
Towards Reversible High-Voltage Multi-Electron Reactions in Alkali-Ion Batteries Using Vanadium Phosphate Positive Electrode Materials

 , and
, and
Abstract
1. Introduction
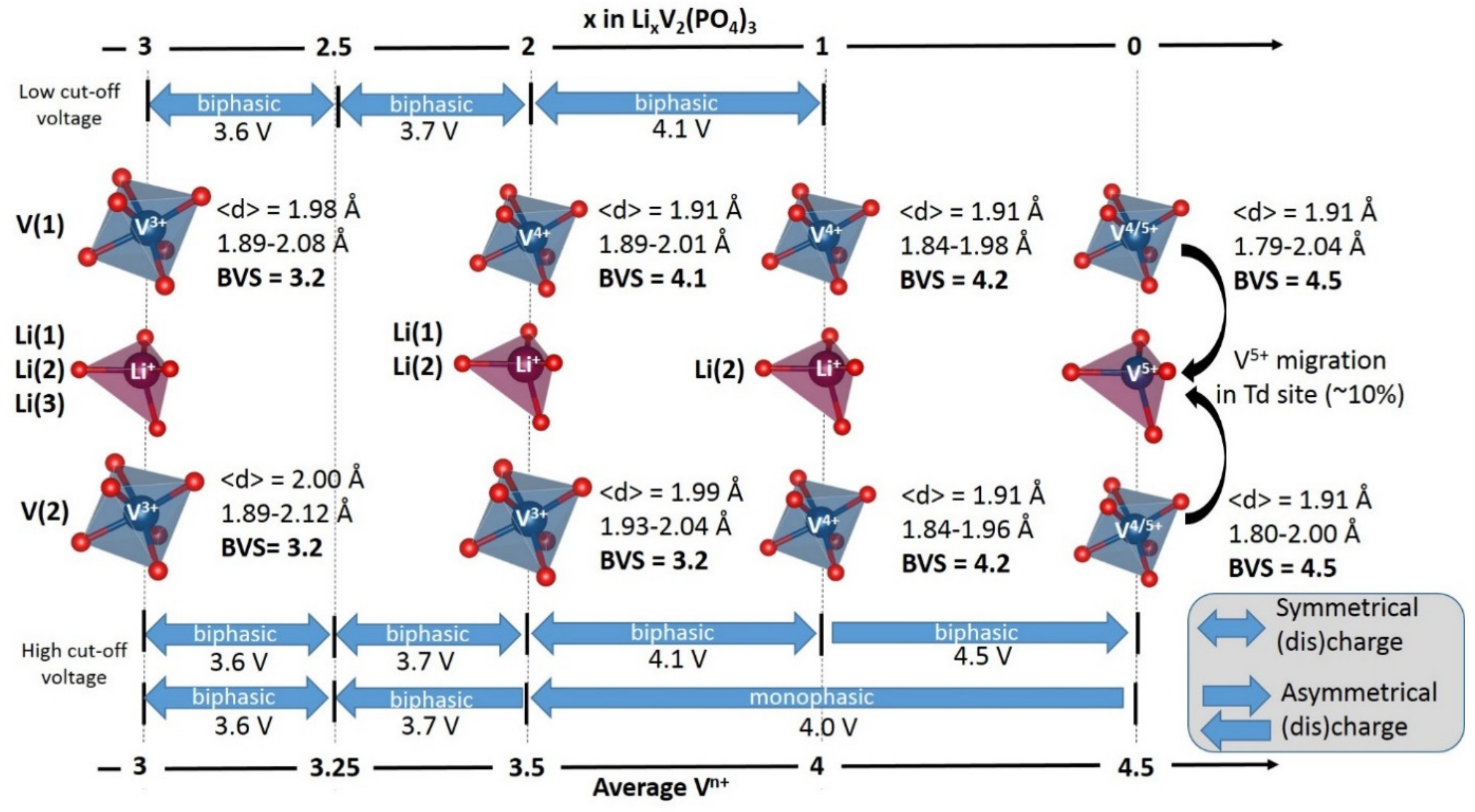
2. Irreversible Multi-Electron Reactions in NASICON and Anti-NASICON AxV2(PO4)3 (A = Li, Na) Structures


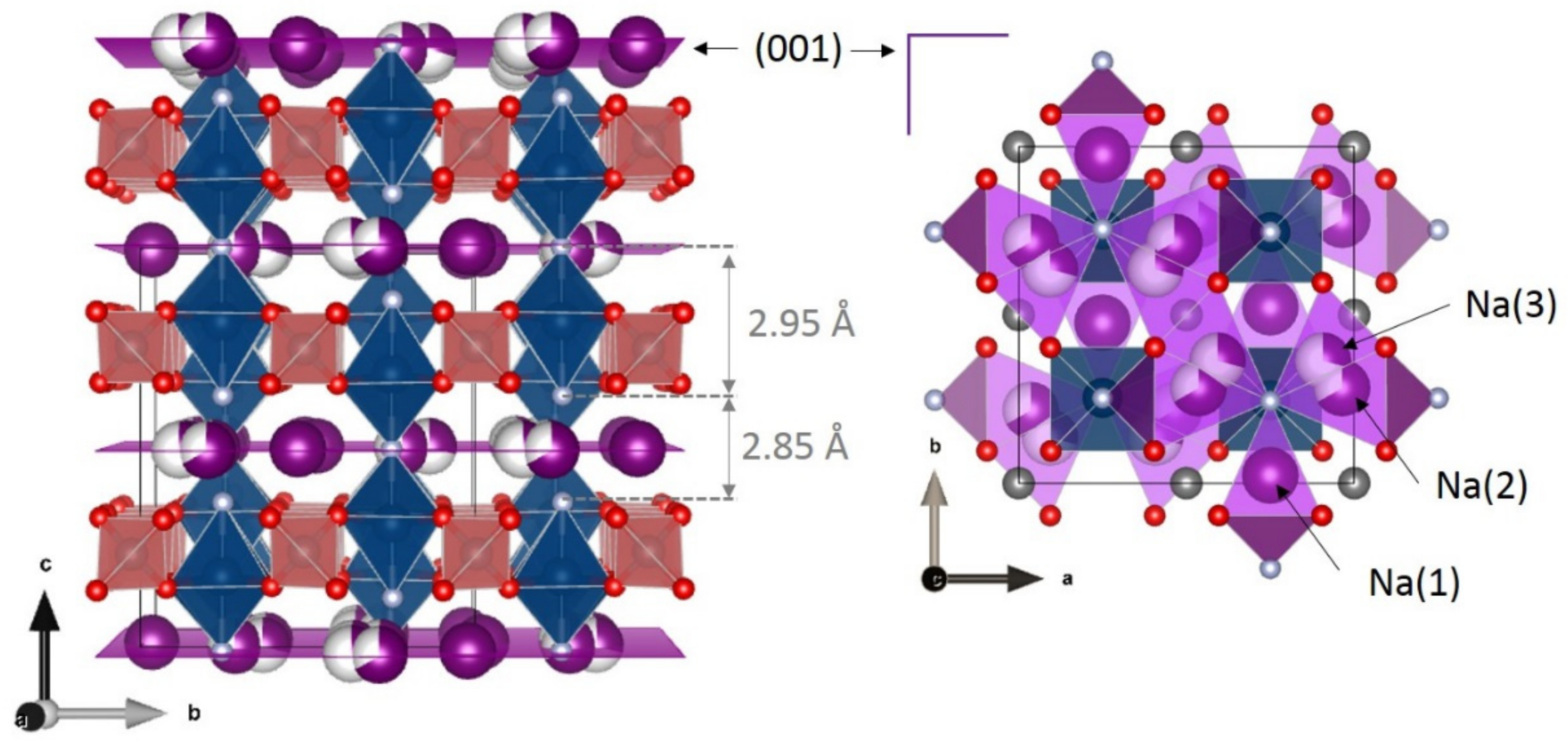
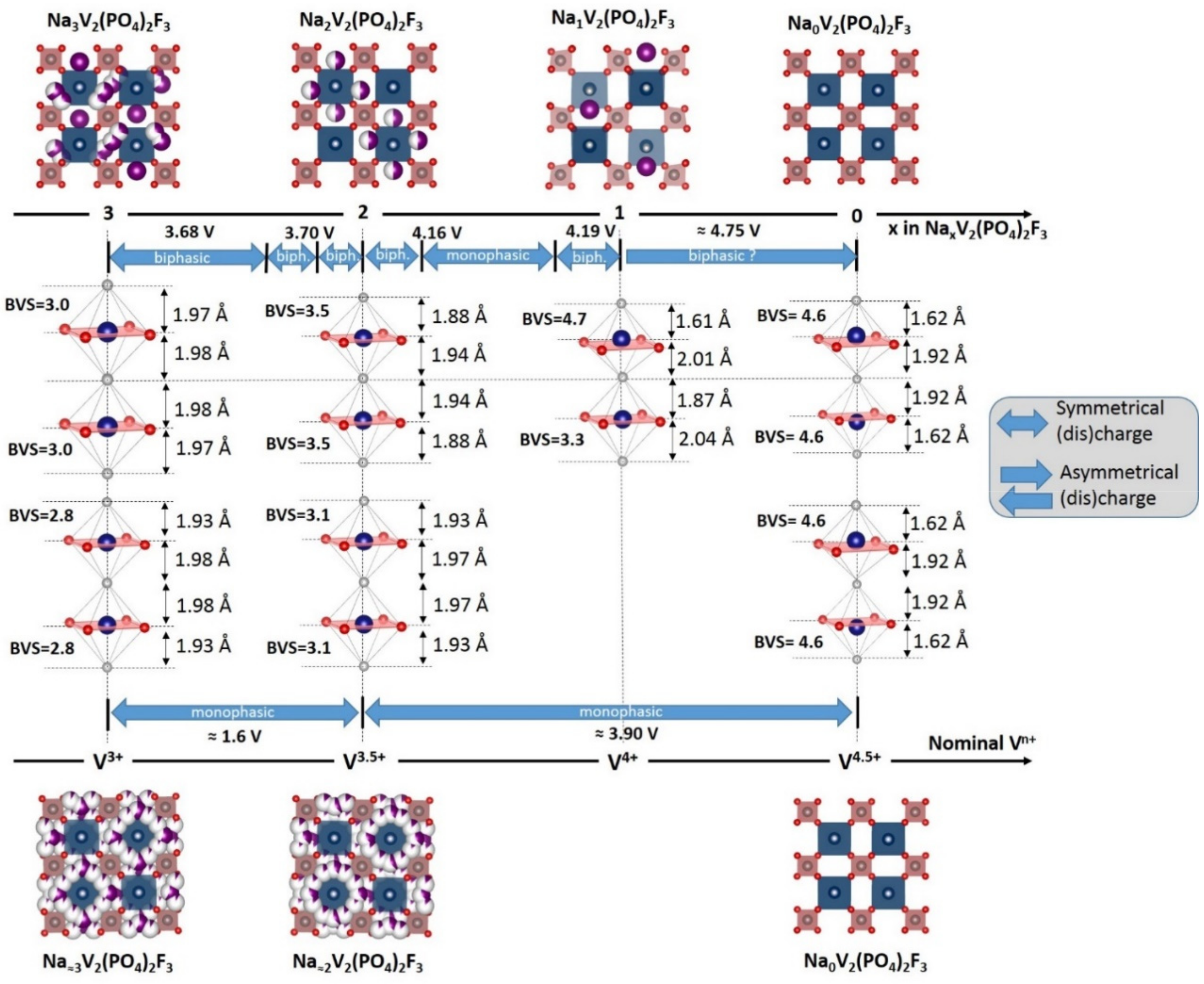
3. Irreversible Multi-Electron Reactions in Na3V2(PO4)2F3
4. Low Voltage Multi-Electron Reactions in Tavorites LixVPO4Y (Y = O, F and/or OH)
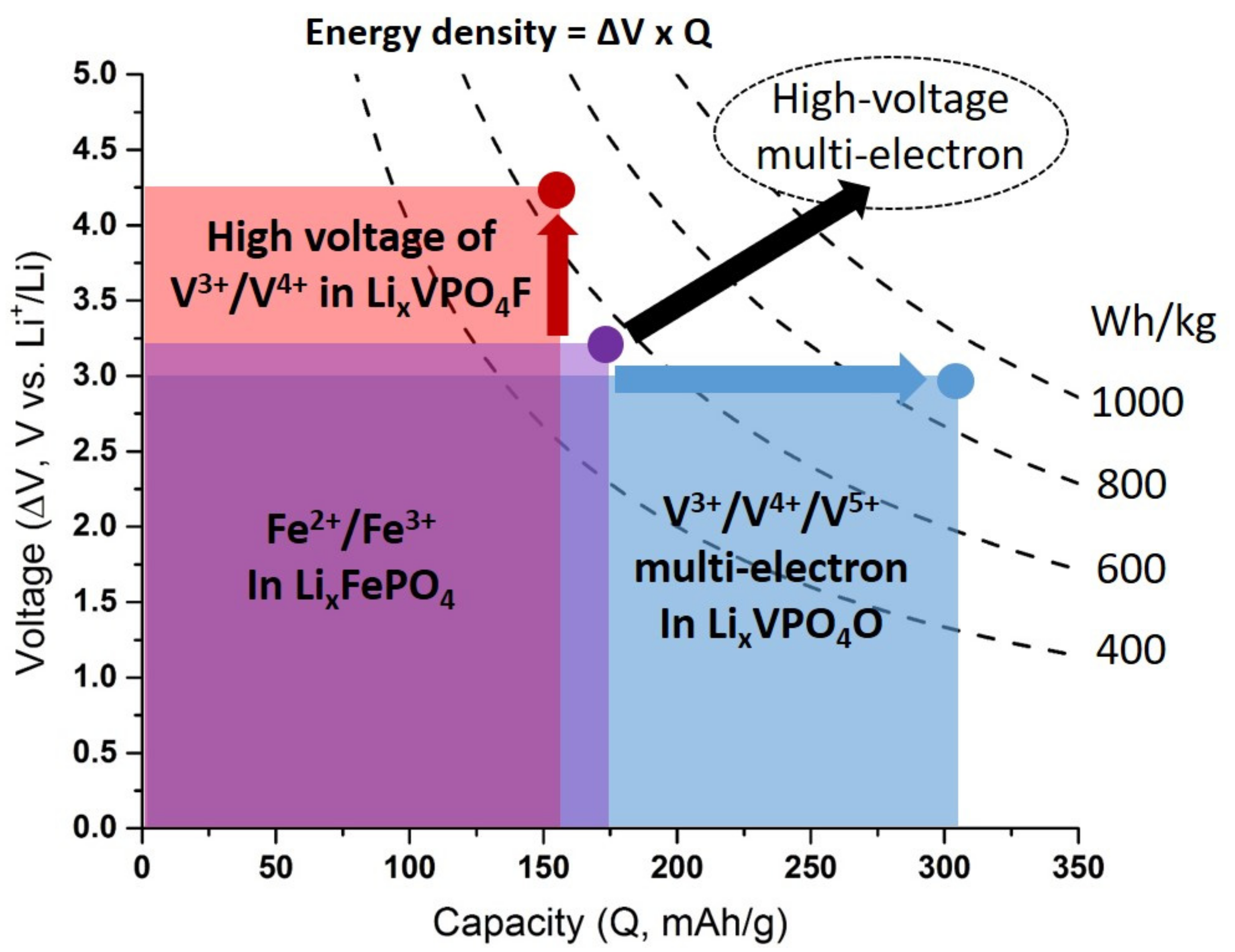
5. Towards Reversible High-Voltage Multi-Electron Reactions
- For type I materials (e.g., Li3V2(PO4)3), in which the vanadyl bond cannot appear due to the involvement of each oxygen atom of VO6 octahedra in a PO4-type entity, the typical evolution of the vanadium environment upon oxidation (from V2+ to V5+) follows a quasi-homogeneous shortening of V-O bonds from V2+ to V4+ and a strong increase in VO6 distortion to reach the V5+ state with corresponding voltages of 1.8 V vs. Li+/Li for V2+/V3+, 3.9 V vs. Li+/Li for V3+/V4+ and 4.4 V vs. Li+/Li for V4+/V5+ redox couples (on average for all the type I materials reported in Table 1).
