
Methylation of Cyanidin-3-O-Glucoside with Dimethyl Carbonate


Abstract
1. Introduction
2. Results and Discussion
2.1. First Observations
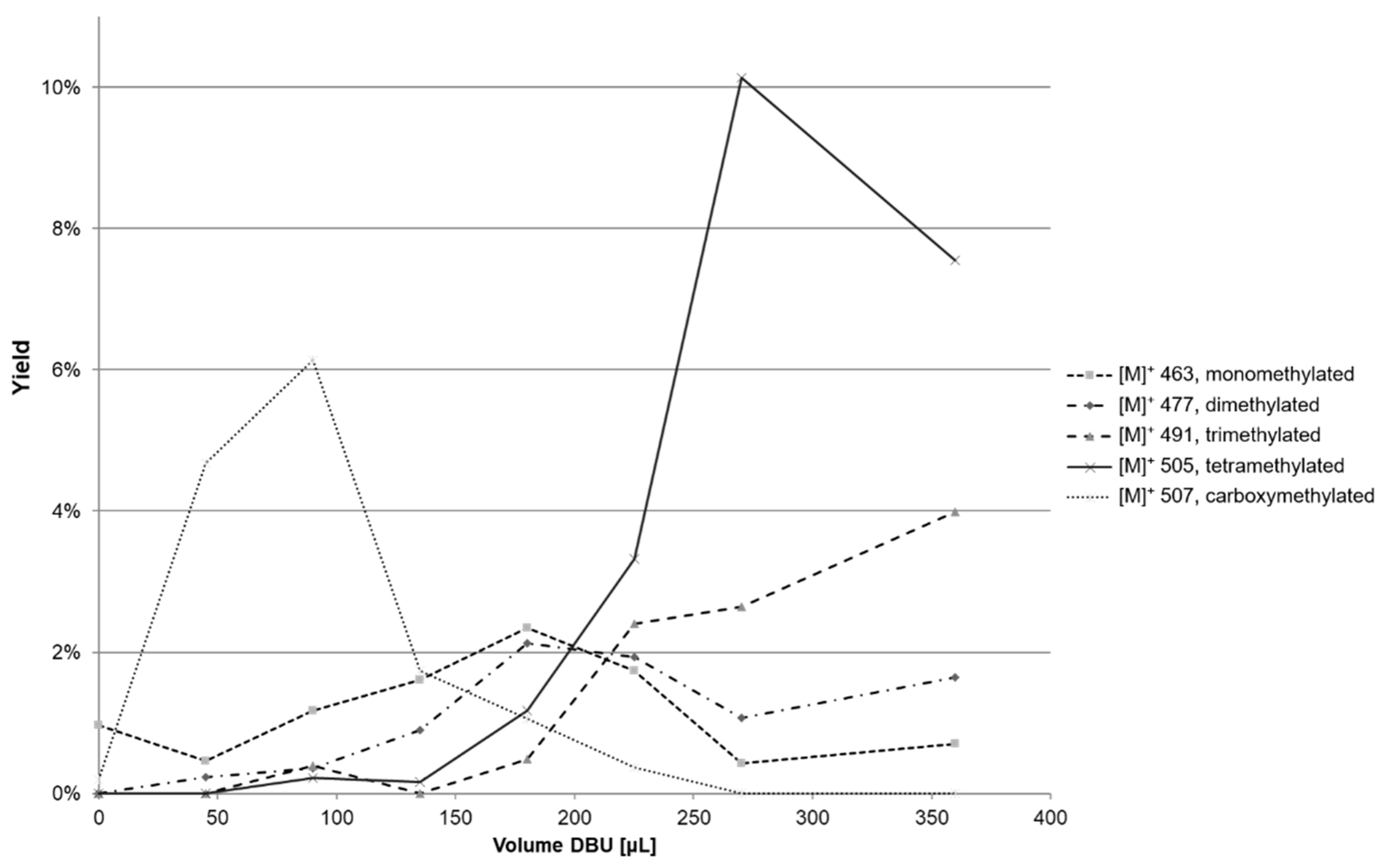
2.2. Experimental Design
2.3. Increase in Solvation of Anthocyanins
2.3.1. Ultrasound
2.3.2. Solvents
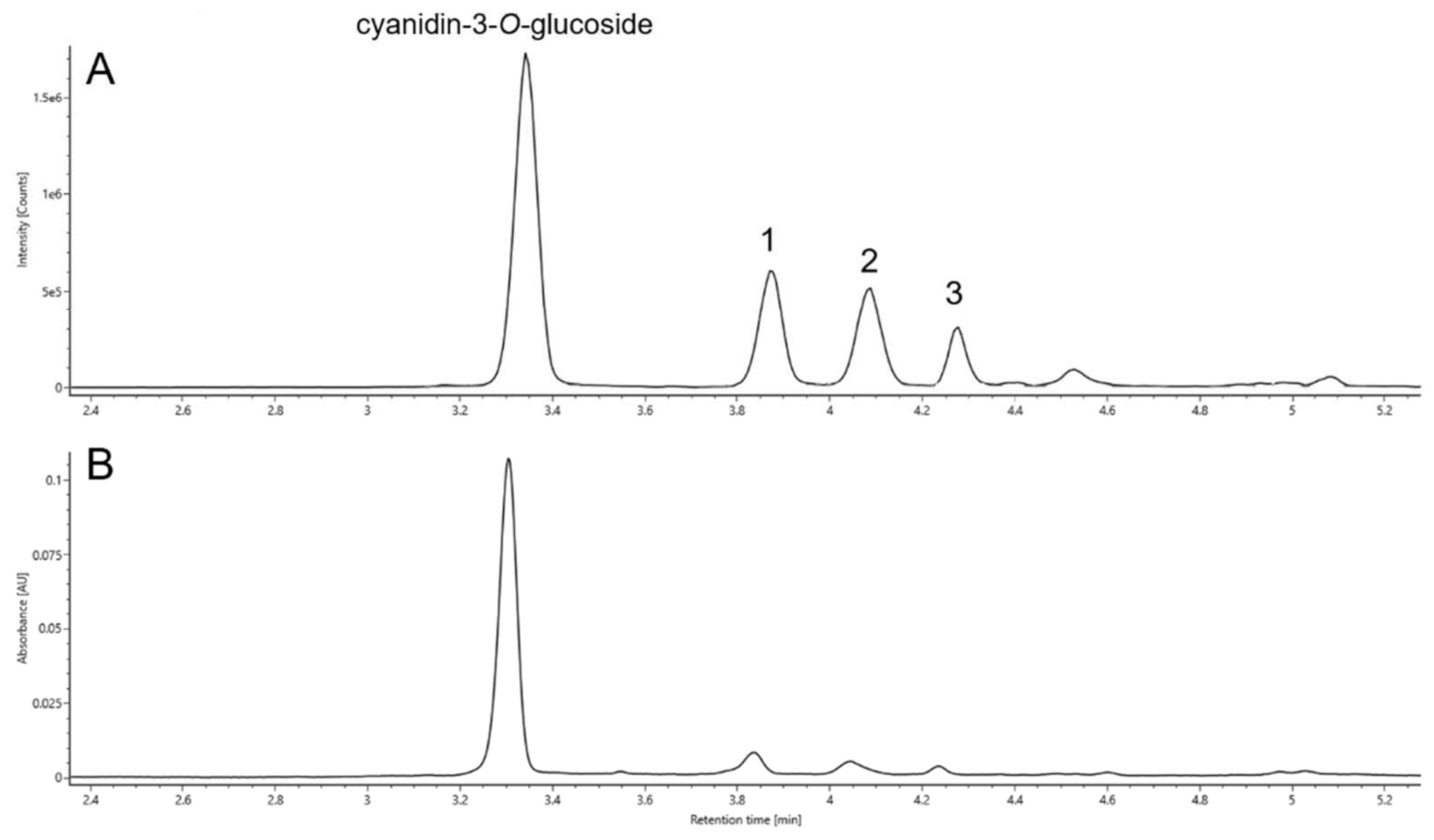
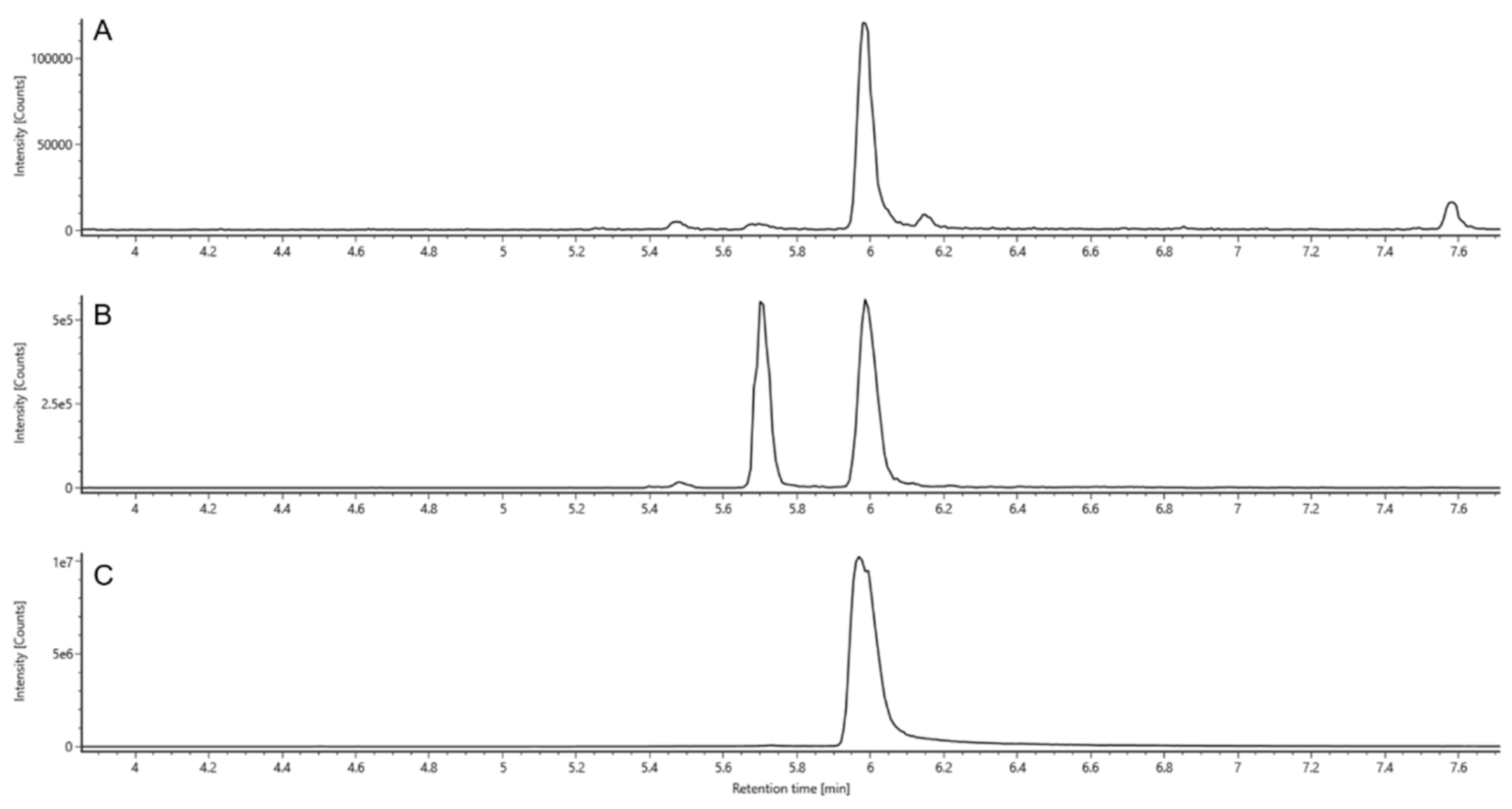
2.4. Identification
2.4.1. Retention Times (RT)
2.4.2. UV Spectra
2.4.3. Mass Spectrometry
2.4.4. Ion Mobility Spectrometry
2.4.5. Views on Reactivity and Order of Methylation
2.4.6. Hydrolysis
3. Materials and Methods
3.1. Chemicals
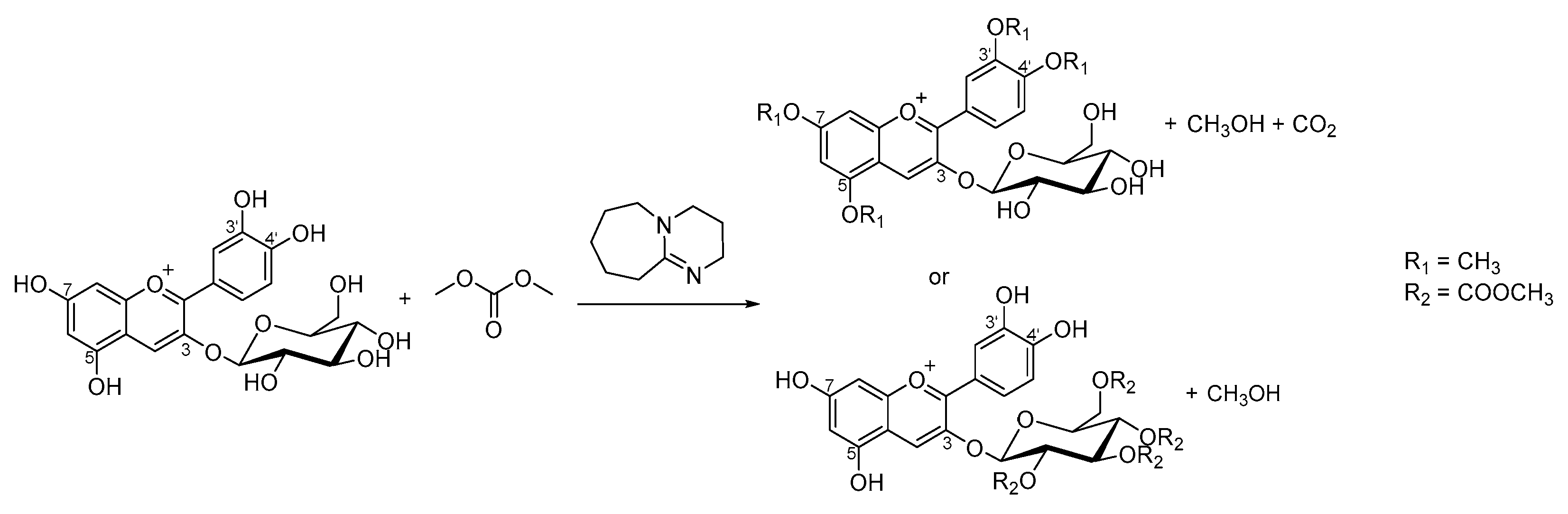
3.2. Hemisynthesis
Sample Preparation
3.3. LC-MS Analysis
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Fernandes, I.; Marques, F.; de Freitas, V.; Mateus, N. Antioxidant and antiproliferative properties of methylated metabolites of anthocyanins. Food Chem. 2013, 141, 2923–2933. [Google Scholar] [CrossRef]
- Mazza, G.J. Anthocyanins and heart health. Ann. Ist. Super. Sanita 2007, 43, 369–374. [Google Scholar]
- Wu, X.; Pittman, H.E.; McKay, S.; Prior, R.L. Aglycones and sugar moieties alter anthocyanin absorption and metabolism after berry consumption in weanling pigs. J. Nutr. 2005, 135, 2417–2424. [Google Scholar] [CrossRef] [PubMed]
- Kamiloglu, S.; Capanoglu, E.; Grootaert, C.; van Camp, J. Anthocyanin Absorption and Metabolism by Human Intestinal Caco-2 Cells—A Review. Int. J. Mol. Sci. 2015, 16, 21555–21574. [Google Scholar] [CrossRef] [PubMed]
