
Therapeutic Targeting of the NRF2 Signaling Pathway in Cancer

 ,
,  , ,
, , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. NRF2 and KEAP1 Signaling Pathway
2.1. The Tumor Suppressive Role of NRF2 Pathway
2.2. The Tumor Suppressive Role of KEAP1
2.3. The Carcinogenic Role of NRF2
2.4. The Carcinogenic Role of KEAP1
3. NRF2 Activation Mechanisms in Cancer
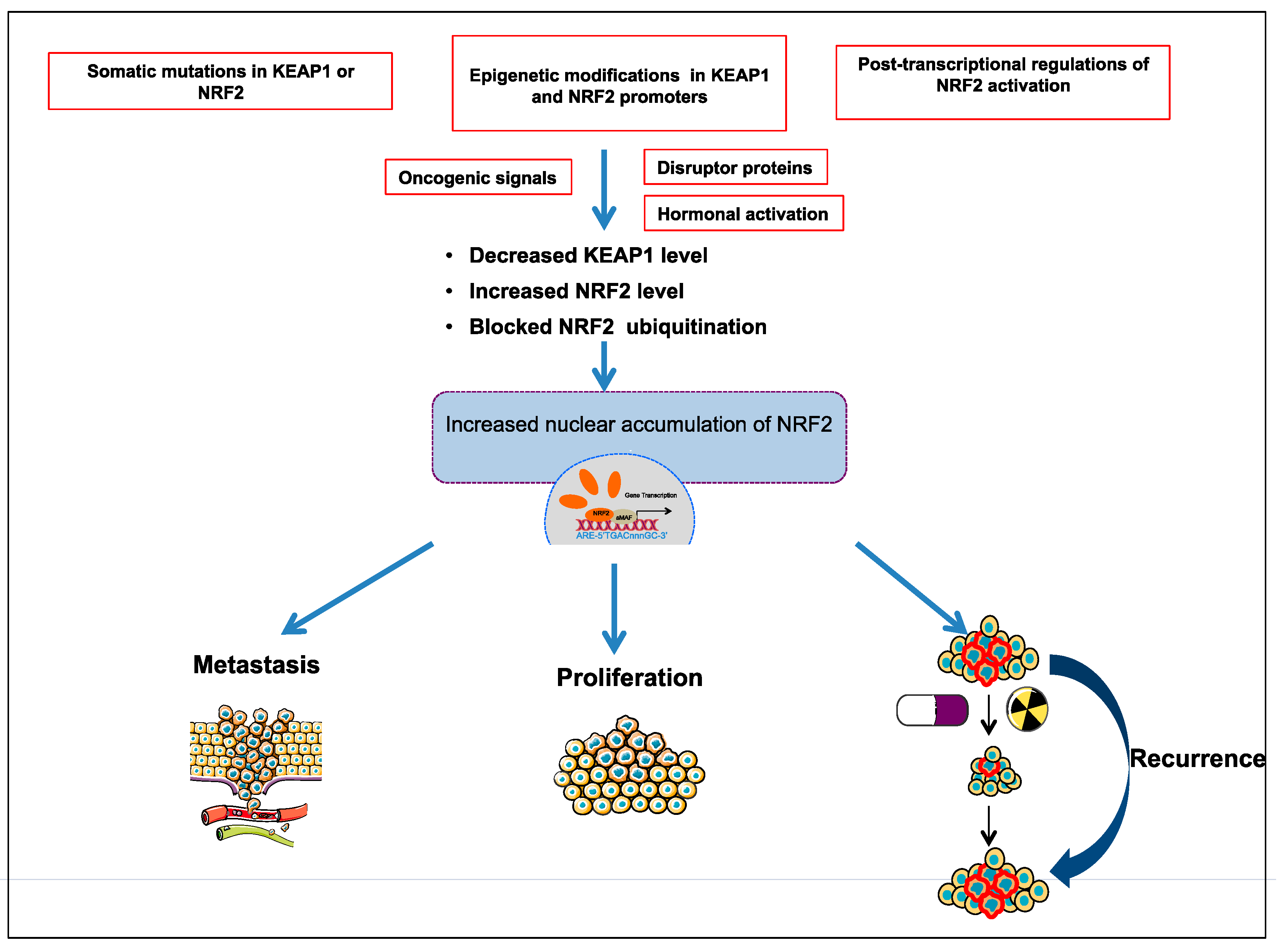
3.1. The Somatic Mutations in KEAP1 or NRF2
3.2. Epigenetic Modifications in KEAP1 and NRF2 Promoters
3.3. Post-Transcriptional Regulation of NRF2 Activation
3.4. Disruptor Proteins
3.5. Oncogenic Signals
3.6. Hormonal Activation
4. NRF2 Modulators for Cancer Therapy
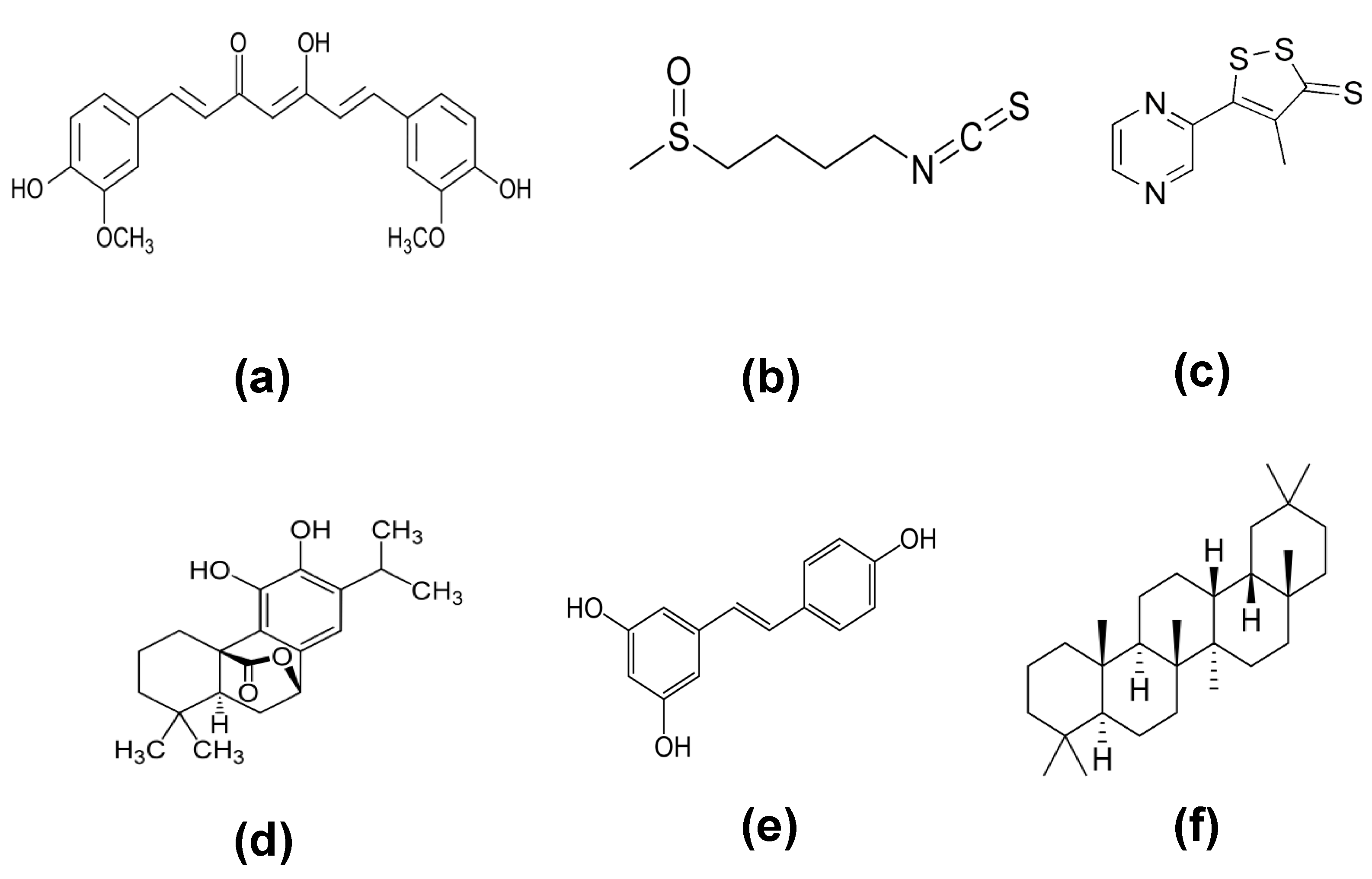
4.1. NRF2 Activators
4.2. NRF2 Inhibitors
5. Conclusions/Perspective
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef]
- Ferlay, J.; Colombet, M.; Soerjomataram, I.; Mathers, C.; Parkin, D.M.; Piñeros, M.; Znaor, A.; Bray, F. Estimating the global cancer incidence and mortality in 2018: GLOBOCAN sources and methods. Int. J. Cancer 2019, 144, 1941–1953. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Hornsveld, M.; Dansen, T.B. The Hallmarks of Cancer from a Redox Perspective. Antioxid. Redox Signal. 2016, 25, 300–325. [Google Scholar] [CrossRef] [PubMed]
- Marengo, B.; Nitti, M.; Furfaro, A.L.; Colla, R.; De Ciucis, C.; Marinari, U.M.; Pronzato, M.A.; Traverso, N.; Domenicotti, C. Redox homeostasis and cellular antioxidant systems: Crucial players in cancer growth and therapy. Oxid. Med. Cell. Longev. 2016, 2016, 6235641. [Google Scholar] [CrossRef] [PubMed]
- Rojo de la Vega, M.; Chapman, E.; Zhang, D.D. NRF2 and the Hallmarks of Cancer. Cancer Cell 2018, 34, 21–43. [Google Scholar] [CrossRef]
- Wu, S.; Lu, H.; Bai, Y. Nrf2 in cancers: A double-edged sword. Cancer Med. 2019, 8, 2252–2267. [Google Scholar] [CrossRef]
- Motohashi, H.; Yamamoto, M. Nrf2–Keap1 defines a physiologically important stress response mechanism. Trends Mol. Med. 2004, 10, 549–557. [Google Scholar] [CrossRef]
- Menegon, S.; Columbano, A.; Giordano, S. The Dual Roles of NRF2 in Cancer. Trends Mol. Med. 2016, 22, 578–593. [Google Scholar] [CrossRef]
- Osburn, W.O.; Karim, B.; Dolan, P.M.; Liu, G.; Yamamoto, M.; Huso, D.L.; Kensler, T.W. Increased colonic inflammatory injury and formation of aberrant crypt foci in Nrf2-deficient mice upon dextran sulfate treatment. Int. J. Cancer 2007, 121, 1883–1891. [Google Scholar] [CrossRef] [PubMed]
- Wakabayashi, N.; Dinkova-Kostova, A.T.; Holtzclaw, W.D.; Kang, M.I.; Kobayashi, A.; Yamamoto, M.; Kensler, T.W.; Talalay, P. Protection against electrophile and oxidant stress by induction of the phase 2 response: Fate of cysteines of the Keap1 sensor modified by inducers. Proc. Natl. Acad. Sci. USA 2004, 101, 2040–2045. [Google Scholar] [CrossRef] [PubMed]
- Rachakonda, G.; Sekhar, K.R.; Jowhar, D.; Samson, P.C.; Wikswo, J.P.; Beauchamp, R.D.; Datta, P.K.; Freeman, M.L. Increased cell migration and plasticity in Nrf2-deficient cancer cell lines. Oncogene 2010, 29, 3703–3714. [Google Scholar] [CrossRef]
- Yamamoto, T.; Yoh, K.; Kobayashi, A.; Ishii, Y.; Kure, S.; Koyama, A.; Sakamoto, T.; Sekizawa, K.; Motohashi, H.; Yamamoto, M. Identification of polymorphisms in the promoter region of the human NRF2 gene. Biochem. Biophys. Res. Commun. 2004, 321, 72–79. [Google Scholar] [CrossRef]
- Suzuki, T.; Shibata, T.; Takaya, K.; Shiraishi, K.; Kohno, T.; Kunitoh, H.; Tsuta, K.; Furuta, K.; Goto, K.; Hosoda, F.; et al. Regulatory Nexus of Synthesis and Degradation Deciphers Cellular Nrf2 Expression Levels. Mol. Cell. Biol. 2013, 33, 2402–2412. [Google Scholar] [CrossRef] [PubMed]
- Panieri, E.; Saso, L. Potential applications of NRF2 inhibitors in cancer therapy. Oxid. Med. Cell. Longev. 2019, 2019. [Google Scholar] [CrossRef]
- Panieri, E.; Telkoparan-Akillilar, P.; Suzen, S.; Saso, L. The nrf2/keap1 axis in the regulation of tumor metabolism: Mechanisms and therapeutic perspectives. Biomolecules 2020, 10, 791. [Google Scholar] [CrossRef]
- Liu, Y.; Lang, F.; Yang, C. NRF2 in human neoplasm: Cancer biology and potential therapeutic target. Pharmacol. Ther. 2020, 107664. [Google Scholar] [CrossRef] [PubMed]
