
In Silico Design and Selection of New Tetrahydroisoquinoline-Based CD44 Antagonist Candidates

 ,
,  ,
,
Abstract
1. Introduction
2. Results
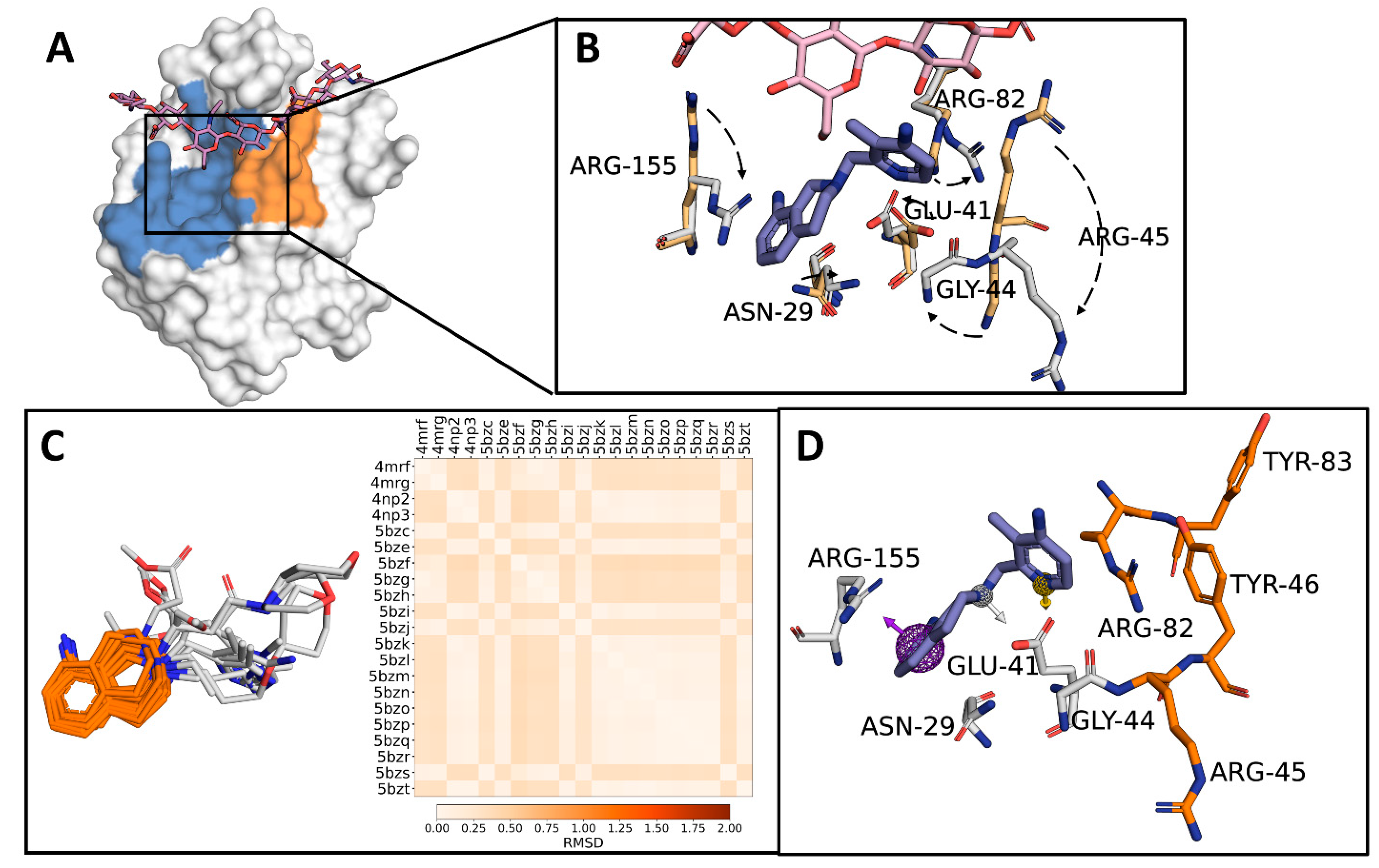
2.1. Identification of a Target Subdomain within the CD44HAbd and Generation of a THQ-Based Pharmacophore
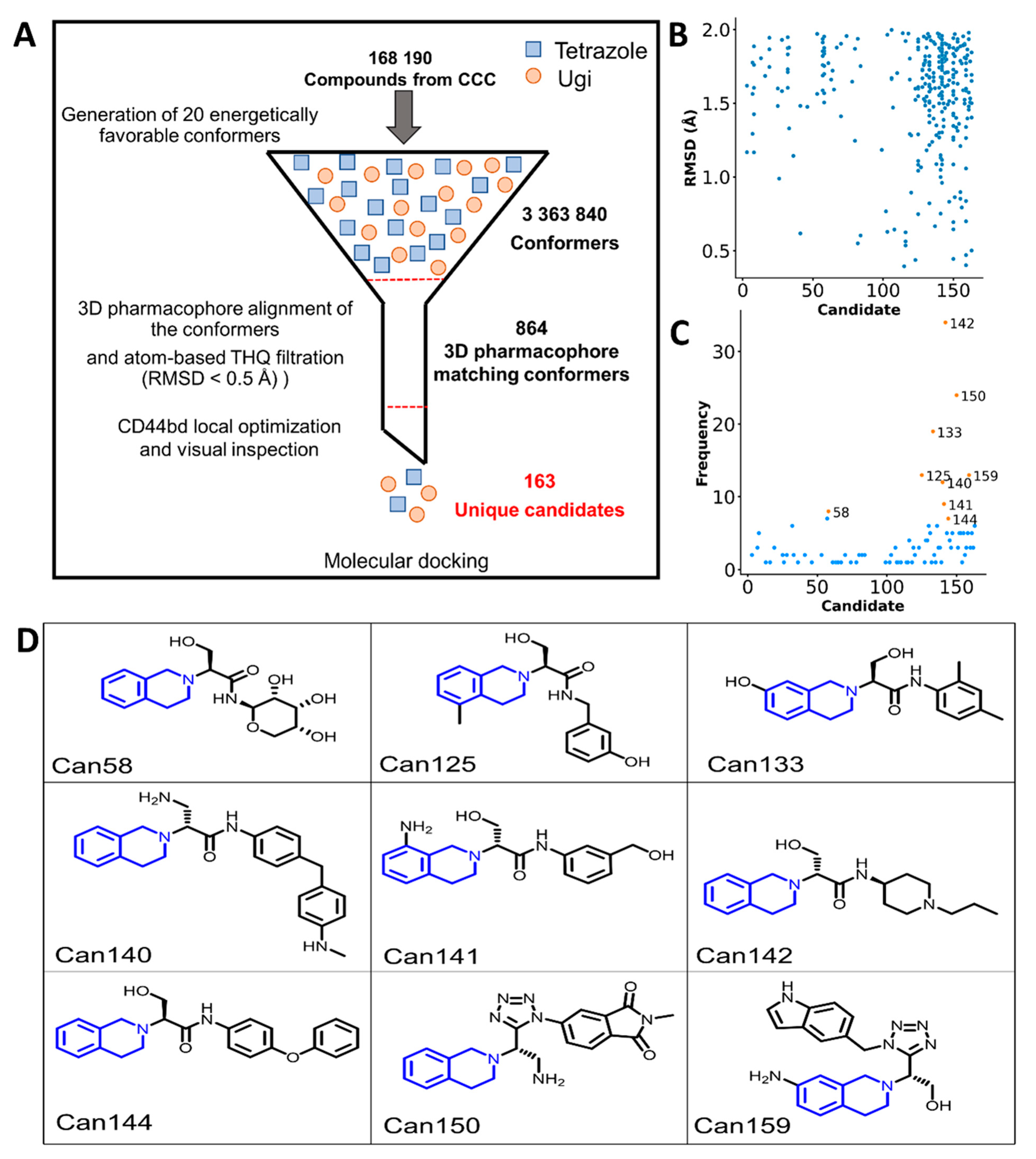
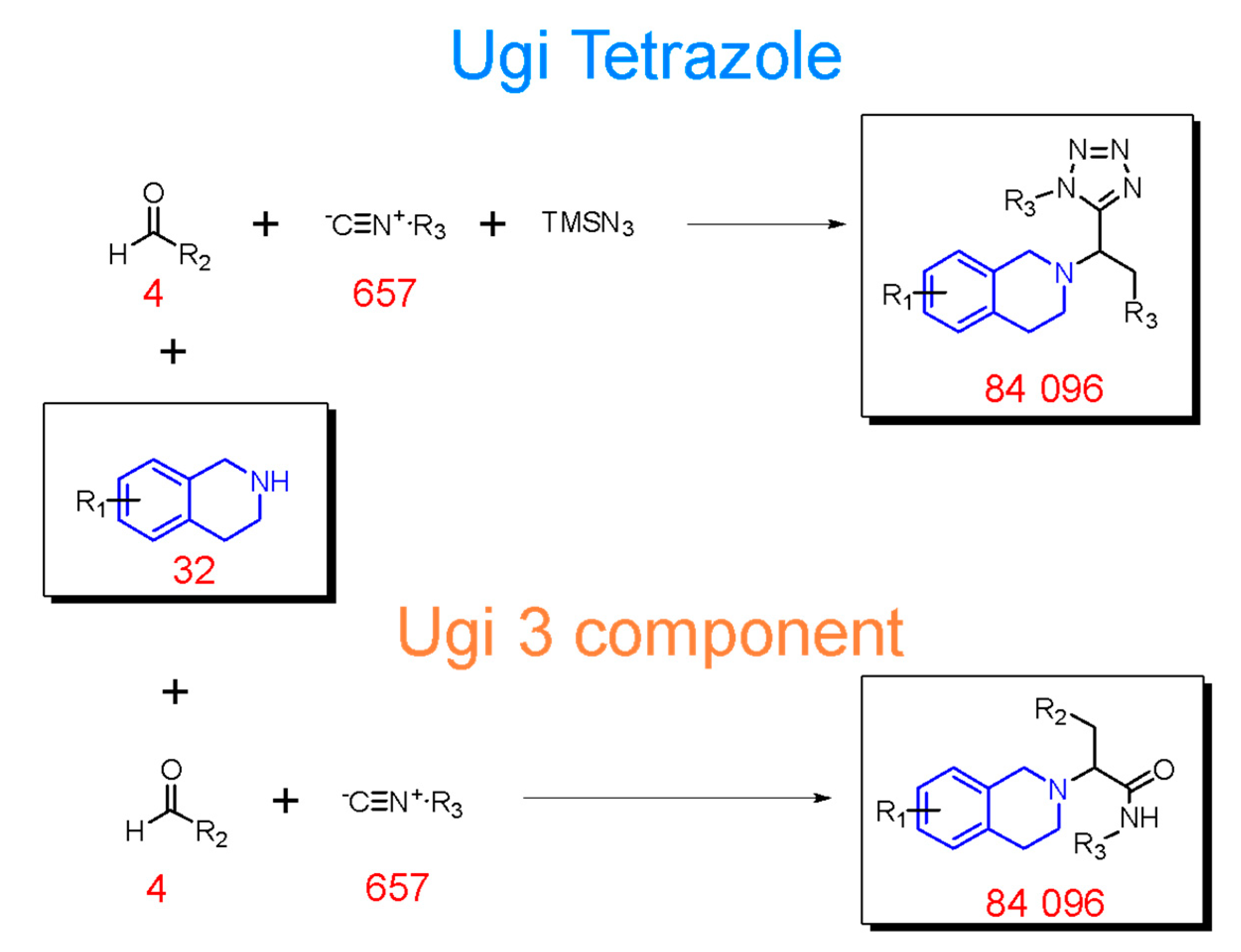
2.2. Generation of THQ-Containing Libraries
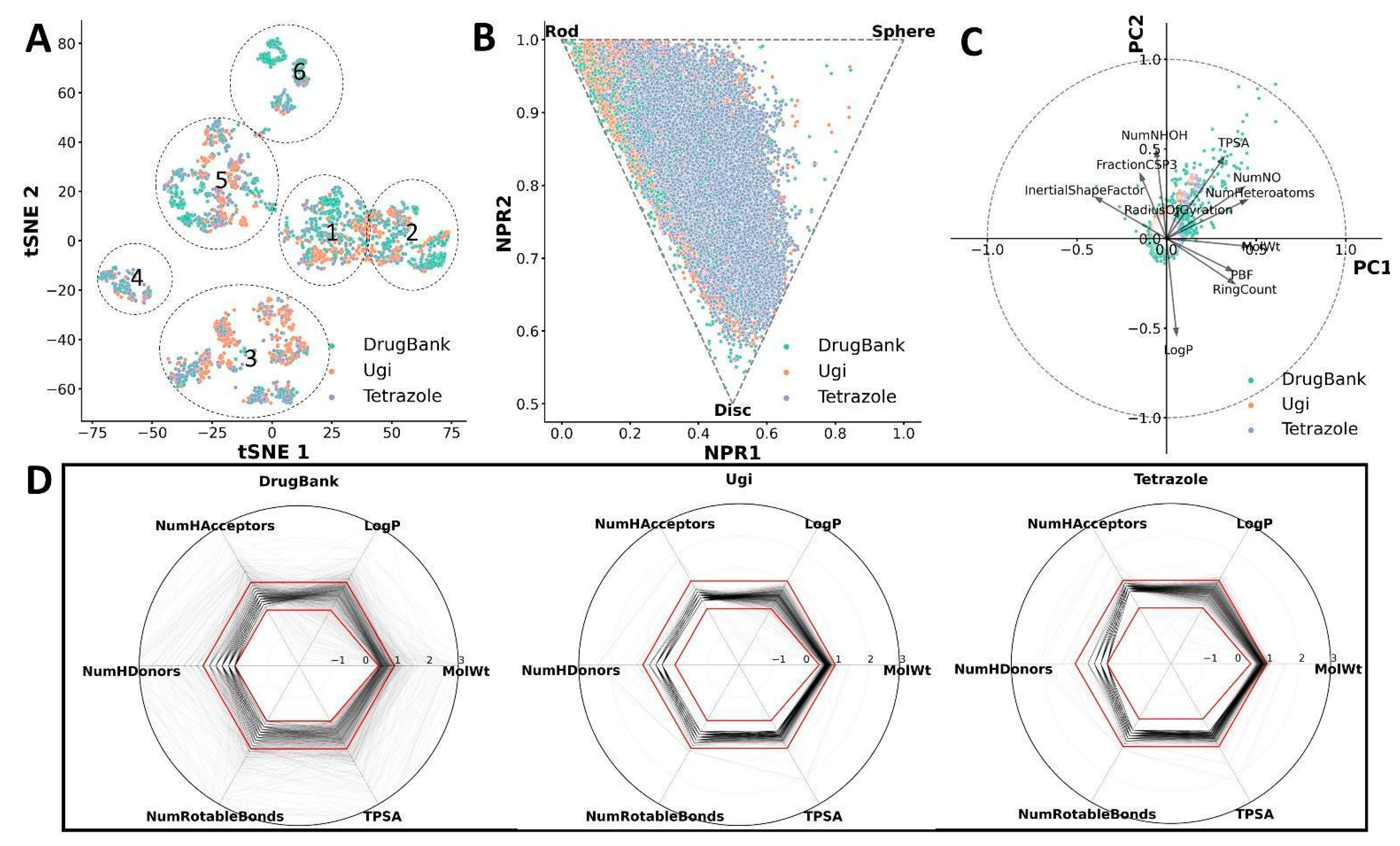
2.3. Cheminformatic Analysis of CCC Compounds
2.4. Virtual Screening
2.5. Molecular Dynamics and Free Energy Calculation
3. Discussion
4. Materials and Methods
4.1. Sequence and Structural Alignments
4.2. The 3D Pharmacophore Modeling
4.3. Combinatorial Computational Chemistry
4.4. Cheminformatic Analysis
4.5. The 3D Pharmacophoric Matching
4.6. Molecular Docking
4.7. Molecular Dynamics
4.8. Interactions Free Energy Calculations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Ponta, H.; Sherman, L.; Herrlich, P.A. CD44: From adhesion molecules to signalling regulators. Nat. Rev. Mol. Cell Biol. 2003, 4, 33–45. [Google Scholar] [CrossRef]
- Zöller, M. CD44: Can a cancer-initiating cell profit from an abundantly expressed molecule? Nat. Rev. Cancer 2011, 11, 254–267. [Google Scholar] [CrossRef]
- Senbanjo, L.T.; Chellaiah, M.A. CD44: A multifunctional cell surface adhesion receptor is a regulator of progression and metastasis of cancer cells. Front. Cell Dev. Biol. 2017, 5. [Google Scholar] [CrossRef]
- Wu, K.; Xu, H.; Yuan, X.; Tian, Y.; Liu, Y.; Liu, Q.; Wu, H.; Wu, G.S. Enrichment of CD44 in basal-type breast cancer correlates with EMT, cancer stem cell gene profile, and prognosis. OTT 2016, 9, 431–444. [Google Scholar] [CrossRef]
- Si, D.; Yin, F.; Peng, J.; Zhang, G. High Expression of CD44 Predicts a Poor Prognosis in Glioblastomas. Cancer Manag. Res. 2020, 12, 769–775. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.; Song, X.; Liu, J.; Li, S.; Gao, W.; Qiu, M.; Yang, C.; Ma, Y.; Chen, Y. Expression of CD44 and the survival in glioma: A meta-analysis. Biosci. Rep. 2020, 40, BSR20200520. [Google Scholar] [CrossRef]
