
Dopamine Self-Polymerization as a Simple and Powerful Tool to Modulate the Viscoelastic Mechanical Properties of Peptide-Based Gels



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
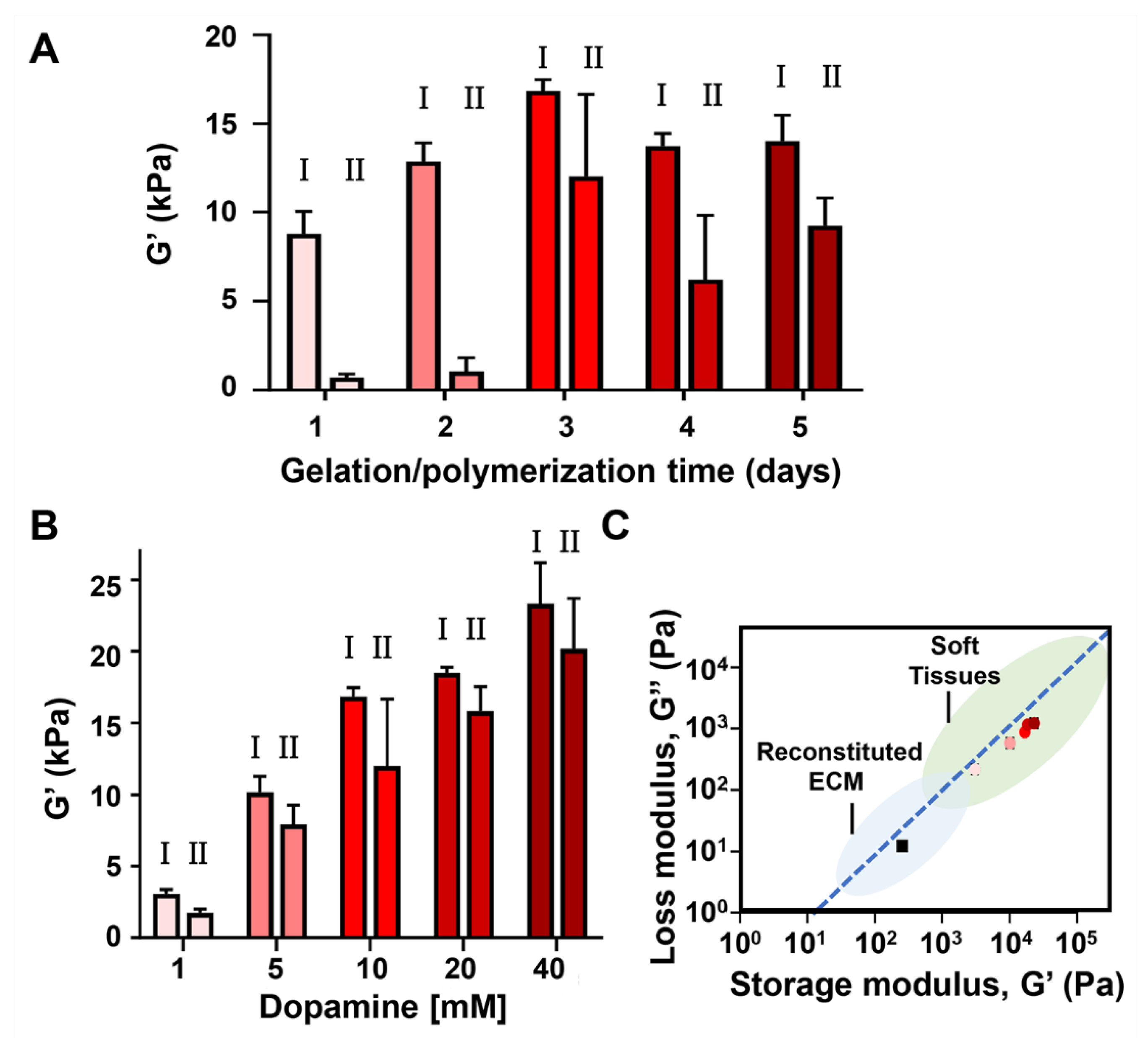
2. Results and Discussion
3. Materials and Methods
3.1. Materials
3.2. Methods
3.2.1. Peptide Synthesis
3.2.2. Gel Preparation
3.2.3. Rheological Studies
3.2.4. Circular Dichroism (CD) Measurements
3.2.5. Transmission Electron Microscopy
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Lee, H.A.; Park, E.; Lee, H. Polydopamine and Its Derivative Surface Chemistry in Material Science: A Focused Review for Studies at KAIST. Adv. Mater. 2020, 32, 1907505. [Google Scholar] [CrossRef]
- Ryu, J.H.; Messersmith, P.B.; Lee, H. Polydopamine Surface Chemistry: A Decade of Discovery. ACS Appl. Mater. Interfaces 2018, 10, 7523–7540. [Google Scholar] [CrossRef]
- Liu, Y.; Ai, K.; Lu, L. Polydopamine and its derivative materials: Synthesis and promising applications in energy, environmental, and biomedical fields. Chem. Rev. 2014, 114, 5057–5115. [Google Scholar] [CrossRef]
- Lee, H.; Dellatore, S.M.; Miller, W.M.; Messersmith, P.B. Mussel-inspired surface chemistry for multifunctional coatings. Science 2007, 318, 426–430. [Google Scholar] [CrossRef] [PubMed]
- Herlinger, E.; Jameson, R.F.; Linert, W. Spontaneous Autoxidation of Dopamine. J. Chem. Soc. Perkin Trans. 1995, 2, 259–263. [Google Scholar] [CrossRef]
- Hong, S.; Wang, Y.; Park, S.Y.; Lee, H. Progressive fuzzy cation-pi assembly of biological catecholamines. Sci. Adv. 2018, 4, eaat7457. [Google Scholar] [CrossRef]
- Cross, E.R.; Coulter, S.M.; Fuentes-Caparros, A.M.; McAulay, K.; Schweins, R.; Laverty, G.; Adams, D.J. Tuning the antimicrobial activity of low molecular weight hydrogels using dopamine autoxidation. Chem. Commun. 2020, 56, 8135–8138. [Google Scholar] [CrossRef] [PubMed]
- Fichman, G.; Gazit, E. Self-assembly of short peptides to form hydrogels: Design of building blocks, physical properties and technological applications. Acta Biomater. 2014, 10, 1671–1682. [Google Scholar] [CrossRef] [PubMed]
- Du, X.; Zhou, J.; Shi, J.; Xu, B. Supramolecular Hydrogelators and Hydrogels: From Soft Matter to Molecular Biomaterials. Chem. Rev. 2015, 115, 13165–13307. [Google Scholar] [CrossRef]
- Chaudhuri, O.; Cooper-White, J.; Janmey, P.A.; Mooney, D.J.; Shenoy, V.B. Effects of extracellular matrix viscoelasticity on cellular behaviour. Nature 2020, 584, 535–546. [Google Scholar] [CrossRef]