- Jung, B.J.; Yoo, H.S.; Shin, S.; Park, Y.J.; Jeon, S.M. Dysregulation of NRF2 in cancer: From molecular mechanisms to therapeutic opportunities. Biomol. Ther. 2018, 26, 57–68. [Google Scholar] [CrossRef]
- Leinonen, H.M.; Kansanen, E.; Pölönen, P.; Heinäniemi, M.; Levonen, A.L. Role of the keap1-Nrf2 Pathway in Cancer. Adv. Cancer Res. 2014, 122, 281–320. [Google Scholar] [PubMed]
- Moi, P.; Chan, K.; Asunis, I.; Cao, A.; Kan, Y.W. Isolation of NF-E2-related factor 2 (Nrf2), a NF-E2-like basic leucine zipper transcriptional activator that binds to the tandem NF-E2/AP1 repeat of the β-globin locus control region. Proc. Natl. Acad. Sci. USA 1994, 91, 9926–9930. [Google Scholar] [CrossRef] [PubMed]
- Motohashi, H.; Katsuoka, F.; Engel, J.D.; Yamamoto, M. Small Maf proteins serve as transcriptional cofactors for keratinocyte differentiation in the Keap1-Nrf2 regulatory pathway. Proc. Natl. Acad. Sci. USA 2004, 101, 6379–6384. [Google Scholar] [CrossRef]
- Zhang, D.D.; Lo, S.; Cross, J.V.; Templeton, D.J.; Hannink, M. Keap1 is aredox-regulated substrate adaptor protein for a Cul3-dependent ubiquitinligase complex. Mol. Cell. Biol. 2004, 24, 10941–10953. [Google Scholar] [CrossRef]
- Nioi, P.; Nguyen, T.; Sherratt, P.J.; Pickett, C.B. The Carboxy-Terminal Neh3 Domain of Nrf2 Is Required for Transcriptional Activation. Mol. Cell. Biol. 2005, 25, 10895–10906. [Google Scholar] [CrossRef]
- Katoh, Y.; Itoh, K.; Yoshida, E.; Miyagishi, M.; Fukamizu, A.; Yamamoto, M. Two domains of Nrf2 cooperatively bind CBP, a CREB binding protein, and synergistically activate transcription. Genes Cells 2001, 6, 857–868. [Google Scholar] [CrossRef]
- Rada, P.; Rojo, A.I.; Chowdhry, S.; McMahon, M.; Hayes, J.D.; Cuadrado, A. SCF/ -TrCP Promotes Glycogen Synthase Kinase 3-Dependent Degradation of the Nrf2 Transcription Factor in a Keap1-Independent Manner. Mol. Cell. Biol. 2011, 31, 1121–1133. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Liu, K.; Geng, M.; Gao, P.; Wu, X.; Hai, Y.; Li, Y.; Li, Y.; Luo, L.; Hayes, J.D.; et al. RXRα inhibits the NRF2-ARE signaling pathway through a direct interaction with the Neh7 domain of NRF2. Cancer Res. 2013, 73, 3097–3108. [Google Scholar] [CrossRef] [PubMed]
- Magesh, S.; Chen, Y.; Hu, L. Small Molecule Modulators of Keap1-Nrf2-ARE Pathway as Potential Preventive and Therapeutic Agents. Med. Res. Rev. 2012, 32, 687–726. [Google Scholar] [CrossRef] [PubMed]
- Zipper, L.M.; Timothy Mulcahy, R. The Keap1 BTB/POZ dimerization function is required to sequester Nrf2 in cytoplasm. J. Biol. Chem. 2002, 277, 36544–36552. [Google Scholar] [CrossRef] [PubMed]
- Dinkova-Kostova, A.T.; Holtzclaw, W.D.; Cole, R.N.; Itoh, K.; Wakabayashi, N.; Katoh, Y.; Yamamoto, M.; Talalay, P. Direct evidence that sulfhydryl groups of Keap1 are the sensors regulating induction of phase 2 enzymes that protect against carcinogens and oxidants. Proc. Natl. Acad. Sci. USA 2002, 99, 11908–11913. [Google Scholar] [CrossRef] [PubMed]
- Um, H.C.; Jang, J.H.; Kim, D.H.; Lee, C.; Surh, Y.J. Nitric oxide activates Nrf2 through S-nitrosylation of Keap1 in PC12 cells. Nitric Oxide 2011. [Google Scholar] [CrossRef]
- Xie, L.; Gu, Y.; Wen, M.; Zhao, S.; Wang, W.; Ma, Y.; Meng, G.; Han, Y.; Wang, Y.; Liu, G.; et al. Hydrogen sulfide induces Keap1 S-sulfhydration and suppresses diabetes-accelerated atherosclerosis via Nrf2 activation. Diabetes 2016. [Google Scholar] [CrossRef] [PubMed]
- Itoh, K.; Wakabayashi, N.; Katoh, Y.; Ishii, T.; Igarashi, K.; Engel, J.D.; Yamamoto, M. Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain. Genes Dev. 1999, 13, 76–86. [Google Scholar] [CrossRef] [PubMed]
- Itoh, K.; Mimura, J.; Yamamoto, M. Discovery of the negative regulator of Nrf2, keap1: A historical overview. Antioxid. Redox Signal. 2010, 13, 1665–1678. [Google Scholar] [CrossRef]
- Ramos-Gomez, M.; Dolan, P.M.; Itoh, K.; Yamamoto, M.; Kensler, T.W. Interactive effects of nrf2 genotype and oltipraz on benzo[a]pyrene-DNA adducts and tumor yield in mice. Carcinogenesis 2003, 24, 461–467. [Google Scholar] [CrossRef] [PubMed]
