
Rate Dependence on Inductive and Resonance Effects for the Organocatalyzed Enantioselective Conjugate Addition of Alkenyl and Alkynyl Boronic Acids to β-Indolyl Enones and β-Pyrrolyl Enones

 ,
,
Abstract
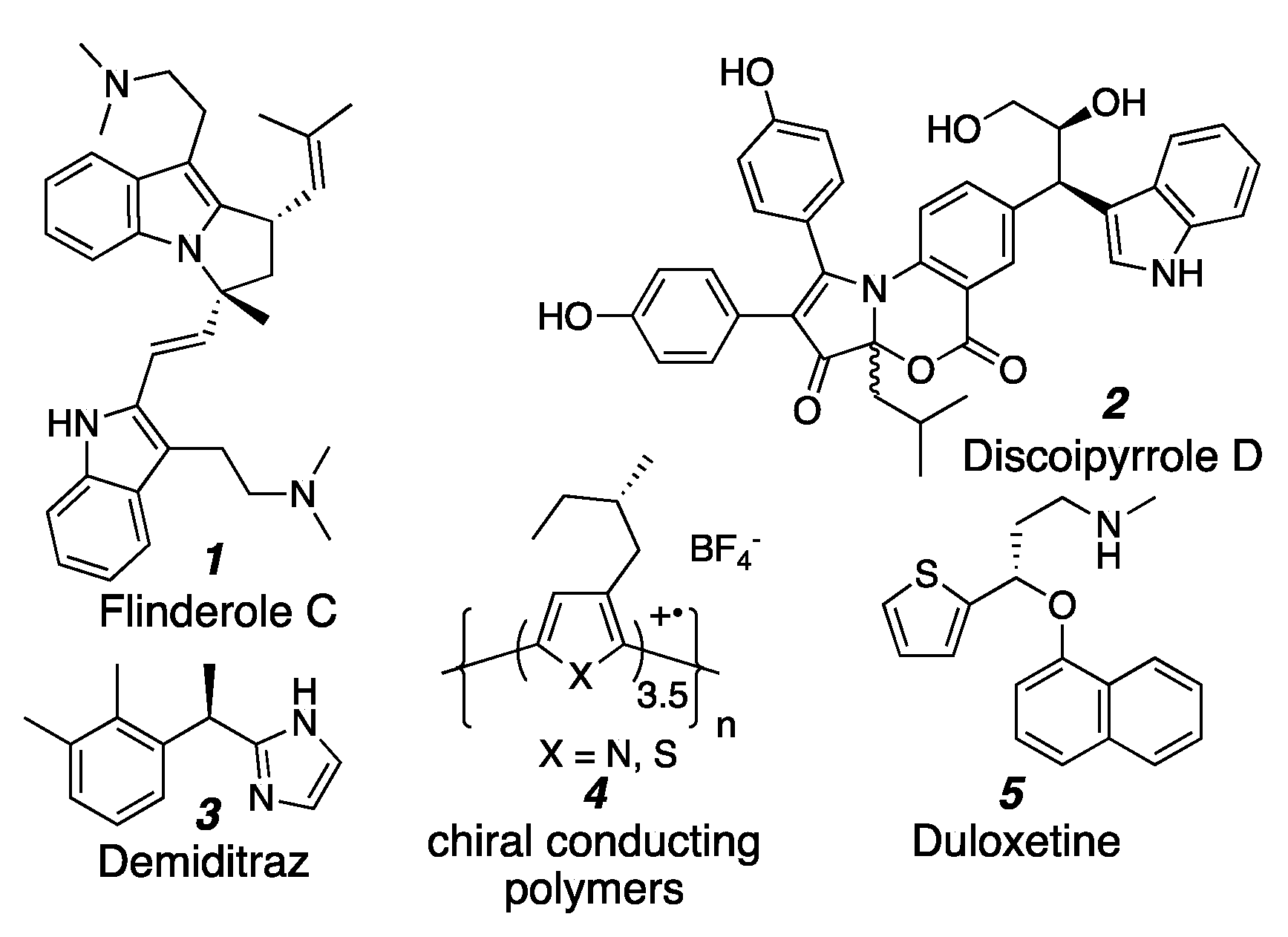
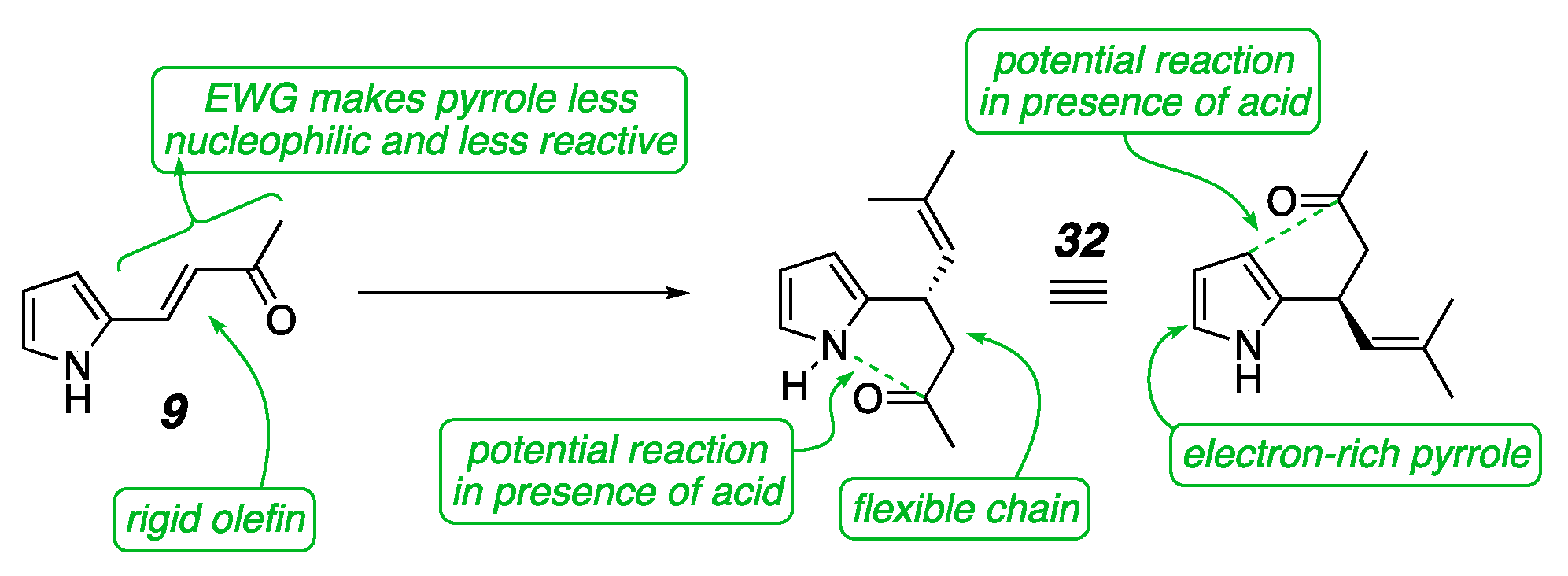
1. Introduction
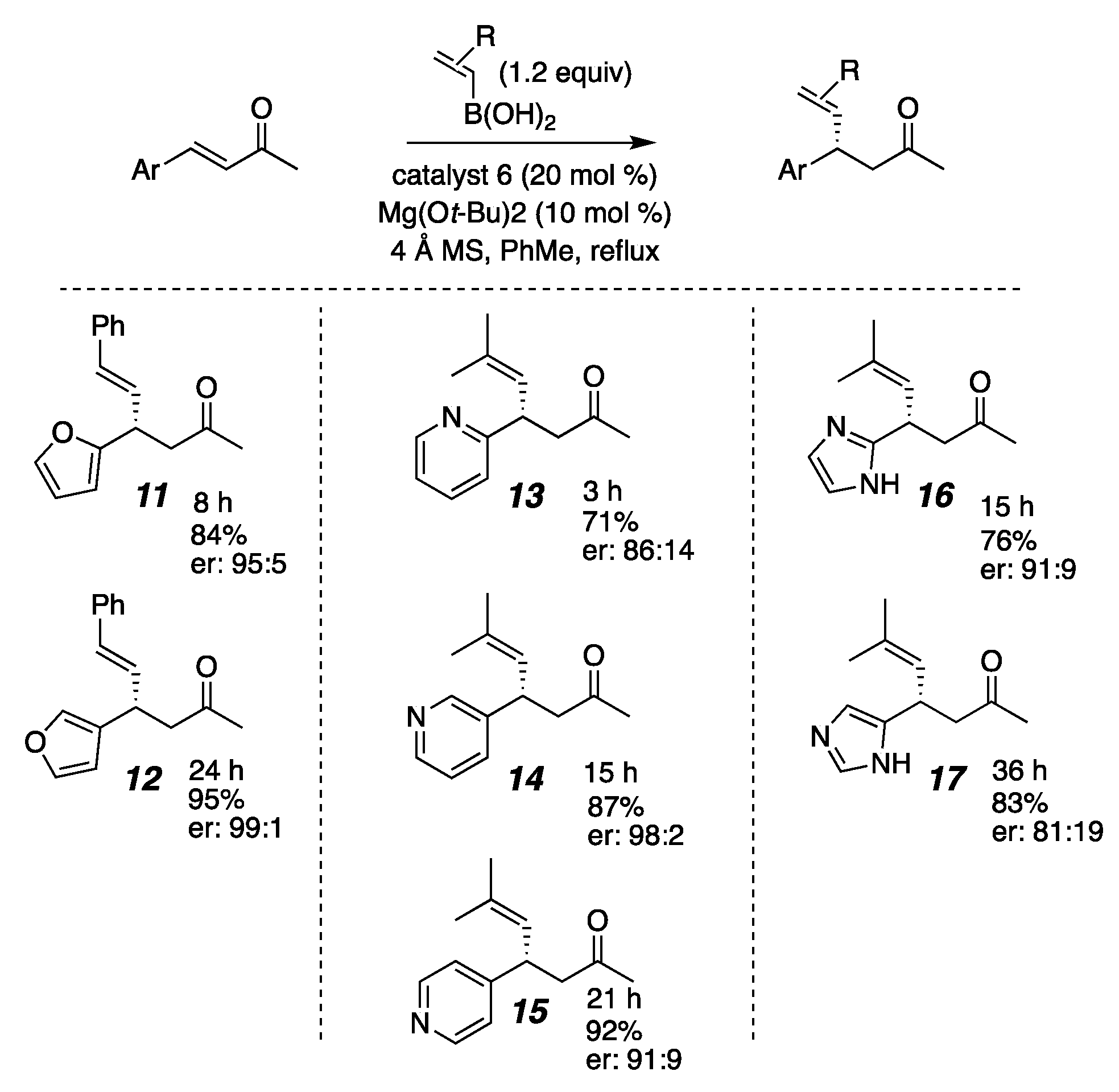
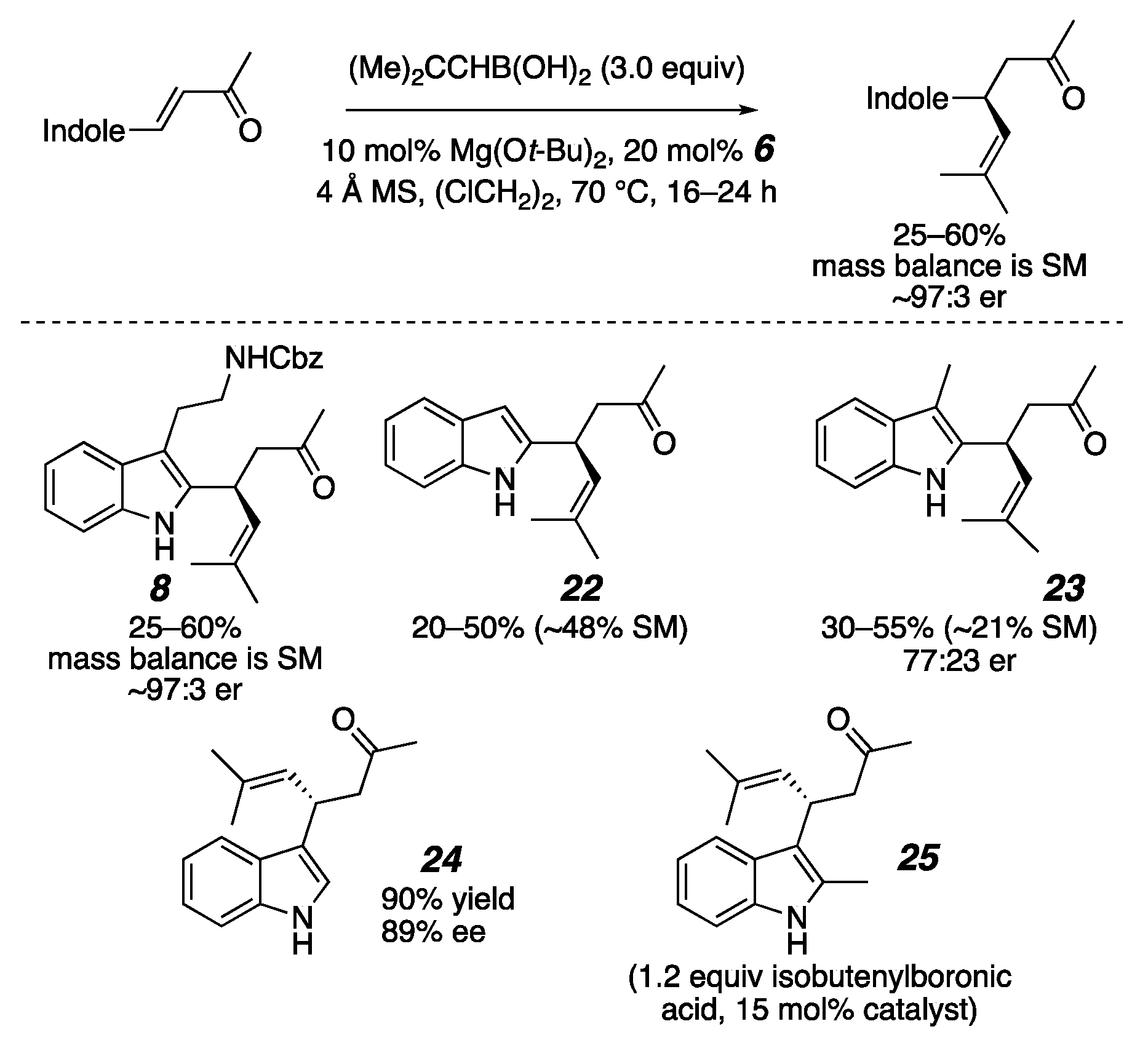
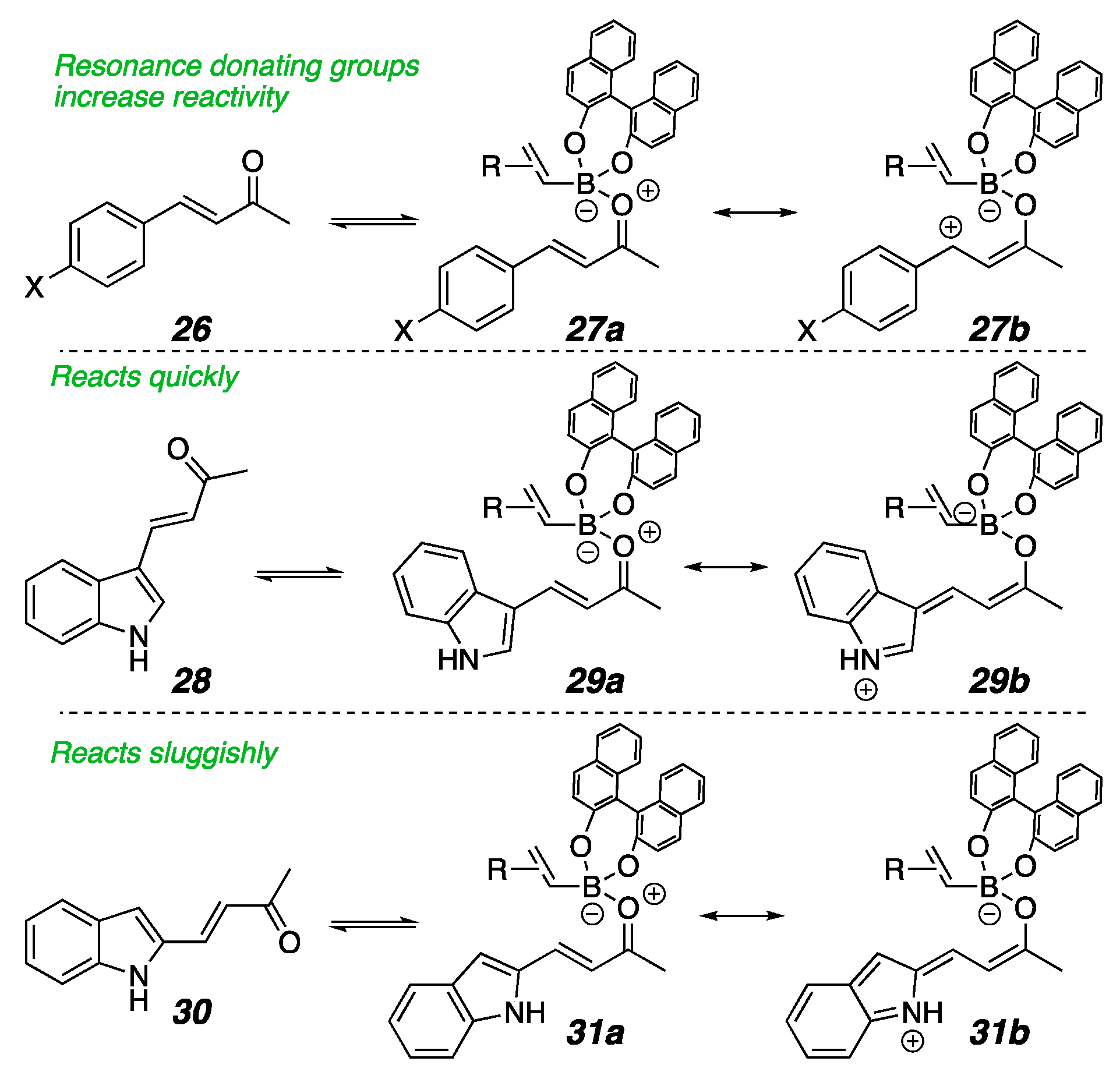
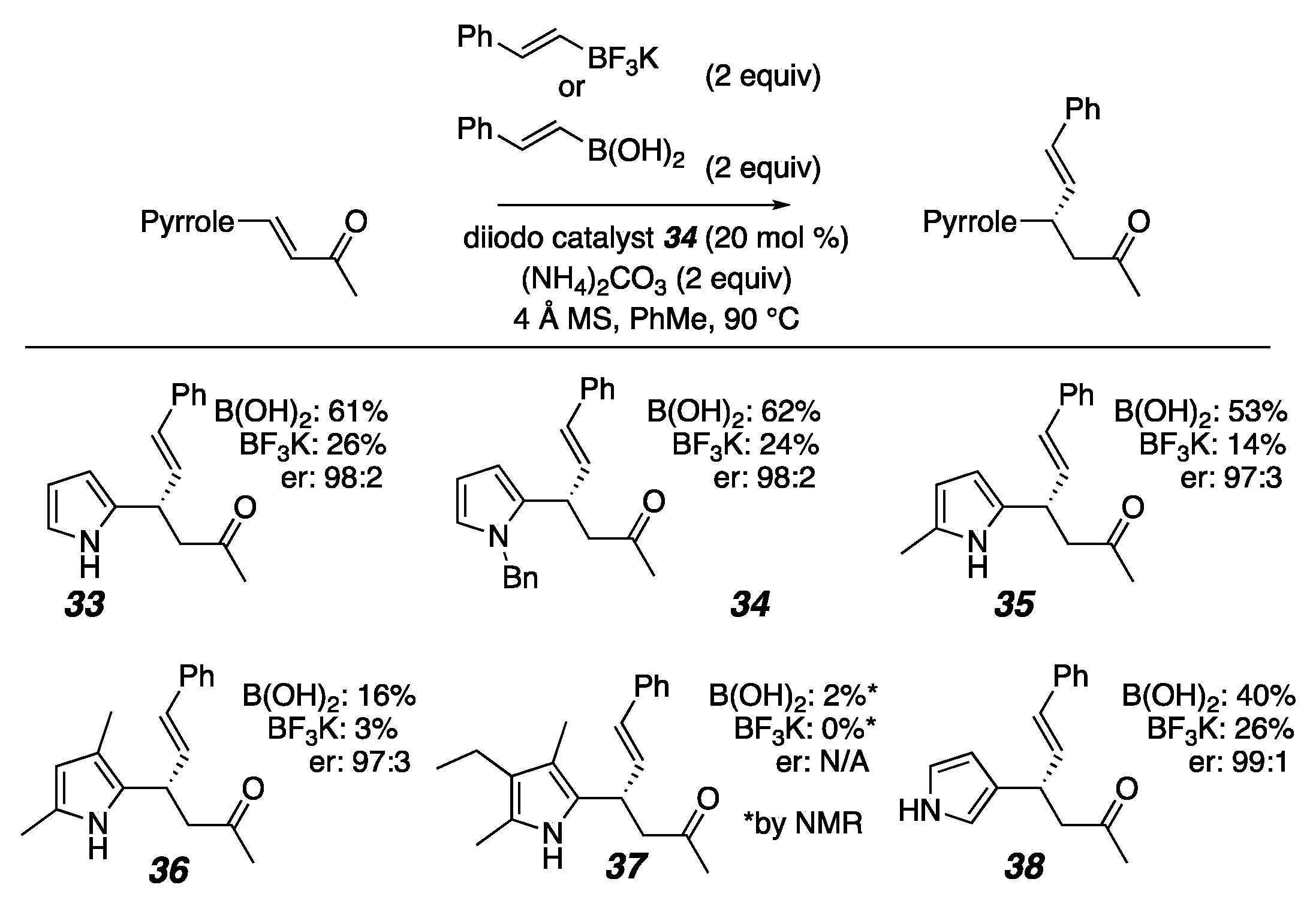
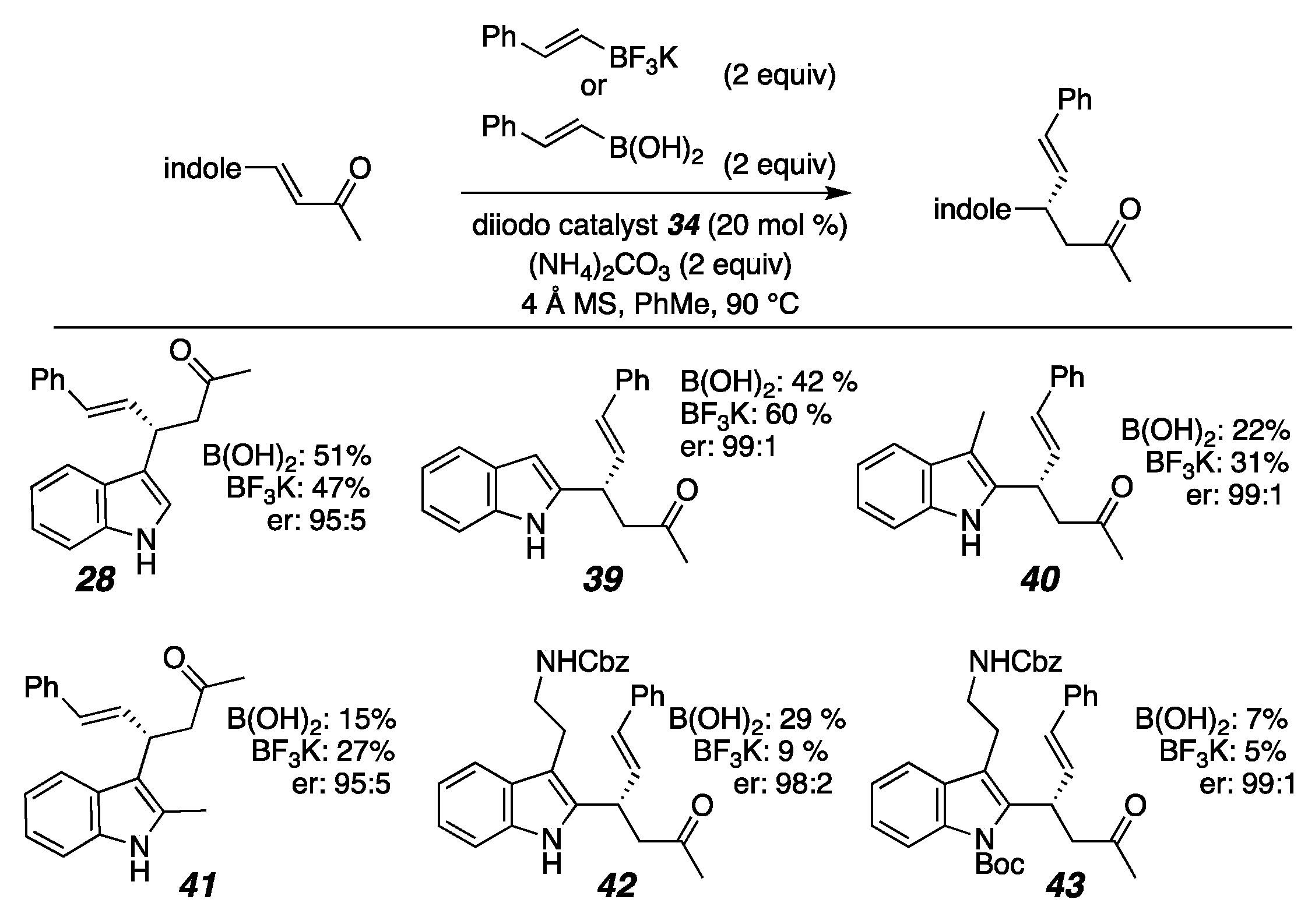
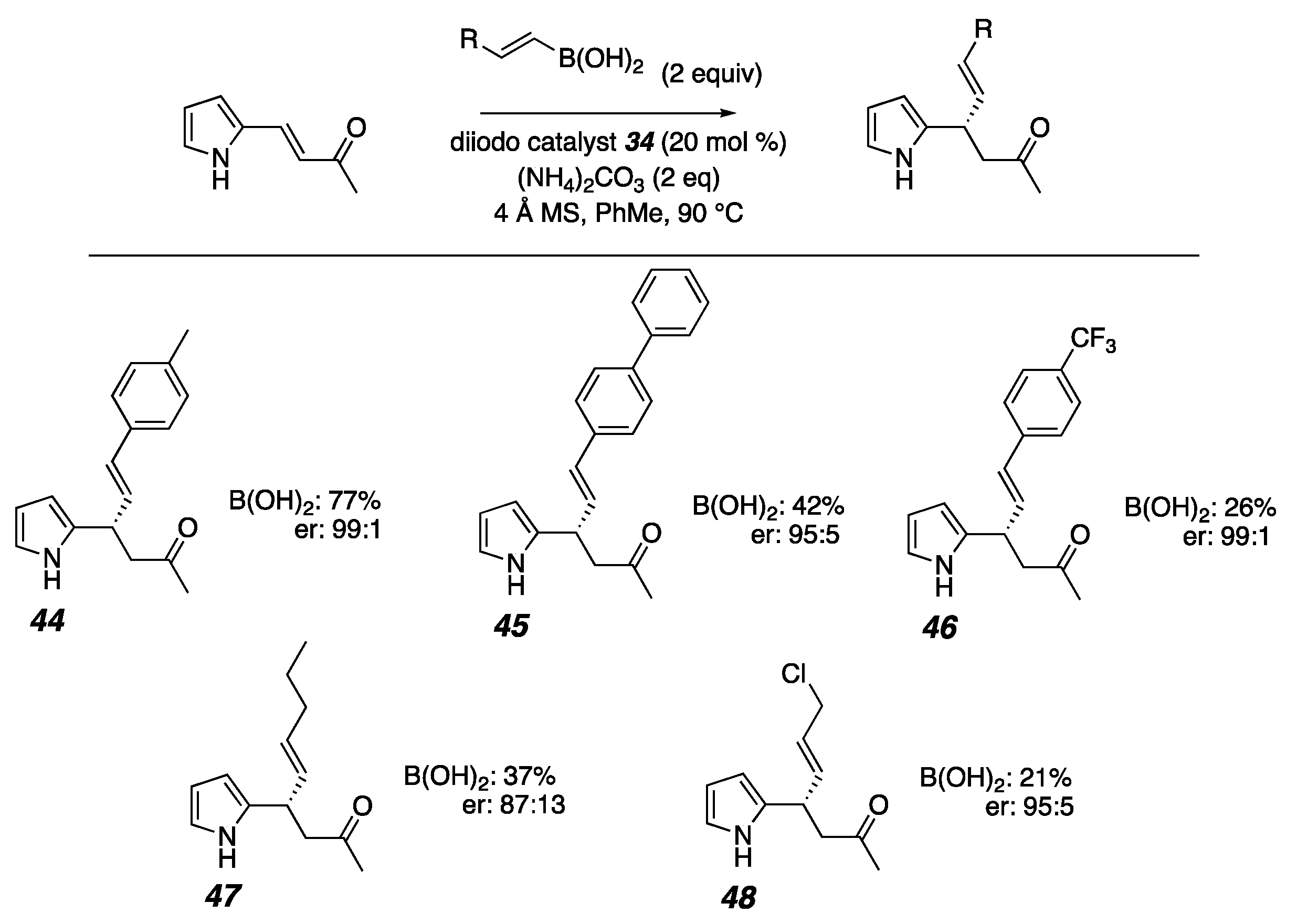
2. Results
3. Conclusions
4. Materials and Methods
4.1. Materials
4.2. General Considerations
- HPLC Columns for Separation of Enantiomers:
4.3. General Procedure for the Synthesis of Starting Materials (Enone)
4.3.1. (E)-4-(5-methyl-1H-pyrrol-2-yl)but-3-en-2-one
4.3.2. (E)-4-(3,5-dimethyl-1H-pyrrol-2-yl)but-3-en-2-one
4.3.3. (E)-4-(4-ethyl-3,5-dimethyl-1H-pyrrol-2-yl)but-3-en-2-one
4.3.4. (E)-4-(3-methyl-1H-indol-2-yl)but-3-en-2-one
4.4. Procedure for Boronic Acid Synthesis: 2-Methylprop-1-Enylboronic Acid
4.5. General Procedure for 1,4-Conjugate Addition (Mg(t-BuO)2 as Additive)
- (S)-6-methyl-4-(1H-pyrrol-2-yl)hept-5-en-2-one (10)
4.6. General Procedure for 1,4-Conjugate Addition ((NH4)2CO3 as an Additive)
- (S,E)-4-(3-methyl-1H-indol-2-yl)-6-phenylhex-5-en-2-one(40)
- (S,E)-4-(2-methyl-1H-indol-3-yl)-6-phenylhex-5-en-2-one(41)
- (S,E)-4-(5-methyl-1H-pyrrol-2-yl)-6-phenylhex-5-en-2-one (35)
- (S,E)-4-(3,5-dimethyl-1H-pyrrol-2-yl)-6-phenylhex-5-en-2-one (36)
- (S,E)-4-(4-ethyl-3,5-dimethyl-1H-pyrrol-2-yl)-6-phenylhex-5-en-2-one(37)
- (S,E)-6-phenyl-4-(1H-pyrrol-3-yl)hex-5-en-2-one (38)
- (S,E)-4-(1H-pyrrol-2-yl)-6-(p-tolyl)hex-5-en-2-one (44)
- (S,E)-6-([1,1′-biphenyl]-4-yl)-4-(1H-pyrrol-2-yl)hex-5-en-2-one (45)
- (S,E)-4-(1H-pyrrol-2-yl)-6-(4-(trifluoromethyl)phenyl)hex-5-en-2-one (46)
- (S,E)-4-(1H-pyrrol-2-yl)non-5-en-2-one (47)
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References and Note
- Riviere, J.E.; Brooks, J.D.; Collard, W.T.; Deng, J.; de Rose, G.; Mahabir, S.P.; Merritt, D.A.; Marchiondo, A.A. Prediction of Formulation Effects on Dermal Absorption of Topically Applied Ectoparasiticides Dosed in Vitro on Canine and Porcine Skin Using a Mixture-Adjusted Quantitative Structure Permeability Relationship. J. Vet. Pharmacol. Ther. 2014, 37, 435–444. [Google Scholar] [CrossRef]
- Wong, D.T.; Bymaster, F.P. Dual serotonin and noradrenaline uptake inhibitor class of antidepressants—Potential for greater efficacy or just hype? In Progress in Drug Research; Glasel, J.A., Kolb, V.M., Skatrud, P.L., Ford, J.W., Stevens, E.B., Treherne, J.M., Packer, J., Bushfield, M., Wong, D.T., Bymaster, F.P., et al., Eds.; Progress in Drug Research; Birkhäuser: Basel, Switzerland, 2002; pp. 169–222. ISBN 978-3-0348-8183-8. [Google Scholar]
- Bertolini, S.; Bon, G.B.; Campbell, L.M.; Farnier, M.; Langan, J.; Mahla, G.; Pauciullo, P.; Sirtori, C.; Egros, F.; Fayyad, R.; et al. Efficacy and Safety of Atorvastatin Compared to Pravastatin in Patients with Hypercholesterolemia. Atherosclerosis 1997, 130, 191–197. [Google Scholar] [CrossRef]
- Lind, T.; Rydberg, L.; Kylebäck, A.; Jonsson, A.; Andersson, T.; Hasselgren, G.; Holmberg, J.; Röhss, K. Esomeprazole Provides Improved Acid Control vs. Omeprazole In Patients with Symptoms of Gastro-Oesophageal Reflux Disease. Aliment. Pharmacol. Ther. 2000, 14, 861–867. [Google Scholar] [CrossRef] [PubMed]
- Moussa, I.; Oetgen, M.; Roubin, G.; Colombo, A.; Wang, X.; Iyer, S.; Maida, R.; Collins, M.; Kreps, E.; Moses, J.W. Effectiveness of Clopidogrel and Aspirin versus Ticlopidine and Aspirin in Preventing Stent Thrombosis after Coronary Stent Implantation. Circulation 1999, 99, 2364–2366. [Google Scholar] [CrossRef]
- Fernandez, L.S.; Buchanan, M.S.; Carroll, A.R.; Feng, Y.J.; Quinn, R.J.; Avery, V.M. Flinderoles A–C: Antimalarial Bis-Indole Alkaloids from Flindersia Species. Org. Lett. 2009, 11, 329–332. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Potts, M.B.; Colosimo, D.; Herrera-Herrera, M.L.; Legako, A.G.; Yousufuddin, M.; White, M.A.; MacMillan, J.B. Discoipyrroles A–D: Isolation, Structure Determination, and Synthesis of Potent Migration Inhibitors from Bacillus hunanensis. J. Am. Chem. Soc. 2013, 135, 13387–13392. [Google Scholar] [CrossRef] [PubMed]