- Hellmund, K.S.; Koksch, B. Self-Assembling Peptides as Extracellular Matrix Mimics to Influence Stem Cell’s Fate. Front. Chem. 2019, 7, 172. [Google Scholar] [CrossRef]
- Ashworth, J.C.; Thompson, J.L.; James, J.R.; Slater, C.E.; Pijuan-Galito, S.; Lis-Slimak, K.; Holley, R.J.; Meade, K.A.; Thompson, A.; Arkill, K.P.; et al. Peptide gels of fully-defined composition and mechanics for probing cell-cell and cell-matrix interactions in vitro. Matrix Biol. 2020, 85–86, 15–33. [Google Scholar] [CrossRef] [PubMed]
- Alakpa, E.V.; Jayawarna, V.; Lampel, A.; Burgess, K.V.; West, C.C.; Bakker, S.C.J.; Roy, S.; Javid, N.; Fleming, S.; Lamprou, D.A.; et al. Tunable Supramolecular Hydrogels for Selection of Lineage-Guiding Metabolites in Stem Cell Cultures. Chem 2016, 1, 298–319. [Google Scholar] [CrossRef]
- Jacob, R.S.; Ghosh, D.; Singh, P.K.; Basu, S.K.; Jha, N.N.; Das, S.; Sukul, P.K.; Patil, S.; Sathaye, S.; Kumar, A.; et al. Self healing hydrogels composed of amyloid nano fibrils for cell culture and stem cell differentiation. Biomaterials 2015, 54, 97–105. [Google Scholar] [CrossRef]
- Li, Y.; Qin, M.; Cao, Y.; Wang, W. Designing the mechanical properties of peptide-based supramolecular hydrogels for biomedical applications. Sci. China Phys. Mech. 2014, 57, 849–858. [Google Scholar] [CrossRef]
- Pashuck, E.T.; Cui, H.G.; Stupp, S.I. Tuning Supramolecular Rigidity of Peptide Fibers through Molecular Structure. J. Am. Chem. Soc. 2010, 132, 6041–6046. [Google Scholar] [CrossRef]
- Koch, F.; Muller, M.; Konig, F.; Meyer, N.; Gattlen, J.; Pieles, U.; Peters, K.; Kreikemeyer, B.; Mathes, S.; Saxer, S. Mechanical characteristics of beta sheet-forming peptide hydrogels are dependent on peptide sequence, concentration and buffer composition. R. Soc. Open Sci. 2018, 5, 171562. [Google Scholar] [CrossRef] [PubMed]
- Geisler, I.M.; Schneider, J.P. Evolution-Based Design of an Injectable Hydrogel. Adv. Funct. Mater. 2012, 22, 529–537. [Google Scholar] [CrossRef]
- Micklitsch, C.M.; Medina, S.H.; Yucel, T.; Nagy-Smith, K.J.; Pochan, D.J.; Schneider, J.P. Influence of Hydrophobic Face Amino Acids on the Hydrogelation of beta-Hairpin Peptide Amphiphiles. Macromolecules 2015, 48, 1281–1288. [Google Scholar] [CrossRef]
- Aulisa, L.; Dong, H.; Hartgerink, J.D. Self-assembly of multidomain peptides: Sequence variation allows control over cross-linking and viscoelasticity. Biomacromolecules 2009, 10, 2694–2698. [Google Scholar] [CrossRef]
- Uchida, N.; Muraoka, T. Current Progress in Cross-Linked Peptide Self-Assemblies. Int. J. Mol. Sci. 2020, 21, 7577. [Google Scholar] [CrossRef] [PubMed]