- Chen, J.; Zhou, J.; Lu, J.; Xiong, H.; Shi, X.; Gong, L. Significance of CD44 expression in head and neck cancer: A systemic review and meta-analysis. BMC Cancer 2014, 14, 15. [Google Scholar] [CrossRef] [PubMed]
- Papadaki, C.; Manolakou, S.; Lagoudaki, E.; Pontikakis, S.; Ierodiakonou, D.; Vogiatzoglou, K.; Messaritakis, I.; Trypaki, M.; Giannikaki, L.; Sfakianaki, M.; et al. Correlation of PKM2 and CD44 protein expression with poor prognosis in platinum-treated epithelial ovarian cancer: A retrospective study. Cancers (Basel) 2020, 12, 1013. [Google Scholar] [CrossRef]
- Bourguignon, L.Y.W.; Spevak, C.C.; Wong, G.; Xia, W.; Gilad, E. Hyaluronan-CD44 interaction with protein kinase Cϵ promotes oncogenic signaling by the stem cell marker nanog and the production of microRNA-21, leading to down-regulation of the tumor suppressor protein PDCD4, anti-apoptosis, and chemotherapy resistance. J. Biol. Chem. 2009, 284, 26533–26546. [Google Scholar] [CrossRef]
- Bourguignon, L.Y.W. Matrix Hyaluronan-CD44 Interaction Activates MicroRNA and LncRNA Signaling Associated With Chemoresistance, Invasion, and Tumor Progression. Front. Oncol. 2019, 9, 492. [Google Scholar] [CrossRef]
- Bourguignon, L.Y.W.; Wong, G.; Shiina, M. Up-regulation of histone methyltransferase, DOT1L, by matrix hyaluronan promotes microRNA-10 expression leading to tumor cell invasion and chemoresistance in cancer stem cells from head and neck squamous cell carcinoma. J. Biol. Chem. 2016, 291, 10571–10585. [Google Scholar] [CrossRef]
- Hill, A.; McFarlane, S.; Mulligan, K.; Gillespie, H.; Draffin, J.E.; Trimble, A.; Ouhtit, A.; Johnston, P.G.; Harkin, D.P.; McCormick, D.; et al. Cortactin underpins CD44-promoted invasion and adhesion of breast cancer cells to bone marrow endothelial cells. Oncogene 2006, 25, 6079–6091. [Google Scholar] [CrossRef] [PubMed]
- Cieply, B.; Koontz, C.; Frisch, S.M. CD44S-hyaluronan interactions protect cells resulting from EMT against anoikis. Matrix Biol. 2015, 48, 55–65. [Google Scholar] [CrossRef]