- Bouktaib, M.; Lebrun, S.; Atmani, A.; Rolando, C. Hemisynthesis of all the O-monomethylated analogues of quercetin including the major metabolites, through selective protection of phenolic functions. Tetrahedron 2002, 58, 10001–10009. [Google Scholar] [CrossRef]
- Blount, J.W.; Redan, B.W.; Ferruzzi, M.G.; Reuhs, B.L.; Cooper, B.R.; Harwood, J.S.; Shulaev, V.; Pasinetti, G.; Dixon, R.A. Synthesis and quantitative analysis of plasma-targeted metabolites of catechin and epicatechin. J. Agric. Food Chem. 2015, 63, 2233–2240. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Pittman, H.E.; Prior, R.L. Pelargonidin is absorbed and metabolized differently than cyanidin after marionberry consumption in pigs. J. Nutr. 2004, 134, 2603–2610. [Google Scholar] [CrossRef]
- Cruz, L.; Basílio, N.; Mateus, N.; Pina, F.; de Freitas, V. Characterization of kinetic and thermodynamic parameters of cyanidin-3-glucoside methyl and glucuronyl metabolite conjugates. J. Phys. Chem. B 2015, 119, 2010–2018. [Google Scholar] [CrossRef]
- Schmitt, S.; Tratzka, S.; Schieber, A.; Passon, M. Hemisynthesis of Anthocyanin Phase II Metabolites by Porcine Liver Enzymes. J. Agric. Food Chem. 2019, 67, 6177–6189. [Google Scholar] [CrossRef]
- Cruz, L.; Mateus, N.; de Freitas, V. First chemical synthesis report of an anthocyanin metabolite with in vivo occurrence: Cyanidin-4′-O-methyl-3-glucoside. Tetrahedron Lett. 2013, 54, 2865–2869. [Google Scholar] [CrossRef]
- Cren-Olivé, C.; Lebrun, S.; Rolando, C. An efficient synthesis of the four mono methylated isomers of (+)-catechin including the major metabolites and of some dimethylated and trimethylated analogues through selective protection of the catechol ring. J. Chem. Soc. Perkin Trans. 2002, 6, 821–830. [Google Scholar] [CrossRef]