- Vallakati, R.; May, J.A. Biomimetic Synthesis of the Antimalarial Flindersial Alkaloids. J. Am. Chem. Soc. 2012, 4. [Google Scholar] [CrossRef]
- Kotkar, D.; Joshi, V.; Ghosh, P.K. Towards Chiral Metals. Synthesis of Chiral Conducting Polymers from Optically Active Thiophene and Pyrrole Derivatives. J. Chem. Soc. Chem. Commun. 1988, 917–918. [Google Scholar] [CrossRef]
- Lovering, F.; Bikker, J.; Humblet, C. Escape from Flatland: Increasing Saturation as an Approach to Improving Clinical Success. J. Med. Chem. 2009, 52, 6752–6756. [Google Scholar] [CrossRef]
- Hung, A.W.; Ramek, A.; Wang, Y.; Kaya, T.; Wilson, J.A.; Clemons, P.A.; Young, D.W. Route to Three-Dimensional Fragments Using Diversity-Oriented Synthesis. Proc. Natl. Acad. Sci. USA 2011, 108, 6799–6804. [Google Scholar] [CrossRef]
- Hajduk, P.J.; Galloway, W.R.J.D.; Spring, D.R. A Question of Library Design. Nature 2011, 470, 42–43. [Google Scholar] [CrossRef]
- Twigg, D.G.; Kondo, N.; Mitchell, S.L.; Galloway, W.R.J.D.; Sore, H.F.; Madin, A.; Spring, D.R. Partially Saturated Bicyclic Heteroaromatics as an Sp3-Enriched Fragment Collection. Angew. Chem. Int. Ed. 2016, 55, 12479–12483. [Google Scholar] [CrossRef] [PubMed]
- Lovering, F. Escape from Flatland 2: Complexity and Promiscuity. MedChemComm 2013, 4, 515–519. [Google Scholar] [CrossRef]
- Liu, Y.; Han, S.-J.; Liu, W.-B.; Stoltz, B.M. Catalytic Enantioselective Construction of Quaternary Stereocenters: Assembly of Key Building Blocks for the Synthesis of Biologically Active Molecules. Acc. Chem. Res. 2015, 48, 740–751. [Google Scholar] [CrossRef]
- Prosser, K.E.; Stokes, R.W.; Cohen, S.M. Evaluation of 3-Dimensionality in Approved and Experimental Drug Space. ACS Med. Chem. Lett. 2020, 11, 1292–1298. [Google Scholar] [CrossRef] [PubMed]
- Guo, C.; Song, J.; Luo, S.-W.; Gong, L.-Z. Enantioselective Oxidative Cross-Coupling Reaction of 3-Indolylmethyl C–H Bonds with 1,3-Dicarbonyls Using a Chiral Lewis Acid-Bonded Nucleophile to Control Stereochemistry. Angew. Chem. Int. Ed. 2010, 49, 5558–5562. [Google Scholar] [CrossRef]
- Ohmura, T.; Awano, T.; Suginome, M. Stereospecific Suzuki-Miyaura Coupling of Chiral α-(Acylamino)Benzylboronic Esters with Inversion of Configuration. J. Am. Chem. Soc. 2010, 132, 13191–13193. [Google Scholar] [CrossRef] [PubMed]
- Taylor, B.L.H.; Swift, E.C.; Waetzig, J.D.; Jarvo, E.R. Stereospecific Nickel-Catalyzed Cross-Coupling Reactions of Alkyl Ethers: Enantioselective Synthesis of Diarylethanes. J. Am. Chem. Soc. 2011, 133, 389–391. [Google Scholar] [CrossRef]
- Zhou, Q.; Srinivas, H.D.; Dasgupta, S.; Watson, M.P. Nickel-Catalyzed Cross-Couplings of Benzylic Pivalates with Arylboroxines: Stereospecific Formation of Diarylalkanes and Triarylmethanes. J. Am. Chem. Soc. 2013, 135, 3307–3310. [Google Scholar] [CrossRef] [PubMed]
- Do, H.-Q.; Chandrashekar, E.R.R.; Fu, G.C. Nickel/Bis(Oxazoline)-Catalyzed Asymmetric Negishi Arylations of Racemic Secondary Benzylic Electrophiles to Generate Enantioenriched 1,1-Diarylalkanes. J. Am. Chem. Soc. 2013, 135, 16288–16291. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.; Potter, B.; Morken, J.P. A Catalytic Enantiotopic-Group-Selective Suzuki Reaction for the Construction of Chiral Organoboronates. J. Am. Chem. Soc. 2014, 136, 6534–6537. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Xin, X.; Wang, Z.; Lu, G.; Ma, Y.; Liu, L. Catalytic Enantioselective Oxidative Coupling of Saturated Ethers with Carboxylic Acid Derivatives. Nat. Commun. 2019, 10, 559. [Google Scholar] [CrossRef]
- Yu, J.; Ying, P.; Wang, H.; Xiang, K.; Su, W. Mechanochemical Asymmetric Cross-Dehydrogenative Coupling Reaction: Liquid-Assisted Grinding Enables Reaction Acceleration and Enantioselectivity Control. Adv. Synth. Catal. 2020, 362, 893–902. [Google Scholar] [CrossRef]
- Schäfer, P.; Palacin, T.; Sidera, M.; Fletcher, S.P. Asymmetric Suzuki-Miyaura Coupling of Heterocycles via Rhodium-Catalysed Allylic Arylation of Racemates. Nat. Commun. 2017, 8, 15762. [Google Scholar] [CrossRef] [PubMed]
- Brambilla, M.; Tredwell, M. Palladium-Catalyzed Suzuki-Miyaura Cross-Coupling of Secondary α-(Trifluoromethyl)Benzyl Tosylates. Angew. Chem. Int. Ed. 2017, 56, 11981–11985. [Google Scholar] [CrossRef] [PubMed]
