- Yau, W.-M.; Wimley, W.C.; Gawrisch, A.K.; White, S.H. The Preference of Tryptophan for Membrane Interfaces. Biochemistry 1998, 37, 14713–14718. [Google Scholar] [CrossRef]
- Wimley, W.C.; White, S.H. Experimentally determined hydrophobicity scale for proteins at membrane interfaces. Nat. Struct. Mol. Biol. 1996, 3, 842–848. [Google Scholar] [CrossRef]
- Strøm, M.B.; Haug, B.E.; Skar, M.L.; Stensen, W.; Stiberg, T.; Svendsen, J.S. The Pharmacophore of Short Cationic Antibacterial Peptides. J. Med. Chem. 2003, 46, 1567–1570. [Google Scholar] [CrossRef]
- Velkov, T.; Thompson, P.E.; Nation, R.L.; Li, J. Structure−Activity Relationships of Polymyxin Antibiotics. J. Med. Chem. 2009, 53, 1898–1916. [Google Scholar] [CrossRef]
- Wang, J.; Chou, S.; Xu, L.; Zhu, X.; Dong, N.; Shan, A.; Chen, Z. High specific selectivity and Membrane-Active Mechanism of the synthetic centrosymmetric α-helical peptides with Gly-Gly pairs. Sci. Rep. 2015, 5, 15963. [Google Scholar] [CrossRef] [PubMed]