- Gudbergsson, J.M.; Christensen, E.; Kostrikov, S.; Moos, T.; Duroux, M.; Kjær, A.; Johnsen, K.B.; Andresen, T.L. Conventional Treatment of Glioblastoma Reveals Persistent CD44+ Subpopulations. Mol. Neurobiol. 2020, 57, 3943–3955. [Google Scholar] [CrossRef]
- Yu, F.; Yao, H.; Zhu, P.; Zhang, X.; Pan, Q.; Gong, C.; Huang, Y.; Hu, X.; Su, F.; Lieberman, J.; et al. Let-7 regulates self renewal and tumorigenicity of breast cancer cells. Cell 2007, 131, 1109–1123. [Google Scholar] [CrossRef]
- Hong, S.P.; Wen, J.; Bang, S.; Park, S.; Song, S.Y. CD44-positive cells are responsible for gemcitabine resistance in pancreatic cancer cells. Int. J. Cancer 2009, 125, 2323–2331. [Google Scholar] [CrossRef] [PubMed]
- Dylla, S.J.; Beviglia, L.; Park, I.-K.; Chartier, C.; Raval, J.; Ngan, L.; Pickell, K.; Aguilar, J.; Lazetic, S.; Smith-Berdan, S.; et al. Colorectal cancer stem cells are enriched in xenogeneic tumors following chemotherapy. PLoS ONE 2008, 3, e2428. [Google Scholar] [CrossRef]
- Wang, L.; Huang, X.; Zheng, X.; Wang, X.; Li, S.; Zhang, L.; Yang, Z.; Xia, Z. Enrichment of prostate cancer stem-like cells from human prostate cancer cell lines by culture in serum-free medium and chemoradiotherapy. Int. J. Biol. Sci. 2013, 9, 472–479. [Google Scholar] [CrossRef]
- Hiraga, T.; Ito, S.; Nakamura, H. Cancer stem-like cell marker CD44 promotes bone metastases by enhancing tumorigenicity, cell motility, and hyaluronan production. Cancer Res. 2013, 73, 4112–4122. [Google Scholar] [CrossRef]
- Su, Y.-J.; Lai, H.-M.; Chang, Y.-W.; Chen, G.-Y.; Lee, J.-L. Direct reprogramming of stem cell properties in colon cancer cells by CD44. EMBO J. 2011, 30, 3186–3199. [Google Scholar] [CrossRef]
- Gao, Y.; Foster, R.; Yang, X.; Feng, Y.; Shen, J.K.; Mankin, H.J.; Hornicek, F.J.; Amiji, M.M.; Duan, Z. Up-regulation of CD44 in the development of metastasis, recurrence and drug resistance of ovarian cancer. Oncotarget 2015, 6, 9313–9326. [Google Scholar] [CrossRef]
- Banerji, S.; Wright, A.J.; Noble, M.; Mahoney, D.J.; Campbell, I.D.; Day, A.J.; Jackson, D.G. Structures of the Cd44-hyaluronan complex provide insight into a fundamental carbohydrate-protein interaction. Nat. Struct. Mol. Biol. 2007, 14, 234–239. [Google Scholar] [CrossRef]
