
Research on Molecular Structure and Electronic Properties of Ln3+ (Ce3+, Tb3+, Pr3+)/Li+ and Eu2+ Co-Doped Sr2Si5N8 via DFT Calculation



Abstract
:1. Introduction
2. Results and Discussion
2.1. Structures Distortion of Doped Models
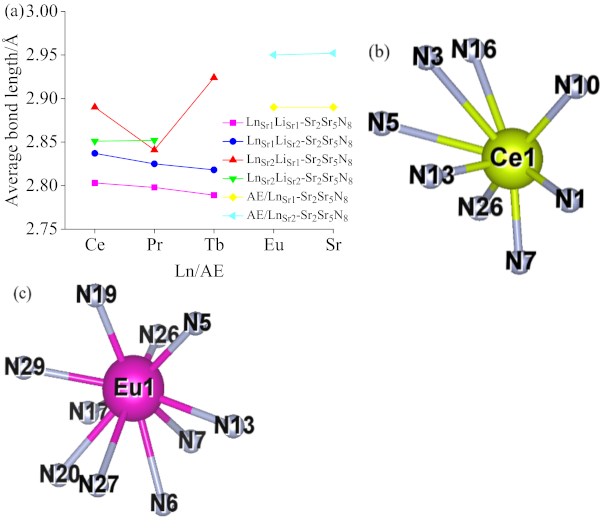
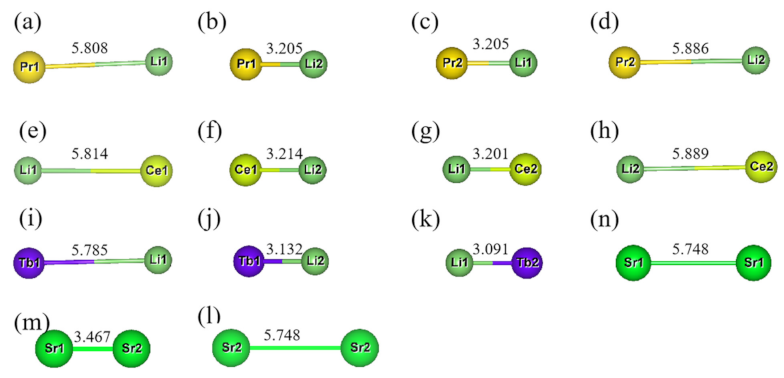
2.1.1. Ln-Li Distance and [SrN] Coordination Polyhedron Parameters of Sr2Si5N8:Ln3+/Li+
2.1.2. Lattice Constant and [LnN] Ligand Parameters of Sr2Si5N8:Ln3+/Li+/Eu2+
2.1.3. Ln-Li-Eu distance and [SrN] Coordination Polyhedron Parameters of Sr2Si5N8:Ln3+/Li+/Eu2+
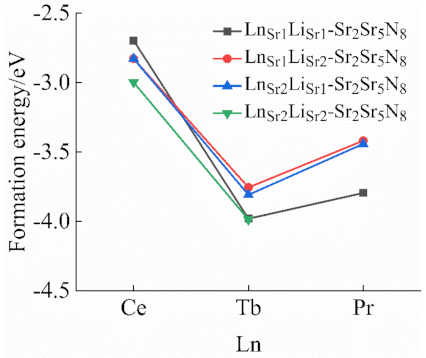
2.2. Formation Energy of Doped Models
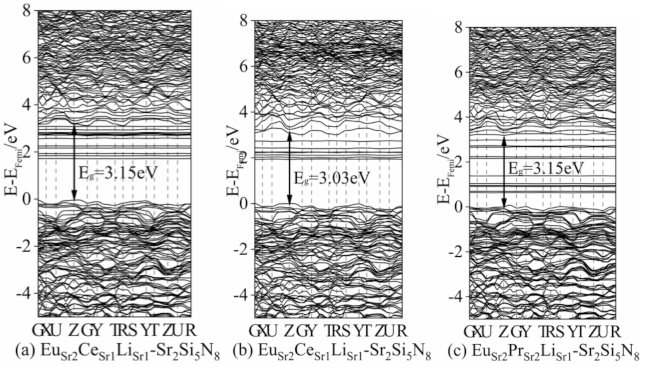
2.3. Band Structures and Density of States
Determination of DFT+U Parameters of Each System
3. Material and Methods
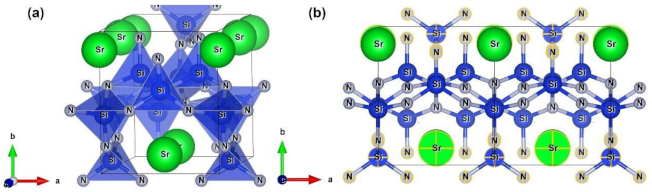
3.1. Theoretical Models
3.2. Computational Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Li, Y.Q.; van Steen, J.E.J.; van Krevel, J.W.H.; Botty, G.; Delsing, A.C.A.; DiSalvo, F.J.; de With, G.; Hintzen, H.T. Luminescence properties of red-emitting M2Si5N8:Eu2+ (M = Ca, Sr, Ba) LED conversion phosphors. J. Alloys Compd. 2006, 417, 273–279. [Google Scholar] [CrossRef]
- Liu, Y.H.; Chen, L.; Zhou, X.F.; Liu, R.H.; Zhuang, W.D. Structure, luminescence and thermal quenching properties of Eu doped Sr2-xBaxSi5N8 red phosphors. J. Solid State Chem. 2016, 246, 145–149. [Google Scholar] [CrossRef]
- Chen, L.; Liu, R.-H.; Zhuang, W.-D.; Liu, Y.-H.; Hu, Y.-S.; Du, F. Effect of Eu2+ concentration on the thermal quenching mechanism of Sr2Si5N8:Eu2+ red phosphors. Chin. J. Lumin. 2015, 36, 371–376. [Google Scholar] [CrossRef]
- Seibald, M.; Rosenthal, T.; Oeckler, O.; Maak, C.; Tücks, A.; Schmidt, P.J.; Wiechert, D.; Schnick, W. New polymorph of the highly efficient LED-phosphor SrSi2O2N2: Eu2+-polytypism of a layered oxonitridosilicate. Chem. Mater. 2013, 25, 1852–1857. [Google Scholar] [CrossRef]
- Chen, L.; Liu, R.H.; Zhuang, W.D.; Liu, Y.H.; Hu, Y.S.; Zhou, X.F.; Ma, X.L. A study on photoluminescence and energy transfer of SrAlSi4N7: Eu2+, Ce3+ phosphors for application in white-light LED. J. Alloys Compd. 2015, 627, 218–221. [Google Scholar] [CrossRef]