- In type II materials (e.g., LiVPO4F), at least one of the ligands around vanadium is unshared with a phosphate group and hence would be available to form the vanadyl bond. However, in that case, the nature of this ligand (F− instead of O2−) inhibits its formation. From V2+ to V4+, the evolution of the vanadium environment follows a similar trend with slightly higher voltages than for type I due to the higher ionicity of V-F versus V-O. For V5+, for instance in deintercalated Na3V2(PO4)2F3, a “vanadyl-like” distortion appears with V-F bond length of 1.6 Å and 1.9 Å. Such an F…V-F sequence has never been reported elsewhere and the precise nature of the V-F bonds formed is still to be clarified.
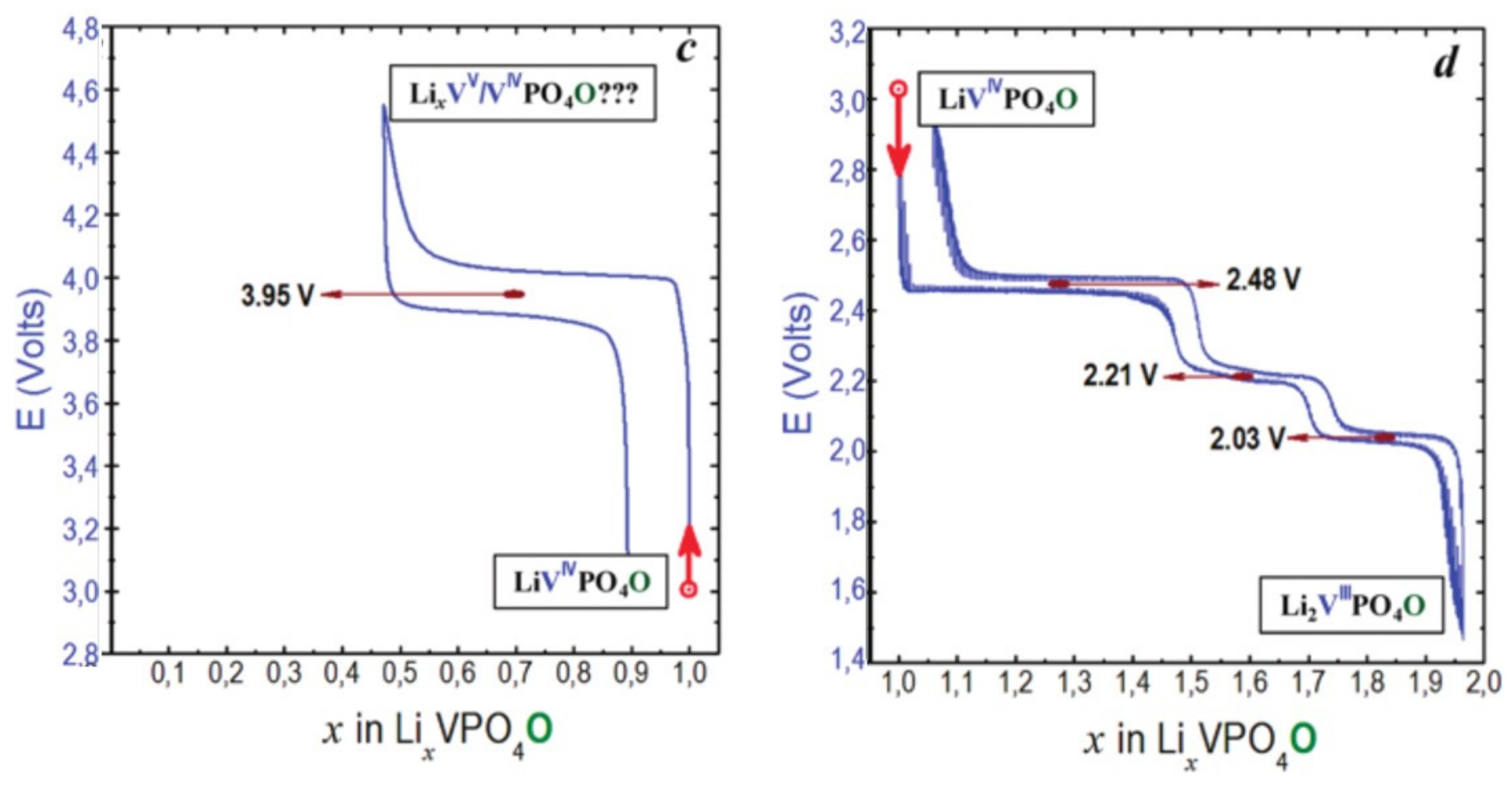
- Type III group (e.g., LiVPO4O) gathers the structures having at least one oxygen belonging to VO6 octahedra available to form the covalent vanadyl bond for vanadium oxidation states higher than 3. In this class of materials, the V3+ environments are quasi undistorted. As the oxidation state of vanadium is increased, vanadium leaves the inversion center of the VO6 octahedra in order to form the vanadyl bond. The formation of a distorted VIVO6 octahedra (with typical distances ranging between 1.6 and 2.4 Å along dz2 and quasi equivalent equatorial distances around 2 Å) and VVO5 pyramids (in which the short V=O bond is about 1.6 Å and a shortening of the equatorial distances is observed around 1.8–1.9 Å) are observed. The corresponding voltages appear completely different to those of type I and type II materials: 2.4 V vs. Li+/Li for the V3+-O/V4+=O and 3.95 V vs. Li+/Li for the V4+=O/V5+=O redox couples.
6. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Padhi, A.K.; Nanjundaswamy, K.S.; Goodenough, J.B. Phospho-Olivines as Positive-Electrode Materials for Rechargeable Lithium Batteries. J. Electrochem. Soc. 1997, 144, 1188–1194. [Google Scholar] [CrossRef]
- Andersson, A.S.; Thomas, J.O.; Kalska, B.; Häggström, L. Thermal Stability of LiFePO4-Based Cathodes. Electrochem. Solid State Lett. 2000, 3, 66–68. [Google Scholar] [CrossRef]
- Andersson, A.S.; Kalska, B.; Haggstrom, L.; Thomas, J.O. Lithium Extraction/Insertion in LiFePO4: An X-Ray Diffraction and Mössbauer Spectroscopy Study. Solid State Ion. 2000, 130, 41–52. [Google Scholar] [CrossRef]
- Yamada, A.; Chung, S.C.; Hinokuma, K. Optimized LiFePO4 for Lithium Battery Cathodes. J. Electrochem. Soc. 2001, 148, 224–229. [Google Scholar] [CrossRef]
- Huang, H.; Yin, S.; Nazar, L.F. Approaching Theoretical Capacity of LiFePO4 at Room Temperature at High Rates. Electrochem. Solid State Lett. 2001, 4, 170–172. [Google Scholar] [CrossRef]
- Recham, N.; Chotard, J.-N.; Jumas, J.-C.; Laffont, L.; Armand, M.; Tarascon, J.-M. Ionothermal Synthesis of Li-Based Fluorophosphates Electrodes. Chem. Mater. 2010, 22, 1142–1148. [Google Scholar] [CrossRef]
- Barpanda, P.; Ati, M.; Melot, B.C.; Rousse, G.; Chotard, J.-N.; Doublet, M.-L.; Sougrati, M.T.; Corr, S.A.; Jumas, J.-C.; Tarascon, J.-M. A 3.90 V Iron-Based Fluorosulphate Material for Lithium-Ion Batteries Crystallizing in the Triplite Structure. Nat. Mater. 2011, 10, 772–779. [Google Scholar] [CrossRef]
- Lv, D.; Bai, J.; Zhang, P.; Wu, S.; Li, Y.; Wen, W.; Jiang, Z.; Mi, J.; Zhu, Z.; Yang, Y. Understanding the High Capacity of Li 2 FeSiO 4: In Situ XRD/XANES Study Combined with First-Principles Calculations. Chem. Mater. 2013, 25, 2014–2020. [Google Scholar] [CrossRef]
- Siu, C.; Seymour, I.D.; Britto, S.; Zhang, H.; Rana, J.; Feng, J.; Omenya, F.O.; Zhou, H.; Chernova, N.A.; Zhou, G.; et al. Enabling Multi-Electron Reaction of ε-VOPO4 to Reach Theoretical Capacity for Lithium-Ion Batteries. Chem. Commun. 2018, No. 54, 7802–7805. [Google Scholar] [CrossRef]
- Shi, Y.; Zhou, H.; Seymour, I.D.; Britto, S.; Rana, J.; Wangoh, L.W.; Huang, Y.; Yin, Q.; Reeves, P.J.; Zuba, M.; et al. Electrochemical Performance of Nanosized Disordered LiVOPO4. ACS Omega 2018, 3, 7310–7323. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Zhou, H.; Britto, S.; Seymour, I.D.; Wiaderek, K.M.; Omenya, F.; Chernova, N.A.; Chapman, K.W.; Grey, C.P.; Whittingham, M.S. A High-Performance Solid-State Synthesized LiVOPO4 for Lithium-Ion Batteries. Electrochem. Commun. 2019, 105, 106491. [Google Scholar] [CrossRef]
- Chung, Y.; Cassidy, E.; Lee, K.; Siu, C.; Huang, Y.; Omenya, F.; Rana, J.; Wiaderek, K.M.; Chernova, N.A.; Chapman, K.W.; et al. Nonstoichiometry and Defects in Hydrothermally Synthesized ϵ-LiVOPO4. ACS Appl. Energy Mater. 2019, 2, 4792–4800. [Google Scholar] [CrossRef]
- Rana, J.; Shi, Y.; Zuba, M.J.; Wiaderek, K.M.; Feng, J.; Zhou, H.; Ding, J.; Tianpin, W.; Cibin, G.; Balasubramanian, M.; et al. Role of Disorder in Limiting the True Multi-Electron Redox in ε-LiVOPO4. J. Mater. Chem. A 2018, 42, 1–10. [Google Scholar] [CrossRef]
- Ateba Mba, J.; Masquelier, C.; Suard, E.; Croguennec, L. Synthesis and Crystallographic Study of Homeotypic LiVPO4F and LiVPO4O. Chem. Mater. 2012, 24, 1223–1234. [Google Scholar] [CrossRef]
- Boudin, S.; Guesdon, A.; Leclaire, A.; Borel, M.M. Review on Vanadium Phosphates with Mono and Divalent Metallic Cations: Syntheses, Structural Relationships and Classification, Properties. Int. J. Inorg. Mater. 2000, 2, 561–579. [Google Scholar] [CrossRef]
- Whittingham, M.S. Ultimate Limits to Intercalation Reactions for Lithium Batteries. Chem. Rev. 2014, 114, 11414–11443. [Google Scholar] [CrossRef] [PubMed]
- Croguennec, L.; Palacin, M.R. Recent Achievements on Inorganic Electrode Materials for Lithium-Ion Batteries. J. Am. Chem. Soc. 2015, 137, 3140–3156. [Google Scholar] [CrossRef]
- Masquelier, C.; Croguennec, L. Polyanionic (Phosphates, Silicates, Sulfates) Frameworks as Electrode Materials for Rechargeable Li (or Na) Batteries. Chem. Rev. 2013, 113, 6552–6591. [Google Scholar] [CrossRef] [PubMed]
- Yabuuchi, N.; Kubota, K.; Dahbi, M.; Komaba, S. Research Development on Sodium-Ion Batteries. Chem. Rev. 2014, 114, 11636–11682. [Google Scholar] [CrossRef] [PubMed]
- Shi, H.-Y.; Jia, Z.; Wu, W.; Zhang, X.; Liu, X.-X.; Sun, X. The Development of Vanadyl Phosphate Cathode Materials for Energy Storage Systems: A Review. Chem. A Eur. J. 2020, 26, 8190–8204. [Google Scholar] [CrossRef] [PubMed]
- Beneš, L.; Melánová, K.; Svoboda, J.; Zima, V. Intercalation Chemistry of Layered Vanadyl Phosphate: A Review. J. Incl. Phenom. Macrocycl. Chem. 2012, 73, 33–53. [Google Scholar] [CrossRef]
- Jian, Z.; Hu, Y.-S.; Ji, X.; Chen, W. NASICON-Structured Materials for Energy Storage. Adv. Mater. 2017, 29, 1601925. [Google Scholar] [CrossRef]
- Anantharamulu, N.; Koteswara Rao, K.; Rambabu, G.; Vijaya Kumar, B.; Radha, V.; Vithal, M. A Wide-Ranging Review on Nasicon Type Materials. J. Mater. Sci. 2011, 46, 2821–2837. [Google Scholar] [CrossRef]
- Manthiram, A.; Goodenough, J.B. Lithium Insertion into Fe2(SO4)3 Frameworks. J. Power Sources 1989, 26, 403–408. [Google Scholar] [CrossRef]
- Huang, H.; Yin, S.C.; Kerr, T.; Taylor, N.; Nazar, L.F. Nanostructured Composites: A High Capacity, Fast Rate Li3V2(PO4)3/Carbon Cathode for Rechargeable Lithium Batteries. Adv. Mater. 2002, 14, 1525–1528. [Google Scholar] [CrossRef]
- Patoux, S.; Wurm, C.; Morcrette, M.; Rousse, G.; Masquelier, C. A Comparative Structural and Electrochemical Study of Monoclinic Li3Fe2(PO4)3 and Li3V2(PO4)3. J. Power Sources 2003, 119, 278–284. [Google Scholar] [CrossRef]
- Yin, S.-C.; Grondey, H.; Strobel, P.; Anne, M.; Nazar, L.F. Electrochemical Property: Structure Relationships in Monoclinic Li3-YV2(PO4)3. J. Am. Chem. Soc. 2003, 125, 10402–10411. [Google Scholar] [CrossRef] [PubMed]