- Bajorath, J.; Greenfield, B.; Munro, S.B.; Day, A.J.; Aruffo, A. Identification of CD44 residues important for hyaluronan binding and delineation of the binding site. J. Biol. Chem. 1998, 273, 338–343. [Google Scholar] [CrossRef]
- Liu, L.K.; Finzel, B.C. Fragment-based identification of an inducible binding site on cell surface receptor CD44 for the design of protein-carbohydrate interaction inhibitors. J. Med. Chem. 2014, 57, 2714–2725. [Google Scholar] [CrossRef]
- Baggio, C.; Barile, E.; Di Sorbo, G.; Kipps, T.J.; Pellecchia, M. The cell surface receptor CD44: NMR-based characterization of putative ligands. ChemMedChem 2016, 11, 1097–1106. [Google Scholar] [CrossRef]
- Aguirre-Alvarado, C.; Segura-Cabrera, A.; Velázquez-Quesada, I.; Hernández-Esquivel, M.A.; García-Pérez, C.A.; Guerrero-Rodríguez, S.L.; Ruiz-Moreno, A.J.; Rodríguez-Moreno, A.; Pérez-Tapia, S.M.; Velasco-Velázquez, M.A. Virtual screening-driven repositioning of etoposide as CD44 antagonist in breast cancer cells. Oncotarget 2016, 7, 23772–23784. [Google Scholar] [CrossRef][Green Version]
- Pustuła, M.; Czub, M.; Łabuzek, B.; Surmiak, E.; Tomala, M.; Twarda-Clapa, A.; Guzik, K.; Popowicz, G.M.; Holak, T.A. NMR fragment-based screening for development of the CD44-binding small molecules. Bioorg Chem. 2019, 82, 284–289. [Google Scholar] [CrossRef]
- Zhang, J.; Patil, P.; Kurpiewskab, K.; Kalinowska, T.; luścikb, J.; Dömling, A. Hydrazine in the Ugi Tetrazole Reaction Synthesis. Synthesis (Stuttg) 2016, 48, 1122–1130. [Google Scholar]
- Neochoritis, C.G.; Zhao, T.; Dömling, A. Tetrazoles via multicomponent reactions. Chem. Rev. 2019, 119, 1970–2042. [Google Scholar] [CrossRef]
- Tripolitsiotis, N.P.; Thomaidi, M.; Neochoritis, C.G. The Ugi three-component reaction; a valuable tool in modern organic synthesis. Eur. J. Org. Chem. 2020, 2020, 6525–6554. [Google Scholar] [CrossRef]
- Wishart, D.S.; Knox, C.; Guo, A.C.; Shrivastava, S.; Hassanali, M.; Stothard, P.; Chang, Z.; Woolsey, J. DrugBank: A comprehensive resource for in silico drug discovery and exploration. Nucleic Acids Res. 2006, 34, D668–D672. [Google Scholar] [CrossRef]