- Itoh, K.; Chiba, T.; Takahashi, S.; Ishii, T.; Igarashi, K.; Katoh, Y.; Oyake, T.; Hayashi, N.; Satoh, K.; Hatayama, I.; et al. An Nrf2/small Maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochem. Biophys. Res. Commun. 1997, 236, 313–322. [Google Scholar] [CrossRef]
- Kwak, M.K.; Egner, P.A.; Dolan, P.M.; Ramos-Gomez, M.; Groopman, J.D.; Itoh, K.; Yamamoto, M.; Kensler, T.W. Role of phase 2 enzyme induction in chemoprotection by dithiolethiones. Mutat. Res. Fundam. Mol. Mech. Mutagen. 2001, 480–481, 305–315. [Google Scholar] [CrossRef]
- Hye-Youn Cho, A.E.J. Role of NRF2 in Protection against Hyperoxic Lung Injury in Mice. Free Radic. Biol. Med. 2002, 26, 75–182. [Google Scholar]
- Wakabayashi, N.; Itoh, K.; Wakabayashi, J.; Motohashi, H.; Noda, S.; Takahashi, S.; Imakado, S.; Kotsuji, T.; Otsuka, F.; Roop, D.R.; et al. Keap1-null mutation leads to postnatal lethality due to constitutive Nrf2 activation. Nat. Genet. 2003, 35, 238–245. [Google Scholar] [CrossRef]
- Chien, M.H.; Lee, W.J.; Hsieh, F.K.; Li, C.F.; Cheng, T.Y.; Wang, M.Y.; Chen, J.S.; Chow, J.M.; Jan, Y.H.; Hsiao, M.; et al. Keap1-Nrf2 interaction suppresses cell motility in lung adenocarcinomas by targeting the S100P protein. Clin. Cancer Res. 2015, 21, 4719–4732. [Google Scholar] [CrossRef]
- Mitsuishi, Y.; Taguchi, K.; Kawatani, Y.; Shibata, T.; Nukiwa, T.; Aburatani, H.; Yamamoto, M.; Motohashi, H. Nrf2 Redirects Glucose and Glutamine into Anabolic Pathways in Metabolic Reprogramming. Cancer Cell 2012, 22, 66–79. [Google Scholar] [CrossRef]
- You, A.; Nam, C.W.; Wakabayashi, N.; Yamamoto, M.; Kensler, T.W.; Kwak, M.K. Transcription factor Nrf2 maintains the basal expression of Mdm2: An implication of the regulation of p53 signaling by Nrf2. Arch. Biochem. Biophys. 2011, 507, 356–364. [Google Scholar] [CrossRef]
- Shim, G.S.; Manandhar, S.; Shin, D.H.; Kim, T.H.; Kwak, M.K. Acquisition of doxorubicin resistance in ovarian carcinoma cells accompanies activation of the NRF2 pathway. Free Radic. Biol. Med. 2009, 47, 1619–1631. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.J.; Sun, Z.; Villeneuve, N.F.; Zhang, S.; Zhao, F.; Li, Y.; Chen, W.; Yi, X.; Zheng, W.; Wondrak, G.T.; et al. Nrf2 enhances resistance of cancer cells to chemotherapeutic drugs, the dark side of Nrf2. Carcinogenesis 2008, 29, 1235–1243. [Google Scholar] [CrossRef]
- Yoshino, H.; Murakami, K.; Nawamaki, M.; Kashiwakura, I. Effects of Nrf2 knockdown on the properties of irradiated cell conditioned medium from A549 human lung cancer cells. Biomed. Reports 2018, 8, 461–465. [Google Scholar] [CrossRef]
- Singh, A.; Misra, V.; Thimmulappa, R.K.; Lee, H.; Ames, S.; Hoque, M.O.; Herman, J.G.; Baylin, S.B.; Sidransky, D.; Gabrielson, E.; et al. Dysfunctional KEAP1-NRF2 interaction in non-small-cell lung cancer. PLoS Med. 2006, 3, 1865–1876. [Google Scholar] [CrossRef] [PubMed]
- Lignitto, L.; LeBoeuf, S.E.; Homer, H.; Jiang, S.; Askenazi, M.; Karakousi, T.R.; Pass, H.I.; Bhutkar, A.J.; Tsirigos, A.; Ueberheide, B.; et al. Nrf2 Activation Promotes Lung Cancer Metastasis by Inhibiting the Degradation of Bach1. Cell 2019. [Google Scholar] [CrossRef]
- Zhang, C.; Wang, H.J.; Bao, Q.C.; Wang, L.; Guo, T.K.; Chen, W.L.; Xu, L.L.; Zhou, H.S.; Bian, J.L.; Yang, Y.R.; et al. NRF2 promotes breast cancer cell proliferation and metastasis by increasing RhoA/ROCK pathway signal transduction. Oncotarget 2016, 7, 73593–73606. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Zhang, C.; Zhang, L.; Yang, Q.; Zhou, S.; Wen, Q.; Wang, J. Nrf2 is a potential prognostic marker and promotes proliferation and invasion in human hepatocellular carcinoma. BMC Cancer 2015. [Google Scholar] [CrossRef]
- Arfmann-Knubel, S.; Struck, B.; Genrich, G.; Helm, O.; Sipos, B.; Sebens, S.; Schafer, H. The Crosstalk between Nrf2 and TGF-β1 in the Epithelial-Mesenchymal Transition of Pancreatic Duct Epithelial Cells. PLoS ONE 2015, 10, e0132978. [Google Scholar] [CrossRef]
- Zhao, Q.; Mao, A.; Guo, R.; Zhang, L.; Yan, J.; Sun, C.; Tang, J.; Ye, Y.; Zhang, Y.; Zhang, H. Suppression of radiation-induced migration of non-small cell lung cancer through inhibition of Nrf2-Notch Axis. Oncotarget 2017, 8, 36603. [Google Scholar] [CrossRef] [PubMed]