- Daicho, H.; Shinomiya, Y.; Enomoto, K.; Nakano, A.; Sawa, H.; Matsuishi, S.; Hosono, H. A novel red-emitting K2Ca(PO4)F:Eu2+ phosphor with a large Stokes shift. Chem. Commun. 2018, 54, 884–887. [Google Scholar] [CrossRef] [PubMed]
- Shang, M.M.; Li, C.X.; Lin, J. ChemInform abstract: How to produce white light in a single-phase host? ChemInform 2014, 43, 1372–1386. [Google Scholar]
- Dorenbos, P. A review on how Lanthanide impurity levels change with chemistry and structure of inorganic compounds. ECS J. Solid State Sci. Technol. 2013, 2, R3001–R3011. [Google Scholar]
- Li, Y.Q.; de With, G.; Hintzen, H.T. The effect of replacement of Sr by Ca on the structural and luminescence properties of the red-emitting Sr2Si5B8:Eu2+LED conversion phosphor. J. Solid State Chem. 2008, 181, 515–524. [Google Scholar]
- Bulloni, C.; García-Fuente, A.; Urland, W.; Daul, C. Effect of Ca2+ codoping on the Eu2+ luminescence properties in the Sr2Si5N8:Eu2+ host lattice: A theoretical approach. Phys. Chem. Chem. Phys. 2015, 17, 24925–24930. [Google Scholar] [CrossRef]
- Chen, L.; Liu, R.H.; Zhuang, W.D.; Liu, Y.H.; Hu, Y.S.; Zhou, X.F.; Gao, W.; Ma, X.L. Structure, photoluminescence, and thermal quenching properties of Eu doped Sr2AlxSi5−xN8−x/3 red phosphors. CrystEngComm 2015, 17, 3687–3694. [Google Scholar] [CrossRef]
- Wang, T.; Zheng, P.; Liu, X.L.; Chen, H.F.; Bian, L.; Liu, Q.L. Effects of replacement of AlO+ for SiN+ on the structure and optical properties of Sr2Si5N8:Eu2+ phosphors. J. Lumin. 2014, 147, 173–178. [Google Scholar] [CrossRef]
- Qiao, J.; Ning, L.; Molokeev, M.S.; Chuang, Y.C.; Zhang, Q.; Poeppelmeier, K.R.; Xia, Z. Site-Selective Occupancy of Eu2+ Toward Blue-Light-Excited Red Emission in a Rb3YSi2O7:Eu Phosphor. Angew. Chem. Int. Ed. 2019, 131, 11645–11650. [Google Scholar]
- Li, H.; Li, W.; Liu, X.; Ren, C.; Miao, X.; Li, X. Engineering of Gd/Er/Lu-triple-doped Bi2MoO6 to synergistically boost the photocatalytic performance in three different aspects: Oxidizability, light absorption and charge separation. Appl. Surf. Sci. 2019, 463, 556–565. [Google Scholar] [CrossRef]