- Polton, D.J. Installation and operational experiences with MACCS (Molecular Access System). Online Rev. 1982, 6, 8. [Google Scholar] [CrossRef]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settingsq. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef]
- Liu, L.K.; Finzel, B. High-resolution crystal structures of alternate forms of the human CD44 hyaluronan-binding domain reveal a site for protein interaction. Acta Crystallogr. Sect. Struct. Biol. Commun. 2014, 70, 1155–1161. [Google Scholar] [CrossRef]
- Zarganes-Tzitzikas, T.; Chandgude, A.L.; Dömling, A. Multicomponent Reactions, Union of MCRs and beyond. Chem. Rec. 2015, 15, 981–996. [Google Scholar] [CrossRef]
- Dömling, A.; Wang, W.; Wang, K. Chemistry and biology of multicomponent reactions. Chem. Rev. 2012, 112, 3083–3135. [Google Scholar] [CrossRef]
- Awale, M.; Reymond, J.-L. Web-based 3D-visualization of the DrugBank chemical space. J. Cheminform. 2016, 8, 25. [Google Scholar] [CrossRef]
- Doak, B.C.; Zheng, J.; Dobritzsch, D.; Kihlberg, J. How beyond rule of 5 drugs and clinical candidates bind to their targets. J. Med. Chem. 2016, 59, 2312–2327. [Google Scholar] [CrossRef]
- Li, J.; Di Lorenzo, V.; Patil, P.; Ruiz-Moreno, A.J.; Kurpiewska, K.; Kalinowska-Tłuścik, J.; Velasco-Velázquez, M.A.; Dömling, A. Scaffolding-Induced Property Modulation of Chemical Space. ACS Comb. Sci. 2020, 22, 356–360. [Google Scholar] [CrossRef]
- Bowman, W.C. Neuromuscular block. Br. J. Pharmacol. 2006, 147 (Suppl 1), S277–S286. [Google Scholar] [CrossRef]
- Doroshyenko, O.; Fuhr, U. Clinical pharmacokinetics and pharmacodynamics of solifenacin. Clin. Pharm. 2009, 48, 281–302. [Google Scholar] [CrossRef]
- Asmar, R.; Sayegh, F.; Tracz, W.; Hlawaty, M.; Olszowska, M.; Jourde, M.; Vincent, M.; Goujoun, B.; Maldonado, J. Reversal of left ventricular hypertrophy with the ACE inhibitor moexipril in patients with essential hypertension. Acta Cardiol. 2002, 57, 31–32. [Google Scholar]