- Greenfield, M.A.; Hoffman, J.R.; de la Cruz, M.O.; Stupp, S.I. Tunable mechanics of peptide nanofiber gels. Langmuir 2010, 26, 3641–3647. [Google Scholar] [CrossRef]
- DiMaio, J.T.M.; Doran, T.M.; Ryan, D.M.; Raymond, D.M.; Nilsson, B.L. Modulating Supramolecular Peptide Hydrogel Viscoelasticity Using Biomolecular Recognition. Biomacromolecules 2017, 18, 3591–3599. [Google Scholar] [CrossRef]
- Bairagi, D.; Biswas, P.; Basu, K.; Hazra, S.; Hermida-Merino, D.; Sinha, D.K.; Hamley, I.W.; Banerjee, A. Self-Assembling Peptide-Based Hydrogel: Regulation of Mechanical Stiffness and Thermal Stability and 3D Cell Culture of Fibroblasts. ACS Appl. Bio. Mater. 2019, 2, 5235–5244. [Google Scholar] [CrossRef]
- Bakota, E.L.; Aulisa, L.; Galler, K.M.; Hartgerink, J.D. Enzymatic Cross-Linking of a Nanofibrous Peptide Hydrogel. Biomacromolecules 2011, 12, 82–87. [Google Scholar] [CrossRef]
- Li, Y.; Ding, Y.; Qin, M.; Cao, Y.; Wang, W. An enzyme-assisted nanoparticle crosslinking approach to enhance the mechanical strength of peptide-based supramolecular hydrogels. Chem. Commun. 2013, 49, 8653–8655. [Google Scholar] [CrossRef] [PubMed]
- Khalily, M.A.; Goktas, M.; Guler, M.O. Tuning viscoelastic properties of supramolecular peptide gels via dynamic covalent crosslinking. Org. Biomol. Chem. 2015, 13, 1983–1987. [Google Scholar] [CrossRef] [PubMed]
- Seow, W.Y.; Hauser, C.A.E. Tunable Mechanical Properties of Ultrasmall Peptide Hydrogels by Crosslinking and Functionalization to Achieve the 3D Distribution of Cells. Adv. Healthc. Mater. 2013, 2, 1219–1223. [Google Scholar] [CrossRef]
- Fichman, G.; Schneider, J.P. Utilizing Frémy’s Salt to Increase the Mechanical Rigidity of Supramolecular Peptide-Based Gel Networks. Front. Bioeng. Biotechnol. 2021, 8, 1491. [Google Scholar] [CrossRef]
- Radvar, E.; Azevedo, H.S. Supramolecular Peptide/Polymer Hybrid Hydrogels for Biomedical Applications. Macromol. Biosci. 2019, 19, e1800221. [Google Scholar] [CrossRef]
- Xing, R.; Li, S.; Zhang, N.; Shen, G.; Mohwald, H.; Yan, X. Self-Assembled Injectable Peptide Hydrogels Capable of Triggering Antitumor Immune Response. Biomacromolecules 2017, 18, 3514–3523. [Google Scholar] [CrossRef]
- Chakraborty, P.; Ghosh, M.; Schnaider, L.; Adadi, N.; Ji, W.; Bychenko, D.; Dvir, T.; Adler-Abramovich, L.; Gazit, E. Composite of Peptide-Supramolecular Polymer and Covalent Polymer Comprises a New Multifunctional, Bio-Inspired Soft Material. Macromol. Rapid Commun. 2019, 40, e1900175. [Google Scholar] [CrossRef]