- Yoo, N.J.; Kim, H.R.; Kim, Y.R.; An, C.H.; Lee, S.H. Somatic mutations of the KEAP1 gene in common solid cancers. Histopathology 2012, 60, 943–952. [Google Scholar] [CrossRef]
- Konstantinopoulos, P.A.; Spentzos, D.; Fountzilas, E.; Francoeur, N.; Sanisetty, S.; Grammatikos, A.P.; Hecht, J.L.; Cannistra, S.A. Keap1 mutations and Nrf2 pathway activation in epithelial ovarian cancer. Cancer Res. 2011, 71, 5081–5089. [Google Scholar] [CrossRef]
- Shibata, T.; Kokubu, A.; Gotoh, M.; Ojima, H.; Ohta, T.; Yamamoto, M.; Hirohashi, S. Genetic Alteration of Keap1 Confers Constitutive Nrf2 Activation and Resistance to Chemotherapy in Gallbladder Cancer. Gastroenterology 2008, 135, 1358–1368. [Google Scholar] [CrossRef]
- Padmanabhan, B.; Tong, K.I.; Ohta, T.; Nakamura, Y.; Scharlock, M.; Ohtsuji, M.; Kang, M.I.; Kobayashi, A.; Yokoyama, S.; Yamamoto, M. Structural basis for defects of Keap1 activity provoked by its point mutations in lung cancer. Mol. Cell 2006, 21, 689–700. [Google Scholar] [CrossRef]
- Ohta, T.; Iijima, K.; Miyamoto, M.; Nakahara, I.; Tanaka, H.; Ohtsuji, M.; Suzuki, T.; Kobayashi, A.; Yokota, J.; Sakiyama, T.; et al. Loss of Keap1 function activates Nrf2 and provides advantages for lung cancer cell growth. Cancer Res. 2008, 68, 1303–1309. [Google Scholar] [CrossRef]
- Gañán-Gómez, I.; Wei, Y.; Yang, H.; Boyano-Adánez, M.C.; García-Manero, G. Oncogenic functions of the transcription factor Nrf2. Free Radic. Biol. Med. 2013, 65, 750–764. [Google Scholar] [CrossRef]
- Panieri, E.; Buha, A.; Telkoparan-akillilar, P.; Cevik, D.; Kouretas, D.; Veskoukis, A.; Skaperda, Z.; Tsatsakis, A.; Wallace, D.; Suzen, S.; et al. Potential applications of NRF2 modulators in cancer therapy. Antioxidants 2020, 9, 193. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, L.D.; Lee, J.; Gnad, F.; Klijn, C.; Schaub, A.; Reeder, J.; Daemen, A.; Bakalarski, C.E.; Holcomb, T.; Shames, D.S.; et al. Recurrent Loss of NFE2L2 Exon 2 Is a Mechanism for Nrf2 Pathway Activation in Human Cancers. Cell Rep. 2016, 16, 2605–2617. [Google Scholar] [CrossRef] [PubMed]
- Lebovitz, C.B.; Robertson, A.G.; Goya, R.; Jones, S.J.; Morin, R.D.; Marra, M.A.; Gorski, S.M. Cross-cancer profiling of molecular alterations within the human autophagy interaction network. Autophagy 2015, 11, 1668–1687. [Google Scholar] [CrossRef]
- Tian, H.; Zhang, B.F.; Di, J.H.; Jiang, G.; Chen, F.F.; Li, H.Z.; Li, L.T.; Pei, D.S.; Zheng, J.N. Keap1: One stone kills three birds Nrf2, IKKβ and Bcl-2/Bcl-xL. Cancer Lett. 2012, 325, 26–34. [Google Scholar] [CrossRef] [PubMed]
- Derks, J.L.; Leblay, N.; Lantuejoul, S.; Dingemans, A.M.C.; Speel, E.J.M.; Fernandez-Cuesta, L. New Insights into the Molecular Characteristics of Pulmonary Carcinoids and Large Cell Neuroendocrine Carcinomas, and the Impact on Their Clinical Management. J. Thorac. Oncol. 2018, 13, 752–766. [Google Scholar] [CrossRef]
- Fernandez-Cuesta, L.; Peifer, M.; Lu, X.; Sun, R.; Ozretić, L.; Danila, S.; Zander, T.; Leenders, F.; George, J.; Müller, C.; et al. Frequent mutations in chromatin-remodeling genes in pulmonary carcinoids. Nat. Commun. 2014, 5, 3518. [Google Scholar] [CrossRef]
- Muscarella, L.A.; Parrella, P.; D’Alessandro, V.; la Torre, A.; Barbano, R.; Fontana, A.; Tancredi, A.; Guarnieri, V.; Balsamo, T.; Coco, M.; et al. Frequent epigenetics inactivation of KEAP1 gene in non-small cell lung cancer. Epigenetics 2011, 6, 710–719. [Google Scholar] [CrossRef]
- Hanada, N.; Takahata, T.; Zhou, Q.; Ye, X.; Sun, R.; Itoh, J.; Ishiguro, A.; Kijima, H.; Mimura, J.; Itoh, K.; et al. Methylation of the KEAP1 gene promoter region in human colorectal cancer. BMC Cancer 2012, 12, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Singh, A.; Yegnasubramanian, S.; Esopi, D.; Bodas, M.; Wu, H.; Bova, G.S.; Biswal, S. Loss of keap 1 Function in prostate Cancer Cells Causes Chemo- and Radio-resistance and Promotes Tumor Growth. Mol. Cancer Ther. 2011, 9, 1–18. [Google Scholar]
- Muscarella, L.A.; Barbano, R.; D’Angelo, V.; Copetti, M.; Coco, M.; Balsamo, T.; la Torre, A.; Notarangelo, A.; Troiano, M.; Parisi, S.; et al. Regulation of KEAP1 expression by promoter methylation in malignant gliomas and association with patient’s outcome. Epigenetics 2011, 6, 317–325. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.Q.; Zhang, Y.F.; Xia, Y.F.; Zhou, Z.M.; Cao, Y.Q. Promoter demethylation of nuclear factor-erythroid 2-related factor 2 gene in drug-resistant colon cancer cells. Oncol. Lett. 2015, 10, 1287–1292. [Google Scholar] [CrossRef] [PubMed]