- Shintani, R.; Narui, R.; Tsutsumi, Y.; Hayashi, S.; Hayashi, T. Design and Synthesis of New Chiral Phosphorus–Olefin Bidentate Ligands and Their Use in the Rhodium-Catalyzed Asymmetric Addition of Organoboroxines to N-Sulfonyl Imines. Chem. Commun. 2011, 47, 6123–6125. [Google Scholar] [CrossRef] [PubMed]
- Shintani, R.; Takeda, M.; Soh, Y.-T.; Ito, T.; Hayashi, T. Rhodium-Catalyzed Asymmetric Addition of Potassium Organotrifluoroborates to N -Sulfonyl Ketimines. Org. Lett. 2011, 13, 2977–2979. [Google Scholar] [CrossRef] [PubMed]
- DeAngelis, A.; Shurtle, V.W. Rhodium(II)-Catalyzed Enantioselective C-H Functionalization of Indoles. J. Am. Chem. Soc. 2011, 4. [Google Scholar] [CrossRef] [PubMed]
- Unaleroglu, C.; Aytac, S.; Temelli, B. Copper Triflate Catalyzed Regioselective Alkylation of Pyrrole: Conversion of 2-Alkylated Pyrroles to Novel Pyrrolizine Derivatives by Self-Cyclization. Heterocycles 2007, 71, 2427. [Google Scholar] [CrossRef]
- Jensen, K.B.; Thorhauge, J.; Hazell, R.G.; Jørgensen, K.A. Catalytic Asymmetric Friedel–Crafts Alkylation of β,γ-Unsaturated α-Ketoesters: Enantioselective Addition of Aromatic C−H Bonds to Alkenes. Angew. Chem. Int. Ed. 2001, 40, 160–163. [Google Scholar] [CrossRef]
- Friis, S.D.; Pirnot, M.T.; Buchwald, S.L. Asymmetric Hydroarylation of Vinylarenes Using a Synergistic Combination of CuH and Pd Catalysis. J. Am. Chem. Soc. 2016, 138, 8372–8375. [Google Scholar] [CrossRef] [PubMed]
- Odachowski, M.; Bonet, A.; Essafi, S.; Conti-Ramsden, P.; Harvey, J.N.; Leonori, D.; Aggarwal, V.K. Development of Enantiospecific Coupling of Secondary and Tertiary Boronic Esters with Aromatic Compounds. J. Am. Chem. Soc. 2016, 138, 9521–9532. [Google Scholar] [CrossRef]
- Zhao, S.; Gensch, T.; Murray, B.; Niemeyer, Z.L.; Sigman, M.S.; Biscoe, M.R. Enantiodivergent Pd-Catalyzed C–C Bond Formation Enabled through Ligand Parameterization. Science 2018, 362, 670–674. [Google Scholar] [CrossRef] [PubMed]
- Myhill, J.A.; Wilhelmsen, C.A.; Zhang, L.; Morken, J.P. Diastereoselective and Enantioselective Conjunctive Cross-Coupling Enabled by Boron Ligand Design. J. Am. Chem. Soc. 2018, 140, 15181–15185. [Google Scholar] [CrossRef] [PubMed]
- Bandini, M.; Melloni, A.; Umani-Ronchi, A. New Catalytic Approaches in the Stereoselective Friedel-Crafts Alkylation Reaction. Angew. Chem. Int. Ed. 2004, 43, 550–556. [Google Scholar] [CrossRef]
- Rowland, G.B.; Rowland, E.B.; Liang, Y.; Perman, J.A.; Antilla, J.C. The Highly Enantioselective Addition of Indoles to N-Acyl Imines with Use of a Chiral Phosphoric Acid Catalyst. Org. Lett. 2007, 9, 2609–2611. [Google Scholar] [CrossRef]
- Li, G.; Rowland, G.B.; Rowland, E.B.; Antilla, J.C. Organocatalytic Enantioselective Friedel-Crafts Reaction of Pyrrole Derivatives with Imines. Org. Lett. 2007, 9, 4065–4068. [Google Scholar] [CrossRef]
- Matsuzawa, H.; Kanao, K.; Miyake, Y.; Nishibayashi, Y. Remarkable Effect of N -Substituent on Enantioselective Ruthenium-Catalyzed Propargylation of Indoles with Propargylic Alcohols. Org. Lett. 2007, 9, 5561–5564. [Google Scholar] [CrossRef]
- Kanao, K.; Matsuzawa, H.; Miyake, Y.; Nishibayashi, Y. Ruthenium-Catalyzed Enantioselective Propargylation of Indoles with Propargylic Alcohols. Synthesis 2008, 2008, 3869–3873. [Google Scholar] [CrossRef]
- You, S.-L.; Cai, Q.; Zeng, M. Chiral Brønsted Acid Catalyzed Friedel–Crafts Alkylation Reactions. Chem. Soc. Rev. 2009, 38, 2190–2201. [Google Scholar] [CrossRef]
- Pathak, T.P.; Gligorich, K.M.; Welm, B.E.; Sigman, M.S. Synthesis and Preliminary Biological Studies of 3-Substituted Indoles Accessed by a Palladium-Catalyzed Enantioselective Alkene Difunctionalization Reaction. J. Am. Chem. Soc. 2010, 132, 7870–7871. [Google Scholar] [CrossRef] [PubMed]
- Cera, G.; Crispino, P.; Monari, M.; Bandini, M. Stereoselective Synthesis of Tetracyclic Indolines Viagold-Catalyzed Cascade Cyclization Reactions. Chem. Commun. 2011, 47, 7803–7805. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Rauniyar, V.; Wang, Z.J.; Burks, H.E.; Toste, F.D. Enantioselective Synthesis of Highly Substituted Furans by a Copper(II)-Catalyzed Cycloisomerization-Indole Addition Reaction. J. Am. Chem. Soc. 2011, 133, 8486–8489. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Lin, M.; Li, Z.; Liang, X.; Li, G.; Antilla, J.C. Direct Synthesis of Chiral 1,2,3,4-Tetrahydropyrrolo[1,2-a]Pyrazines via a Catalytic Asymmetric Intramolecular Aza-Friedel-Crafts Reaction. Org. Lett. 2011, 13, 4490–4493. [Google Scholar] [CrossRef]