- Nadernezhad, A.; Forster, L.; Netti, F.; Adler-Abramovich, L.; Tessmar, J.; Groll, J. Rheological analysis of the interplay between the molecular weight and concentration of hyaluronic acid in formulations of supramolecular HA/FmocFF hybrid hydrogels. Polym. J. 2020, 52, 1007–1012. [Google Scholar] [CrossRef]
- Schneider, J.P.; Pochan, D.J.; Ozbas, B.; Rajagopal, K.; Pakstis, L.; Kretsinger, J. Responsive hydrogels from the intramolecular folding and self-assembly of a designed peptide. J. Am. Chem. Soc. 2002, 124, 15030–15037. [Google Scholar] [CrossRef]
- Yucel, T.; Micklitsch, C.M.; Schneider, J.P.; Pochan, D.J. Direct observation of early-time hydrogelation in beta-hairpin peptide self-assembly. Macromolecules 2008, 41, 5763–5772. [Google Scholar] [CrossRef]
- Veerman, C.; Rajagopal, K.; Palla, C.S.; Pochan, D.J.; Schneider, J.P.; Furst, E.M. Gelation kinetics of beta-hairpin peptide hydrogel networks. Macromolecules 2006, 39, 6608–6614. [Google Scholar] [CrossRef]
- Ozbas, B.; Rajagopal, K.; Schneider, J.P.; Pochan, D.J. Semiflexible chain networks formed via self-assembly of beta-hairpin molecules. Phys. Rev. Lett. 2004, 93, 268106. [Google Scholar] [CrossRef]
- Branco, M.C.; Pochan, D.J.; Wagner, N.J.; Schneider, J.P. Macromolecular diffusion and release from self-assembled beta-hairpin peptide hydrogels. Biomaterials 2009, 30, 1339–1347. [Google Scholar] [CrossRef]
- Nagy-Smith, K.; Moore, E.; Schneider, J.; Tycko, R. Molecular structure of monomorphic peptide fibrils within a kinetically trapped hydrogel network. Proc. Natl. Acad. Sci. USA 2015, 112, 9816–9821. [Google Scholar] [CrossRef]
- Rajagopal, K.; Ozbas, B.; Pochan, D.J.; Schneider, J.P. Probing the importance of lateral hydrophobic association in self-assembling peptide hydrogelators. Eur. Biophys. J. 2006, 35, 162–169. [Google Scholar] [CrossRef]
- Della Vecchia, N.F.; Avolio, R.; Alfe, M.; Errico, M.E.; Napolitano, A.; d’Ischia, M. Building-Block Diversity in Polydopamine Underpins a Multifunctional Eumelanin-Type Platform Tunable Through a Quinone Control Point. Adv. Funct. Mater. 2013, 23, 1331–1340. [Google Scholar] [CrossRef]
- Ji, Y.L.; Ang, M.B.M.Y.; Hung, H.C.; Huang, S.H.; An, Q.F.; Lee, K.R.; Lai, J.Y. Bio-inspired deposition of polydopamine on PVDF followed by interfacial cross-linking with trimesoyl chloride as means of preparing composite membranes for isopropanol dehydration. J. Membr. Sci. 2018, 557, 58–66. [Google Scholar] [CrossRef]
