
A Systematic Study of the In Vitro Pharmacokinetics and Estimated Human In Vivo Clearance of Indole and Indazole-3-Carboxamide Synthetic Cannabinoid Receptor Agonists Detected on the Illicit Drug Market

Abstract
:1. Introduction
2. Results and Discussion
2.1. Lipophilicity
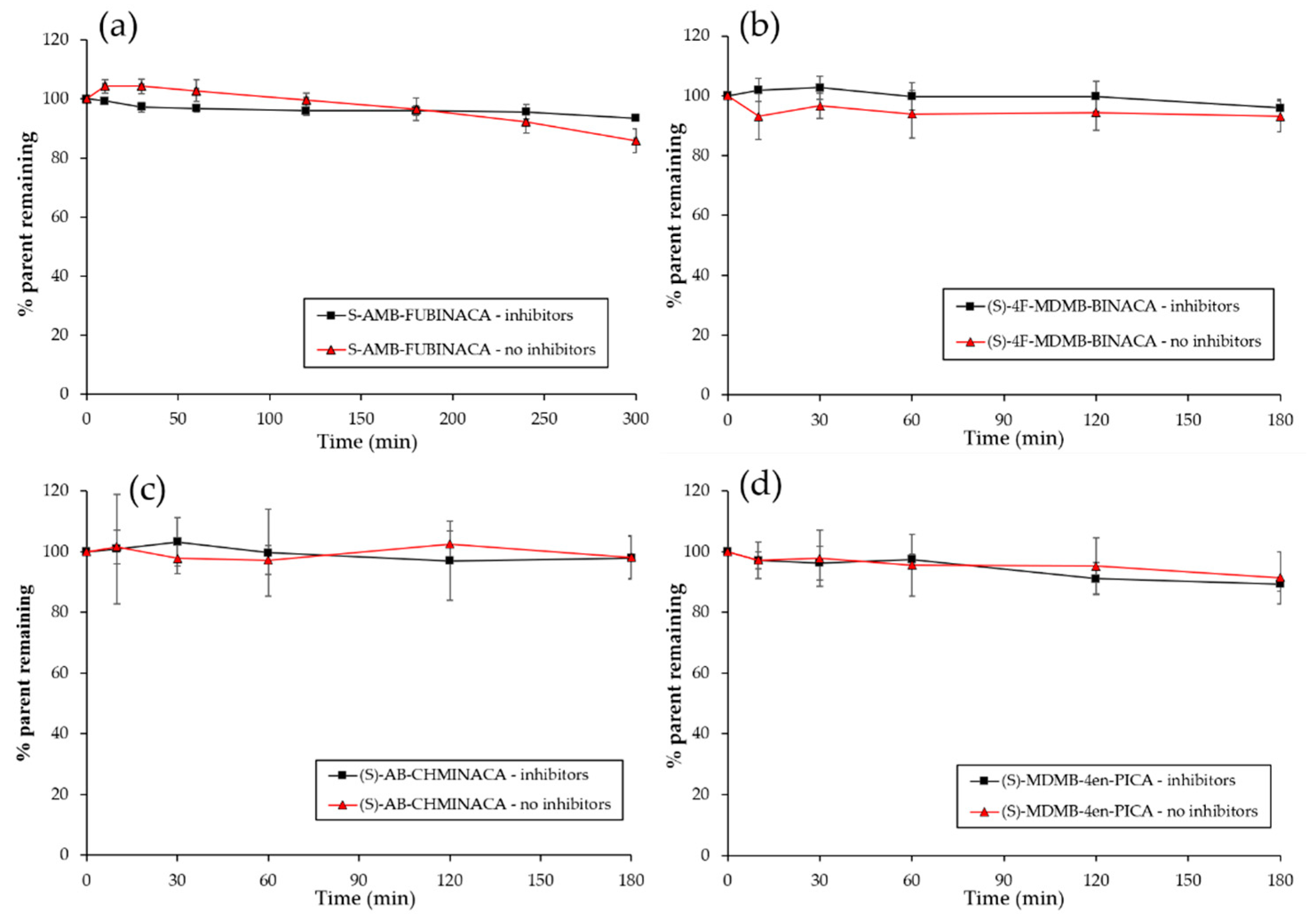
2.2. Plasma Stability Studies
2.3. Plasma Protein Binding (PPB)
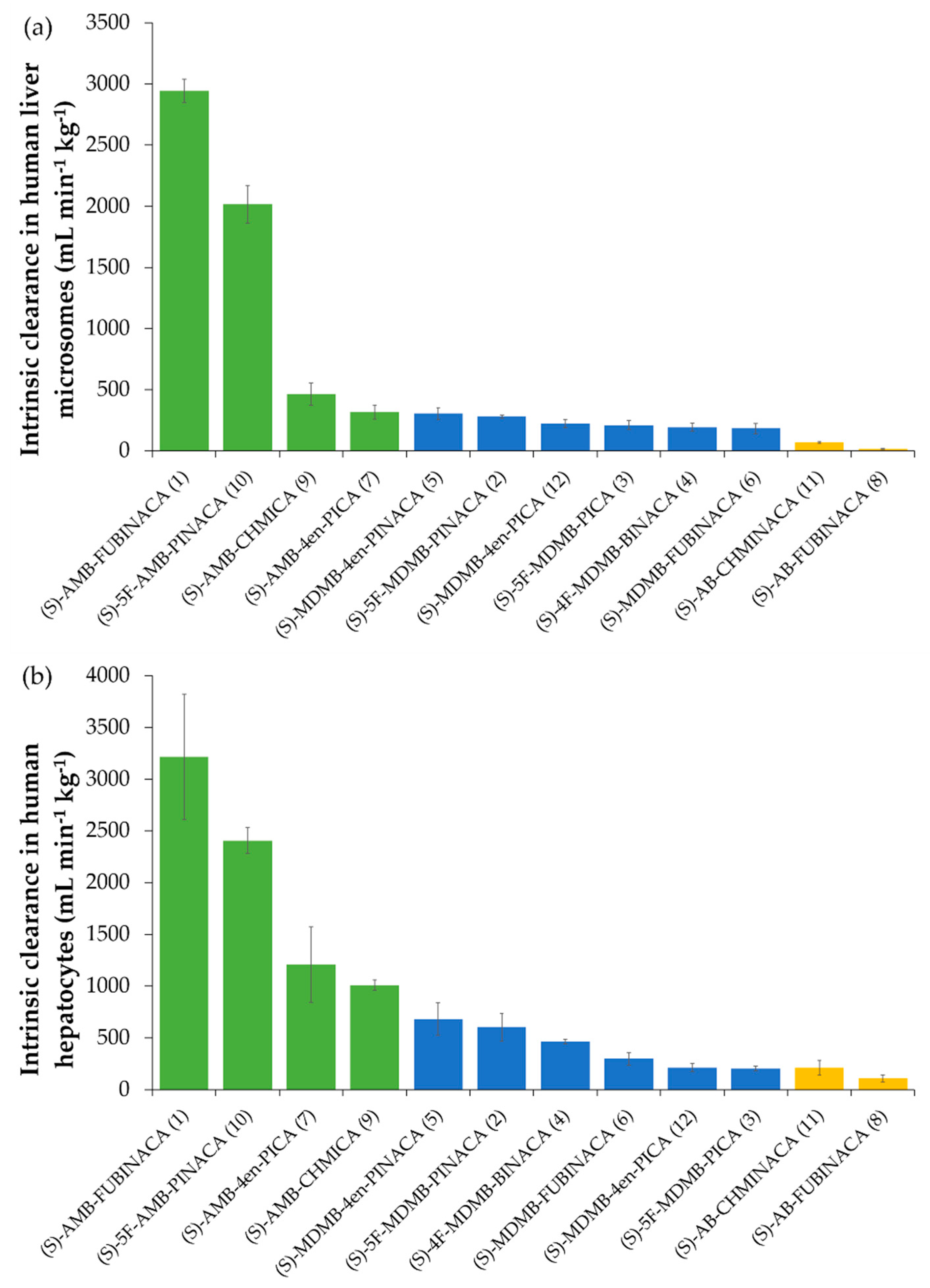
2.4. In Vitro Intrinsic Clearance
2.4.1. Comparison of SCRA (S)-Enantiomer Intrinsic Clearance Rates and Half-Lives
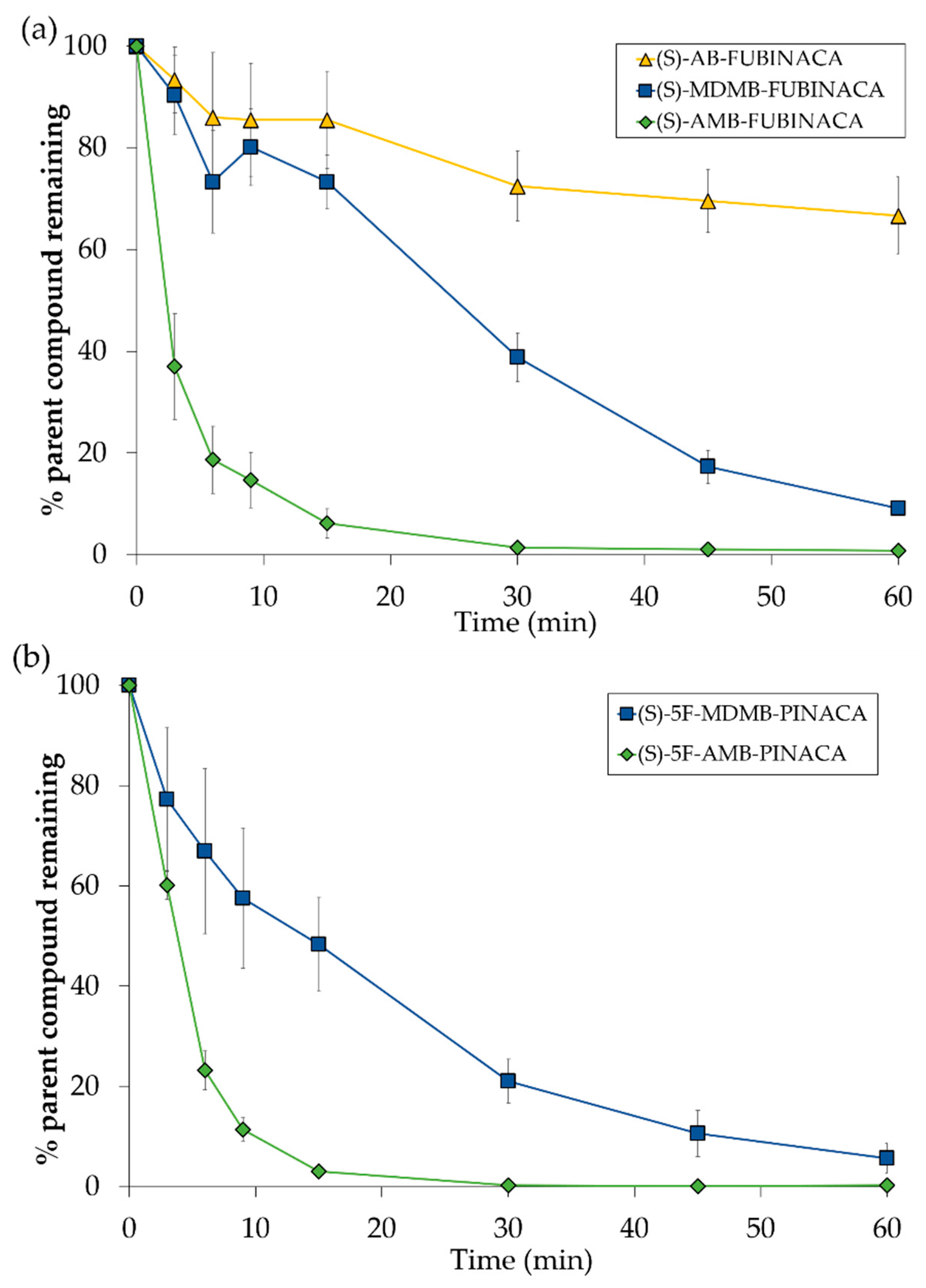
2.4.2. The Influence of Chirality on Intrinsic Clearance Rates and Half-Lives
2.4.3. Comparison of Intrinsic Clearance Calculated from pHLM and pHHeps
2.5. Prediction of Human In Vivo Clearance
2.6. Study Limitations

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | HLM/HHeps * | Reference | ||
|---|---|---|---|---|
| CLint micr (mL min−1 mg−1) | CLint (mL min−1 kg−1) | t1/2 (min) | ||
| JWH-122 * | - | 1350 | 0.95 | [13] |
| MAM-2201 * | - | 1408 | 0.88 | |
| Cl-JWH-122 * | - | 502 | 2.46 | |
| Br-JWH-122 * | - | 497 | 2.49 | |
| I-JWH-122 * | - | 235 | 5.26 | |
| AM1220 * | - | 169 | 3.7 | [105] |
| THJ-018 | 0.036 | 34.2 | 19.2 | [14] |
| THJ-2201 | 0.064 | 60.8 | 10.8 | |
| NNEI | 0.300 | 350 | 2.29 | [15] |
| MN-18 | 0.410 | 469 | 1.71 | |
| NM-2201 | 0.088 | 81.6 | 8.0 | [106] |
| BIM-2201 | 0.142 | 134.1 | 4.9 | [107] |
| Cumyl-PICA | 0.12 | - | 5.92 | [95] |
| 5F-Cumyl-PICA | 0.39 | - | 1.77 | |
| STS-135 | 0.222 | 209 | 3.1 | [108] |
| AMB-4en-PICA (7) | - | 291 | 2.1 | [57] |
| AB-PINACA | 0.037 | 35 | 18.7 | [16] |
| 5F-AB-PINACA | 0.019 | 18 | 35.9 | |
| AMB-PINACA | - | 1.1 | [73] | |