- Cera, G.; Chiarucci, M.; Mazzanti, A.; Mancinelli, M.; Bandini, M. Enantioselective Gold-Catalyzed Synthesis of Polycyclic Indolines. Org. Lett. 2012, 14, 1350–1353. [Google Scholar] [CrossRef]
- Zhuo, M.-H.; Jiang, Y.-J.; Fan, Y.-S.; Gao, Y.; Liu, S.; Zhang, S. Enantioselective Synthesis of Triarylmethanes by Chiral Imidodiphosphoric Acids Catalyzed Friedel–Crafts Reactions. Org. Lett. 2014, 16, 1096–1099. [Google Scholar] [CrossRef]
- Lundy, B.J.; Jansone-Popova, S.; May, J.A. Enantioselective Conjugate Addition of Alkenylboronic Acids to Indole-Appended Enones. Org. Lett. 2011, 13, 4958–4961. [Google Scholar] [CrossRef]
- Miyazaki, Y.; Zhou, B.; Tsuji, H.; Kawatsura, M. Nickel-Catalyzed Asymmetric Friedel-Crafts Propargylation of 3-Substituted Indoles with Propargylic Carbonates Bearing an Internal Alkyne Group. Org. Lett. 2020, 22, 2049–2053. [Google Scholar] [CrossRef]
- Uchikura, T.; Suzuki, R.; Suda, Y.; Akiyama, T. Enantioselective Synthesis of 2-Substituted Indoles Bearing Trifluoromethyl Moiety by the Friedel-Crafts Alkylation Reaction of 4,7-Dihydroindole with N−H Trifluoromethyl Ketimines. ChemCatChem 2020, 12, 4784–4787. [Google Scholar] [CrossRef]
- Wang, C.-J.; Yang, Q.-Q.; Wang, M.-X.; Shang, Y.-H.; Tong, X.-Y.; Deng, Y.-H.; Shao, Z. Catalytic Asymmetric 1,4-Type Friedel–Crafts (Hetero)Arylations of 1-Azadienes: The Highly Enantioselective Syntheses of Chiral Hetero-Triarylmethanes. Org. Chem. Front. 2020, 7, 609–616. [Google Scholar] [CrossRef]
- Gao, Y.; Wang, X.; Wei, Z.; Cao, J.; Liang, D.; Lin, Y.; Duan, H. Asymmetric Synthesis of Spirooxindole-Pyranoindole Products via Friedel–Crafts Alkylation/Cyclization of the Indole Carbocyclic Ring. New J. Chem. 2020, 44, 9788–9792. [Google Scholar] [CrossRef]
- Deng, X.; Wang, Y.; Zhang, S.; Li, L.; Li, G.; Zhao, G.; Tang, Z. An Organocatalytic Asymmetric Friedel–Crafts Reaction of 2-Substituted Indoles with Aldehydes: Enantioselective Synthesis of α-Hydroxyl Ketones by Low Loading of Chiral Phosphoric Acid. Chem. Commun. 2020, 56, 2499–2502. [Google Scholar] [CrossRef]
- Kim, Y.; Lee, J.; Jung, J.; Kim, S.-G. Chiral Brønsted Acid-Catalyzed Friedel–Crafts Reaction of 3-Indolylsulfamidates with Indoles: Synthesis of Enantioenriched Bisindolylmethane Sulfamates. Tetrahedron Lett. 2019, 60, 1625–1630. [Google Scholar] [CrossRef]
- Sato, R.; Tosaka, T.; Masu, H.; Arai, T. Catalytic Asymmetric Synthesis of Chiral Bis(Indolyl)Methanes Using a Ts-PyBidine–Nickel Complex. J. Org. Chem. 2019, 84, 14248–14257. [Google Scholar] [CrossRef]
- Huang, T.; Zhao, Y.; Meng, S.; Chan, A.S.C.; Zhao, J. C7-Functionalization of Indoles via Organocatalytic Enantioselective Friedel-Crafts Alkylation of 4-Amino-Indoles with 2-Butene-1,4-Diones and 3-Aroylacrylates. Adv. Synth. Catal. 2019, 361, 3632–3638. [Google Scholar] [CrossRef]
- Zhu, W.-J.; Gong, J.-F.; Song, M.-P. Synthesis of Chiral Bis(3-Indolyl)Methanes Bearing a Trifluoromethylated All-Carbon Quaternary Stereocenter via Nickel-Catalyzed Asymmetric Friedel–Crafts Alkylation Reaction. J. Org. Chem. 2020, 85, 9525–9537. [Google Scholar] [CrossRef]
- Zhao, Y.; Cai, L.; Huang, T.; Meng, S.; Chan, A.S.C.; Zhao, J. Solvent-Mediated C3/C7 Regioselective Switch in Chiral Phosphoric Acid-Catalyzed Enantioselective Friedel-Crafts Alkylation of Indoles with α-Ketiminoesters. Adv. Synth. Catal. 2020, 362, 1309–1316. [Google Scholar] [CrossRef]
- Schlegel, M.; Coburger, P.; Schneider, C. A Novel Sc(OTf)3-Catalyzed (2 + 2 + 1)-Cycloannulation/Aza-Friedel-Crafts Alkylation Sequence toward Multicyclic 2-Pyrrolines. Chem. Eur. J. 2018, 24, 14207–14212. [Google Scholar] [CrossRef]
- Yonesaki, R.; Kondo, Y.; Akkad, W.; Sawa, M.; Morisaki, K.; Morimoto, H.; Ohshima, T. 3-Mono-Substituted BINOL Phosphoric Acids as Effective Organocatalysts in Direct Enantioselective Friedel–Crafts-Type Alkylation of N-Unprotected α-Ketiminoester. Chem. Eur. J. 2018, 24, 15211–15214. [Google Scholar] [CrossRef] [PubMed]
- Staleva, P.; Hernández, J.G.; Bolm, C. Mechanochemical Copper-Catalyzed Asymmetric Michael-Type Friedel–Crafts Alkylation of Indoles with Arylidene Malonates. Chem. Eur. J. 2019, 25, 9202–9205. [Google Scholar] [CrossRef] [PubMed]