- Pochan, D.J.; Schneider, J.P.; Kretsinger, J.; Ozbas, B.; Rajagopal, K.; Haines, L. Thermally reversible hydrogels via intramolecular folding and consequent self-assembly of a de novo designed peptide. J. Am. Chem. Soc. 2003, 125, 11802–11803. [Google Scholar] [CrossRef]
- Rughani, R.V.; Schneider, J.P. Molecular Design of beta-Hairpin Peptides for Material Construction. MRS Bull. 2008, 33, 530–535. [Google Scholar] [CrossRef]
- Jiang, J.; Zhu, L.; Zhu, L.; Zhu, B.; Xu, Y. Surface characteristics of a self-polymerized dopamine coating deposited on hydrophobic polymer films. Langmuir 2011, 27, 14180–14187. [Google Scholar] [CrossRef] [PubMed]
- O’Connor, N.A.; Syed, A.; Wong, M.; Hicks, J.; Nunez, G.; Jitianu, A.; Siler, Z.; Peterson, M. Polydopamine Antioxidant Hydrogels for Wound Healing Applications. Gels 2020, 6, 39. [Google Scholar] [CrossRef]
- d’Ischia, M.; Napolitano, A.; Ball, V.; Chen, C.T.; Buehler, M.J. Polydopamine and Eumelanin: From Structure-Property Relationships to a Unified Tailoring Strategy. Acc. Chem. Res. 2014, 47, 3541–3550. [Google Scholar] [CrossRef]
- Yan, C.; Altunbas, A.; Yucel, T.; Nagarkar, R.P.; Schneider, J.P.; Pochan, D.J. Injectable solid hydrogel: Mechanism of shear-thinning and immediate recovery of injectable beta-hairpin peptide hydrogels. Soft Matter 2010, 6, 5143–5156. [Google Scholar] [CrossRef]
- Ball, V.; Del Frari, D.; Toniazzo, V.; Ruch, D. Kinetics of polydopamine film deposition as a function of pH and dopamine concentration: Insights in the polydopamine deposition mechanism. J. Colloid Interface Sci. 2012, 386, 366–372. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fichman, G.; Schneider, J.P. Dopamine Self-Polymerization as a Simple and Powerful Tool to Modulate the Viscoelastic Mechanical Properties of Peptide-Based Gels. Molecules 2021, 26, 1363. https://doi.org/10.3390/molecules26051363
Fichman G, Schneider JP. Dopamine Self-Polymerization as a Simple and Powerful Tool to Modulate the Viscoelastic Mechanical Properties of Peptide-Based Gels. Molecules. 2021; 26(5):1363. https://doi.org/10.3390/molecules26051363
Chicago/Turabian StyleFichman, Galit, and Joel P. Schneider. 2021. "Dopamine Self-Polymerization as a Simple and Powerful Tool to Modulate the Viscoelastic Mechanical Properties of Peptide-Based Gels" Molecules 26, no. 5: 1363. https://doi.org/10.3390/molecules26051363
APA StyleFichman, G., & Schneider, J. P. (2021). Dopamine Self-Polymerization as a Simple and Powerful Tool to Modulate the Viscoelastic Mechanical Properties of Peptide-Based Gels. Molecules, 26(5), 1363. https://doi.org/10.3390/molecules26051363