- Yin, S.C.; Strobel, P.S.; Grondey, H.; Nazar, L.F. Li2.5V2(PO4)3: A Room-Temperature Analogue to the Fast-Ion Conducting High-Temperature γ-Phase of Li3V2(PO4)3. Chem. Mater. 2004, 16, 1456–1465. [Google Scholar] [CrossRef]
- Rui, X.; Yan, Q.; Skyllas-Kazacos, M.; Lim, T.M. Li3V2(PO4)3 Cathode Materials for Lithium-Ion Batteries: A Review. J. Power Sources 2014, 258, 19–38. [Google Scholar] [CrossRef]
- Kang, J.; Mathew, V.; Gim, J.; Kim, S.; Song, J.; Im, W.B.; Han, J.; Lee, J.Y.; Kim, J. Pyro-Synthesis of a High Rate Nano-Li3V2 (PO4)3/C Cathode with Mixed Morphology for Advanced Li-Ion Batteries. Sci. Rep. 2014, 4, 1–9. [Google Scholar] [CrossRef]
- Saïdi, M.Y.; Barker, J.; Huang, H.; Swoyer, J.L.; Adamson, G. Performance Characteristics of Lithium Vanadium Phosphate as a Cathode Material for Lithium-Ion Batteries. J. Power Sources 2003, 119, 266–272. [Google Scholar] [CrossRef]
- Kim, S.; Zhang, Z.; Wang, S.; Yang, L.; Cairns, E.J.; Penner-Hahn, J.E.; Deb, A. Electrochemical and Structural Investigation of the Mechanism of Irreversibility in Li3V2(PO4)3 Cathodes. J. Phys. Chem. C 2016, 120, 7005–7012. [Google Scholar] [CrossRef]
- Rui, X.H.; Yesibolati, N.; Chen, C.H. Li3V2(PO4)3/C Composite as an Intercalation-Type Anode Material for Lithium-Ion Batteries. J. Power Sources 2011, 196, 2279–2282. [Google Scholar] [CrossRef]
- Gaubicher, J.; Wurm, C.; Goward, G.; Masquelier, C.; Nazar, L. Rhombohedral Form of Li3V2(PO4)3 as a Cathode in Li-Ion Batteries. Chem. Mater. 2000, 12, 3240–3242. [Google Scholar] [CrossRef]
- Delmas, C.; Olazcuaga, R.; Cherkaoui, F.; Brochu, R.; Leflem, G. New Family of Phosphates with Formula Na3M2(PO4)3 (M= Ti,V,Cr,Fe). C. R. Seances Acad. Sci. 1978, 287, 169–171. [Google Scholar]
- Chotard, J.-N.; Rousse, G.; David, R.; Mentré, O.; Courty, M.; Masquelier, C. Discovery of a Sodium-Ordered Form of Na3V2(PO4)3 below Ambient Temperature. Chem. Mater. 2015, 27, 5982–5987. [Google Scholar] [CrossRef]
- Jian, Z.; Yuan, C.; Han, W.; Lu, X.; Gu, L.; Xi, X.; Hu, Y.; Li, H.; Chen, W.; Chen, D.; et al. Atomic Structure and Kinetics of NASICON NaxV2(PO4)3 Cathode for Sodium-Ion Batteries. Adv. Funct. Mater. 2014, 24, 4265–4272. [Google Scholar] [CrossRef]
- Golalakrishnan, J.; Kasthuri Rangan, K. NASICON-Type Vanadium Phosphate Synthesized by Oxidative Deintercalation of Sodium from Sodium Vanadium Phosphate (Na3V2(PO4)3). Chem. Mater. 1992, 4, 745–747. [Google Scholar] [CrossRef]
- Lalère, F.; Seznec, V.; Courty, M.; David, R.; Chotard, J.N.; Masquelier, C. Improving the Energy Density of Na3V2(PO4)3-Based Positive Electrodes through V/Al Substitution. J. Mater. Chem. A 2015, 3, 16198–16205. [Google Scholar] [CrossRef]
- Mason, C.W.; Gocheva, I.; Hoster, H.E.; Dennis, Y.W. Activating Vanadium’s Highest Oxidation State in the NASICON Structure. ECS Trans. 2014, 58, 41–46. [Google Scholar] [CrossRef]
- Aragón, M.J.; Lavela, P.; Ortiz, G.F.; Tirado, J.L. Benefits of Chromium Substitution in Na3V2(PO4)3 as a Potential Candidate for Sodium-Ion Batteries. ChemElectroChem 2015, 2, 995–1002. [Google Scholar] [CrossRef]
- Liu, R.; Xu, G.; Li, Q.; Zheng, S.; Zheng, G.; Gong, Z.; Li, Y.; Kruskop, E.; Fu, R.; Chen, Z.; et al. Exploring Highly Reversible 1.5-Electron Reactions (V3+/V4+/V5+) in Na3VCr(PO4)3 Cathode for Sodium-Ion Batteries. ACS Appl. Mater. Interfaces 2017, 9, 43632–43639. [Google Scholar] [CrossRef]
- Liu, R.; Zheng, S.; Yuan, Y.; Yu, P.; Liang, Z.; Zhao, W.; Shahbazian-Yassar, R.; Ding, J.; Lu, J.; Yang, Y. Counter-Intuitive Structural Instability Aroused by Transition Metal Migration in Polyanionic Sodium Ion Host. Adv. Energy Mater. 2020, 2003256, 1–9. [Google Scholar] [CrossRef]
- Zhou, W.; Xue, L.; Gao, H.; Li, Y.; Xin, S.; Fu, G.; Cui, Z.; Zhu, Y.; Goodenough, J.B. MV(PO4)3 (M= Mn, Fe, Ni) Structure and Properties for Sodium Extraction. Nano Lett. 2016, 16, 7836–7841. [Google Scholar] [CrossRef]
- Zakharkin, M.V.; Drozhzhin, O.A.; Tereshchenko, I.V.; Chernyshov, D.; Abakumov, A.M.; Antipov, E.V.; Stevenson, K.J. Enhancing Na+ Extraction Limit through High Voltage Activation of the NASICON-Type Na4MnV(PO4)3 Cathode. ACS Appl. Energy Mater. 2018, 1, 5842–5846. [Google Scholar] [CrossRef]
- Zakharkin, M.V.; Drozhzhin, O.A.; Ryazantsev, S.V.; Chernyshov, D.; Kirsanova, M.A.; Mikheev, I.V.; Pazhetnov, E.M.; Antipov, E.V.; Stevenson, K.J. Electrochemical Properties and Evolution of the Phase Transformation Behavior in the NASICON-Type Na3+xMnxV2-x(PO4)3 (0≤x≤1) Cathodes for Na-Ion Batteries. J. Power Sources 2020, 470, 1–8. [Google Scholar] [CrossRef]
- Chen, F.; Kovrugin, V.M.; David, R.; Mentré, O.; Fauth, F.; Chotard, J.N.; Masquelier, C. A NASICON-Type Positive Electrode for Na Batteries with High Energy Density: Na4MnV(PO4)3. Small Methods 2019, 3, 1–9. [Google Scholar] [CrossRef]
- Le Meins, J.; Crosnier-Lopez, M.-P.; Hemon-Ribaud, A.; Courbion, G. Phase Transitions in the Na3M2(PO4)2F3 Family (M= Al3+,V3+,Cr3+,Fe3+,Ga3+): Synthesis, Thermal, Structural, and Magnetic Studies. J. Solid State Chem. 1999, 148, 260–277. [Google Scholar] [CrossRef]
- Bianchini, M.; Brisset, N.; Fauth, F.; Weill, F.; Elkaim, E.; Suard, E.; Masquelier, C.; Croguennec, L. Na3V2(PO4)2F3 Revisited: A High-Resolution Diffraction Study. Chem. Mater. 2014, 26, 4238–4247. [Google Scholar] [CrossRef]
- Shakoor, R.A.; Seo, D.-H.; Kim, H.; Park, Y.-U.; Kim, J.; Kim, S.-W.; Gwon, H.; Lee, S.; Kang, K. A Combined First Principles and Experimental Study on Na3V2(PO4)2F3 for Rechargeable Na Batteries. J. Mater. Chem. 2012, 22, 20535. [Google Scholar] [CrossRef]
- Yan, G.; Mariyappan, S.; Rousse, G.; Jacquet, Q.; Deschamps, M.; David, R.; Mirvaux, B.; Freeland, J.W.; Tarascon, J.M. Higher Energy and Safer Sodium Ion Batteries via an Electrochemically Made Disordered Na3V2(PO4)2F3 Material. Nat. Commun. 2019, 10, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Bianchini, M.; Fauth, F.; Brisset, N.; Weill, F.; Suard, E.; Masquelier, C.; Croguennec, L. Comprehensive Investigation of the Na3V2(PO4)2F3−NaV2(PO4)2F3 System by Operando High Resolution Synchrotron X-ray Diffraction. Chem. Mater. 2015, 27, 3009–3020. [Google Scholar] [CrossRef]