- De Luca, L.; Gitto, R.; Barreca, M.L.; Caruso, R.; Quartarone, S.; Citraro, R.; De Sarro, G.; Chimirri, A. 3D pharmacophore models for 1,2,3,4-tetrahydroisoquinoline derivatives acting as anticonvulsant agents. Arch. Pharm. Chem. Life Sci. 2006, 339, 388–400. [Google Scholar] [CrossRef]
- Zhang, L.Y.; Gallicchio, E.; Friesner, R.A.; Levy, R.M. Solvent models for protein–ligand binding: Comparison of implicit solvent poisson and surface generalized born models with explicit solvent simulations. J. Comput. Chem. 2001, 22, 591–607. [Google Scholar] [CrossRef]
- Amaral, M.; Kokh, D.B.; Bomke, J.; Wegener, A.; Buchstaller, H.P.; Eggenweiler, H.M.; Matias, P.; Sirrenberg, C.; Wade, R.C.; Frech, M. Protein conformational flexibility modulates kinetics and thermodynamics of drug binding. Nat. Commun. 2017, 8, 2276. [Google Scholar] [CrossRef] [PubMed]
- Ferina, J.; Daggett, V. Visualizing protein folding and unfolding. J. Mol. Biol. 2019, 431, 1540–1564. [Google Scholar] [CrossRef]
- Naor, D.; Sionov, R.V.; Ish-Shalom, D. CD44: Structure, function and association with the malignant process. In Advances in Cancer Research; Vande Woude, G.F., Klein, G., Eds.; Academic Press: Cambridge, MA, USA, 1997; Volume 71, pp. 241–319. [Google Scholar]
- Shi, X.; Leng, L.; Wang, T.; Wang, W.; Du, X.; Li, J.; McDonald, C.; Chen, Z.; Murphy, J.W.; Lolis, E.; et al. CD44 is the signaling component of the macrophage migration inhibitory factor-CD74 receptor complex. Immunity 2006, 25, 595–606. [Google Scholar] [CrossRef]
- Fujimoto, T.; Kawashima, H.; Tanaka, T.; Hirose, M.; Toyama-Sorimachi, N.; Matsuzawa, Y.; Miyasaka, M. CD44 binds a chondroitin sulfate proteoglycan, aggrecan. Int. Immunol. 2001, 13, 359–366. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- Cock, P.J.A.; Antao, T.; Chang, J.T.; Chapman, B.A.; Cox, C.J.; Dalke, A.; Friedberg, I.; Hamelryck, T.; Kauff, F.; Wilczynski, B.; et al. Biopython: Freely available Python tools for computational molecular biology and bioinformatics. Bioinformatics 2009, 25, 1422–1423. [Google Scholar] [CrossRef]
- Sunseri, J.; Koes, D.R. Pharmit: Interactive exploration of chemical space. Nucleic Acids Res. 2016, 44, W442–W448. [Google Scholar] [CrossRef]