- Esquela-Kerscher, A.; Slack, F.J. Oncomirs—MicroRNAs with a role in cancer. Nat. Rev. Cancer 2006, 6, 259–269. [Google Scholar] [CrossRef]
- Yamamoto, S.; Inoue, J.; Kawano, T.; Kozaki, K.I.; Omura, K.; Inazawa, J. The impact of miRNA-based molecular diagnostics and treatment of NRF2-stabilized tumors. Mol. Cancer Res. 2014, 12, 58–68. [Google Scholar] [CrossRef] [PubMed]
- Zimta, A.A.; Cenariu, D.; Irimie, A.; Magdo, L.; Nabavi, S.M.; Atanasov, A.G.; Berindan-Neagoe, I. The role of Nrf2 activity in cancer development and progression. Cancers 2019, 11, 1755. [Google Scholar] [CrossRef]
- Shi, L.; Wu, L.; Chen, Z.; Yang, J.; Chen, X.; Yu, F.; Zheng, F.; Lin, X. MiR-141 activates Nrf2-dependent antioxidant pathway via down-regulating the expression of keap1 conferring the resistance of hepatocellular carcinoma cells to 5-fluorouracil. Cell. Physiol. Biochem. 2015, 35, 2333–2348. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.; Zhang, Y.; Luo, Y.; Wang, Z.; Bi, S.; Song, D.; Dai, Y.; Wang, T.; Qiu, L.; Wen, L.; et al. Aldose reductase regulates miR-200a-3p/141-3p to coordinate Keap1-Nrf2, Tgfβ1/2, and Zeb1/2 signaling in renal mesangial cells and the renal cortex of diabetic mice. Free Radic. Biol. Med. 2014, 67, 91–102. [Google Scholar] [CrossRef]
- Copple, I.M.; Lister, A.; Obeng, A.D.; Kitteringham, N.R.; Jenkins, R.E.; Layfield, R.; Foster, B.J.; Goldring, C.E.; Park, B.K. Physical and functional interaction of sequestosome 1 with Keap1 regulates the Keap1-Nrf2 cell defense pathway. J. Biol. Chem. 2010, 285, 16782–16788. [Google Scholar] [CrossRef]
- Jain, A.; Lamark, T.; Sjøttem, E.; Larsen, K.B.; Awuh, J.A.; Øvervatn, A.; McMahon, M.; Hayes, J.D.; Johansen, T. p62/SQSTM1 is a target gene for transcription factor NRF2 and creates a positive feedback loop by inducing antioxidant response element-driven gene transcription. J. Biol. Chem. 2010, 285, 22576–22591. [Google Scholar] [CrossRef] [PubMed]
- Komatsu, M.; Kurokawa, H.; Waguri, S.; Taguchi, K.; Kobayashi, A.; Ichimura, Y.; Sou, Y.S.; Ueno, I.; Sakamoto, A.; Tong, K.I.; et al. The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat. Cell Biol. 2010, 12, 213–223. [Google Scholar] [CrossRef]
- Lau, A.; Wang, X.-J.; Zhao, F.; Villeneuve, N.F.; Wu, T.; Jiang, T.; Sun, Z.; White, E.; Zhang, D.D. A Noncanonical Mechanism of Nrf2 Activation by Autophagy Deficiency: Direct Interaction between Keap1 and p62. Mol. Cell. Biol. 2010, 30, 3275–3285. [Google Scholar] [CrossRef] [PubMed]
- Genschik, P.; Sumara, I.; Lechner, E. The emerging family of CULLIN3-RING ubiquitin ligases (CRL3s): Cellular functions and disease implications. EMBO J. 2013, 32, 2307–2320. [Google Scholar] [CrossRef]
- Inami, Y.; Waguri, S.; Sakamoto, A.; Kouno, T.; Nakada, K.; Hino, O.; Watanabe, S.; Ando, J.; Iwadate, M.; Yamamoto, M.; et al. Persistent activation of Nrf2 through p62 in hepatocellular carcinoma cells. J. Cell Biol. 2011, 193, 275–284. [Google Scholar] [CrossRef] [PubMed]
- Umemura, A.; He, F.; Taniguchi, K.; Nakagawa, H.; Yamachika, S.; Font-Burgada, J.; Zhong, Z.; Subramaniam, S.; Raghunandan, S.; Duran, A.; et al. p62, Upregulated during Preneoplasia, Induces Hepatocellular Carcinogenesis by Maintaining Survival of Stressed HCC-Initiating Cells. Cancer Cell 2016, 29, 935–948. [Google Scholar] [CrossRef]
- Chen, W.; Sun, Z.; Wang, X.J.; Jiang, T.; Huang, Z.; Fang, D.; Zhang, D.D. Direct Interaction between Nrf2 and p21Cip1/WAF1 Upregulates the Nrf2-Mediated Antioxidant Response. Mol. Cell 2009, 34, 663–673. [Google Scholar] [CrossRef]
- Camp, N.D.; James, R.G.; Dawson, D.W.; Yan, F.; Davison, J.M.; Houck, S.A.; Tang, X.; Zheng, N.; Major, M.B.; Moon, R.T. Wilms Tumor Gene on X Chromosome (WTX) Inhibits Degradation of NRF2 Protein through Competitive Binding to KEAP1 Protein. J. Biol. Chem. 2012, 287, 6539–6550. [Google Scholar] [CrossRef]