- Kozłowska, J.; Potaniec, B.; Żarowska, B.; Anioł, M. Synthesis and Biological Activity of Novel O-Alkyl Derivatives of Naringenin and Their Oximes. Molecules 2017, 22, 1485. [Google Scholar] [CrossRef]
- Tundo, P.; Musolino, M.; Aricò, F. The reactions of dimethyl carbonate and its derivatives. Green Chem. 2018, 20, 28–85. [Google Scholar] [CrossRef]
- Tundo, P.; Memoli, S.; Hérault, D.; Hill, K. Synthesis of methylethers by reaction of alcohols with dimethylcarbonate. Green Chem. 2004, 6, 609–612. [Google Scholar] [CrossRef]
- Lee, Y.; Shimizu, I. Convenient O-Methylation of Phenols with Dimethyl Carbonate. Synlett 1998, 1998, 1063–1064. [Google Scholar] [CrossRef]
- Ouk, S.; Thiebaud, S.; Borredon, E.; Legars, P.; Lecomte, L. O-Methylation of phenolic compounds with dimethyl carbonate under solid/liquid phase transfer system. Tetrahedron Lett. 2002, 43, 2661–2663. [Google Scholar] [CrossRef]
- Ouk, S.; Thiébaud, S.; Borredon, E.; Le Gars, P. High performance method for O-methylation of phenol with dimethyl carbonate. Appl. Catal. A 2003, 241, 227–233. [Google Scholar] [CrossRef]
- Rein, M. Copigmentation Reactions and Color Stability of Berry Anthocyanins; University of Helsinki: Helsinki, Finland, 2005; ISBN 952-10-2293-0. [Google Scholar]
- Shieh, W.C.; Dell, S.; Repic, O. 1,8-Diazabicyclo5.4.0undec-7-ene (DBU) and microwave-accelerated green chemistry in methylation of phenols, indoles, and benzimidazoles with dimethyl carbonate. Org. Lett. 2001, 3, 4279–4281. [Google Scholar] [CrossRef] [PubMed]