- Yarlagadda, S.; Sridhar, B.; Subba Reddy, B.V. Oxidative Asymmetric Aza-Friedel-Crafts Alkylation of Indoles with 3-Indolinone-2-Carboxylates Catalyzed by a BINOL Phosphoric Acid and Promoted by DDQ. Chem. Asian J. 2018, 13, 1327–1334. [Google Scholar] [CrossRef]
- Yang, Z.-T.; Yang, W.-L.; Chen, L.; Sun, H.; Deng, W.-P. Organocatalytic Enantioselective Aza-Friedel-Crafts Reactions of Pyrazolinone Ketimines with Hydroxyindoles and Electron-Rich Phenols. Adv. Synth. Catal. 2018, 360, 2049–2054. [Google Scholar] [CrossRef]
- Rahman, A.; Xie, E.; Lin, X. Organocatalytic Asymmetric Synthesis of Benzazepinoindole Derivatives with Trifluoromethylated Quaternary Stereocenters by Chiral Phosphoric Acid Catalysts. Org. Biomol. Chem. 2018, 16, 1367–1374. [Google Scholar] [CrossRef]
- Hatano, M.; Mochizuki, T.; Nishikawa, K.; Ishihara, K. Enantioselective Aza-Friedel-Crafts Reaction of Indoles with Ketimines Catalyzed by Chiral Potassium Binaphthyldisulfonates. ACS Catal. 2018, 8, 349–353. [Google Scholar] [CrossRef]
- Ma, J.; Kass, S.R. Asymmetric Arylation of 2,2,2-Trifluoroacetophenones Catalyzed by Chiral Electrostatically-Enhanced Phosphoric Acids. Org. Lett. 2018, 20, 2689–2692. [Google Scholar] [CrossRef]
- Glavač, D.; Zheng, C.; Dokli, I.; You, S.-L.; Gredičak, M. Chiral Brønsted Acid Catalyzed Enantioselective Aza-Friedel-Crafts Reaction of Cyclic α-Diaryl N-Acyl Imines with Indoles. J. Org. Chem. 2017, 82, 8752–8760. [Google Scholar] [CrossRef] [PubMed]
- Arai, T.; Tsuchida, A.; Miyazaki, T.; Awata, A. Catalytic Asymmetric Synthesis of Chiral 2-Vinylindole Scaffolds by Friedel–Crafts Reaction. Org. Lett. 2017, 19, 758–761. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.-X.; Zhang, Y.; Zhang, Y.; He, X.; Zhang, Z.-W.; Liang, H.; He, W.; Jiang, X.; Chen, X.; Qiu, L. Synthesis of Six-Membered Spirooxindoles via a Chiral Brønsted Acid-Catalyzed Asymmetric Intramolecular Friedel-Crafts Reaction. RSC Adv. 2018, 8, 37035–37039. [Google Scholar] [CrossRef]
- Xun, W.; Xu, B.; Chen, B.; Meng, S.; Chan, A.S.C.; Qiu, F.G.; Zhao, J. Regio and Enantioselective Organocatalytic Friedel-Crafts Alkylation of 4-Aminoindoles at the C7-Position. Org. Lett. 2018, 20, 590–593. [Google Scholar] [CrossRef]
- Zhao, Y.; Wang, L.; Zhao, J. Chiral Phosphoric Acid Catalyzed Aza-Friedel-Crafts Alkylation of Indoles with Cyclic Aryl α-Ketimino Esters. Tetrahedron Lett. 2017, 58, 213–217. [Google Scholar] [CrossRef]
- Stemper, J.; Isaac, K.; Ghosh, N.; Lauwick, H.; Le Duc, G.; Retailleau, P.; Voituriez, A.; Betzer, J.-F.; Marinetti, A. Silyl-Substituted Planar Chiral Phosphoric Acids with Ferrocene-Bridged Paracyclophane Frameworks: Synthesis, Characterization, and Uses in Enantioselective Aza-Friedel-Crafts Reactions. Adv. Synth. Catal. 2017, 359, 519–526. [Google Scholar] [CrossRef]
- Cheng, H.-G.; Miguélez, J.; Miyamura, H.; Yoo, W.-J.; Kobayashi, S. Integration of Aerobic Oxidation and Intramolecular Asymmetric Aza-Friedel-Crafts Reactions with a Chiral Bifunctional Heterogeneous Catalyst. Chem. Sci. 2017, 8, 1356–1359. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Rahman, A.; Lin, X. Enantioselective Synthesis of Cyclic Quaternary α-Amino Acid Derivatives by Chiral Phosphoric Acid Catalysis. Org. Biomol. Chem. 2017, 15, 6033–6041. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.; Kass, S.R. Enantioselective Friedel-Crafts Alkylation between Nitroalkenes and Indoles Catalyzed by Charge Activated Thiourea Organocatalysts. J. Org. Chem. 2017, 82, 13288–13296. [Google Scholar] [CrossRef]
- Xu, W.; Shen, X.; Ma, Q.; Gong, L.; Meggers, E. Restricted Conformation of a Hydrogen Bond Mediated Catalyst Enables the Highly Efficient Enantioselective Construction of an All-Carbon Quaternary Stereocenter. ACS Catal. 2016, 6, 7641–7646. [Google Scholar] [CrossRef]
- Chang, C.-H.; Han, J.-L. Organocatalytic Enantioselective Friedel-Crafts Reaction of Sesamol and Electron-Rich Phenols with Alkylideneindolenine Intermediates Generated from Arylsulfonyl Indoles. ChemistrySelect 2016, 1, 5628–5632. [Google Scholar] [CrossRef]
- Lou, H.; Wang, Y.; Jin, E.; Lin, X. Organocatalytic Asymmetric Synthesis of Dihydrobenzoxazinones Bearing Trifluoromethylated Quaternary Stereocenters. J. Org. Chem. 2016, 81, 2019–2026. [Google Scholar] [CrossRef]