- Tang, W.; Ding, S.; Yuan, Y.; Xie, G. Effect of Ce3+ and Tb3+ codoping on luminescence properties of Na3SrMg11(PO4)9:Eu2+ phosphors. Opt. Mater. 2020, 106, 110003. [Google Scholar] [CrossRef]
- Peng, X.; Li, S.-X.; Liu, X.-J.; Huang, Y.-H.; Huang, Z.-R.; Li, H.-L. Syntheses and photoluminescence properties of Eu2+/Tb3+ doped Sr2Si5N8 phosphors. J. Inorg. Mater. 2015, 30, 397–400. [Google Scholar]
- Li, H.L.; Liu, X.J.; Huang, L.P. Luminescent properties of Luag: Ce phosphors with different ce contents prepared by a sol-gel combustion method. Opt. Mater. 2007, 29, 1138–1142. [Google Scholar] [CrossRef]
- Zorenko, Y.; Gorbenko, V.; Voznyak, T.; Zorenko, T.; Kuklinski, B.; Turos-Matysyak, R.; Grinberg, M. Luminescence properties of phosphors based on Tb3Al5O12 (TbAG) terbium-aluminum garnet. Opt. Spectrosc. 2009, 106, 365–374. [Google Scholar] [CrossRef]
- Zhong, J.Y.; Zhao, W.R.; Du, F.; Wen, J.; Zhuang, W.D.; Liu, R.H.; Duan, C.-K.; Wang, L.G.; Lin, K. Identifying the emission centers and probing the mechanism for highly efficient and thermally stable luminescence in the La3Si6N11: Ce3+Phosphor. J. Phys. Chem. C 2018, 122, 7849–7858. [Google Scholar] [CrossRef]
- Li, Y.Q.; de With, D.; Hintzen, H.T. Luminescence properties of Ce3+-activated alkaline earth silicon nitride M2Si5N8(M=Ca, Sr, Ba) materials. J. Lumin. 2006, 116, 107–116. [Google Scholar]
- Pust, P.; Wochnik, A.S.; Baumann, E.; Schmidt, P.J.; Wiechert, D.; Scheu, C.; Schnick, W. Ca[LiAl3N4]: Eu2+—A narrow-band red-emitting nitridolithoaluminate. Chem. Mater. 2014, 26, 3544–3549. [Google Scholar]
- Wilhelm, D.; Baumann, D.; Seibald, M.; Wurst, K.; Heymann, G.; Huppertz, H. Narrow-band red emission in the nitridolithoaluminate Sr4[LiAl11N14]: Eu2+. Chem. Mater. 2017, 29, 1204–1209. [Google Scholar] [CrossRef]
- Strobel, P.; Weiler, V.; Hecht, C.; Schmidt, P.J.; Schnick, W. Luminescence of the narrow-band red emitting nitridomagnesosilicate Li2(Ca1-xSrx)2[Mg2Si2N6]: Eu2+(x = 0 − 0.06). Chem. Mater. 2017, 29, 1377–1383. [Google Scholar] [CrossRef]
- Fang, C.M.; Hintzen, H.T.; de With, G.; de Groot, R.A. Electronic structure of the alkaline-earth silicon nitrides M2Si5N8(M=Ca and Sr) obtained from first-principles calculations and optical reflectance spectra. J. Phys. Condens. Matter 2001, 13, 67–76. [Google Scholar]