- Tatsuzaki, J.; Ohwada, T.; Otani, Y.; Inagi, R.; Ishikawa, T. A simple and effective preparation of quercetin pentamethyl ether from quercetin. Beilstein J. Org. Chem. 2018, 14, 3112–3121. [Google Scholar] [CrossRef]
- Bernini, R.; Crisante, F.; Ginnasi, M.C. A convenient and safe O-methylation of flavonoids with dimethyl carbonate (DMC). Molecules 2011, 16, 1418–1425. [Google Scholar] [CrossRef] [PubMed]
- Ouk, S.; Thiébaud, S.; Borredon, E.; Le Gars, P. Dimethyl carbonate and phenols to alkyl aryl ethers via clean synthesis. Green Chem. 2002, 4, 431–435. [Google Scholar] [CrossRef]
- Mason, T.J. Ultrasound in synthetic organic chemistry. Chem. Soc. Rev. 1997, 26, 443. [Google Scholar] [CrossRef]
- Riesz, P.; Berdahl, D.; Christman, C.L. Free radical generation by ultrasound in aqueous and nonaqueous solutions. Environ. Health Perspect. 1985, 64, 233–252. [Google Scholar] [CrossRef] [PubMed]
- González-Manzano, S.; González-Paramás, A.; Santos-Buelga, C.; Dueñas, M. Preparation and characterization of catechin sulfates, glucuronides, and methylethers with metabolic interest. J. Agric. Food Chem. 2009, 57, 1231–1238. [Google Scholar] [CrossRef] [PubMed]
- Cren-Olivé, C.; Déprez, S.; Lebrun, S.; Coddeville, B.; Rolando, C. Characterization of methylation site of monomethylflavan-3-ols by liquid chromatography/electrospray ionization tandem mass spectrometry. Rapid Commun. Mass Spectrom. 2000, 14, 2312–2319. [Google Scholar] [CrossRef]
- Donovan, J.L.; Luthria, D.L.; Stremple, P.; Waterhouse, A.L. Analysis of (+)-catechin, (−)-epicatechin and their 3′- and 4′-O-methylated analogs. J. Chromatogr. B Biomed. Sci. Appl. 1999, 726, 277–283. [Google Scholar] [CrossRef]