| 5F-AMB-PINACA (10) | 0.67 | - | 1.0 | |
| 5F-MDMB-PINACA (2) | 0.271 | 256.2 | 3.1 | [54] |
| 5F-APP-PICA | 0.046 | - | 15.1 | [109] |
| 5F-APP-PINACA | 0.202 | - | 3.4 | |
| APP-CHMINACA | 0.133 | - | 5.2 | |
| AB-FUBINACA (8) | 0.011 | 10.5 | 62.6 | [69] |
| ADB-FUBINACA | 0.018 | 16.5 | 39.7 | [17] |
| FDU-PB-22 | 0.056 | 52.7 | 12.4 | [97] |
| FUB-PB-22 | 0.060 | 57.1 | 11.5 | |
3. Materials and Methods
3.1. Chemicals and Reagents
3.2. Instrumentation
3.3. In Silico Log P Prediction
3.4. Experimental Log D7.4
3.5. Plasma Stability Studies
3.6. Plasma Protein Binding
3.7. Metabolic Stability—Pooled Human Liver Microsome Incubations
3.8. Metabolic Stability—Pooled Human Cryopreserved Hepatocyte Incubations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Antonides, L.H.; Cannaert, A.; Norman, C.; Vives, L.; Harrison, A.; Costello, A.; NicDaeid, N.; Stove, C.P.; Sutcliffe, O.B.; McKenzie, C. Enantiospecific synthethis, chiral separation, and biological activity of four indazole-3-carboxamide-type synthetic cannabinoid receptor agonists and their detection in seized drug samples. Front. Chem. 2019, 7, 321. [Google Scholar] [CrossRef]
- Sachdev, S.; Vemuri, K.; Banister, S.D.; Longworth, M.; Kassiou, M.; Santiago, M.; Makriyannis, A.; Connor, M. In vitro determination of the efficacy of illicit synthetic cannabinoids at CB1 receptors. Br. J. Pharmacol. 2019, 176, 4653–4665. [Google Scholar] [CrossRef]
- European Monitoring Centre for Drugs and Drug Addiction. European Drug Report 2019: Trends and Developments; Publications Office of the European Union: Luxembourg, 2020; Available online: https://www.emcdda.europa.eu/publications/edr/trends-developments/2019 (accessed on 1 November 2020).