- Broux, T.; Bamine, T.; Simonelli, L.; Stievano, L.; Fauth, F.; Ménétrier, M.; Carlier, D.; Masquelier, C.; Croguennec, L. VIV Disproportionation Upon Sodium Extraction From Na3V2(PO4)2F3 Observed by Operando X-ray Absorption Spectroscopy and State NMR. J. Phys. Chem. C 2017, 121, 4103–4111. [Google Scholar] [CrossRef]
- Broux, T.; Bamine, T.; Fauth, F.; Simonelli, L.; Olszewski, W.; Marini, C.; Ménétrier, M.; Carlier, D.; Masquelier, C.; Croguennec, L. Strong Impact of the Oxygen Content in Na3V2(PO4)2F3-YOy (0 ≦ y ≦ 0.5) on Its Structural and Electrochemical Properties. Chem. Mater. 2016, 28, 7683–7692. [Google Scholar] [CrossRef]
- Park, Y.U.; Seo, D.H.; Kim, H.; Kim, J.; Lee, S.; Kim, B.; Kang, K. A Family of High-Performance Cathode Materials for Na-Ion Batteries, Na3(VO1-XPO4)2 F1+2x (0 ≤ x ≤ 1): Combined First-Principles and Experimental Study. Adv. Funct. Mater. 2014, 24, 4603–4614. [Google Scholar] [CrossRef]
- Mariyappan, S.; Wang, Q.; Tarascon, J.M. Will Sodium Layered Oxides Ever Be Competitive for Sodium Ion Battery Applications? J. Electrochem. Soc. 2018, 165, 3714–3722. [Google Scholar] [CrossRef]
- Park, Y.U.; Seo, D.H.; Kwon, H.S.; Kim, B.; Kim, J.; Kim, H.; Kim, I.; Yoo, H.I.; Kang, K. A New High-Energy Cathode for a Na-Ion Battery with Ultrahigh Stability. J. Am. Chem. Soc. 2013, 135, 13870–13878. [Google Scholar] [CrossRef]
- Serras, P.; Alonso, J.; Sharma, N.; Miguel, J.; Kubiak, P.; Gubieda, M.-L.; Rojo, T. Electrochemical Na Extraction/Insertion of Na3V2O2x(PO4)2F3−2x. Chem. Mater. 2013, 25, 4917–4925. [Google Scholar] [CrossRef]
- Morimoto, H.; Ito, D.; Ogata, Y.; Suzuki, T.; Sakamaki, K.; Tsuji, T.; Hirukawa, M.; Matsumoto, A.; Tobishima, S. Charge/Discharge Behavior of Triclinic LiTiOPO4 Anode Materials for Lithium Secondary Batteries. Electrochem. Soc. Jpn. 2016, 84, 878–881. [Google Scholar] [CrossRef]
- Barker, J.; Saidi, M.Y.; Swoyer, J.L. Electrochemical Insertion Properties of the Novel Lithium Vanadium Fluorophosphate, LiVPO4F. J. Electrochem. Soc. 2003, 150, 1394–1398. [Google Scholar] [CrossRef]
- Badraoui, A.E.; Pivan, J.-Y.; Maunaye, M.; Pena, O.; Louer, M.; Louer, D. Order-Disorder Phenomena in Vanadium Phosphates. Structures and Properties of Tetragonal and Monoclinic VPO4·H2O. Ann. Chim. Sci. Matériaux 1998, 23, 97–101. [Google Scholar] [CrossRef]
- Lii, K.H.; Li, H.; Cheng, C.; Wang, S. Synthesis and Structural Characterization of Sodium Vanadyl (IV) Orthophosphate NaVOPO4. Z. Krist. Cryst. Mater. 2010, 197, 67–73. [Google Scholar] [CrossRef]
- Bianchini, M.; Ateba-Mba, J.M.; Dagault, P.; Bogdan, E.; Carlier, D.; Suard, E.; Masquelier, C.; Croguennec, L. Multiple Phases in the ε-VPO4O–LiVPO4O–Li2VPO4O System: A Combined Solid State Electrochemistry and Diffraction Structural Study. J. Mater. Chem. A 2014, 2, 10182–10192. [Google Scholar] [CrossRef]
- Song, Y.; Zavalij, P.Y.; Whittingham, M.S. ε-VOPO4: Electrochemical Synthesis and Enhanced Cathode Behavior. J. Electrochem. Soc. 2005, 152, 721–728. [Google Scholar] [CrossRef]
- Boivin, E.; Chotard, J.-N.; Ménétrier, M.; Bourgeois, L.; Bamine, T.; Carlier, D.; Fauth, F.; Suard, E.; Masquelier, C.; Croguennec, L. Structural and Electrochemical Studies of a New Tavorite Composition LiVPO4OH. J. Mater. Chem. A 2016, 4, 11030–11045. [Google Scholar] [CrossRef]
- Lee Harrison, K.; Bridges, C.; Segre, C.; Varnado, D.; Applestone, D.; Bielawski, C.; Paranthaman, M.; Manthiram, A. Chemical and Electrochemical Lithiation of LiVOPO4 Cathodes for Lithium-Ion Batteries. Chem. Mater. 2014, 26, 3849–3861. [Google Scholar] [CrossRef]
- Lin, Y.; Wen, B.; Wiaderek, K.; Sallis, S.; Liu, H.; Lapidus, S.; Borkiewicz, O.; Quackenbush, N.; Chernova, N.; Karki, K.; et al. Thermodynamics, Kinetics and Structural Evolution of ε -LiVOPO4 over Multiple Lithium Intercalation. Chem. Mater. 2016, 28, 1794–1805. [Google Scholar] [CrossRef]
- Barker, J.; Saidi, M.Y.; Swoyer, J.L. A Comparative Investigation of the Li Insertion Properties of the Novel Fluorophosphate Phases, NaVPO4F and LiVPO4F. J. Electrochem. Soc. 2004, 151, 1670–1677. [Google Scholar] [CrossRef]
- Barker, J.; Gover, R.K.B.; Burns, P.; Bryan, A.; Saidi, M.Y.; Swoyer, J.L. Structural and Electrochemical Properties of Lithium Vanadium Fluorophosphate, LiVPO4F. J. Power Sources 2005, 146, 516–520. [Google Scholar] [CrossRef]
- Ellis, B.L.; Ramesh, T.N.; Davis, L.J.M.; Goward, G.R.; Nazar, L.F. Structure and Electrochemistry of Two-Electron Redox Couples in Lithium Metal Fluorophosphates Based on the Tavorite Structure. Chem. Mater. 2011, 23, 5138–5148. [Google Scholar] [CrossRef]
- Boivin, E. Crystal Chemistry of Vanadium Phosphates as Positive Electrode Materials for Li-Ion and Na-Ion Batteries. Ph.D. Thesis, University of Picardie Jules Verne, Amiens, France, 2017. [Google Scholar]
- Ateba Mba, J.-M.; Croguennec, L.; Basir, N.I.; Barker, J.; Masquelier, C. Lithium Insertion or Extraction from/into Tavorite-Type LiVPO4F: An In Situ X-Ray Diffraction Study. J. Electrochem. Soc. 2012, 159, 1171–1175. [Google Scholar] [CrossRef]
- Piao, Y.; Qin, Y.; Ren, Y.; Heald, S.M.; Sun, C.; Zhou, D.; Polzin, B.J.; Trask, S.E.; Amine, K.; Wei, Y.; et al. A XANES Study of LiVPO4F: A Factor Analysis Approach. Phys. Chem. Chem. Phys. 2014, 16, 3254–3260. [Google Scholar] [CrossRef] [PubMed]
- Zhong, S.; Chen, W.; Li, Y.; Zou, Z.; Liu, C. Synthesis of LiVPO4F with High Electrochemical Performance by Sol-Gel Route. Trans. Nonferrous Met. Soc. China 2010, 20, 275–278. [Google Scholar] [CrossRef]
- Rangaswamy, P.; Shetty, V.R.; Suresh, G.S.; Mahadevan, K.M.; Nagaraju, D.H. Enhanced Electrochemical Performance of LiVPO4F/f-Graphene Composite Electrode Prepared via Ionothermal Process. J. Appl. Electrochem. 2017, 47. [Google Scholar] [CrossRef]