- Weng, J.-Q.; Fan, R.-J.; Deng, Q.-M.; Liu, R.-R.; Gao, J.-R.; Jia, Y.-X. Enantioselective Friedel–Crafts Alkylation Reactions of 3-Substituted Indoles with Electron-Deficient Alkenes. J. Org. Chem. 2016, 81, 3023–3030. [Google Scholar] [CrossRef] [PubMed]
- Ge, C.; Liu, R.-R.; Gao, J.-R.; Jia, Y.-X. Cu(I)-Catalyzed Enantioselective Friedel–Crafts Alkylation of Indoles with 2-Aryl- N -Sulfonylaziridines as Alkylating Agents. Org. Lett. 2016, 18, 3122–3125. [Google Scholar] [CrossRef]
- Yang, G.-J.; Du, W.; Chen, Y.-C. Construction of Furan Derivatives with a Trifluoromethyl Stereogenic Center: Enantioselective Friedel–Crafts Alkylations via Formal Trienamine Catalysis. J. Org. Chem. 2016, 81, 10056–10061. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; Lin, L.; Dai, L.; Yuan, X.; Liu, X.; Feng, X. Chiral N,N’-Dioxide–Scandium(III) Complex-Catalyzed Asymmetric Friedel–Crafts Alkylation Reaction of Ortho-Hydroxybenzyl Alcohols with C3-Substituted N-Protected Indoles. Chem. Eur. J. 2016, 22, 18254–18258. [Google Scholar] [CrossRef]
- Wu, Q.; Ma, C.; Du, X.; Chen, Y.; Huang, T.; Shi, X.; Tu, S.; Cai, P. Chiral Brønsted Acid-Catalyzed Alkylation of C3-Substituted Indoles with o-Hydroxybenzyl Alcohols: Highly Enantioselective Synthesis of Diarylindol-2-Ylmethanes and Evaluation on Their Cytotoxicity. Tetrahedron Asymmetry 2016, 27, 307–316. [Google Scholar] [CrossRef]
- Huang, K.; Ma, Q.; Shen, X.; Gong, L.; Meggers, E. Metal-Templated Asymmetric Catalysis: (Z)-1-Bromo-1-Nitrostyrenes as Versatile Substrates for Friedel-Crafts Alkylation of Indoles. Asian J. Org. Chem. 2016, 5, 1198–1203. [Google Scholar] [CrossRef]
- Li, N.-K.; Kong, L.-P.; Qi, Z.-H.; Yin, S.-J.; Zhang, J.-Q.; Wu, B.; Wang, X.-W. Friedel-Crafts Reaction of Indoles with Isatin-Derived β,γ-Unsaturated α-Keto Esters Using a BINOL-Derived Bisoxazoline (BOX)/Copper(II) Complex as Catalyst. Adv. Synth. Catal. 2016, 358, 3100–3112. [Google Scholar] [CrossRef]
- Zhuo, M.-H.; Liu, G.-F.; Song, S.-L.; An, D.; Gao, J.; Zheng, L.; Zhang, S. Chiral Imidodiphosphoric Acids-Catalyzed Friedel-Crafts Reactions of Indoles/Pyrroles with 3-Hydroxy-Indolyloxindoles: Enantioselective Synthesis of 3,3-Diaryloxindoles. Adv. Synth. Catal. 2016, 358, 808–815. [Google Scholar] [CrossRef]
- Le, P.Q.; Nguyen, T.S.; May, J.A. A General Method for the Enantioselective Synthesis of α-Chiral Heterocycles. Org. Lett. 2012, 14, 6104–6107. [Google Scholar] [CrossRef] [PubMed]
- Vallakati, R.; Lundy, B.J.; Jansone-Popova, S.; May, J.A. Biomimetic Synthesis and Studies Toward Enantioselective Synthesis of Flindersial Alkaloids. Chirality 2015, 27, 14–17. [Google Scholar] [CrossRef] [PubMed]
- Shih, J.-L.; Nguyen, T.S.; May, J.A. Organocatalyzed Asymmetric Conjugate Addition of Heteroaryl and Aryl Trifluoroborates: A Synthetic Strategy for Discoipyrrole, D. Angew. Chem. Int. Ed. 2015, 54, 9931–9935. [Google Scholar] [CrossRef] [PubMed]
- Xing, J.; Chen, G.; Cao, P.; Liao, J. Rhodium-Catalyzed Asymmetric Addition of Arylboronic Acids to Indolylnitroalkenes. Eur. J. Org. Chem. 2012, 2012, 1230–1236. [Google Scholar] [CrossRef]
- Dabrowski, J.A.; Villaume, M.T.; Hoveyda, A.H. Enantioselective Synthesis of Quaternary Carbon Stereogenic Centers through Copper-Catalyzed Conjugate Additions of Aryl- and Alkylaluminum Reagents to Acyclic Trisubstituted Enones. Angew. Chem. Int. Ed. 2013, 52, 8156–8159. [Google Scholar] [CrossRef] [PubMed]
- Huo, H.; Fu, C.; Harms, K.; Meggers, E. Asymmetric Catalysis with Substitutionally Labile yet Stereochemically Stable Chiral-at-Metal Iridium(III) Complex. J. Am. Chem. Soc. 2014, 136, 2990–2993. [Google Scholar] [CrossRef]
- Austin, J.F.; MacMillan, D.W.C. Enantioselective Organocatalytic Indole Alkylations. Design of a New and Highly Effective Chiral Amine for Iminium Catalysis. J. Am. Chem. Soc. 2002, 124, 1172–1173. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; MacMillan, D.W.C. Organocatalytic Vinyl and Friedel-Crafts Alkylations with Trifluoroborate Salts. J. Am. Chem. Soc. 2007, 129, 15438–15439. [Google Scholar] [CrossRef] [PubMed]
- Ganesh, M.; Seidel, D. Catalytic Enantioselective Additions of Indoles to Nitroalkenes. J. Am. Chem. Soc. 2008, 130, 16464–16465. [Google Scholar] [CrossRef]