- Shen, T.-Y.; Wu, Y.; Shen, Y.-F.; Zou, Z.-G.; Zhang, F.; Long, Z.-J. Photophysical properties and electronic structure of Sr2Si5N8:Eu2+. J. Synth. Cryst. 2014, 43, 1212–1216. [Google Scholar]
- Xi, Q. Research on Ionic Radius. J. Hainan Norm. Univ. 2001, 14, 68–75. [Google Scholar]
- Zhang, L.G.; Jin, H.; Yang, W.Y.; Xie, Z.P.; Miao, H.Z.; An, L. Optical properties of single-crystalline alpha-Si3N4 nanobelts. Appl. Phys. Lett. 2005, 86, 061908. [Google Scholar] [CrossRef]
- Sikander, A.; Muhammad, I.; Ayaz, K.S.; Ali, Z.; Kityk, I.V.; Muhammad, S.; Al-Sehemi, A.G. Doping induced effect on optical and band structure properties of Sr2Si5N8based phosphors: DFT approach. J. Alloy. Compound. 2019, 771, 1072–1079. [Google Scholar] [CrossRef]
- Ten Kate, O.M.; Zhang, Z.; Dorenbos, P.; Hintzen, H.T.; Van der Kolk, E. 4f and 5d energy levels of the divalent and trivalent lanthanide ions in M2Si5N8 (M=Ca, Sr, Ba). J. Solid State Chem. 2013, 197, 209–217. [Google Scholar] [CrossRef]
- Chawla, S.; Kumar, N.; Chander, H. Broad yellow orange emission from SrAl2O4:Pr3+phosphor with blue excitation for application to white LEDs. J. Lumin. 2009, 129, 114–118. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficiency of ab-initio total energy calcutions for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 1996, 6, 15–50. [Google Scholar] [CrossRef]
- Dufek, P.; Blaha, P.; Sliwko, V.; Schwarz, K. Generalized-gradient-approximation description of band splittings in transition-metal oxides and fluorides. Phys. Rev. B Condens. Matter 1994, 49, 10170–10175. [Google Scholar] [CrossRef] [PubMed]
- Dudarev, S.L.; Botton, G.A.; Savrasov, S.Y.; Humphreys, C.J.; Sutton, A.P. Electron-energy-loss spectra and the structural stability of nickel oxide: An LSDA+U study. Phys. Rev. B 1998, 57, 1505–1509. [Google Scholar] [CrossRef]
- Guo, H.; Wu, Q. The calculation method of effective coordination number. J. Shenyang Univ. Chem. Technol. 1991, 4, 245. [Google Scholar]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 2 × 2 × 1 | Cell Parameters | Volume (Å3) | [LnN] Polyhedral Volume (Å3) | [LnN] Distortion Index (Å) | [LnN] Effective Coordination Number | |||