- Feuereisen, M.M.; Hoppe, J.; Zimmermann, B.F.; Weber, F.; Schulze-Kaysers, N.; Schieber, A. Characterization of phenolic compounds in Brazilian pepper (Schinus terebinthifolius Raddi) exocarp. J. Agric. Food Chem. 2014, 62, 6219–6226. [Google Scholar] [CrossRef]
- Day, A.J.; Bao, Y.; Morgan, M.R.; Williamson, G. Conjugation position of quercetin glucuronides and effect on biological activity. Free Radic. Biol. Med. 2000, 29, 1234–1243. [Google Scholar] [CrossRef]
- Toki, K.; Saito, N.; Irie, Y.; Tatsuzawa, F.; Shigihara, A.; Honda, T. 7-O-Methylated anthocyanidin glycosides from Catharanthus roseus. Phytochemistry 2008, 69, 1215–1219. [Google Scholar] [CrossRef]
- Hvattum, E.; Ekeberg, D. Study of the collision-induced radical cleavage of flavonoid glycosides using negative electrospray ionization tandem quadrupole mass spectrometry. J. Mass Spectrom. 2003, 38, 43–49. [Google Scholar] [CrossRef] [PubMed]
- Barnes, J.S.; Schug, K.A. Structural characterization of cyanidin-3,5-diglucoside and pelargonidin-3,5-diglucoside anthocyanins: Multi-dimensional fragmentation pathways using high performance liquid chromatography-electrospray ionization-ion trap-time of flight mass spectrometry. Int. J. Mass Spectrom. 2011, 308, 71–80. [Google Scholar] [CrossRef]
- Dangles, O.; Fenger, J.A. The Chemical Reactivity of Anthocyanins and Its Consequences in Food Science and Nutrition. Molecules 2018, 23, 1970. [Google Scholar] [CrossRef] [PubMed]
- Rao, K.V.; Owoyale, J.A. Partial methylation of quercetin: Direct synthesis of tamarixetin, ombuin and ayanin. J. Heterocycl. Chem. 1976, 13, 1293–1295. [Google Scholar] [CrossRef]