- Ma, J.; Cai, H.; Wu, T.; Sobhian, B.; Huo, Y.; Alcivar, A.; Mehta, M.; Cheung, K.L.; Ganesan, S.; Kong, A.-N.T.; et al. PALB2 Interacts with KEAP1 To Promote NRF2 Nuclear Accumulation and Function. Mol. Cell. Biol. 2012, 32, 1506–1517. [Google Scholar] [CrossRef] [PubMed]
- Lu, K.; Alcivar, A.L.; Ma, J.; Foo, T.K.; Zywea, S.; Mahdi, A.; Huo, Y.; Kensler, T.W.; Gatza, M.L.; Xia, B. NRF2 induction supporting breast cancer cell survival is enabled by oxidative stress-induced DPP3-KEAP1 interaction. Cancer Res. 2017, 77, 2881–2892. [Google Scholar] [CrossRef]
- Denicola, G.M.; Karreth, F.A.; Humpton, T.J.; Gopinathan, A.; Wei, C.; Frese, K.; Mangal, D.; Yu, K.H.; Yeo, C.J.; Calhoun, E.S.; et al. Oncogene-induced Nrf2 transcription promotes ROS detoxification and tumorigenesis. Nature 2011, 475, 106–110. [Google Scholar] [CrossRef] [PubMed]
- Rojo, A.I.; Rada, P.; Mendiola, M.; Ortega-Molina, A.; Wojdyla, K.; Rogowska-Wrzesinska, A.; Hardisson, D.; Serrano, M.; Cuadrado, A. The PTEN/NRF2 axis promotes human carcinogenesis. Antioxid. Redox Signal. 2014, 21, 2498–2514. [Google Scholar] [CrossRef]
- Tao, S.; Wang, S.; Moghaddam, S.J.; Ooi, A.; Chapman, E.; Wong, P.K.; Zhang, D.D. Oncogenic KRAS Confers Chemoresistance by Upregulating NRF2. Cancer Res. 2014, 74, 7430–7441. [Google Scholar] [CrossRef] [PubMed]
- Chowdhry, S.; Zhang, Y.; Mcmahon, M. Nrf2 is controlled by two distinct β -TrCP recognition motifs in its Neh6 domain, one of which can be modulated by GSK-3 activity. Oncogene 2014, 32, 3765–3781. [Google Scholar] [CrossRef]
- Liao, H.; Zhou, Q.; Zhang, Z.; Wang, Q.; Sun, Y.; Yi, X.; Feng, Y. NRF2 is overexpressed in ovarian epithelial carcinoma and is regulated by gonadotrophin and sex-steroid hormones. Oncol. Rep. 2012, 27, 1918–1924. [Google Scholar]
- Zhang, Z.; Wang, Q.; Ma, J.; Yi, X.; Zhu, Y.; Xi, X.; Feng, Y.; Jin, Z. AntiReactive oxygen species regulate FSH-induced expression of vascular endothelial growth factor via Nrf2 and HIF1a signaling in human epithelial ovarian cancer. Oncol. Rep. 2013, 29, 1429–1434. [Google Scholar] [CrossRef] [PubMed]
- Tong, K.I.; Katoh, Y.; Kusunoki, H.; Itoh, K.; Tanaka, T.; Yamamoto, M. Keap1 Recruits Neh2 through Binding to ETGE and DLG Motifs: Characterization of the Two-Site Molecular Recognition Model. Mol. Cell. Biol. 2006, 26, 2887–2900. [Google Scholar] [CrossRef]
- Kobayashi, A.; Kang, M.-I.; Watai, Y.; Tong, K.I.; Shibata, T.; Uchida, K.; Yamamoto, M. Oxidative and Electrophilic Stresses Activate Nrf2 through Inhibition of Ubiquitination Activity of Keap1. Mol. Cell. Biol. 2006, 26, 221–229. [Google Scholar] [CrossRef] [PubMed]
- Jasinski, P.; Zwolak, P.; Terai, K.; Vogel, R.I.; Borja-Cacho, D.; Dudek, A.Z. MT477 acts in tumor cells as an AURKA inhibitor and strongly induces NRF-2 signaling. Anticancer Res. 2011, 31, 1181–1187. [Google Scholar]
- Kuang, Y.; Sechi, M.; Nurra, S.; Ljungman, M.; Neamati, N. Design and Synthesis of Novel Reactive Oxygen Species Inducers for the Treatment of Pancreatic Ductal Adenocarcinoma. J. Med. Chem. 2018, 61, 1576–1594. [Google Scholar] [CrossRef]
- Qin, J.J.; Cheng, X.D.; Zhang, J.; Zhang, W.D. Dual roles and therapeutic potential of Keap1-Nrf2 pathway in pancreatic cancer: A systematic review. Cell Commun. Signal. 2019, 17, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Kong, B.; Qia, C.; Erkan, M.; Kleeff, J.; Michalski, C.W. Overview on how oncogenic Kras promotes pancreatic carcinogenesis by inducing low intracellular ROS levels. Front. Physiol. 2013, 4, 246. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Jiang, Z.; Zhou, C.; Chen, K.; Li, X.; Wang, Z.; Wu, Z.; Ma, J.; Ma, Q.; Duan, W. Activation of Nrf2 by Sulforaphane Inhibits High Glucose-Induced Progression of Pancreatic Cancer via AMPK Dependent Signaling. Cell. Physiol. Biochem. 2018, 50, 1201–1215. [Google Scholar] [CrossRef] [PubMed]
- Tong, Y.-H.; Zhang, B.; Fan, Y.; Lin, N.-M. Keap1–Nrf2 pathway: A promising target towards lung cancer prevention and therapeutics. Chronic Dis. Transl. Med. 2015, 1, 175–186. [Google Scholar] [CrossRef]
- Tsai, T.F.; Chen, P.C.; Lin, Y.C.; Chou, K.Y.; Chen, H.E.; Ho, C.Y.; Lin, J.F.; Hwang, T.I.S. Miconazole contributes to NRF2 activation by noncanonical P62-keap1 pathway in bladder cancer cells. Drug Des. Dev. Ther. 2020, 14, 1209. [Google Scholar] [CrossRef]