- Potts, A.J.; Cano, C.; Thomas, S.H.L.; Hill, S.L. Synthetic cannabinoid receptor agonists: Classification and nomenclature. Clin. Toxicol. 2020, 58, 82–98. [Google Scholar] [CrossRef]
- Luethi, D.; Liechti, M.E. Designer drugs: Mechanism of action and adverse effects. Arch. Toxicol. 2020, 94, 1085–1133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giorgetti, A.; Busardò, F.P.; Tittarelli, R.; Auwärter, V.; Giorgetti, R. Post-mortem toxicology: A systematic review of death cases involving synthetic cannabinoid receptor agonists. Front. Psychiatry 2020, 11, 464. [Google Scholar] [CrossRef]
- Trecki, J.; Gerona, R.R.; Schwartz, M.D. Synthetic cannabinoid-related illnesses and deaths. N. Engl. J. Med. 2015, 373, 103–107. [Google Scholar] [CrossRef]
- Adams, A.J.; Banister, S.D.; Irizarry, L.; Trecki, J.; Schwartz, M.; Gerona, R. “Zombie” outbreak caused by the synthetic cannabinoid AMB-FUBINACA in New York. N. Engl. J. Med. 2017, 376, 235–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sherpa, D.; Paudel, B.M.; Subedi, B.H.; Chow, R.D. Synthetic cannabinoids: The multi-organ failure and metabolic derancements associated with getting high. J. Community Hosp. Intern. Med. Perspect. 2015, 5, 27540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kolla, N.J.; Mishra, A. The endocannabinoid system, aggression, and the violence of synthetic cannabinoid use, borderline personality disorder, antisocial personality disorder, and other psychiatric disorders. Front. Behav. Neurosci. 2018, 12, 41. [Google Scholar] [CrossRef] [PubMed]
- Oliveira da Cruz, J.F.; Ioannidou, C.; Zottola, A.C.P.; Muguruza, C.; Gomez-Sotres, P.; Fernandez, M.; Callado, L.F.; Marsicano, G.; Busquets-Garcia, A. Sex-dependent pharmacological profiles of the synthetic cannabinoid MMB-Fubinaca. Addict. Biol. 2020, e12940. [Google Scholar] [CrossRef]
- Castaneto, M.S.; Wohlfarth, A.; Desrosiers, N.A.; Hartman, R.L.; Gorelick, D.A.; Huestis, M.A. Synthetic cannabinoids pharmacokinetics and detection methods in biological matrices. Drug Metab. Rev. 2015, 47, 124–174. [Google Scholar] [CrossRef]
- Davidsen, A.B.; Mardal, M.; Linnet, K. In vitro metabolism and hepatic intrinsic clearance of the synthetic cannabinoid receptor agonist JWH-122 and its four ω-halogenated analogues. AAPS J. 2019, 21, 1–9. [Google Scholar] [CrossRef]
- Diao, X.; Wohlfarth, A.; Pang, S.; Scheidweiler, K.B.; Huestis, M.A. High-resolution mass spectrometry for characterizing the metabolism of synthetic cannabinoid THJ-018 and its 5-fluoro analog THJ-2201 after incubation in human hepatocytes. Clin. Chem. 2016, 62, 157–169. [Google Scholar] [CrossRef]
- Kevin, R.C.; Lefever, T.W.; Snyder, R.W.; Patel, P.R.; Gamage, T.F.; Fennell, T.R.; Wiley, J.L.; McGregor, I.S.; Thomas, B.F. Kinetic and metabolic profiles of synthetic cannabinoids NNEI and MN-18. Drug Test. Anal. 2018, 10, 137–147. [Google Scholar] [CrossRef] [Green Version]
- Wohlfarth, A.; Castaneto, M.S.; Zhu, M.; Pang, S.; Scheidweiler, K.B.; Kronstrand, R.; Huestis, M.A. Pentylindole/pentylindazole synthetic cannabinoids and their 5-fluoro analogs produce different primary metabolites: Metabolite profiling for AB-PINACA and 5F-AB-PINACA. AAPS J. 2015, 17, 660–677. [Google Scholar] [CrossRef] [Green Version]
- Carlier, J.; Diao, X.; Wohlfarth, A.; Scheidweiler, K.; Huestis, M.A. In vitro metabolite profiling of ADB-FUBINACA, a new synthetic cannabinoid. Curr. Neuropharmacol. 2017, 15, 682–691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hess, C.; Schoeder, C.T.; Pillaiyar, T.; Madea, B.; Müller, C.E. Pharmacological evaluation of synthetic cannabinoids identified as constituents of spice. Forensic Toxicol. 2016, 34, 329–343. [Google Scholar] [CrossRef] [Green Version]
- Kakehashi, H.; Shima, N.; Ishikawa, A. Effects of lipophilicity and functional groups of synthetic cannabinoids on their blood concentrations and urinary excretion. Forensic Sci. Int. 2020, 307, 110106. [Google Scholar] [CrossRef] [PubMed]
- Schaefer, N.; Kröll, A.K.; Körbel, C.; Laschke, M.W.; Menger, M.D.; Maurer, H.H.; Meyer, M.R.; Schmidt, P.H. Distribution of the (synthetic) cannabinoids JWH-210, RCS-4, as well as ∆9-tetrahydrocannabinol following pulmonary administration to pigs. Arch. Toxicol. 2019, 93, 2211–2218. [Google Scholar] [CrossRef] [PubMed]
- Saito, K.; Kokaji, Y.; Muranaka, Y.; Ito, R. Simultaneous determination of synthetic cannabinoids in illegal herbal products and blood by LC/TOF-MS, and linear regression analysis of retention time using log Pow. Forensic Chem. 2020, 17, 100202. [Google Scholar] [CrossRef]
- Norman, C.; Walker, G.; McKirdy, B.; McDonald, C.; Fletcher, D.; Antonides, L.H.; Sutcliffe, O.B.; NicDaéid, N.; McKenzie, C. Detection and quantitation of synthetic cannabinoid receptor agonists in infused papers from prisons in a constantly evolving illicit market. Drug Test. Anal. 2020, 12, 538–544. [Google Scholar] [CrossRef] [Green Version]
- Schoeder, C.T.; Hess, C.; Madea, B.; Meiler, J.; Müller, C.E. Pharmacological evaluation of new constituents of “Spice”: Synthetic cannabinoids based on indole, indazole, benzimidazole and carbazole scaffolds. Forensic Toxicol. 2018, 36, 385–403. [Google Scholar] [CrossRef] [Green Version]
- Al-Matrouk, A.; Algallaf, M.; AlShemmeri, A.; BoJbarah, H. Identification of synthetic cannabinoids that were seized, consumed, or associated with deaths in Kuwait in 2018 using GC-MS and LC-MS-MS analysis. Forensic Sci. Int. 2019, 303, 109960. [Google Scholar] [CrossRef]
- Somerville, R.F.; Hassan, V.R.; Kolbe, E.; Partington, H.K.; Walsh, K.A.J.; Kappatos, D.C.; Johnson, C.S. The identification and quantification of synthetic cannabinoids seized in New Zealand in 2017. Forensic Sci. Int. 2019, 300, 19–27. [Google Scholar] [CrossRef] [PubMed]
- Seely, K.A.; Lapoint, J.; Moran, J.H.; Fattore, L. Spice drugs are more than harmless herbal blends: A review of the pharmacology and toxicology of synthetic cannabinoids. Prog. Neuropsychopharmacol. Biol. Psychiatry 2012, 39, 234–243. [Google Scholar] [CrossRef] [Green Version]
- Münster-Müller, S.; Matzenbach, I.; Knepper, T.; Zimmermann, R.; Pütz, M. Profiling of synthesis-related impurities of the synthetic cannabinoid Cumyl-5F-PINACA in seized samples of e-liquids via multivariate analysis of UHPLC−MSn data. Drug Test. Anal. 2019, 12, 119–126. [Google Scholar] [CrossRef]
- Peace, M.R.; Krakowiak, R.I.; Wolf, C.E.; Poklis, A.; Poklis, J.L. Identification of MDMB-FUBINACA in commercially available e-liquid formulations sold for use in electronic cigarettes. Forensic Sci. Int. 2017, 271, 92–97. [Google Scholar] [CrossRef] [Green Version]
- Doi, T.; Asada, A.; Takeda, A.; Tagami, T.; Katagi, M.; Kamata, H.; Sawabe, Y. Enantioseparation of the carboxamide-type synthetic cannabinoids N-(1-amino-3-methyl-1-oxobutan-2-yl)-1-(5-fluoropentyl)-1H-indazole-3-carboxamide and methyl [1-(5-fluoropentyl)-1H-indazole-3-carbonyl]-valinate in illicit herbal products. J. Chromatogr. A 2016, 1473, 83–89. [Google Scholar] [CrossRef] [PubMed]
- Doi, T.; Tagami, T.; Takeda, A.; Asada, A.; Sawabe, Y. Evaluation of carboxamide-type synthetic cannabinoids as CB1/CB2 receptor agonists: Difference between the enantiomers. Forensic Toxicol. 2018, 36, 51–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Antonides, L.H.; Cannaert, A.; Norman, C.; NicDáeid, N.; Sutcliffe, O.B.; Stove, C.P.; McKenzie, C. Shape matters: The application of chiral profiling and activity based assays to the evaluation of synthetic cannabinoid receptor agonists in infused papers from prisons. Drug Test. Anal. 2020, 1–20. [Google Scholar] [CrossRef]
- Millar, S.A.; Stone, N.L.; Yates, A.S.; O’Sullivan, S.E. A systematic review on the pharmacokinetics of cannabidiol in humans. Front. Pharmacol. 2018, 9, 1365. [Google Scholar] [CrossRef] [PubMed]
- Huestis, M.A. Human cannabinoid pharmacokinetics. Chem. Biodivers. 2007, 4, 1770–1804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohlsson, A.; Lindgren, J.E.; Wahlen, A.; Agurell, S.; Hollister, L.E.; Gillespie, H.K. Plasma delta-9 tetrahydrocannabinol concentrations and clinical effects after oral and intravenous administration and smoking. Clin. Pharmacol. Ther. 1980, 28, 409–416. [Google Scholar] [CrossRef] [PubMed]
- Huestis, M.A. Pharmacokinetics and metabolism of the plant cannabinoids, delta9-tetrahydrocannabinol, cannabidiol and cannabinol. Handb. Exp. Pharmacol. 2005, 168, 657–690. [Google Scholar] [CrossRef]
- Grotenhermen, F. Pharmacokinetics and pharmacodynamics of cannabinoids. Clin. Pharmacokin. 2003, 42, 327–360. [Google Scholar] [CrossRef]
- Franz, F.; Jechle, H.; Wilde, M.; Angerer, V.; Huppertz, L.M.; Longworth, M.; Kassiou, M.; Jung, M.; Auwärter, V. Structure-metabolism relationships of valine and tert-leucine-derived synthetic cannabinoid receptor agonists: A systematic comparison of the in vitro phase I metabolism using pooled human liver microsomes and high-resolution mass spectrometry. Forensic Toxicol. 2019, 37, 316–329. [Google Scholar] [CrossRef]
- Diao, X.; Huestis, M.A. New synthetic cannabinoids metabolism and strategies to best identify optimal marker metabolites. Front. Chem. 2019, 7, 1–9. [Google Scholar] [CrossRef]