- Fleschhut, J.; Kratzer, F.; Rechkemmer, G.; Kulling, S.E. Stability and biotransformation of various dietary anthocyanins in vitro. Eur. J. Nutr. 2006, 45, 7–18. [Google Scholar] [CrossRef] [PubMed]
- Tomaz, I.; Šikuten, I.; Preiner, D.; Andabaka, Ž.; Huzanić, N.; Lesković, M.; Karoglan Kontić, J.; Ašperger, D. Stability of polyphenolic extracts from red grape skins after thermal treatments. Chem. Pap. 2019, 73, 195–203. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound Number | [M]+ (m/z) Observed | [M]+ (m/z) Calculated | Mass Error (mDa) | RT (min) | CCS Value [M]+ (Å2) | Fragment Ions (m/z) | Yield (%) a | λmax (nm) |
|---|---|---|---|---|---|---|---|---|
| Reference Substances | ||||||||
| cyanidin-3-O-glucoside | 21 b | |||||||
| 449.1078 | 449.1078 | ±0.0 | 3.42 | 202.8 ± 0.4 | 287.0551 | 518 | ||
| peonidin-3-O-glucoside | ||||||||
| 463.1234 | 463.1235 | −0.1 | 4.19 | 207.0 ± 0.3 | 286.0471, 301.0707 | 519 | ||
| Methylated Compounds | ||||||||
| monomethylated cyanidin-3-O-glucoside | 8 | |||||||
| 1 | 463.1230 | 463.1235 | −0.5 | 3.95 | 207.2 ± 0.2 | 286.0470, 301.0707 | 520 | |
| 2 | 463.1228 | 463.1235 | −0.7 | 4.16 | 208.1 ± 0.3 | 286.0473, 301.0704 | 517 | |
| 3 | 463.1231 | 463.1235 | −0.4 | 4.36 | 207.3 ± 0.3 | 286.0471, 301.0706 | 517 | |
| dimethylated cyanidin-3-O-glucoside | 3 | |||||||
| 6 | 477.1386 | 477.1391 | −0.5 | 4.60 | 213.5 ± 0.4 | 300.0624, 315.0861 | 516 | |
| 7 | 477.1397 | 477.1391 | +0.6 | 4.99 | 212.9 ± 0.5 | 315.0862 | 518 | |
| 8 | 477.1390 | 477.1391 | −0.1 | 5.17 | 212.3 ± 0.1 | 300.0638, 315.0858 | 516 | |
| trimethylated cyanidin-3-O-glucoside | 1 | |||||||
| 9 | 491.1547 | 491.1548 | −0.1 | 5.38 | 218.3 ± 0.2 | 329.1019 | 520 | |