{kind=link}
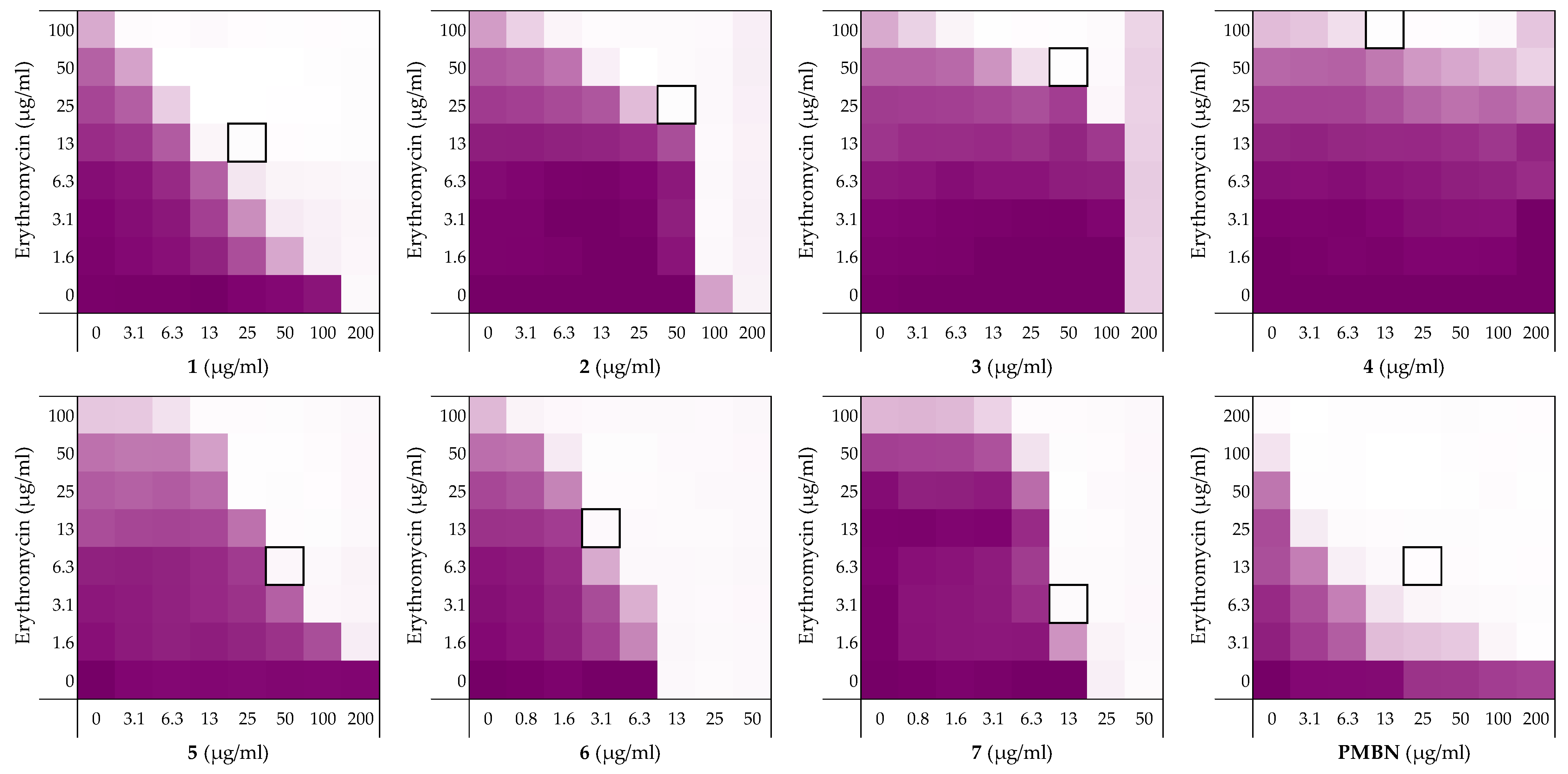{kind=link}
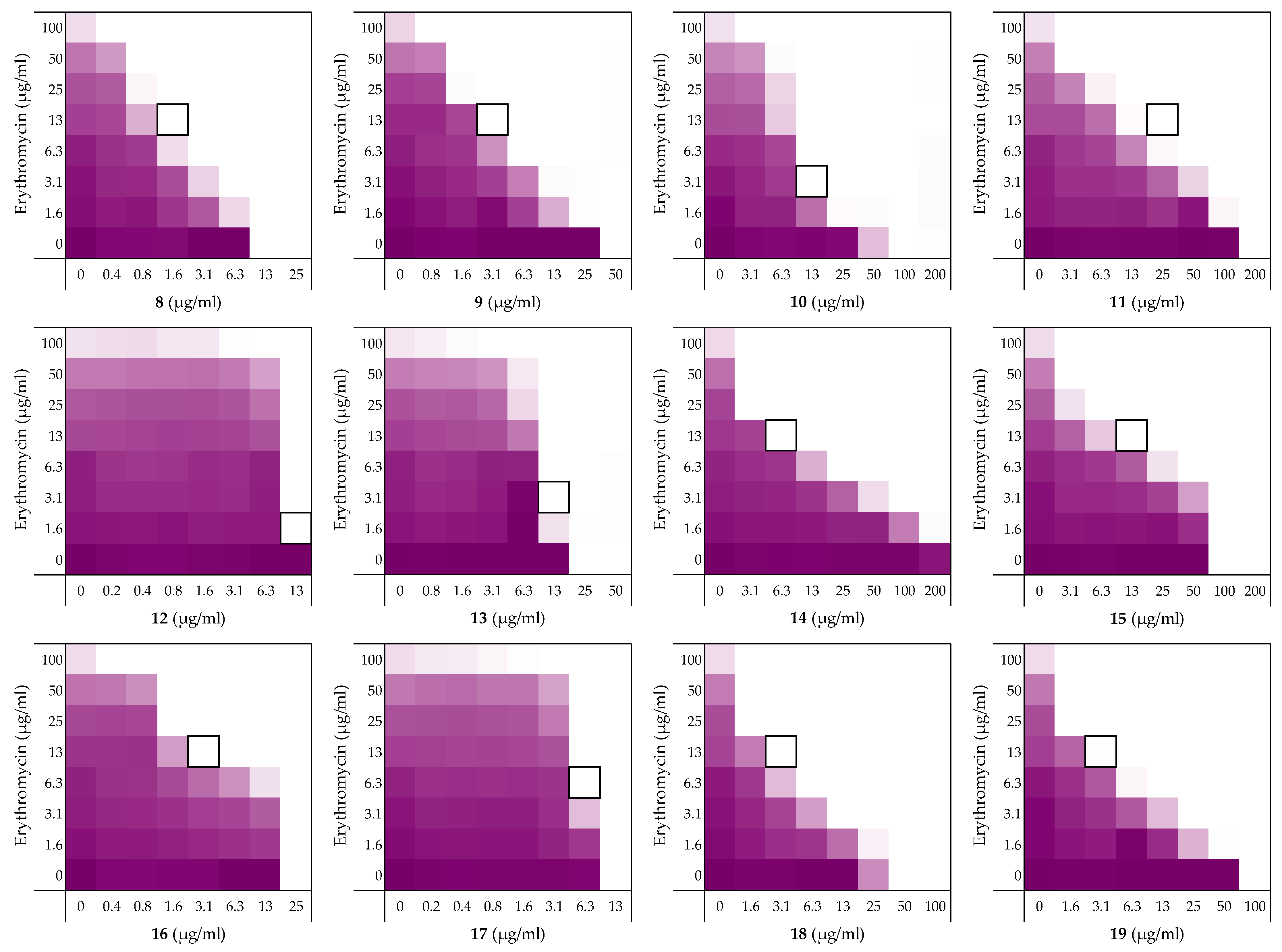{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
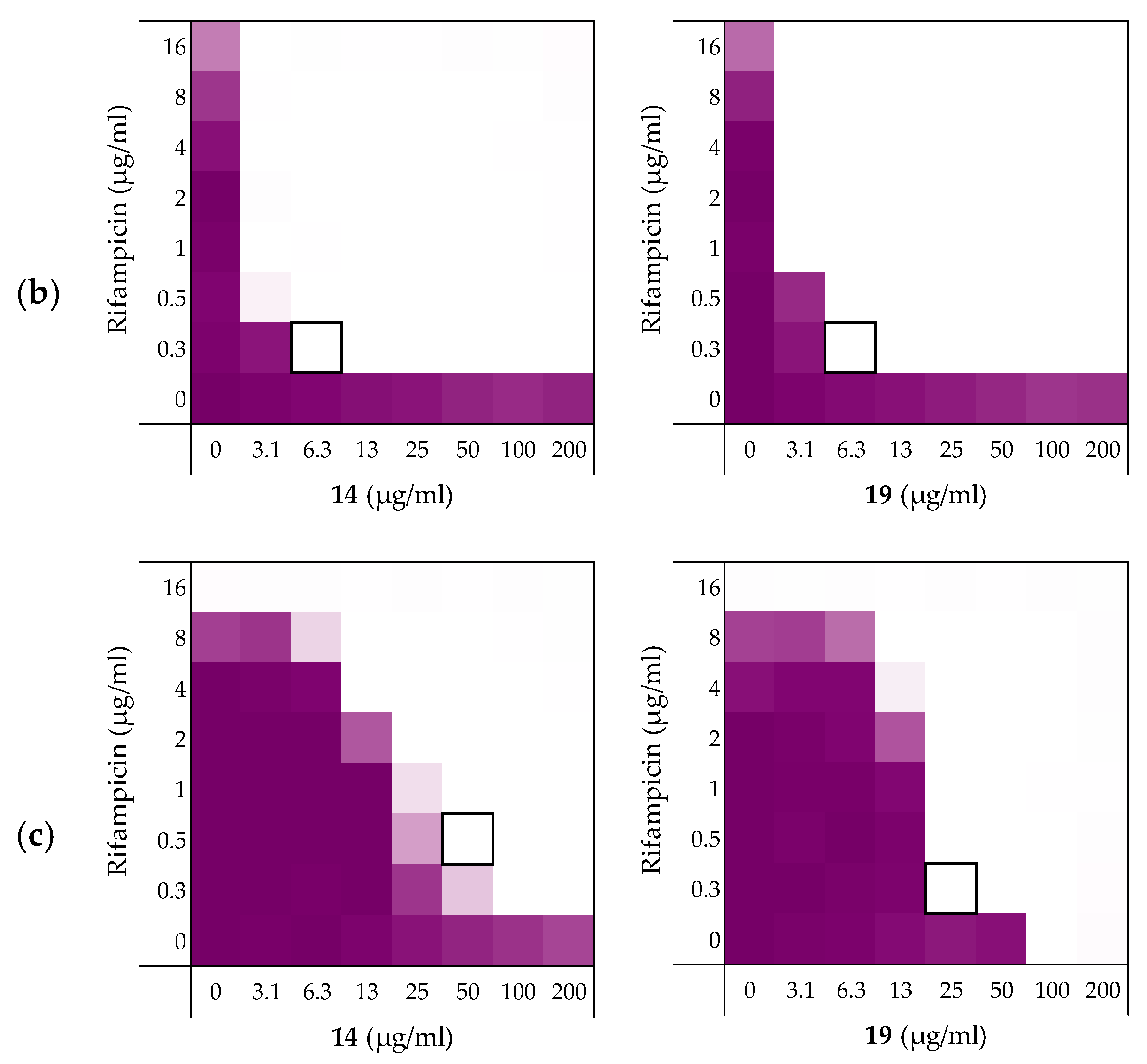{kind=link}
{kind=link}
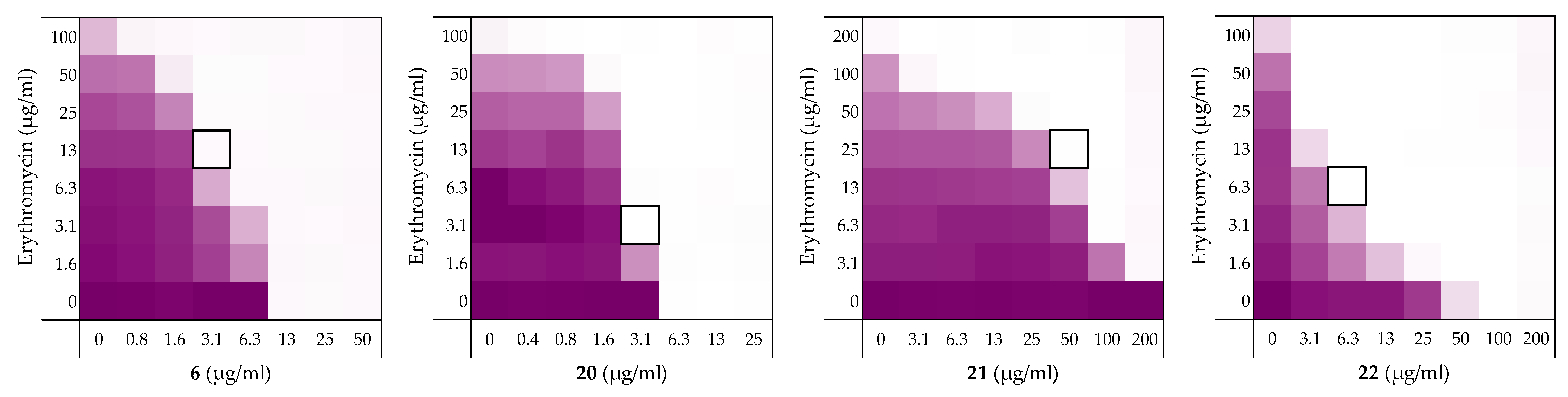{kind=link}
| Peptide Sequence | MIC a | MSCpeptide b | FICi c | Hemolysis d | |
|---|---|---|---|---|---|
| 1 | H2N-VFRLKKWIQKVI-OH | 200 | 25 | 0.188 | 0% |
| 2 | H2N-HVFRLKKWIQKVIDQFGE-OH | 200 | 50 | 0.375 | 9% |
| 3 | H2N-FYTHVFRLKKWIQKVIDQFGE-OH | 200 | 50 | 0.500 | 43% |
| 4 | H2N-GKYGFYTHVFRLKKWIQKVIDQFGE-OH | >200 | 12.5 | >0.5 | 70% |
| 5 | Ac-VFRLKKWIQKVI-OH | >200 | 50 | 0.156 | 0% |
| 6 | H2N-VFRLKKWIQKVI-CONH2 | 12.5 | 3.13 | 0.313 | 4% |
| 7 | Ac-VFRLKKWIQKVI-CONH2 | 50 | 12.5 | 0.266 | 5% |
| PMBN | >200 | 25 | 0.125 | - |
| Peptide Sequence | MIC a | FICi b | Hemolysis c | |
|---|---|---|---|---|
| 6 | H2N-VFRLKKWIQKVI-NH2 | 12.5 | 0.313 | 4% |
| 8 | H2N-AFRLKKWIQKVI-NH2 | 12.5 | 0.188 | 2% |