- Saddawi-Konefka, R.; Seelige, R.; Gross, E.T.E.; Levy, E.; Searles, S.C.; Washington, A.; Santosa, E.K.; Liu, B.; O’Sullivan, T.E.; Harismendy, O.; et al. Nrf2 Induces IL-17D to Mediate Tumor and Virus Surveillance. Cell Rep. 2016, 16, 2348–2358. [Google Scholar] [CrossRef]
- Cooke, M.S.; Evans, M.D.; Dizdaroglu, M.; Lunec, J. Oxidative DNA damage: Mechanisms, mutation, and disease. FASEB J. 2003, 17, 1195–1214. [Google Scholar] [CrossRef]
- Tao, S.; Park, S.L.; De La Vega, M.R.; Zhang, D.D.; Wondrak, G.T. Systemic administration of the apocarotenoid bixin protects skin against solar UV-induced damage through activation of NRF2. Free Radic. Biol. Med. 2015, 89, 690–700. [Google Scholar] [CrossRef] [PubMed]
- Al-Sawaf, O.; Clarner, T.; Fragoulis, A.; Kan, Y.W.; Pufe, T.; Streetz, K.; Wruck, C.J. Nrf2 in health and disease: Current and future clinical implications. Clin. Sci. 2015, 129, 989–999. [Google Scholar] [CrossRef]
- Panieri, E.; Saso, L. Inhibition of the NRF2/KEAP1 Axis: A Promising Therapeutic Strategy to Alter Redox Balance of Cancer Cells. Antioxid. Redox Signal. 2021. [Google Scholar] [CrossRef]
- Zarei, M.; Du, H.; Nassar, A.H.; Yan, R.E.; Giannikou, K.; Johnson, S.H.; Lam, H.C.; Henske, E.P.; Wang, Y.; Zhang, T.; et al. Tumors with TSC mutations are sensitive to CDK7 inhibition through NRF2 and glutathione depletion. J. Exp. Med. 2019, 216, 2635–2652. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.-S.; Lee, J.; Nam, L.B.; Yoo, O.-K.; Pham, K.-T.; Duong, T.-H.-M.; Keum, Y.-S. Homoharringtonine stabilizes secondary structure of guanine-rich sequence existing in the 5′-untranslated region of Nrf2. Bioorg. Med. Chem. Lett. 2019, 29, 2189–2196. [Google Scholar] [CrossRef]
- Gao, A.-M.; Zhang, X.-Y.; Ke, Z.-P. Apigenin sensitizes BEL-7402/ADM cells to doxorubicin through inhibiting miR-101/Nrf2 pathway. Oncotarget 2017, 8, 82085–82091. [Google Scholar] [CrossRef] [PubMed]
- Gambardella, V.; Gimeno-Valiente, F.; Tarazona, N.; Ciarpaglini, C.M.; Roda, D.; Fleitas, T.; Tolosa, P.; Cejalvo, J.M.; Huerta, M.; Roselló, S.; et al. NRF2 through RPS6 Activation Is Related to Anti-HER2 Drug Resistance in HER2 -Amplified Gastric Cancer. Clin. Cancer Res. 2019, 25, 1639–1649. [Google Scholar] [CrossRef] [PubMed]
- El-Naggar, A.M.; Somasekharan, S.P.; Wang, Y.; Cheng, H.; Negri, G.L.; Pan, M.; Wang, X.Q.; Delaidelli, A.; Rafn, B.; Cran, J.; et al. Class I HDAC inhibitors enhance YB-1 acetylation and oxidative stress to block sarcoma metastasis. EMBO Rep. 2019, 20, e48375. [Google Scholar] [CrossRef]
- Xiang, Y.; Ye, W.; Huang, C.; Yu, D.; Chen, H.; Deng, T.; Zhang, F.; Lou, B.; Zhang, J.; Shi, K.; et al. Brusatol enhances the chemotherapy efficacy of gemcitabine in pancreatic cancer via the Nrf2 signalling pathway. Oxid. Med. Cell. Longev. 2018, 2018, 2360427. [Google Scholar] [CrossRef]
- Evans, J.P.; Winiarski, B.K.; Sutton, P.A.; Jones, R.P.; Ressel, L.; Duckworth, C.A.; Pritchard, D.M.; Lin, Z.-X.; Fretwell, V.L.; Tweedle, E.M.; et al. The Nrf2 inhibitor brusatol is a potent antitumour agent in an orthotopic mouse model of colorectal cancer. Oncotarget 2018, 9, 27104–27116. [Google Scholar] [CrossRef]
- Karathedath, S.; Rajamani, B.M.; Musheer Aalam, S.M.; Abraham, A.; Varatharajan, S.; Krishnamurthy, P.; Mathews, V.; Velayudhan, S.R.; Balasubramanian, P. Role of NF-E2 related factor 2 (Nrf2) on chemotherapy resistance in acute myeloid leukemia (AML) and the effect of pharmacological inhibition of Nrf2. PLoS ONE 2017, 12, e0177227. [Google Scholar] [CrossRef] [PubMed]
- Tsuchida, K.; Tsujita, T.; Hayashi, M.; Ojima, A.; Keleku-Lukwete, N.; Katsuoka, F.; Otsuki, A.; Kikuchi, H.; Oshima, Y.; Suzuki, M.; et al. Halofuginone enhances the chemo-sensitivity of cancer cells by suppressing NRF2 accumulation. Free Radic. Biol. Med. 2017, 103, 236–247. [Google Scholar] [CrossRef] [PubMed]