- Messinger, R.J.; Ménétrier, M.; Salager, E.; Boulineau, A.; Duttine, M.; Carlier, D.; Ateba Mba, J.-M.; Croguennec, L.; Masquelier, C.; Massiot, D.; et al. Revealing Defects in Crystalline Lithium-Ion Battery Electrodes by Solid-State NMR: Applications to LiVPO4F. Chem. Mater. 2015, 27, 5212–5221. [Google Scholar] [CrossRef]
- Bamine, T.; Boivin, E.; Boucher, F.; Messinger, R.J.; Salager, E.; Deschamps, M.; Masquelier, C.; Croguennec, L.; Ménétrier, M.; Carlier, D. Understanding Local Defects in Li-Ion Battery Electrodes through Combined DFT/NMR Studies: Application to LiVPO4F. J. Phys. Chem. C 2017, 121, 3219–3227. [Google Scholar] [CrossRef]
- Kim, M.; Lee, S.; Kang, B. Fast-Rate Capable Electrode Material with Higher Energy Density than LiFePO4: 4.2 V LiVPO4F Synthesized by Scalable Single-Step Solid-State Reaction. Adv. Sci. 2015, 3, 1500366. [Google Scholar] [CrossRef]
- Kim, M.; Lee, S.; Kang, B. High Energy Density Polyanion Electrode Material: LiVPO4O1-XFx (x ≈ 0.25) with Tavorite Structure. Chem. Mater. 2017, 29, 4690–4699. [Google Scholar] [CrossRef]
- Boivin, E.; David, R.; Chotard, J.N.; Bamine, T.; Iadecola, A.; Bourgeois, L.; Suard, E.; Fauth, F.; Carlier, D.; Masquelier, C.; et al. LiVPO4F1-YOy Tavorite-Type Compositions: Influence of the Vanadyl-Type Defects’ Concentration on the Structure and Electrochemical Performance. Chem. Mater. 2018, 30, 5682–5693. [Google Scholar] [CrossRef]
- Boivin, E.; Iadecola, A.; Fauth, F.; Chotard, J.N.; Masquelier, C.; Croguennec, L. Redox Paradox of Vanadium in Tavorite LiVPO4F1-YOy. Chem. Mater. 2019, 31, 7367–7376. [Google Scholar] [CrossRef]
- Wurm, C.; Morcrette, M.; Rousse, G.; Dupont, L.; Masquelier, C. Lithium Insertion/Extraction into/from LiMX2O7 Compositions (M = Fe, V.; X = P, As) Prepared via a Solution Method. Chem. Mater. 2002, 14, 2701–2710. [Google Scholar] [CrossRef]
- Barker, J.; Gover, R.K.B.; Burns, P.; Bryan, A. LiVP2O7: A Viable Lithium-Ion Cathode Material? Electrochem. Solid State Lett. 2005, 8, 446–448. [Google Scholar] [CrossRef]
- Deng, C.; Zhang, S. 1D Nanostructured Na7V4(P2O7)4(PO4) as High-Potential and Superior-Performance Cathode Material for Sodium-Ion Batteries. Appl. Mater. Interfaces 2014, 6, 9111–9117. [Google Scholar] [CrossRef] [PubMed]
- Kovrugin, V.; Chotard, J.-N.; Fauth, F.; Jamali, A.; David, R.; Christian, M. Structural and Electrochemical Studies of Novel Batteries. J. Mater. Chem. A 2017, 5, 14365–14376. [Google Scholar] [CrossRef]
- Kim, J.; Yoon, G.; Kim, H.; Park, Y.U.; Kang, K. Na3V(PO4)2: A New Layered-Type Cathode Material with High Water Stability and Power Capability for Na-Ion Batteries. Chem. Mater. 2018, 30, 3683–3689. [Google Scholar] [CrossRef]
- Liu, R.; Liang, Z.; Xiang, Y.; Zhao, W.; Liu, H.; Chen, Y. Recognition of V3+/V4+/V5+ Multielectron Reactions in Na3V(PO4)2: A Potential High Energy Density Cathode for Sodium-Ion Batteries. Molecules 2020, 25, 1000. [Google Scholar] [CrossRef]
- Sandineni, P.; Madria, P.; Ghosh, K.; Choudhury, A. A Square Channel Vanadium Phosphite Framework as High Voltage Cathode for Li- and Na- Ion Batteries. Mater. Adv. 2020. [Google Scholar] [CrossRef]
- Kuang, Q.; Xu, J.; Zhao, Y.; Chen, X.; Chen, L. Layered Monodiphosphate Li9V3(P2O7)3(PO4)2: A Novel Cathode Material for Lithium-Ion Batteries. Electrochim. Acta 2011, 56, 2201–2205. [Google Scholar] [CrossRef]
- Deng, C.; Zhang, S.; Zhao, B. First Exploration of Ultra Fine Na7V3(P2O7)4 as a High-Potential Cathode Material for Sodium-Ion Battery. Energy Storage Mater. 2016, 4, 71–78. [Google Scholar] [CrossRef]
- Reynaud, M.; Wizner, A.; Katcho, N.A.; Loaiza, L.C.; Galceran, M.; Carrasco, J.; Rojo, T.; Armand, M.; Casas-Cabanas, M. Sodium Vanadium Nitridophosphate Na3V(PO3)3N as a High-Voltage Positive Electrode Material for Na-Ion and Li-Ion Batteries. Electrochem. Commun. 2017, 84, 14–18. [Google Scholar] [CrossRef]
- Boivin, E.; Chotard, J.-N.; Bamine, T.; Carlier, D.; Serras, P.; Veronica, P.; Rojo, T.; Iadecola, A.; Dupont, L.; Bourgeois, L.; et al. Vanadyl-Type Defects in Tavorite-like NaVPO4F: From the Average Long Range Structure to Local Environments. J. Mater. Chem. A 2017, 5, 25044–25055. [Google Scholar] [CrossRef]
- Fedotov, S.S.; Khasanova, N.R.; Samarin, A.S.; Drozhzhin, O.A.; Batuk, D.; Karakulina, O.M.; Hadermann, J.; Abakumov, A.M.; Antipov, E.V. AVPO4F (A = Li, K): A 4 V Cathode Material for High-Power Rechargeable Batteries. Chem. Mater. 2016, 28, 411–415. [Google Scholar] [CrossRef]
- Zhang, B.; Dugas, R.; Rousse, G.; Rozier, P.; Abakumov, A.M.; Tarascon, J.M. Insertion Compounds and Composites Made by Ball Milling for Advanced Sodium-Ion Batteries. Nat. Commun. 2016, 7, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Makimura, Y.; Cahill, L.S.; Iriyama, Y.; Goward, G.R.; Nazar, L.F. Layered Lithium Vanadium Fluorophosphate, Li 5 V(PO 4 ) 2 F 2: A 4 V Class Positive Electrode Material for Lithium-Ion Batteries. Chem. Mater. 2008, 20, 4240–4248. [Google Scholar] [CrossRef]
- Liang, Z.; Zhang, X.; Liu, R.; Ortiz, G.F.; Zhong, G.; Xiang, Y.; Chen, S.; Mi, J.; Wu, S.; Yang, Y. New Dimorphs of Na5V(PO4)2F2 as an Ultrastable Cathode Material for Sodium-Ion Batteries. ACS Appl. Energy Mater. 2020, 3, 1181–1189. [Google Scholar] [CrossRef]
- Barker, J.; Saidi, M.Y.; Swoyer, J.L. Electrochemical Properties of β-LiVOPO4 Prepared by Carbothermal Reduction. J. Electrochem. Soc. 2004, 151, 796–800. [Google Scholar] [CrossRef]
- He, G.; Kan, W.; Manthiram, A. β-NaVOPO4 Obtained by a Low-Temperature Synthesis Process: A New 3.3 V Cathode for Sodium-Ion Batteries. Chem. Mater. 2016, 28, 1503–1512. [Google Scholar] [CrossRef]
- He, G.; Bridges, C.A.; Manthiram, A. Crystal Chemistry of Electrochemically and Chemically Lithiated Layered α-LiVOPO4. Chem. Mater. 2015, 27, 6699–6707. [Google Scholar] [CrossRef]