- Schaefer, N.; Nordmeier, F.; Kröll, A.K.; Körbel, C.; Laschke, M.W.; Menger, M.D.; Maurer, H.H.; Meyer, M.R.; Schmidt, P.H. Is adipose tissue suitable for detection of (synthetic) cannabinoids? A comparative study analyzing antemortem and postmortem specimens following pulmonary administration of JWH-210, RCS-4, as well as ∆9-tetrahydrocannabinol to pigs. Arch. Toxicol. 2020, 94, 3421–3431. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Murthy, P.; Bharath, M.M.S. Chemistry, metabolism, and toxicology of cannabis: Clinical implications. Iran J Psychiatry 2012, 7, 149–156. [Google Scholar] [PubMed]
- Saito, T.; Namera, A.; Miura, N.; Ohta, S.; Miyazaki, S.; Osawa, M.; Inokuchi, S. A fatal case of MAM-2201 poisoning. Forensic Toxicol. 2013, 31, 333–337. [Google Scholar] [CrossRef]
- Sasaki, C.; Saito, T.; Shinozuka, T.; Irie, W.; Murakami, C.; Maeda, K.; Nakamaru, N.; Oishi, M.; Nakamura, S.; Kurihara, K. A case of death caused by abuse of a synthetic cannabinoid N-1-naphthalenyl-1-pentyl-1H-indole-3-carboxamide. Forensic Toxicol. 2014, 33, 165–169. [Google Scholar] [CrossRef]
- Franz, F.; Haschimi, B.; King, L.A.; Auwärter, V. Extraordinary long detection window of a synthetic cannabinoid metabolite in human urine—Potential impact on therapeutic decisions. Drug Test. Anal. 2020, 12, 391–396. [Google Scholar] [CrossRef] [PubMed]
- Mühlebach, S.; Wyss, P.A.; Bickel, M.H. Comparative adipose tissue kinetics of thiopental, DDE and 2,4,5,2′,4′,5′-hexachlorobiphenyl in the rat. Xenobiotica 1985, 15, 485–491. [Google Scholar] [CrossRef]
- Betschart, H.R.; Jondorf, W.R.; Bickel, M.H. Differences in adipose tissue distribution of basic lipophilic drugs between intraperitoneal and other routes of administration. Xenobiotica 1988, 18, 113–121. [Google Scholar] [CrossRef]
- Moor, M.J.; Steiner, S.H.; Jachertz, G.; Bicker, M.H. Adipose tissue distribution and chemical structure of basic lipophilic drugs: Desipramine, N-acetyl desipramine, and haloperidol. Pharmacol. Toxicol. 1992, 70, 121–124. [Google Scholar] [CrossRef] [PubMed]
- Bickel, M.H. The role of adipose tissue in the distribution and storage of drugs. Prog. Drug Res. 1984, 28, 273–303. [Google Scholar] [CrossRef]
- Hutter, M.; Moosmann, B.; Kneisel, S.; Auwärter, V. Characteristics of the designer drug and synthetic cannabinoid receptor agonist AM-2201 regarding its chemistry and metabolism. J. Mass Spectrom. 2013, 48, 885–894. [Google Scholar] [CrossRef]
- Toennes, S.W.; Geraths, A.; Pogoda, W.; Paulke, A.; Wunder, C.; Theunissen, E.L.; Ramaekers, J.G. Pharmacokinetic properties of the synthetic cannabinoid JWH-018 and of its metabolites in serum after inhalation. J. Pharm. Biomed. Anal. 2017, 140, 215–222. [Google Scholar] [CrossRef] [PubMed]
- Giorgetti, A.; Mogler, L.; Haschimi, B.; Halter, S.; Franz, F.; Westphal, F.; Fischmann, S.; Riedel, J.; Pütz, M.; Auwärter, V. Detection and phase I metabolism of the 7-azaindole-derived synthetic cannabinoid 5F-AB-P7AICA including a preliminary pharmacokinetic evaluation. Drug Test. Anal. 2020, 12, 78–91. [Google Scholar] [CrossRef]
- Tai, S.; Fantegrossi, W.E. Pharmacological and toxicological effects of synthetic cannabinoids and their metabolites. Curr. Top. Behav. Neurosci. 2017, 32, 249–262. [Google Scholar] [CrossRef]
- Tai, S.; Fantegrossi, W.E. Synthetic cannabinoids: Pharmacology, behavioral effects, and abuse potential. Curr. Addict. Rep. 2014, 1, 129–136. [Google Scholar] [CrossRef] [PubMed]
- Finlay, D.B.; Manning, J.J.; Ibsen, M.S.; Macdonald, C.E.; Patel, M.; Javitch, J.A.; Banister, S.D.; Glass, M. Do toxic synthetic cannabinoid receptor agonists have signature in vitro activity profiles? A case study of AMB-FUBINACA. ACS Chem. Neurosci. 2019, 10, 4350–4360. [Google Scholar] [CrossRef]
- Presley, B.C.; Castaneto, M.S.; Logan, B.K.; Jansen-Varnum, S.A. Metabolic profiling of synthetic cannabinoid 5F-ADB and identification of metabolites in authentic human blood samples via human liver microsome incubation and ultra-high performance liquid chromatography/high-resolution mass spectrometry. Rapid Commun. Mass Spectrom. 2020, 34, e8908. [Google Scholar] [CrossRef]
- Wagmann, L.; Frankenfeld, F.; Park, Y.M.; Herrmann, J.; Fischmann, S.; Westphal, F.; Müller, R.; Flockerzi, V.; Meyer, M.R. How to study the metabolism of new psychoactive substances for the purpose of toxicological screenings—A follow-up study comparing pooled human liver S9, HepaRG cells, and zebrafish larvae. Front. Chem. 2020, 8, 539. [Google Scholar] [CrossRef]
- Watanabe, S.; Vikingsson, S.; Åstrand, A.; Gréen, H.; Kronstrand, R. Biotransformation of the new synthetic cannabinoid with an alkene, MDMB-4en-PINACA, by human hepatocytes, human liver microsomes, human urine and blood. AAPS J. 2020, 22, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, S.; Wu, X.; Dahlen, J.; Konradsson, P.; Vikingsson, S.; Kronstrand, R.; Gréen, H. Metabolism of MMB022 and identification of dihydrodiol formation in vitro using synthesized standards. Drug Test. Anal. 2020, 12, 1432–1441. [Google Scholar] [CrossRef]
- Obach, R.S. Cytochrome P450-catalyzed metabolism of ezlopitant alkene (CJ-12,458), a pharmacologically active metabolite of ezlopitant: Enzyme kinetics and mechanism of an alkene hydration reaction. Drug Metab. Dispos. 2001, 29, 1057–1067. [Google Scholar]
- De Costa, K.S.; Black, S.R.; Thomas, B.F.; Burgess, J.P.; Mathews, J.M. Metabolism and disposition of α-methylstyrene in rats. Drug Metab. Dispos. 2001, 29, 166–171. [Google Scholar] [PubMed]
- Presley, B.C.; Logan, B.K.; Jansen-Varnum, S.A. In vitro phase I metabolism of indazole carboxamide synthetic cannabinoid MDMB-CHMINACA via human liver microsome incubation and high-resolution mass spectrometry. Drug Test. Anal. 2019, 11, 1264–1276. [Google Scholar] [CrossRef]
- Thomsen, R.; Nielsen, L.M.; Holm, N.B.; Rasmussen, H.B.; Linnet, K. Synthetic cannabimimetic agents metabolized by carboxylesterases. Drug Test. Anal. 2015, 7, 565–576. [Google Scholar] [CrossRef] [PubMed]
- Kong, T.Y.; Kim, J.H.; Kim, D.K.; Lee, H.S. Synthetic cannabinoids are substrates and inhibitors of multiple drug-metabolizing enzymes. Arch. Pharm. Res. 2018, 41, 691–710. [Google Scholar] [CrossRef] [PubMed]
- Krotulski, A.J.; Bishop-Freeman, S.C.; Mohr, A.L.A.; Logan, B.K. Evaluation of synthetic cannabinoid metabolites in human blood in the absence of parent compounds: A stability assessment. J. Anal. Toxicol. 2021, 45, 60–68. [Google Scholar] [CrossRef]
- Kavanagh, P.; Grigoryev, A.; Krupina, N. Detection of metabolites of two synthetic cannabimimetics, MDMB-FUBINACA and ADB-FUBINACA, in authentic human urine specimens by accurate mass LC-MS: A comparison of intersecting metabolic patterns. Forensic Toxicol. 2017, 35, 284–300. [Google Scholar] [CrossRef]
- Hess, C.; Krueger, L.; Unger, M.; Madea, B. Freeze-thaw stability and long-term stability of 84 synthetic cannabinoids in serum. Drug Test. Anal. 2017, 9, 1506–1511. [Google Scholar] [CrossRef]
- Li, B.; Sedlacek, M.; Manoharan, I.; Boopathy, R.; Duysen, E.G.; Masson, P.; Lockridge, O. Butyrylcholinesterase, paraoxonase, and albumin esterase, but not carboxylesterase, are present in human plasma. Biochem. Pharmacol. 2015, 70, 1673–1684. [Google Scholar] [CrossRef] [PubMed]
- Di, L. The impact of carboxylesterases in drug metabolism and pharmacokinetics. Curr. Drug Metab. 2018, 19, 91–102. [Google Scholar] [CrossRef]
- Fu, J.; Pacyniak, E.; Leed, M.G.D.; Sadgrove, M.P.; Marson, L.; Jay, M. Interspecies differences in the metabolism of a multi-ester prodrug by carboxylesterases. J. Pharm. Sci. 2016, 105, 989–995. [Google Scholar] [CrossRef] [Green Version]
- Castaneto, M.S.; Wohlfarth, A.; Pang, S.; Zhu, M.; Scheidweiler, K.B.; Kronstrand, R.; Huestis, M.A. Identification of AB-FUBINACA metabolites in human hepatocytes and urine using high-resolution mass spectrometry. Forensic Toxicol. 2015, 33, 295–310. [Google Scholar] [CrossRef]
- Mogler, L.; Franz, F.; Rentsch, D.; Angerer, V.; Weinfurtner, G.; Longworth, M.; Banister, S.D.; Kassiou, M.; Moosmann, B.; Auwärter, V. Detection of the recently emerged synthetic cannabinoid 5F-MDMB-PICA in ‘legal high’ products and human urine samples. Drug Test. Anal. 2017, 10, 196–205. [Google Scholar] [CrossRef] [PubMed]
- Haschimi, B.; Mogler, L.; Halter, S.; Giorgetti, A.; Schwarze, B.; Westphal, F.; Fischmann, S.; Auwärter, V. Detection of the recently emerged synthetic cannabinoid 4F-MDMB-BINACA in “legal high” products and human urine specimens. Drug Test. Anal. 2019, 11, 1377–1386. [Google Scholar] [CrossRef]
- Yeter, O.; Ozturk, Y.E. Metabolic profiling of synthetic cannabinoid 5F-ADB by human liver microsome incubations and urine samples using high-resolution mass spectrometry. Drug Test. Anal. 2018, 11, 847–858. [Google Scholar] [CrossRef] [PubMed]
- Andersson, M.; Diao, X.; Wohlfarth, A.; Scheidweiler, K.B.; Huestis, M.A. Metabolic profiling of new synthetic cannabinoids AMB and 5F-AMB by human hepatocyte and liver microsome incubations and high-resolution mass spectrometry. Rapid Commun. Mass Spectrom. 2016, 30, 1067–1078. [Google Scholar] [CrossRef] [PubMed]
- Hartman, D.A. Determination of the stability of drugs in plasma. Curr. Protoc. Pharmacol. 2003, 19, 7.6.1–7.6.8. [Google Scholar] [CrossRef]
- Cyprotex. Plasma Stability Assay Product Sheet. 2020. Available online: https://www.cyprotex.com/product_sheets/Cyprotex_Plasma_Stability_Product_Sheet.pdf (accessed on 3 June 2020).