- Duong, H.Q.; Yi, Y.W.; Kang, H.J.; Hong, Y.B.; Tang, W.; Wang, A.; Seong, Y.S.; Bae, I. Inhibition of NRF2 by PIK-75 augments sensitivity of pancreatic cancer cells to gemcitabine. Int. J. Oncol. 2014, 44, 959–969. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.X.T.; Troadec, E.; Kalvala, A.; Kumar, B.; Hoang, D.H.; Viola, D.; Zhang, B.; Nguyen, D.Q.; Aldoss, I.; Ghoda, L.; et al. The Bcl-2 inhibitor venetoclax inhibits Nrf2 antioxidant pathway activation induced by hypomethylating agents in AML. J. Cell. Physiol. 2019, 234, 14040–14049. [Google Scholar] [CrossRef]
- Lee, J.; Kang, J.-S.; Nam, L.B.; Yoo, O.-K.; Keum, Y.-S. Suppression of NRF2/ARE by convallatoxin sensitises A549 cells to 5-FU-mediated apoptosis. Free Radic. Res. 2018, 52, 1416–1423. [Google Scholar] [CrossRef]
- Arlt, A.; Sebens, S.; Krebs, S.; Geismann, C.; Grossmann, M.; Kruse, M.-L.; Schreiber, S.; Schäfer, H. Inhibition of the Nrf2 transcription factor by the alkaloid trigonelline renders pancreatic cancer cells more susceptible to apoptosis through decreased proteasomal gene expression and proteasome activity. Oncogene 2013, 32, 4825–4835. [Google Scholar] [CrossRef]
- Tang, X.; Wang, H.; Fan, L.; Wu, X.; Xin, A.; Ren, H.; Wang, X.J. Luteolin inhibits Nrf2 leading to negative regulation of the Nrf2/ARE pathway and sensitization of human lung carcinoma A549 cells to therapeutic drugs. Free Radic. Biol. Med. 2011, 50, 1599–1609. [Google Scholar] [CrossRef]
- Yang, H.; Liu, B.; Xie, F.; Yang, W.; Cao, N. Luteolin induces mitochondrial apoptosis in HT29 cells by inhibiting the Nrf2/ARE signaling pathway. Exp. Ther. Med. 2020, 19, 2179–2187. [Google Scholar] [CrossRef]
- Singh, A.; Venkannagari, S.; Oh, K.H.; Zhang, Y.-Q.; Rohde, J.M.; Liu, L.; Nimmagadda, S.; Sudini, K.; Brimacombe, K.R.; Gajghate, S.; et al. Small Molecule Inhibitor of NRF2 Selectively Intervenes Therapeutic Resistance in KEAP1-Deficient NSCLC Tumors. ACS Chem. Biol. 2016, 11, 3214–3225. [Google Scholar] [CrossRef]
- Furfaro, A.L.; Piras, S.; Domenicotti, C.; Fenoglio, D.; De Luigi, A.; Salmona, M.; Moretta, L.; Marinari, U.M.; Pronzato, M.A.; Traverso, N.; et al. Role of Nrf2, HO-1 and GSH in neuroblastoma cell resistance to bortezomib. PLoS ONE 2016, 11, e0152465. [Google Scholar] [CrossRef]
- Valenzuela, M.; Glorieux, C.; Stockis, J.; Sid, B.; Sandoval, J.M.; Felipe, K.B.; Kviecinski, M.R.; Verrax, J.; Calderon, P.B. Retinoic acid synergizes ATO-mediated cytotoxicity by precluding Nrf2 activity in AML cells. Br. J. Cancer 2014, 111, 874–882. [Google Scholar] [CrossRef]
- Xiu, J.W.; Hayes, J.D.; Henderson, C.J.; Wolf, C.R. Identification of retinoic acid as an inhibitor of transcription factor Nrf2 through activation of retinoic acid receptor alpha. Proc. Natl. Acad. Sci. USA 2007, 104, 19589–19594. [Google Scholar] [CrossRef]
- Sova, M.; Saso, L. Design and development of Nrf2 modulators for cancer chemoprevention and therapy: A review. Drug Des. Dev. Ther. 2018, 12, 3181. [Google Scholar] [CrossRef] [PubMed]
- Schieber, M.; Chandel, N.S. ROS function in redox signaling and oxidative stress. Curr. Biol. 2014, 24, R453–R462. [Google Scholar] [CrossRef]
- Suzuki, M.; Otsuki, A.; Keleku-Lukwete, N.; Yamamoto, M. Overview of redox regulation by Keap1–Nrf2 system in toxicology and cancer. Curr. Opin. Toxicol. 2016, 1, 29–36. [Google Scholar] [CrossRef]
- Kensler, T.W.; Wakabayashi, N.; Biswal, S. Cell survival responses to environmental stresses via the Keap1-Nrf2-ARE pathway. Annu. Rev. Pharmacol. Toxicol. 2007, 47, 89–116. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Telkoparan-Akillilar, P.; Panieri, E.; Cevik, D.; Suzen, S.; Saso, L. Therapeutic Targeting of the NRF2 Signaling Pathway in Cancer. Molecules 2021, 26, 1417. https://doi.org/10.3390/molecules26051417
Telkoparan-Akillilar P, Panieri E, Cevik D, Suzen S, Saso L. Therapeutic Targeting of the NRF2 Signaling Pathway in Cancer. Molecules. 2021; 26(5):1417. https://doi.org/10.3390/molecules26051417
Chicago/Turabian StyleTelkoparan-Akillilar, Pelin, Emiliano Panieri, Dilek Cevik, Sibel Suzen, and Luciano Saso. 2021. "Therapeutic Targeting of the NRF2 Signaling Pathway in Cancer" Molecules 26, no. 5: 1417. https://doi.org/10.3390/molecules26051417
APA StyleTelkoparan-Akillilar, P., Panieri, E., Cevik, D., Suzen, S., & Saso, L. (2021). Therapeutic Targeting of the NRF2 Signaling Pathway in Cancer. Molecules, 26(5), 1417. https://doi.org/10.3390/molecules26051417