- Pedregosa, F.; Varoquaux, G.; Gramfort, A.; Michel, V.; Thirion, B.; Grisel, O.; Blondel, M.; Prettenhofer, P.; Weiss, R.; Dubourg, V.; et al. Scikit-learn: Machine Learning in Python. J. Mach. Learn. Res. 2011, 12, 2825–2830. [Google Scholar]
- Gerber, P.R.; Müller, K. MAB, a generally applicable molecular force field for structure modelling in medicinal chemistry. J. Comput. Aided. Mol. Des. 1995, 9, 251–268. [Google Scholar] [CrossRef]
- Gerber, P.R. Topological pharmacophore description of chemical structures using MAB-Force-Field-Derived data and corresponding similarity measures. In Fundamentals of Molecular Similarity; Carbó-Dorca, R., Gironés, X., Mezey, P.G., Eds.; Mathematical and Computational Chemistry; Springer US: Boston, MA, USA, 2001; pp. 67–81. ISBN 978-1-4757-3273-3. [Google Scholar]
- Le Guilloux, V.; Schmidtke, P.; Tuffery, P. Fpocket: An open source platform for ligand pocket detection. BMC Bioinform. 2009, 10, 168. [Google Scholar] [CrossRef]
- Jones, G.; Willett, P.; Glen, R.C.; Leach, A.R.; Taylor, R. Development and validation of a genetic algorithm for flexible docking. J. Mol. Biol. 1997, 267, 727–748. [Google Scholar] [CrossRef]
- Virtanen, P.; Gommers, R.; Oliphant, T.E.; Haberland, M.; Reddy, T.; Cournapeau, D.; Burovski, E.; Peterson, P.; Weckesser, W.; Bright, J.; et al. SciPy 1.0: Fundamental algorithms for scientific computing in Python. Nat. Methods 2020, 17, 261–272. [Google Scholar] [CrossRef]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef]
- Jo, S.; Kim, T.; Iyer, V.G.; Im, W. CHARMM-GUI: A web-based graphical user interface for CHARMM. J. Comput. Chem. 2008, 29, 1859–1865. [Google Scholar] [CrossRef]
- Gowers, R.J.; Linke, M.; Barnoud, J.; Reddy, T.J.E.; Melo, M.N.; Seyler, S.L.; Domański, J.; Dotson, D.L.; Buchoux, S.; Kenney, I.M.; et al. MDAnalysis: A python package for the rapid analysis of molecular dynamics simulations. In Proceedings of the 15th Python in Science Conference (SciPy 2016), Austin, TX, USA, 11–17 July 2016; pp. 98–105. [Google Scholar]