- Presley, B.C.; Castaneto, M.S.; Logan, B.K.; Jansen-Varnum, S.A. Assessment of synthetic cannabinoid FUB-AMB and its ester hydrolysis metabolite in human liver microsomes and human blood samples by UHPLC-MS/MS. Biomed. Chrom. 2020, 34, e4884. [Google Scholar] [CrossRef]
- Sands, C.D.; Chan, E.S.; Welty, T.E. Revisiting the significance of warfarin protein-binding displacement interactions. Ann. Pharmacother. 2002, 36, 1642–1644. [Google Scholar] [CrossRef]
- Nguyen, L.P.; Gerstein, N.S. Cardiovascular Pharmacology in Noncardiac Surgery. In Essentials of Cardiac Anesthesia for Noncardiac Surgery; Kaplan, J.A., Cronin, B., Maus, T.M., Eds.; Elsevier: Amsterdam, The Netherlands, 2019. [Google Scholar] [CrossRef]
- Miller, R.R.; Madeira, M.; Wood, H.B.; Geissler, W.M.; Raab, C.E.; Martin, I.J. Integrating the impact of lipophilicity on potency and pharmacokinetic parameters enables the use of diverse chemical space during small molecule drug optimization. J. Med. Chem. 2020, 63, 12156–12170. [Google Scholar] [CrossRef]
- Di, L.; Umland, J.P.; Trapa, P.E.; Maurer, T.S. Impact of recovery on fraction unbound using equilibrium dialysis. J. Pharm. Sci. 2012, 101, 1327–1335. [Google Scholar] [CrossRef]
- Coleman, M.D. Factors Affecting Drug Metabolism. In Human Drug Metabolism: An Introduction; Coleman, M.D., Ed.; John Wiley & Sons: Hoboken, NJ, USA, 2010. [Google Scholar] [CrossRef]
- Stringer, R.; Nicklin, P.L.; Houston, J.B. Reliability of human cryopreserved hepatocytes and liver microsomes as in vitro systems to predict metabolomic clearance. Xenobiotica 2008, 38, 1313–1329. [Google Scholar] [CrossRef] [PubMed]
- McGinnity, D.F.; Soars, M.G.; Urbanowicz, R.A.; Riley, R.J. Evaluation of fresh and cryopreserved hepatocytes as in vitro drug metabolism tools for the prediction of metabolic clearance. Drug Metab. Dispos. 2004, 32, 1247–1253. [Google Scholar] [CrossRef] [Green Version]
- Lau, Y.Y.; Sapidou, E.; Cui, X.; White, R.E.; Cheng, K.C. Development of a novel in vitro model to predict hepatic clearance using fresh, cryopreserved, and sandwich-cultured hepatocytes. Drug Metab. Dispos. 2002, 30, 1446–1454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riley, R.J.; McGinnity, D.F.; Austin, R.P. A unified model for predicting human hepatic, metabolic clearance from in vitro intrinsic clearance data in hepatocytes and microsomes. Drug Metab. Dispos. 2005, 33, 1304–1311. [Google Scholar] [CrossRef] [Green Version]
- Wood, F.L.; Houston, J.B.; Hallifax, D. Clearance prediction methodology needs fundamental improvement: Trends common to rat and human hepatocytes/microsomes and implications for experimental methodology. Drug Metab. Dispos. 2017, 45, 1178–1188. [Google Scholar] [CrossRef] [Green Version]
- Erratico, C.; Negreira, N.; Norouzizadeh, H.; Covaci, A.; Neels, H.; Maudens, K.; van Nuijs, A.L. In vitro and in vivo human metabolism of the synthetic cannabinoid AB-CHMINACA. Drug Test. Anal. 2015, 7, 866–876. [Google Scholar] [CrossRef]
- Shen, Z.; Lv, C.; Zeng, S. Significance and challenges of stereoselectivity assessing methods in drug metabolism. J. Pharm. Anal. 2016, 6, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di, L.; Keefer, C.; Scott, D.O.; Strelevitz, T.J.; Chang, G.; Bi, Y.A.; Lai, Y.; Duckworth, J.; Fenner, K.; Troutman, M.D.; et al. Mechanistic insights from comparing intrinsic clearance values between human liver microsomes and hepatocytes to guide drug design. Eur. J. Med. Chem. 2012, 57, 441–448. [Google Scholar] [CrossRef] [PubMed]
- Truver, M.T.; Watanabe, S.; Åstrand, A.; Vikingsson, S.; Green, H.; Swortwood, M.J.; Kronstrand, R. 5F-MDMB-PICA metabolite identification and cannabinoid receptor activity. Drug Test. Anal. 2020, 12, 127–135. [Google Scholar] [CrossRef]
- Wang, D.; Zou, L.; Jin, Q.; Hou, J.; Ge, G.; Yang, L. Human carboxylesterases: A comprehensive review. Acta Pharm. Sin. B. 2018, 8, 699–712. [Google Scholar] [CrossRef] [PubMed]
- Kilford, P.J.; Gertz, M.; Houston, J.B.; Galetin, A. Hepatocellular binding of drugs: Correction for unbound fraction in hepatocyte incubations using microsomal binding or drug lipophilicity data. Drug Metab. Dispos. 2008, 36, 1194–1197. [Google Scholar] [CrossRef] [Green Version]
- Hallifax, D.; Houston, J.B. Binding of drugs to hepatic microsomes: Comment and assessment of current prediction methodology with recommendation for improvement. Drug Metab. Dispos. 2006, 34, 724–726. [Google Scholar] [CrossRef] [Green Version]
- Ménochet, K.; Kenworthy, K.E.; Houston, J.B.; Galetin, A. Use of mechanistic modeling to assess interindividual variability and interspecies differences in active uptake in human and rat hepatocytes. Drug Metabol. Dispos. 2012, 40, 1744–1756. [Google Scholar] [CrossRef] [Green Version]
- Kevin, R.C.; Lefever, T.W.; Snyder, R.W.; Patel, P.R.; Fennell, T.R.; Wiley, J.L.; McGregor, I.S.; Thomas, B.F. In vitro and in vivo pharmacokinetics and metabolism of synthetic cannabinoids CUMYL-PICA and 5F-CUMYL-PICA. Forensic Toxicol. 2017, 35, 333–347. [Google Scholar] [CrossRef] [Green Version]
- Baranczewski, P.; Stanczak, A.; Sundberg, K.; Svensson, R.; Wallin, A.; Jansson, J.; Garberg, P.; Postlind, H. Introduction to in vitro estimation of metabolic stability and drug interactions of new chemical entities in drug discovery and development. Pharmacol. Rep. 2006, 58, 453–472. [Google Scholar] [PubMed]