- Dobish, M.C.; Johnston, J.N. Chiral Brønsted Base-Promoted Nitroalkane Alkylation: Enantioselective Synthesis of Sec.-Alkyl-3-Substituted Indoles. Org. Lett. 2010, 12, 5744–5747. [Google Scholar] [CrossRef]
- Luan, Y.; Schaus, S.E. Enantioselective Addition of Boronates to o-Quinone Methides Catalyzed by Chiral Biphenols. J. Am. Chem. Soc. 2012, 134, 19965–19968. [Google Scholar] [CrossRef]
- Hara, S.; Hyuga, S.; Aoyama, M.; Sato, M.; Suzuki, A. Bf3 Etherate Mediated 1,4-Addition of 1-Alkenyldialkoxyboranes to α,β-Unsaturated Ketones. A Stereoselective Synthesis of γ, δ-Unsaturated Ketones. Tetrahedron Lett. 1990, 31, 247–250. [Google Scholar] [CrossRef]
- Zhu, H.; Yin, L.; Chang, Z.; Wang, Y.; Dou, X. Rhodium-Catalyzed Asymmetric Conjugate Addition of Organoboronic Acids to Carbonyl-Activated Alkenyl Azaarenes. Adv. Synth. Catal. 2020, 362, 3142–3147. [Google Scholar] [CrossRef]
- Han, J.-L.; Liao, Y.-T.; Chang, C.-H. Asymmetric Organocatalytic Conjugate Addition of Electron-Rich Phenols and 1,3-Dicarbonyls to Arylsulfonyl Indoles in an Oil-Water Biphasic System. Eur. J. Org. Chem. 2019, 2019, 5815–5823. [Google Scholar] [CrossRef]
- Wang, J.-R.; Jiang, X.-L.; Hang, Q.-Q.; Zhang, S.; Mei, G.-J.; Shi, F. Catalytic Asymmetric Conjugate Addition of Indoles to Para -Quinone Methide Derivatives. J. Org. Chem. 2019, 84, 7829–7839. [Google Scholar] [CrossRef]
- Li, S.-W.; Wan, Q.; Kang, Q. Chiral-at-Metal Rh(III) Complex-Catalyzed Michael Addition of Pyrazolones with α,β-Unsaturated 2-Acyl Imidazoles. Org. Lett. 2018, 20, 1312–1315. [Google Scholar] [CrossRef]
- Liang, W.; Yin, W.; Wang, T.; Qiu, F.G.; Zhao, J. Organocatalytic Stereoselective Conjugate Addition of 3-Substituted Oxindoles with in Situ Generated Ortho-Quinone Methides. Tetrahedron Lett. 2018, 59, 1742–1747. [Google Scholar] [CrossRef]
- Correia, J.T.M.; List, B.; Coelho, F. Catalytic Asymmetric Conjugate Addition of Indolizines to α,β-Unsaturated Ketones. Angew. Chem. Int. Ed. 2017, 56, 7967–7970. [Google Scholar] [CrossRef]
- Metz, T.L.; Evans, J.; Stanley, L.M. Catalytic Conjugate Addition of Electron-Rich Heteroarenes to β,β-Disubstituted Enones. Org. Lett. 2017, 19, 3442–3445. [Google Scholar] [CrossRef]
- Ma, H.-L.; Xie, L.; Zhang, Z.; Li, J.-Q.; Qin, Z.-H.; Fu, B. Enantioselective Copper(II)-Catalyzed Conjugate Addition of Indoles to β-Substituted Unsaturated Acyl Phosphonates. Adv. Synth. Catal. 2016, 358, 1011–1016. [Google Scholar] [CrossRef]
- Fujishima, H.; Takada, E.; Kara, S.; Suzuki, A. Boron Trifluoride Etherate Mediated 1,4-Addition of (1-Alkynyl)Diisopropoxyboranes to α,β-Unsaturated Ketones. A Convenient New Route to 3-Alkynyl Ketone Synthesis. Chem. Lett. 2006, 21. [Google Scholar] [CrossRef]
- Takada, E.; Hara, S.; Suzuki, A. A Stereoselective Synthesis of Γδ-Unsaturated Ketones Possessing Perfluoroalkyl Groups by Trifluoroborane Etherate Mediated 1,4-Addition Reaction of Alkenyldiisopropoxyboranes to α,β-Unsaturated Ketones. Tetrahedron Lett. 1993, 34, 7067–7070. [Google Scholar] [CrossRef]
- Hara, S.; Ishimura, S.; Suzuki, A. Cyanuric Fluoride-Induced 1,4-Addition Reaction of Alkenylboronic Acids to α,β-Unsaturated Ketones. Stereoselective Synthesis of γ,δ-Unsaturated Ketones Having Functionalities. Synlett 1996, 1996, 993–994. [Google Scholar] [CrossRef]
- Hara, S.; Shudoh, H.; Ishimura, S.; Suzuki, A. The Regioselective 1,4-Addition Reaction of Alkenylboronic Acids to α,β,A′,Β′-Unsaturated Ketones. Bull. Chem. Soc. Jpn. 1998, 71, 2403–2408. [Google Scholar] [CrossRef]
- Yoshida, K.; Hayashi, T. Rhodium-Catalyzed additions of boronic acids to Alkenes and Carbonyl compounds. In Boronic Acids; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2006; pp. 171–203. ISBN 978-3-527-60654-2. [Google Scholar]
- Chong, J.M.; Shen, L.; Taylor, N.J. Asymmetric Conjugate Addition of Alkynylboronates to Enones. J. Am. Chem. Soc. 2000, 122, 1822–1823. [Google Scholar] [CrossRef]
- Wu, T.R.; Chong, J.M. Ligand-Catalyzed Asymmetric Alkynylboration of Enones: A New Paradigm for Asymmetric Synthesis Using Organoboranes. J. Am. Chem. Soc. 2005, 127, 3244–3245. [Google Scholar] [CrossRef]
- Wu, T.R.; Chong, J.M. Asymmetric Conjugate Alkenylation of Enones Catalyzed by Chiral Diols. J. Am. Chem. Soc. 2007, 129, 4908–4909. [Google Scholar] [CrossRef]