| 9 | H2N-VARLKKWIQKVI-NH2 | 50 | 0.125 | 0% |
| 10 | H2N-VFALKKWIQKVI-NH2 | 100 | 0.141 | 4% |
| 11 | H2N-VFRAKKWIQKVI-NH2 | 200 | 0.188 | 1% |
| 12 | H2N-VFRLAKWIQKVI-NH2 | 25 | >0.5 | 30% |
| 13 | H2N-VFRLKAWIQKVI-NH2 | 25 | >0.5 | 19% |
| 14 | H2N-VFRLKKAIQKVI-NH2 | >200 | 0.078 | 1% |
| 15 | H2N-VFRLKKWAQKVI-NH2 | 100 | 0.188 | 1% |
| 16 | H2N-VFRLKKWIAKVI-NH2 | 25 | 0.188 | 4% |
| 17 | H2N-VFRLKKWIQAVI-NH2 | 12.5 | >0.5 | 21% |
| 18 | H2N-VFRLKKWIQKAI-NH2 | 50 | 0.125 | 2% |
| 19 | H2N-VFRLKKWIQKVA-NH2 | 100 | 0.094 | 1% |
| Peptides | Rifampicin | Novobiocin | Vancomycin |
|---|---|---|---|
| 6 | 0.156 | 0.188 | 0.188 |
| 10 | 0.141 | 0.078 | 0.156 |
| 14 | 0.141 | 0.031 | 0.250 |
| 19 | 0.078 | 0.039 | 0.078 |
| PMBN | 0.063 | 0.047 | 0.156 |
| Pathogen | 6 | 10 | 14 | 19 |
|---|---|---|---|---|
| E. coli BW25113 | 0.156 | 0.141 | 0.141 | 0.078 |
| E. coli ATCC25922TM | 0.047 | 0.031 | 0.031 | 0.031 |
| E. coli W3110 | >0.5 b | >0.5 a | 0.188 | 0.078 |