|---|---|---|---|---|---|---|---|---|
| Supercells | a (Å) | b (Å) | c (Å) | α,β,γ/(°) | ||||
| Sr2Si5N8(Sr2) | 11.498 | 6.881 | 9.405 | α = β = γ = 90.0 | 744.074 | 48.866 | 0.062 | 6.707 |
| Sr2Si5N8(Sr1) | 11.498 | 6.881 | 9.405 | α = β = γ = 90.0 | 744.074 | 32.657 | 0.075 | 5.448 |
| EuSi2-Sr2Si5N8 | 11.498 | 6.876 | 9.403 | α = 89.9, β = γ = 90.0 | 743.131 | 48.545 | 0.072 | 5.771 |
| EuSr1-Sr2Si5N8 | 11.493 | 6.876 | 9.402 | α = 90.1, β = γ = 90.0 | 743.046 | 32.379 | 0.090 | 4.728 |
| CeSr1LiSr1-Sr2Si5N8 | 11.492 | 6.874 | 9.373 | α = 90.4, β = γ = 90.0 | 740.450 | 27.921 | 0.119 | 4.919 |
| CeSr1LiSr2-Sr2Si5N8 | 11.505 | 6.869 | 9.376 | α = 90.1, β = γ = 90 | 741.010 | 27.857 | 0.110 | 4.776 |
| CeSr2LiSr1-Sr2Si5N8 | 11.485 | 6.876 | 9.370 | α = 89.9, β = γ = 90 | 739.910 | 45.191 | 0.092 | 5.550 |
| CeSr2LiSr2-Sr2Si5N8 | 11.504 | 6.863 | 9.380 | α = 89.7, β = γ = 90 | 740.700 | 44.354 | 0.090 | 5.597 |
| PrSr1LiSr1-Sr2Si5N8 | 11.487 | 6.873 | 9.372 | α = 90.4, β = γ = 90 | 739.830 | 27.797 | 0.123 | 4.849 |
| PrSr1LiSr2-Sr2Si5N8 | 11.501 | 6.872 | 9.372 | α = 90.1, β = γ = 90.0 | 740.680 | 27.689 | 0.114 | 4.645 |
| PrSr2LiSr1-Sr2Si5N8 | 11.498 | 6.881 | 9.405 | α = β = γ = 90.0 | 739.340 | 45.213 | 0.101 | 5.420 |
| PrSr2LiSr2-Sr2Si5N8 | 11.502 | 6.863 | 9.380 | α = 89.7, β = γ = 90.0 | 740.450 | 44.345 | 0.093 | 5.337 |
| TbSr1LiSr1-Sr2Si5N8 | 11.470 | 6.863 | 9.360 | α = 90.5, β = γ = 90.0 | 736.970 | 27.321 | 0.154 | 4.676 |
| TbSr1LiSr2-Sr2Si5N8 | 11.493 | 6.859 | 9.370 | α = 90.1, β = γ = 90.0 | 738.970 | 27.300 | 0.147 | 4.496 |
| TbSr2LiSr1-Sr2Si5N8 | 11.479 | 6.850 | 9.376 | α = 89.9, β = γ = 90.0 | 737.230 | 44.902 | 0.150 | 4.724 |
| 2 × 2 × 1 Supercell | [SrN] Average Bond Length (Å) | [SrN] Polyhedral Volume (Å3) | [SrN] Distortion Index (Å) | [SrN] Effective Coordination Number | [SrN] Coordination Number |
|---|---|---|---|---|---|
| CeSr1LiSr1-Sr2Si5N8 | 2.994 | 49.700 | 0.0723 | 7.000 | 10 |
| 2.942 | 48.317 | 0.0720 | 6.620 | 10 | |
| 2.886 | 32.405 | 0.0757 | 5.420 | 8 | |
| CeSr1LiSr2-Sr2Si5N8 | 2.925 | 47.655 | 0.063 | 7.070 | 10 |
| 2.954 | 48.845 | 0.073 | 6.358 | 10 | |
| 2.907 | 35.144 | 0.086 | 5.052 | 8 | |