- Salentin, S.; Schreiber, S.; Haupt, V.J.; Adasme, M.F.; Schroeder, M. PLIP: Fully automated protein–ligand interaction profiler. Nucleic Acids Res. 2015, 43, W443–W447. [Google Scholar] [CrossRef]
- Genheden, S.; Ryde, U. The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin. Drug Discov. 2015, 10, 449–461. [Google Scholar] [CrossRef]
- Kumari, R.; Kumar, R.; Lynn, A. g_mmpbsa—A GROMACS tool for high-throughput MM-PBSA calculations. J. Chem. Inf. Model. 2014, 54, 1951–1962. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Van der Waals Energy | Electrostatic Energy | Polar Solvation Energy | SASA Energy | Binding Energy |
|---|---|---|---|---|---|
| Can58 | −33.871 ± 35.190 | −18.447 ± 29.906 | 47.592 ± 66.887 | −4.667 ± 4.853 | −9.393 ± 41.581 |
| Can125 | −116.512 ± 15.988 | −52.356 ± 23.296 | 123.555 ± 27.579 | −13.962 ± 1.252 | −59.274 ± 17.744 |
| Can140 | −96.931 ± 36.947 | −128.983 ± 66.843 | 106.522 ± 90.455 | −12.078 ± 4.022 | −131.470 ± 41.310 |
| Can159 | −99.395 ± 18.056 | −34.928 ± 25.315 | 112.733 ± 40.928 | −11.318 ± 1.938 | −32.908 ± 17.750 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ruiz-Moreno, A.J.; Reyes-Romero, A.; Dömling, A.; Velasco-Velázquez, M.A. In Silico Design and Selection of New Tetrahydroisoquinoline-Based CD44 Antagonist Candidates. Molecules 2021, 26, 1877. https://doi.org/10.3390/molecules26071877
Ruiz-Moreno AJ, Reyes-Romero A, Dömling A, Velasco-Velázquez MA. In Silico Design and Selection of New Tetrahydroisoquinoline-Based CD44 Antagonist Candidates. Molecules. 2021; 26(7):1877. https://doi.org/10.3390/molecules26071877
Chicago/Turabian StyleRuiz-Moreno, Angel J., Atilio Reyes-Romero, Alexander Dömling, and Marco A. Velasco-Velázquez. 2021. "In Silico Design and Selection of New Tetrahydroisoquinoline-Based CD44 Antagonist Candidates" Molecules 26, no. 7: 1877. https://doi.org/10.3390/molecules26071877
APA StyleRuiz-Moreno, A. J., Reyes-Romero, A., Dömling, A., & Velasco-Velázquez, M. A. (2021). In Silico Design and Selection of New Tetrahydroisoquinoline-Based CD44 Antagonist Candidates. Molecules, 26(7), 1877. https://doi.org/10.3390/molecules26071877