| E. coli mcr-1 | 0.141 | 0.078 | 0.125 | 0.125 |
| E. coli EQASmcr-1 | 0.078 | 0.078 | 0.094 | 0.094 |
| E. coli EQASmcr-2 | 0.094 | 0.141 | 0.094 | 0.125 |
| E. coli EQASmcr-3 | 0.078 | 0.078 | 0.047 | 0.031 |
| Pathogen | 6 | 10 | 14 | 19 |
|---|---|---|---|---|
| A. baumannii ATCC17978 | 0.125 | 0.125 | 0.078 | 0.094 |
| K. pneumoniae ATCC13883 | 0.063 | 0.063 | 0.023 | 0.023 |
| P. aeruginosa ATCC27853 | >0.5 b | 0.250 | 0.156 | 0.266 |
| Peptide Sequence | MIC | MSCpeptide | FICi a | Hemolysis b | |
|---|---|---|---|---|---|
| 6 | H2-VFRLKKWIQKVI-NH2 | 12.5 | 3.13 | 0.313 | 4% |
| 20 | H2N-vfrlkkwiqkvi-NH2 | 6.25 | 3.13 | >0.5 a | 3% |
| 21 | H2N-IVKQIWKKLRFV-NH2 | >200 | 50 | 0.250 | 2% |
| 22 | H2-ivkqiwkklrfv-NH2 | 100 | 6.25 | 0.094 | 3% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wesseling, C.M.J.; Wood, T.M.; Slingerland, C.J.; Bertheussen, K.; Lok, S.; Martin, N.I. Thrombin-Derived Peptides Potentiate the Activity of Gram-Positive-Specific Antibiotics against Gram-Negative Bacteria. Molecules 2021, 26, 1954. https://doi.org/10.3390/molecules26071954
Wesseling CMJ, Wood TM, Slingerland CJ, Bertheussen K, Lok S, Martin NI. Thrombin-Derived Peptides Potentiate the Activity of Gram-Positive-Specific Antibiotics against Gram-Negative Bacteria. Molecules. 2021; 26(7):1954. https://doi.org/10.3390/molecules26071954
Chicago/Turabian StyleWesseling, Charlotte M. J., Thomas M. Wood, Cornelis J. Slingerland, Kristine Bertheussen, Samantha Lok, and Nathaniel I. Martin. 2021. "Thrombin-Derived Peptides Potentiate the Activity of Gram-Positive-Specific Antibiotics against Gram-Negative Bacteria" Molecules 26, no. 7: 1954. https://doi.org/10.3390/molecules26071954
APA StyleWesseling, C. M. J., Wood, T. M., Slingerland, C. J., Bertheussen, K., Lok, S., & Martin, N. I. (2021). Thrombin-Derived Peptides Potentiate the Activity of Gram-Positive-Specific Antibiotics against Gram-Negative Bacteria. Molecules, 26(7), 1954. https://doi.org/10.3390/molecules26071954