- Satya Kishore, M.; Pralong, V.; Caignaert, V.; Malo, S.; Hebert, S.; Varadaraju, U.V.; Raveau, B. Topotactic Insertion of Lithium in the Layered Structure Li4VO(PO4)2: The Tunnel Structure Li5VO(PO4)2. J. Solid State Chem. 2008, 181, 976–982. [Google Scholar] [CrossRef]
- Kim, J.; Kim, H.; Lee, S. High Power Cathode Material Na4VO(PO4)2 with Open Framework for Na Ion Batteries. Chem. Mater. 2017, 29, 3363–3366. [Google Scholar] [CrossRef]
- Kishore, M.S.; Pralong, V.; Caignaert, V.; Varadaraju, U.V.; Raveau, B. A New Lithium Vanadyl Diphosphate Li2VOP2O7: Synthesis and Electrochemical Study. Solid State Sci. 2008, 10, 1285–1291. [Google Scholar] [CrossRef]
- Manthiram, A.; Goodenough, J.B. Lithium Insertion into Fe2(MO4)3 Frameworks: Comparison of M = W with M = MO. J. Solid State Chem. 1987, 71, 349–360. [Google Scholar] [CrossRef]
- Ben Yahia, M.; Lemoigno, F.; Rousse, G.; Boucher, F.; Tarascon, J.-M.; Doublet, M.-L. Origin of the 3.6 V to 3.9 V Voltage Increase in the LiFeSO4F Cathodes for Li-Ion Batteries. Energy Environ. Sci. 2012, 5, 9584. [Google Scholar] [CrossRef]
- Parapari, S.S.; Ateba Mba, J.M.; Tchernychova, E.; Mali, G.; Arčon, I.; Kapun, G.; Gülgün, M.A.; Dominko, R. Effects of a Mixed O/F Ligand in the Tavorite-Type LiVPO4O Structure. Chem. Mater. 2020, 32, 262–272. [Google Scholar] [CrossRef]
- Boivin, E.; Chotard, J.N.; Ménétrier, M.; Bourgeois, L.; Bamine, T.; Carlier, D.; Fauth, F.; Masquelier, C.; Croguennec, L. Oxidation under Air of Tavorite LiVPO4F: Influence of Vanadyl-Type Defects on Its Electrochemical Properties. J. Phys. Chem. C 2016, 120, 26187–26198. [Google Scholar] [CrossRef]
- Nguyen, L.H.B.; Iadecola, A.; Belin, S.; Olchowka, J.; Masquelier, C.; Carlier, D.; Croguennec, L. A Combined Operando Synchrotron X-ray Absorption Spectroscopy and First-Principles Density Functional Theory Study to Unravel the Vanadium Redox Paradox in the Na3V2(PO4)2F3−Na3V2(PO4)2FO2 Compositions. J. Phys. Chem. C 2020, 124, 23511–23522. [Google Scholar] [CrossRef]
- Bamine, T.; Boivin, E.; Masquelier, C.; Croguennec, L.; Salager, E.; Carlier, D. Local Atomic and Electronic Structure in the LiVPO4(F,O) Tavorite-Type Materials from Solid State NMR Combined with DFT Calculations. Magn. Reson. Chem. 2020, 58, 1109–1117. [Google Scholar] [CrossRef]
- Ni, S.; Liu, J.; Chao, D.; Mai, L. Vanadate-Based Materials for Li-Ion Batteries: The Search for Anodes for Practical Applications. Adv. Energy Mater. 2019, 9, 1803324. [Google Scholar] [CrossRef]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| As Synthetized Compositions | Initial Vn+ | M/P Ratio | V2+/V3+ | V3+/V4+ | V4+/V5+ | Ref. | |||
|---|---|---|---|---|---|---|---|---|---|
| E (V vs. Li+/Li) | Capacity (mAh/g) | E (V vs. Li+/Li) | Capacity (mAh/g) | E (V vs. Li+/Li) | Capacity (mAh/g) | ||||
| Type I Materials | |||||||||
| Na3V2(PO4)3 | V3+ | 0.67 | 1.9 * | 59 | 3.7 * | 118 | / | / | [39] |
| Na3V1.5Al0.5(PO4)3 | V3+ | 0.67 | 1.9 * | 60 | 3.7 * | 85 | 4.3 * | 28 | [39] |
| Na3VCr(PO4)3 | V3+ | 0.67 | / | / | 3.7 * | 60 | 4.4 * | 50 | [42] |
| Na4VMn(PO4)3 | V3+ | 0.67 | / | / | 3.7 * | 60 | 4.2 * | 50 | [47] |
| r-Li3V2(PO4)3 | V3+ | 0.67 | / | / | 3.7 | 131 | / | / | [34] |
| m-Li3V2(PO4)3 | V3+ | 0.67 | 1.8 | 131 | 3.9 | 131 | 4.5 | 33 | [27] |
| LiVP2O7 | V3+ | 0.5 | 2 | 116 | 4.3 | 95 | / | / | [82,83] |
| Na7V4(P2O7)3(PO4)2 | V3+ | 0.5 | / | / | 4.2 * | 90 | / | / | [84] |
| Na7V3Al1(P2O7)3(PO4)2 | V3+ | 0.5 | / | / | 4.2 * | 77 | 4.5 | 46 | [85] |
| Na3V(PO4)2 | V3+ | 0.5 | / | / | 3.8 * | 90 | 4.4 * | 20 | [86,87] |
| LiV(HPO3)2 | V3+ | 0.5 | / | / | 4.1 | 75 | / | / | [88] |
| Li9V3(P2O7)3(PO4)2 | V3+ | 0.375 | / | / | 3.7 | 55 | 4.5 | 55 | [89] |
| Na7V3(P2O7)4 | V3+ | 0.375 | / | / | 4.3 * | 80 | / | / | [90] |
| Na3V(PO3)3N | V3+ | 0.33 | / | / | 4.3 * | 74 | / | / | [91] |
| Type II Materials | |||||||||
| LiVPO4F | V3+ | 1 | 1.8 | 156 | 4.2 | 156 | / | / | [14] |
| NaVPO4F | V3+ | 1 | / | / | ≈4.2 * | 20 | / | / | [92] |
| LiVPO4OH | V3+ | 1 | 1.4 | 155 | / | / | / | / | [65] |
| (Li,K)VPO4F | V3+ | 1 | / | / | 4.0 | 110 | / | / | [93] |
| Na3V2(PO4)2F3 | V3+ | 0.67 | 1.5 * | 64 | 4.0 * | 64 | ≈ 4.8 * | 64 | [51,53,94] |
| Li5V(PO4)2F2 | V3+ | 0.5 | / | / | 4.15 | 85 | 4.7 | 85 | [95] |
| t-Na5V(PO4)2F2 | V3+ | 0.5 | / | / | 3.7 * | 62 | / | / | [96] |
| o-Na5V(PO4)2F2 | V3+ | 0.5 | / | / | 3.9 * | 65 | / | / | [96] |
| Type III Materials | |||||||||
| α-LiVPO4O | V4+ | 1 | / | / | 2.4 | 155 | 3.95 | 150 | [14] |
| β-LiVPO4O | V4+ | 1 | / | / | 2.2 | 155 | 4 | 130 | [97] |
| β-NaVPO4O | V4+ | 1 | / | / | / | / | 3.6 * | 58 | [98] |
| γ-LiVPO4O | V4+ | 1 | / | / | 2 | 80 | 4 | 150 | [99] |
| Li4VO(PO4)2 | V4+ | 0.5 | / | / | 2 | 94 | 4.1 | 94 | [100] |
| Na4VO(PO4)2 | V4+ | 0.5 | / | / | / | / | 3.8 * | 77 | [101] |
| Li2VOP2O7 | V4+ | 0.5 | / | / | / | / | 4.1 | 64 | [102] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Boivin, E.; Chotard, J.-N.; Masquelier, C.; Croguennec, L. Towards Reversible High-Voltage Multi-Electron Reactions in Alkali-Ion Batteries Using Vanadium Phosphate Positive Electrode Materials. Molecules 2021, 26, 1428. https://doi.org/10.3390/molecules26051428
Boivin E, Chotard J-N, Masquelier C, Croguennec L. Towards Reversible High-Voltage Multi-Electron Reactions in Alkali-Ion Batteries Using Vanadium Phosphate Positive Electrode Materials. Molecules. 2021; 26(5):1428. https://doi.org/10.3390/molecules26051428
Chicago/Turabian StyleBoivin, Edouard, Jean-Noël Chotard, Christian Masquelier, and Laurence Croguennec. 2021. "Towards Reversible High-Voltage Multi-Electron Reactions in Alkali-Ion Batteries Using Vanadium Phosphate Positive Electrode Materials" Molecules 26, no. 5: 1428. https://doi.org/10.3390/molecules26051428
APA StyleBoivin, E., Chotard, J.-N., Masquelier, C., & Croguennec, L. (2021). Towards Reversible High-Voltage Multi-Electron Reactions in Alkali-Ion Batteries Using Vanadium Phosphate Positive Electrode Materials. Molecules, 26(5), 1428. https://doi.org/10.3390/molecules26051428