| CeSr2LiSr1-Sr2Si5N8 | 2.956 | 49.440 | 0.066 | 6.211 | 10 |
| 2.963 | 45.359 | 0.088 | 5.889 | 10 | |
| 2.879 | 32.349 | 0.081 | 5.741 | 8 | |
| CeSr2LiSr2-Sr2Si5N8 | 2.975 | 49.291 | 0.075 | 5.989 | 10 |
| 2.870 | 34.093 | 0.080 | 5.350 | 8 | |
| 2.909 | 33.711 | 0.067 | 5.899 | 8 | |
| PrSr1LiSr1-Sr2Si5N8 | 2.940 | 48.289 | 0.072 | 6.646 | 10 |
| 2.994 | 49.734 | 0.073 | 7.022 | 10 | |
| 2.884 | 32.335 | 0.076 | 5.434 | 8 | |
| PrSr1LiSr2-Sr2Si5N8 | 2.923 | 47.601 | 0.062 | 7.148 | 10 |
| 2.956 | 48.950 | 0.072 | 6.378 | 10 | |
| 2.907 | 35.144 | 0.085 | 5.085 | 8 | |
| PrSr1LiSr2-Sr2Si5N8 | 2.923 | 47.601 | 0.062 | 7.148 | 10 |
| 2.907 | 35.144 | 0.085 | 5.085 | 8 | |
| 2.955 | 48.950 | 0.072 | 6.378 | 10 | |
| PrSr2LiSr2-Sr2Si5N8 | 2.970 | 49.192 | 0.073 | 6.087 | 10 |
| 2.872 | 34.160 | 0.081 | 5.321 | 8 | |
| 2.908 | 33.695 | 0.068 | 5.886 | 8 | |
| TbSr1LiSr1-Sr2Si5N8 | 2.937 | 48.233 | 0.071 | 6.874 | 10 |
| 2.999 | 49.821 | 0.075 | 6.968 | 10 | |
| 2.877 | 32.096 | 0.075 | 5.467 | 8 | |
| TbSr1LiSr2-Sr2Si5N8 | 2.923 | 47.602 | 0.061 | 7.231 | 10 |
| 2.963 | 49.304 | 0.073 | 6.116 | 10 | |
| 2.899 | 34.905 | 0.083 | 5.159 | 8 | |
| TbSr2LiSr1-Sr2Si5N8 | 2.883 | 32.733 | 0.086 | 5.729 | 8 |
| 2.958 | 49.914 | 0.079 | 5.940 | 10 | |
| 2.955 | 45.191 | 0.085 | 6.112 | 10 | |
| EuSr1-Sr2Si5N8 | 2.947 | 48.690 | 0.061 | 6.735 | 10 |
| 2.952 | 48.914 | 0.060 | 6.962 | 10 | |
| 2.888 | 32.658 | 0.074 | 5.449 | 8 | |
| EuSr2-Sr2Si5N8 | 2.951 | 48.904 | 0.061 | 6.701 | 10 |
| 2.888 | 32.706 | 0.072 | 5.625 | 8 | |
| 2.889 | 32.641 | 0.074 | 5.453 | 8 | |
| 2.942 | 38.657 | 0.085 | 5.536 | 8 |
| 2 × 2 × 1 | Cell Parameters | Volume (Å3) | [LnN] Polyhedral Volume (Å3) | [LnN] Distortion Index (Å) | [LnN] Effective Coordination Number | |||
|---|---|---|---|---|---|---|---|---|
| Supercells | a (Å) | b (Å) | c (Å) | α,β,γ (°) | ||||
| EuSr1CeSr1LiSr2-Sr2Si5N8(Eu) | 11.490 | 6.874 | 9.375 | α = 90.3, β = γ = 90.0 | 740.470 | 32.042 | 0.0947 | 4.654 |
| EuSr1CeSr1LiSr2-Sr2Si5N8(Ce) | 11.490 | 6.874 | 9.375 | α = 90.3, β = γ = 90.0 | 740.470 | 28.112 | 0.1219 | 4.926 |
| EuSr2CeSr1LiSr2-Sr2Si5N8(Eu) | 11.492 | 6.867 | 9.370 | α = 90.3, β = γ = 90.0 | 739.470 | 48.094 | 0.0613 | 6.455 |