| 10 | 491.1543 | 491.1548 | −0.5 | 5.97 | 216.5 ± 0.4 | 329.1026 | 518 | |
| 11 | 491.1548 | 491.1548 | ±0.0 | 6.08 | 218.3 ± 0.3 | 329.1019 | 517 | |
| tetramethylated cyanidin-3-O-glucoside | 1 | |||||||
| 12 | 505.1686 | 505.1704 | −1.8 | 6.97 | 223.3 ± 0.2 | 343.1201 | 518 | |
| carboxymethylated cyanidin-3-O-glucoside | 4 | |||||||
| 13 | 507.1132 | 507.1133 | −0.1 | 4.45 | 215.7 ± 0.3 | 287.0544 | 515 | |
| 14 | 507.1134 | 507.1133 | +0.1 | 4.56 | 212.2 ± 0.3 | 287.0549 | 515 | |
| 15 | 507.1134 | 507.1133 | +0.1 | 5.00 | 212.8 ± 0.6 | 287.0551 | 521 | |
| Compound Number | [M]+ (m/z) Observed | [M]+ (m/z) Calculated | Mass Error (mDa) | RT (min) | CCS Value [M]+ (Å2) | Fragment Ions MS (m/z) | λmax (nm) | |
|---|---|---|---|---|---|---|---|---|
| Reference Substances | ||||||||
| cyanidin | ||||||||
| 287.0548 | 287.0550 | −0.2 | 4.86 | 154.7 ± 0.2 | 213.0545, 231.06493, 241.04919, 269.04454 | 527 | ||
| peonidin | ||||||||
| 301.0711 | 301.0707 | +0.4 | 5.98 | 159.1 ± 0.1 | 201.05474, 229.04958, 258.05195, 286.0471 | 527 | ||
| 7-O-methylcyanidin | ||||||||
| 301.0702 | 301.0707 | −0.5 | 5.99 | 160.3 ± 0.5 | 286.04672, 258.05171 | 524 | ||
| Methylated Compounds | ||||||||
| monomethylated cyanidin | ||||||||
| 16 | 301.0706 | 301.0707 | −0.1 | 5.48 | 160.6 ± 0.5 | 258.05262, 269.04454, 286.04732 | n.d. | |
| 17 | 301.0704 | 301.0707 | −0.3 | 5.71 | 159.4 ± 0.2 | 258.05262, 286.04732 | 528 | |
| 18 | 301.0701 | 301.0707 | −0.6 | 5.99 | 160.3 ± 0.5 | 201.05407, 213.05477, 229.04907, 258.05165, 286.04683 | 524 | |
| dimethylated cyanidin | ||||||||