- Turner, H.M.; Patel, J.; Niljianskul, N.; Chong, J.M. Binaphthol-Catalyzed Asymmetric Conjugate Arylboration of Enones. Org. Lett. 2011, 13, 5796–5799. [Google Scholar] [CrossRef]
- Sugiura, M.; Tokudomi, M.; Nakajima, M. Enantioselective Conjugate Addition of Boronic Acids to Enones Catalyzed by O-Monoacyltartaric Acids. Chem. Commun. 2010, 46, 7799–7800. [Google Scholar] [CrossRef] [PubMed]
- Kotani, S.; Osakama, K.; Sugiura, M.; Nakajima, M. A Tertiary Amine as A Hydride Donor: Trichlorosilyl Triflate-Mediated Conjugate Reduction of Unsaturated Ketones. Org. Lett. 2011, 13, 3968–3971. [Google Scholar] [CrossRef] [PubMed]
- Grimblat, N.; Sugiura, M.; Pellegrinet, S.C. A Hydrogen Bond Rationale for the Enantioselective β-Alkenylboration of Enones Catalyzed by O-Monoacyltartaric Acids. J. Org. Chem. 2014, 79, 6754–6758. [Google Scholar] [CrossRef] [PubMed]
- Sugiura, M.; Kinoshita, R.; Nakajima, M. O-Monoacyltartaric Acid Catalyzed Enantioselective Conjugate Addition of a Boronic Acid to Dienones: Application to the Synthesis of Optically Active Cyclopentenones. Org. Lett. 2014, 16, 5172–5175. [Google Scholar] [CrossRef]
- Inokuma, T.; Takasu, K.; Sakaeda, T.; Takemoto, Y. Hydroxyl Group-Directed Organocatalytic Asymmetric Michael Addition of α,β-Unsaturated Ketones with Alkenylboronic Acids. Org. Lett. 2009, 11, 2425–2428. [Google Scholar] [CrossRef]
- Pellegrinet, S.C.; Goodman, J.M. Asymmetric Conjugate Addition of Alkynylboronates to Enones: Rationale for the Intriguing Catalysis Exerted by Binaphthols. J. Am. Chem. Soc. 2006, 128, 3116–3117. [Google Scholar] [CrossRef]
- Paton, R.S.; Goodman, J.M.; Pellegrinet, S.C. Theoretical Study of the Asymmetric Conjugate Alkenylation of Enones Catalyzed by Binaphthols. J. Org. Chem. 2008, 73, 5078–5089. [Google Scholar] [CrossRef]
- Nguyen, T.S.; Yang, M.S.; May, J.A. Experimental Mechanistic Insight into the BINOL-Catalyzed Enantioselective Conjugate Addition of Boronates to Enones. Tetrahedron Lett. 2015, 56, 3337–3341. [Google Scholar] [CrossRef]
- We Also Examined the Use of an Additional Lewis Acid as Reported by Yu and Mao.
- Wang, J.-F.; Meng, X.; Zhang, C.-H.; Yu, C.-M.; Mao, B. Organocatalytic Enantioselective Conjugate Alkynylation of β-Aminoenones: Access to Chiral β-Alkynyl-β-Amino Carbonyl Derivatives. Org. Lett. 2020, 22, 7427–7432. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

| Entry | Additive | Yield |
|---|---|---|
| 1 | Mg(Ot-Bu)2 | 10% |
| 2 | (NH4)2CO3 | 64% |
| 3 | K2CO3 | 53% |
| 4 | Cs2CO3 | 35% |
| 5 | Li2CO3 | 29% |
| 6 | Na2CO3 | 4% |
| 7 | K3PO4 | 34% |
| 8 | NaHMDS | 13% |
| 9 | LiHMDS | 6% |
| 10 | KOH | 5% |
| 11 | NaOH | 4% |
| 12 | KOt-Bu | 4% |
| 13 | NaOt-Bu | 3% |
| 14 | LiOt-Bu | 0% |
| 15 | NH4Cl | 2% |
| 16 | NH4HSO4 | 0% |
| 17 | DBU | 3% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Boylan, A.; Nguyen, T.S.; Lundy, B.J.; Li, J.-Y.; Vallakati, R.; Sundstrom, S.; May, J.A. Rate Dependence on Inductive and Resonance Effects for the Organocatalyzed Enantioselective Conjugate Addition of Alkenyl and Alkynyl Boronic Acids to β-Indolyl Enones and β-Pyrrolyl Enones. Molecules 2021, 26, 1615. https://doi.org/10.3390/molecules26061615
Boylan A, Nguyen TS, Lundy BJ, Li J-Y, Vallakati R, Sundstrom S, May JA. Rate Dependence on Inductive and Resonance Effects for the Organocatalyzed Enantioselective Conjugate Addition of Alkenyl and Alkynyl Boronic Acids to β-Indolyl Enones and β-Pyrrolyl Enones. Molecules. 2021; 26(6):1615. https://doi.org/10.3390/molecules26061615
Chicago/Turabian StyleBoylan, Amy, Thien S. Nguyen, Brian J. Lundy, Jian-Yuan Li, Ravikrishna Vallakati, Sasha Sundstrom, and Jeremy A. May. 2021. "Rate Dependence on Inductive and Resonance Effects for the Organocatalyzed Enantioselective Conjugate Addition of Alkenyl and Alkynyl Boronic Acids to β-Indolyl Enones and β-Pyrrolyl Enones" Molecules 26, no. 6: 1615. https://doi.org/10.3390/molecules26061615
APA StyleBoylan, A., Nguyen, T. S., Lundy, B. J., Li, J.-Y., Vallakati, R., Sundstrom, S., & May, J. A. (2021). Rate Dependence on Inductive and Resonance Effects for the Organocatalyzed Enantioselective Conjugate Addition of Alkenyl and Alkynyl Boronic Acids to β-Indolyl Enones and β-Pyrrolyl Enones. Molecules, 26(6), 1615. https://doi.org/10.3390/molecules26061615