| EuSr2CeSr1LiSr2-Sr2Si5N8(Ce) | 11.492 | 6.867 | 9.370 | α = 90.3, β = γ = 90.0 | 739.470 | 28.046 | 0.1254 | 5.026 |
| EuSr2PrSr2LiSr2-Sr2Si5N8(Eu) | 11.480 | 6.8607 | 9.377 | α = 90.3, β = γ = 90.0 | 738.566 | 48.574 | 0.0943 | 5.849 |
| EuSr2PrSr2LiSr2-Sr2Si5N8(Pr) | 11.480 | 6.8607 | 9.377 | α = 90.3, β = γ = 90.0 | 738.566 | 45.876 | 0.0880 | 4.752 |
| 2 × 2 × 1 Supercell | [SrN] Average Bond Length (Å) | [SrN] Polyhedral Volume (Å3) | [SrN] Distortion Index (Å) | [SrN] Effective Coordination Number | [SrN] Coordination Number |
|---|---|---|---|---|---|
| EuSr1CeSr1LiSr1 | 2.941 | 48.361 | 0.0666 | 6.884 | 10 |
| 2.989 | 49.546 | 0.0721 | 7.040 | 10 | |
| 2.887 | 32.507 | 0.0755 | 5.406 | 8 | |
| EuSr2CeSr11LiSr1 | 2.945 | 48.444 | 0.0718 | 6.590 | 10 |
| 2.991 | 49.691 | 0.0705 | 7.039 | 10 | |
| 2.880 | 32.241 | 0.0755 | 5.427 | 8 | |
| EuSr2PrSr21LiSr1 | 2.960 | 49.248 | 0.0646 | 6.260 | 10 |
| 2.863 | 31.895 | 0.0825 | 5.680 | 8 | |
| 2.889 | 32.623 | 0.0753 | 5.360 | 8 |
| Ueff (eV) | Eu2+ 4f—CBM(eV) | |
|---|---|---|
| Eu12+ | Eu22+ | |
| 0 | 0.84 | 0.84 |
| 2 | 1.48 | 1.50 |
| 4 | 2.22 | 2.23 |
| 6 | 2.86 | 2.93 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yin, Z.; Li, M.; Zhang, J.; Shen, Q. Research on Molecular Structure and Electronic Properties of Ln3+ (Ce3+, Tb3+, Pr3+)/Li+ and Eu2+ Co-Doped Sr2Si5N8 via DFT Calculation. Molecules 2021, 26, 1849. https://doi.org/10.3390/molecules26071849
Yin Z, Li M, Zhang J, Shen Q. Research on Molecular Structure and Electronic Properties of Ln3+ (Ce3+, Tb3+, Pr3+)/Li+ and Eu2+ Co-Doped Sr2Si5N8 via DFT Calculation. Molecules. 2021; 26(7):1849. https://doi.org/10.3390/molecules26071849
Chicago/Turabian StyleYin, Ziqian, Meijuan Li, Jianwen Zhang, and Qiang Shen. 2021. "Research on Molecular Structure and Electronic Properties of Ln3+ (Ce3+, Tb3+, Pr3+)/Li+ and Eu2+ Co-Doped Sr2Si5N8 via DFT Calculation" Molecules 26, no. 7: 1849. https://doi.org/10.3390/molecules26071849
APA StyleYin, Z., Li, M., Zhang, J., & Shen, Q. (2021). Research on Molecular Structure and Electronic Properties of Ln3+ (Ce3+, Tb3+, Pr3+)/Li+ and Eu2+ Co-Doped Sr2Si5N8 via DFT Calculation. Molecules, 26(7), 1849. https://doi.org/10.3390/molecules26071849