- Diao, X.; Scheidweiler, K.B.; Wohlfarth, A.; Pang, S.; Kronstrand, R.; Huestis, M.A. In vitro and in vivo human metabolism of synthetic cannabinoids FDU-PB-22 and FUB-PB-22. AAPS J. 2016, 18, 455–464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hasegawa, K.; Wurita, A.; Minakata, K.; Gonmori, K.; Nozawa, H.; Yamagishi, I.; Watanabe, K.; Suzuki, O. Postmortem distribution of AB-CHMINACA, 5-fluoro-AMB, and diphenidine in body fluids and solid tissues in a fatal poisoning case: Usefulness of adipose tissue for detection of the drugs in unchanged forms. Forensic Toxicol. 2015, 33, 45–53. [Google Scholar] [CrossRef]
- Kneisel, S.; Teske, J.; Auwärter, V. Analysis of synthetic cannabinoids in abstinence control: Long drug detection windows in serum and implications for practitioners. Drug Test. Anal. 2014, 6, 135–136. [Google Scholar] [CrossRef] [PubMed]
- Gottardo, R.; Bertaso, A.; Pascali, J.; Sorio, D.; Musile, G.; Trapani, E.; Seri, C.; Serpelloni, G.; Tagliaro, F. Micellar electrokinetic chromatography: A new simple tool for the analysis of synthetic cannabinoids in herbal blends and for the rapid estimation of their log P values. J. Chromatgr. A 2012, 1267, 198–205. [Google Scholar] [CrossRef]
- Schaefer, N.; Kettner, M.; Laschke, M.W.; Schaefer, N.; Kettner, M.; Laschke, M.W.; Schlote, J.; Ewald, A.H.; Menger, M.D.; Maurer, H.H.; et al. Distribution of synthetic cannabinoids JWH-210, RCS-4 and ∆ 9-tetrahydrocannabinol after intravenous administration to pigs. Curr. Neuropharmacol. 2017, 15, 713–723. [Google Scholar] [CrossRef] [Green Version]
- Laizure, S.C.; Herring, V.; Hu, Z.; Witbrodt, K.; Parker, R.B. The role of human carboxylesterases in drug metabolism: Have we overlooked their importance? J. Hum. Pharmacol. Drug Ther. 2013, 33, 210–222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.Y.; Wang, Y.; Prakash, C. Xenobiotic-metabolizing enzymes in human lung. Curr. Drug Metab. 2006, 7, 939–948. [Google Scholar] [CrossRef] [PubMed]
- Somers, G.I.; Lindsay, N.; Lowdon, B.M.; Jones, A.E.; Freathy, C.; Ho, S.; Woodrooffe, A.J.; Bayliss, M.K.; Manchee, G.R. A comparison of the expression and metabolizing activities of phase I and II enzymes in freshly isolated human lung parenchymal cells and cryopreserved human hepatocytes. Drug Metab. Dispos. 2007, 35, 1797–1805. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, S.; Kuzhiumparambil, U.; Shanlin, F. In vitro metabolism of synthetic cannabinoid AM1220 by human liver microsomes and Cunninghamella elegans using liquid chromatography coupled with high resolution mass spectrometry. Forensic Toxicol. 2018, 36, 435–446. [Google Scholar] [CrossRef] [Green Version]
- Diao, X.; Carlier, J.; Zhu, M.; Pang, S.; Kronstrand, R.; Scheidweiler, K.B.; Huestis, M.A. In vitro and in vivo human metabolism of a new synthetic cannabinoid NM-2201 (CBL-2201). Forensic Toxicol. 2017, 35, 20–32. [Google Scholar] [CrossRef] [PubMed]
- Diao, X.; Scheidweiler, K.B.; Wohlfarth, A.; Zhu, M.; Pang, S.; Huestis, M.A. Strategies to distinguish new synthetic cannabinoid FUBIMINA (BIM-2201) intake from its isomer THJ-2201: Metabolism of FUBIMINA in human hepatocytes. Forensic Toxicol. 2016, 34, 256–267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gandhi, A.S.; Wohlfarth, A.; Zhu, M.; Pang, S.; Castaneto, M.; Scheidweiler, K.B.; Huestis, M.A. High-resolution mass spectrometric metabolite profiling of a novel synthetic designer drug, N-(adamantan-1-yl)-1-(5-fluoropentyl)-1H-indole-3-carboxamide (STS-135), using cryopreserved human hepatocytes and assessment of metabolic stability with human liver microsomes. Drug Test. Anal. 2015, 7, 187–198. [Google Scholar] [CrossRef]
- Cooman, T.; Bell, S. In vitro metabolism of the synthetic cannabinoids PX-1, PX-2, and PX-3 by high-resolution mass spectrometry and their clearance rates in human liver microsomes. Rapid Commun. Mass Spec. 2019, 33, 1816–1825. [Google Scholar] [CrossRef]
- ThermoFisher Scientific. Certificate of Analysis: Pooled Human Liver Microsomes, Lot Number PL050E-A. 2019. Available online: https://www.thermofisher.com/document-connect/document-connect.html?url=https%3A%2F%2Fassets.thermofisher.com%2FTFS-Assets%2FLSG%2Fcertificate%2FCertificates-of-Analysis%2FHMMCPL%2520PL050E-A.pdf&title=UEwwNTBFLUE= (accessed on 25 February 2021).
- ThermoFisher Scientific. Certificate of Analysis: Pooled Human Liver Microsomes, Lot Number PL050E-B. 2019. Available online: https://www.thermofisher.com/document-connect/document-connect.html?url=https%3A%2F%2Fassets.thermofisher.com%2FTFS-Assets%2FLSG%2Fcertificate%2FCertificates-of-Analysis%2FHMMCPL%2520PL050E-B.pdf&title=UEwwNTBFLUI= (accessed on 25 February 2021).
- ThermoFisher Scientific. Certificate of Analysis: Primary Human Hepatocytes Cryopreserved Suspension, Lot Number HUE50-N. 2016. Available online: https://www.thermofisher.com/document-connect/document-connect.html?url=https%3A%2F%2Fassets.thermofisher.com%2FTFS-Assets%2FLSG%2Fcertificate%2FCertificates%2520of%2520Analysis%2FHue50N_HMCS50.pdf&title=SHVlNTBO (accessed on 25 February 2021).