| 19 | 315.0860 | 315.0863 | −0.3 | 6.67 | 164.4 ± 0.3 | 272.06712, 300.06231 | 527 | |
| 20 | 315.0860 | 315.0863 | −0.3 | 6.74 | 164.7 ± 0.6 | 257.04412, 201.05023, 272.06728, 301.06719, | 528 | |
| 21 | 315.0858 | 315.0863 | −0.5 | 6.97 | 164.9 ± 0.3 | 257.04467, 271.06016, 300.06297 | 523 | |
| trimethylated cyanidin a | ||||||||
| 22 | 329.1011 | 329.1020 | −0.9 | 7.78 | 170.0 ± 0.4 | 314.07906, 286.08402, 271.06265, 257.08148 | 526 | |
| 23 | 329.1017 | 329.1020 | −0.3 | 7.94 | 170.0 ± 0.2 | 314.07861, 286.08251, 271.06018, 257.08116 | 524 | |
| 24 | 329.1014 | 329.1020 | −0.6 | 8.09 | 170.9 ± 0.4 | 313.07039, 314.0769, 285.07596, 268.07533 | 524 | |
| tetramethylated cyanidin | ||||||||
| 25 | 343.1177 | 343.1176 | +0.1 | 8.80 | 176.8 ± 0.2 | 327.08619, 312.06264, 299.09129, 285.07529 | 522 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Straßmann, S.; Brehmer, T.; Passon, M.; Schieber, A. Methylation of Cyanidin-3-O-Glucoside with Dimethyl Carbonate. Molecules 2021, 26, 1342. https://doi.org/10.3390/molecules26051342
Straßmann S, Brehmer T, Passon M, Schieber A. Methylation of Cyanidin-3-O-Glucoside with Dimethyl Carbonate. Molecules. 2021; 26(5):1342. https://doi.org/10.3390/molecules26051342
Chicago/Turabian StyleStraßmann, Sarah, Tillman Brehmer, Maike Passon, and Andreas Schieber. 2021. "Methylation of Cyanidin-3-O-Glucoside with Dimethyl Carbonate" Molecules 26, no. 5: 1342. https://doi.org/10.3390/molecules26051342
APA StyleStraßmann, S., Brehmer, T., Passon, M., & Schieber, A. (2021). Methylation of Cyanidin-3-O-Glucoside with Dimethyl Carbonate. Molecules, 26(5), 1342. https://doi.org/10.3390/molecules26051342