- ThermoFisher Scientific. Certificate of Analysis: Primary Human Hepatocytes Cryopreserved Suspension, Lot Number HUE50-P. 2020. Available online: https://www.thermofisher.com/document-connect/document-connect.html?url=https%3A%2F%2Fassets.thermofisher.com%2FTFS-Assets%2FLSG%2Fcertificate%2FCertificates-of-Analysis%2FHMCS50%2520HUE50-P.pdf&title=SFVFNTAtUA== (accessed on 25 February 2021).
- Wang, R.; Fu, Y.; Lai, L. A new atom-additive method for calculating partition coefficients. J. Chem. Inf. Comp. Sci. 1997, 37, 615–621. [Google Scholar] [CrossRef]
- Manchester, K.R.; Maskell, P.D.; Waters, L. Experimental versus theoretical log D7.4, pKa and plasma protein binding values for benzodiazepines appearing as new psychoactive substances. Drug Test. Anal. 2018, 10, 1258–1269. [Google Scholar] [CrossRef] [Green Version]
- Obach, R.S.; Baxter, J.G.; Liston, T.E.; Silber, B.M.; Jones, B.C.; MacIntyre, F.; Range, D.J.; Wastall, P. The prediction of human pharmacokinetic parameters from preclinical and in vitro metabolism data. J. Pharmacol. Exp. Ther. 1997, 283, 46–58. [Google Scholar]
- Rane, A.; Wilkinson, G.R.; Shand, D.G. Prediction of hepatic extraction ratio from in vitro measurement of intrinsic clearance. J. Pharmacol. Exp. Ther. 1977, 200, 420–424. [Google Scholar] [PubMed]
- Obach, R.S. Prediction of human clearance of twenty-nine drugs from hepatic microsomal intrinsic clearance data- an examination of in vitro half-life approach and nonspecific binding to microsomes. Drug Metab. Dispos. 1999, 27, 1350–1359. [Google Scholar]
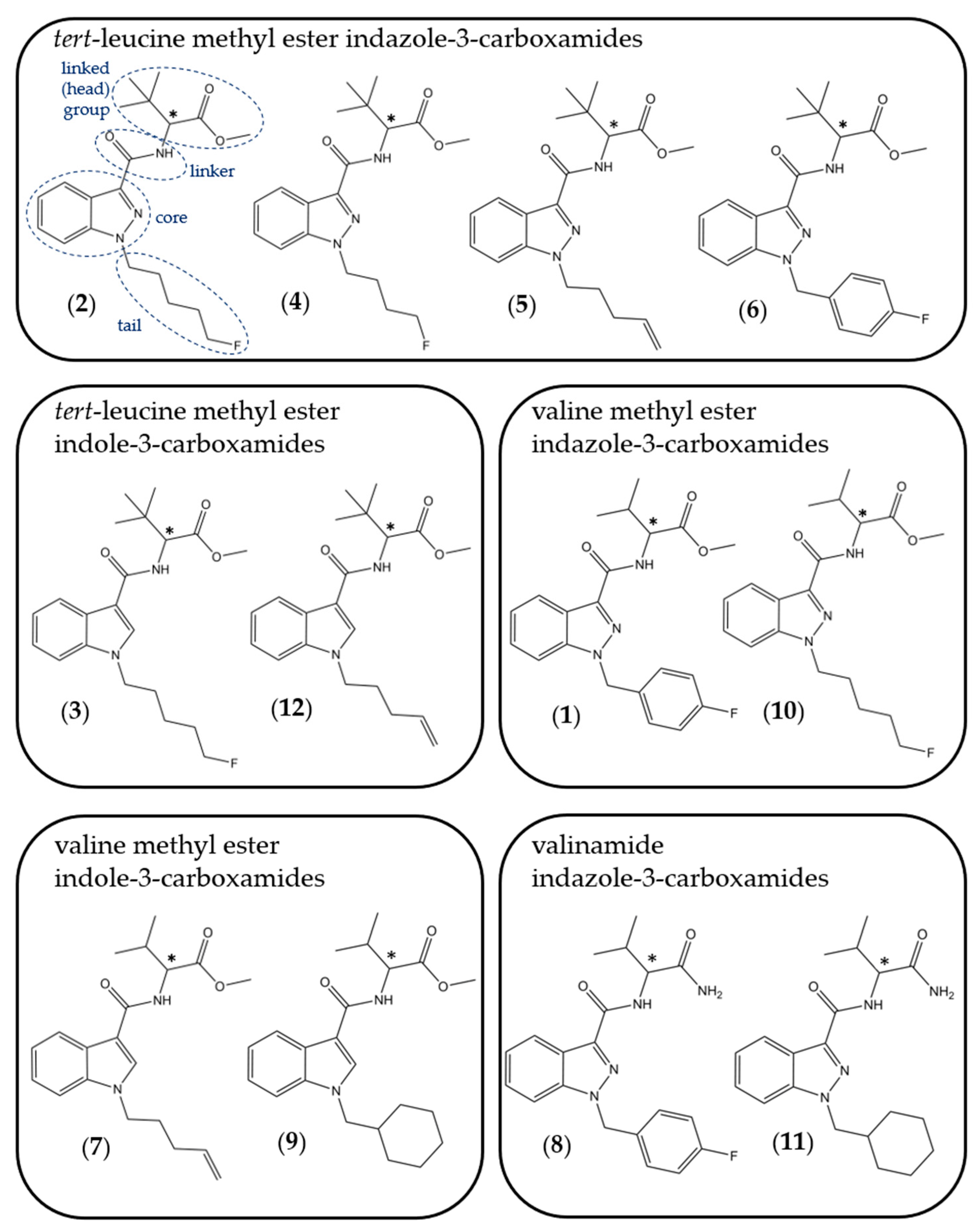
| Compound | Experimental Log D7.4 | SwissADME Predicted Log P | Log P Range from Other Software Packages |
|---|---|---|---|
| MDMB-4en-PINACA (5) | 4.95 | 3.41 | 3.61–4.00 |
| AMB-CHMICA (9) | 4.77 | 3.84 | 4.30–5.51 |
| MDMB-FUBINACA (6) | 4.69 | 3.83 | 4.08–4.24 |
| 5F-MDMB-PINACA (2) | 4.50 | 3.63 | 3.76–3.90 |
| MDMB-4en-PICA (12) | 4.40 | 3.77 | 3.98–4.98 |
| AMB-FUBINACA (1) | 4.28 | 3.50 | 3.75–4.09 |
| 4F-MDMB-BINACA (4) | 4.18 | 3.39 | 3.33–3.40 |
| 5F-AMB-PINACA (10) | 4.07 | 3.41 | 3.31–3.64 |
| 5F-MDMB-PICA (3) | 4.06 | 3.98 | 4.10–4.90 |
| AMB-4en-PICA (7) | 3.97 | 3.53 | 3.50–4.83 |
| AB-CHMINACA (11) | 3.71 | 2.91 | 3.10–3.55 |
| AB-FUBINACA (8) | 2.81 | 2.80 | 2.66–3.24 |
| 5F-AB-P7AICA | - | 2.45 | 1.57–2.74 |
| Compound | Parent Compound Remaining (%) (3 h; a 5 h) | |||
|---|---|---|---|---|
| No Esterase Inhibitors | n = | Esterase Inhibitors | n = | |
| Procaine (control) | 0.03 ± 0.02; a 0.08 ± 0.11 | 5 | 98.3 ± 8.0; a 91.0 ± 1.7 | 5 |
| (S)-AB-FUBINACA (8) | 104.4 ± 1.1 | 3 | 102.7 ± 2.5 | 3 |
| (S)-AB-CHMINACA (11) | 98.2 ± 7.2 | 3 | 97.9 ± 7.0 | 3 |
| (S)-5F-MDMB-PICA (3) | 97.3 ± 6.4 | 3 | 99.5 ± 5.8 | 3 |
| (R)-5F-MDMB-PICA (3) | 103.2 ± 14.5 | 3 | 97.7 ± 5.6 | 3 |
| (S)-AMB-FUBINACA (1) | 96.6 ± 3.8; a 85.9 ± 4.0 | 4 | 96.1 ± 1.6; a 93.5 ± 0.1 | 3 |
| (S)-MDMB-4en-PINACA (5) | 94.8 ± 0.7 | 3 | 89.0 ± 3.3 | 3 |
| (R)-MDMB-4en-PINACA (5) | 87.2 ± 6.9 | 3 | 89.4 ± 3.4 | 3 |
| (S)-MDMB-FUBINACA (6) | 94.2 ± 6.3 | 3 | 91.6 ± 2.2 | 3 |
| (S)-AMB-CHMICA (9) | 93.9 ± 9.3 | 3 | 90.5 ± 3.4 | 3 |
| (S)-4F-MDMB-BINACA (4) | 93.1 ± 5.1 | 3 | 96.0 ± 2.9 | 3 |
| (R)-4F-MDMB-BINACA (4) | 91.1 ± 6.8 | 3 | 96.0 ± 1.7 | 3 |
| (S)-5F-AMB-PINACA (10) | 93.0 ± 6.1 | 3 | 89.8 ± 1.5 | 3 |
| (S)-AMB-4en-PICA (7) | 92.6 ± 8.3 | 3 | 95.6 ± 6.8 | 3 |
| (R)-AMB-4en-PICA (7) | 91.7 ± 1.9 | 3 | 95.5 ± 2.9 | 3 |
| (S)-5F-MDMB-PINACA (2) | 92.5 ± 10.2 | 7 | 97.0 ± 1.1 | 3 |
| (R)-5F-MDMB-PINACA (2) | 95.9 ± 6.7 | 6 | 100.1 ± 7.0 | 7 |
| (S)-MDMB-4en-PICA (12) | 91.5 ± 8.6 | 3 | 89.4 ± 2.3 | 3 |
| (R)-MDMB-4en-PICA (12) | 90.3 ± 5.4 | 3 | 87.7 ± 11.0 | 3 |
| Compound | PPB (%) | Fraction Unbound (fu) | n = |
|---|---|---|---|
| (S)-MDMB-FUBINACA (6) | 99.5 ± 0.08 | 0.005 ± 0.0008 | 3 |
| (S)-MDMB-4en-PINACA (5) | 99.0 ± 0.01 | 0.010 ± 0.0001 | 3 |
| (R)-MDMB-4en-PINACA (5) | 98.1 ± 0.71 | 0.019 ± 0.0071 | 3 |
| (S)-AMB-CHMICA (9) | 98.8 ± 0.06 | 0.012 ± 0.0006 | 3 |
| (S)-AMB-FUBINACA (1) | 98.1 ± 0.08 | 0.019 ± 0.0008 | 3 |
| (S)-AB-FUBINACA (8) | 97.9 ± 0.44 | 0.021 ± 0.0044 | 3 |
| (S)-5F-MDMB-PINACA (2) | 97.8 ± 0.19 | 0.022 ± 0.0019 | 3 |
| (R)-5F-MDMB-PINACA (2) | 96.0 ± 0.58 | 0.040 ± 0.0058 | 4 |
| (S)-AB-CHMINACA (11) | 97.2 ± 2.19 | 0.028 ± 0.0219 | 3 |
| (S)-MDMB-4en-PICA (12) | 96.5 ± 0.32 | 0.035 ± 0.0031 | 4 |
| (R)-MDMB-4en-PICA (12) | 94.7 ± 1.11 | 0.053 ± 0.0111 | 4 |
| (S)-AMB-4en-PICA (7) | 94.7 ± 0.37 | 0.053 ± 0.0037 | 3 |
| (R)-AMB-4en-PICA (7) | 94.1 ± 0.10 | 0.059 ± 0.0010 | 3 |
| (S)-5F-AMB-PINACA (10) | 94.2 ± 0.08 | 0.058 ± 0.0008 | 3 |
| (S)-4F-MDMB-BINACA (4) | 93.9 ± 0.28 | 0.061 ± 0.0028 | 3 |
| (R)-4F-MDMB-BINACA (4) | 88.9 ± 0.49 | 0.111 ± 0.0049 | 4 |
| (S)-5F-MDMB-PICA (3) | 93.8 ± 0.07 | 0.062 ± 0.0007 | 3 |
| (R)-5F-MDMB-PICA (3) | 93.8 ± 0.15 | 0.062 ± 0.0015 | 3 |
| Compound | T1/2 (min) | Microsomal Intrinsic Clearance, CLint micr (mL min−1 mg Microsomal Protein−1) | Intrinsic Clearance, CLint (mL min−1 kg−1) | Predicted In Vivo Hepatic Clearance, CLH (mL min−1 kg−1) | Hepatic Extraction Ratio, EH | n |
|---|---|---|---|---|---|---|
| (S)-AMB-FUBINACA (1) | 0.6 ± 0.02 | 2.182 ± 0.071 | 2944 ± 95.9 | 15.27 ± 0.14 | 0.73 ± 0.006 | 3 |
| (R)-AMB-FUBINACA (1) | 5.9 ± 0.48 | 0.237 ± 0.020 | 320 ± 26.8 | - | - | 6 |
| (S)-5F-AMB-PINACA (10) | 0.9 ± 0.07 | 1.494 ± 0.115 | 2016 ± 155.7 | 17.79 ± 0.20 | 0.85 ± 0.010 | 6 |
| (R)-5F-AMB-PINACA (10) | 6.5 ± 0.42 | 0.213 ± 0.014 | 288 ± 19.1 | - | - | 6 |
| (S)-AMB-CHMICA (9) | 4.1 ± 0.75 | 0.343 ± 0.069 | 463 ± 92.9 | 4.37 ± 0.68 | 0.21 ± 0.032 | 3 |
| (R)-AMB-CHMICA (9) | 8.4 ± 1.0 | 0.167 ± 0.019 | 226 ± 25.4 | - | - | 3 |
| (S)-AMB-4en-PICA (7) | 6.0 ± 1.0 | 0.236 ± 0.042 | 318 ± 56.6 | 9.29 ± 0.89 | 0.44 ± 0.042 | 10 |
| (R)-AMB-4en-PICA (7) | 4.1 ± 0.75 | 0.348 ± 0.068 | 469 ± 92.2 | 11.8 ± 0.97 | 0.56 ± 0.046 | 11 |
| (S)-MDMB-4en-PINACA (5) | 6.3 ± 1.0 | 0.226 ± 0.037 | 305 ± 49.3 | 2.66 ± 0.37 | 0.13 ± 0.018 | 7 |
| (R)-MDMB-4en-PINACA (5) | 5.5 ± 0.59 | 0.253 ± 0.026 | 341 ± 34.6 | 4.94 ± 0.39 | 0.24 ± 0.019 | 7 |
| (S)-5F-MDMB-PINACA (2) | 6.7 ± 0.31 | 0.207 ± 0.009 | 280 ± 12.8 | 4.76 ± 0.17 | 0.23 ± 0.008 | 5 |
| (R)-5F-MDMB-PINACA (2) | 8.0 ± 1.1 | 0.176 ± 0.024 | 237 ± 31.7 | 6.52 ± 0.61 | 0.31 ± 0.029 | 5 |
| (S)-5F-MDMB-PICA (3) | 9.0 ± 1.4 | 0.156 ± 0.027 | 211 ± 36.9 | 8.01 ± 0.84 | 0.38 ± 0.04 | 3 |
| (R)-5F-MDMB-PICA (3) | 11 ± 0.11 | 0.132 ± 0.001 | 178 ± 1.86 | 7.22 ± 0.05 | 0.34 ± 0.002 | 3 |
| (S)-MDMB-4en-PICA (12) | 8.5 ± 1.3 | 0.166 ± 0.024 | 224 ± 32.4 | 5.69 ± 0.61 | 0.27 ± 0.029 | 11 |
| (R)-MDMB-4en-PICA (12) | 5.4 ± 0.74 | 0.260 ± 0.040 | 350 ± 53.3 | 9.81 ± 0.75 | 0.47 ± 0.036 | 9 |
| (S)-4F-MDMB-BINACA (4) | 9.9 ± 1.9 | 0.143 ± 0.025 | 193 ± 33.8 | 7.51 ± 0.88 | 0.36 ± 0.042 | 3 |
| (R)-4F-MDMB-BINACA (4) | 14 ± 0.68 | 0.096 ± 0.005 | 130 ± 6.26 | 8.55 ± 0.24 | 0.41 ± 0.012 | 3 |
| (S)-MDMB-FUBINACA (6) | 11 ± 2.4 | 0.135 ± 0.030 | 183 ± 40.2 | 0.87 ± 0.18 | 0.04 ± 0.009 | 6 |
| (R)-MDMB-FUBINACA (6) | 20 ± 2.5 | 0.071 ± 0.009 | 95.4 ± 12.3 | - | - | 6 |
| (S)-AB-CHMINACA (11) | 27 ± 2.9 | 0.051 ± 0.005 | 69.1 ± 7.35 | 1.77 ± 0.17 | 0.08 ± 0.008 | 3 |
| (R)-AB-CHMINACA (11) | 43 ± 5.0 | 0.033 ± 0.004 | 44.1 ± 4.89 | - | - | 3 |
| (S)-AB-FUBINACA (8) | 118 ± 28 | 0.012 ± 0.003 | 16.4 ± 4.24 | 0.34 ± 0.09 | 0.02 ± 0.004 | 3 |
| (R)-AB-FUBINACA (8) | 145 ± 45 | 0.010 ± 0.003 | 13.7 ± 4.06 | - | - | 3 |
| Compound | T1/2 (min) | Intrinsic Clearance, CLint (mL min−1 kg−1) | Predicted In Vivo Hepatic Clearance, CLH (mL min−1 kg−1) | Hepatic Extraction Ratio, EH | n |
|---|---|---|---|---|---|
| (S)-AMB-FUBINACA (1) | 2.5 ± 0.55 | 3216 ± 607 | 15.52 ± 0.85 | 0.74 ± 0.041 | 8 |
| (R)-AMB-FUBINACA (1) | 3.1 ± 0.57 | 2557 ± 463 | - | - | 8 |
| (S)-5F-AMB-PINACA (10) | 3.2 ± 0.16 | 2404 ± 125 | 18.25 ± 0.12 | 0.87 ± 0.006 | 3 |
| (R)-5F-AMB-PINACA (10) | 4.7 ± 0.26 | 1623 ± 87.0 | - | - | 3 |
| (S)-AMB-4en-PICA (7) | 6.9 ± 2.1 | 1205 ± 363 | 15.56 ± 1.24 | 0.74 ± 0.059 | 8 |
| (R)-AMB-4en-PICA (7) | 5.5 ± 2.1 | 1589 ± 616 | 16.79 ± 1.33 | 0.80 ± 0.063 | 8 |
| (S)-AMB-CHMICA (9) | 7.6 ± 0.35 | 1011 ± 48.5 | 7.68 ± 0.22 | 0.37 ± 0.011 | 3 |
| (R)-AMB-CHMICA (9) | 8.3 ± 0.35 | 918 ± 38.2 | - | - | 3 |
| (S)-MDMB-4en-PINACA (5) | 12 ± 2.6 | 683 ± 158 | 5.11 ± 0.89 | 0.24 ± 0.042 | 7 |
| (R)-MDMB-4en-PINACA (5) | 22 ± 3.6 | 363 ± 66.5 | 5.16 ± 0.70 | 0.25 ± 0.033 | 7 |
| (S)-5F-MDMB-PINACA (2) | 13 ± 3.3 | 604 ± 135 | 8.05 ± 1.15 | 0.38 ± 0.055 | 12 |
| (R)-5F-MDMB-PINACA (2) | 23 ± 1.8 | 334 ± 25.5 | 8.16 ± 0.38 | 0.39 ± 0.018 | 6 |
| (S)-4F-MDMB-BINACA (4) | 16 ± 0.74 | 466 ± 21.6 | 12.08 ± 0.23 | 0.58 ± 0.011 | 3 |
| (R)-4F-MDMB-BINACA (4) | 58 ± 17.2 | 139 ± 37.4 | 8.77 ± 1.43 | 0.42 ± 0.068 | 4 |
| (S)-MDMB-FUBINACA (6) | 26 ± 5.0 | 298 ± 62.7 | 1.39 ± 0.27 | 0.07 ± 0.013 | 3 |
| (R)-MDMB-FUBINACA (6) | 32 ± 5.4 | 239 ± 40.7 | - | - | 3 |
| (S)-MDMB-4en-PICA (12) | 37 ± 6.9 | 213 ± 41.6 | 5.46 ± 0.78 | 0.26 ± 0.037 | 7 |
| (R)-MDMB-4en-PICA (12) | 44 ± 6.3 | 176 ± 24.0 | 6.42 ± 0.62 | 0.31 ± 0.029 | 7 |
| (S)-5F-MDMB-PICA (3) | 38 ± 4.6 | 204 ± 23.2 | 7.87 ± 0.58 | 0.37 ± 0.027 | 3 |
| (R)-5F-MDMB-PICA (3) | 54 ± 9.4 | 145 ± 24.1 | 6.27 ± 0.74 | 0.30 ± 0.035 | 3 |
| (S)-AB-CHMINACA (11) | 39 ± 8.8 | 210 ± 72.9 | 4.52 ± 1.12 | 0.22 ± 0.053 | 6 |
| (R)-AB-CHMINACA (11) | 38 ± 8.7 | 213 ± 54.3 | - | - | 5 |
| (S)-AB-FUBINACA (8) | 76 ± 24 | 110 ± 34.5 | 2.06 ± 0.58 | 0.10 ± 0.028 | 8 |
| (R)-AB-FUBINACA (8) | 69 ± 11 | 113 ± 15.6 | - | - | 8 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brandon, A.M.; Antonides, L.H.; Riley, J.; Epemolu, O.; McKeown, D.A.; Read, K.D.; McKenzie, C. A Systematic Study of the In Vitro Pharmacokinetics and Estimated Human In Vivo Clearance of Indole and Indazole-3-Carboxamide Synthetic Cannabinoid Receptor Agonists Detected on the Illicit Drug Market. Molecules 2021, 26, 1396. https://doi.org/10.3390/molecules26051396
Brandon AM, Antonides LH, Riley J, Epemolu O, McKeown DA, Read KD, McKenzie C. A Systematic Study of the In Vitro Pharmacokinetics and Estimated Human In Vivo Clearance of Indole and Indazole-3-Carboxamide Synthetic Cannabinoid Receptor Agonists Detected on the Illicit Drug Market. Molecules. 2021; 26(5):1396. https://doi.org/10.3390/molecules26051396
Chicago/Turabian StyleBrandon, Andrew M., Lysbeth H. Antonides, Jennifer Riley, Ola Epemolu, Denise A. McKeown, Kevin D. Read, and Craig McKenzie. 2021. "A Systematic Study of the In Vitro Pharmacokinetics and Estimated Human In Vivo Clearance of Indole and Indazole-3-Carboxamide Synthetic Cannabinoid Receptor Agonists Detected on the Illicit Drug Market" Molecules 26, no. 5: 1396. https://doi.org/10.3390/molecules26051396