
Synthesis of Bis(Carboranyl)amides 1,1′-μ-(CH2NH(O)C(CH2)n-1,2-C2B10H11)2 (n = 0, 1) and Attempt of Synthesis of Gadolinium Bis(Dicarbollide)

 ,
,  , and
, and 
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
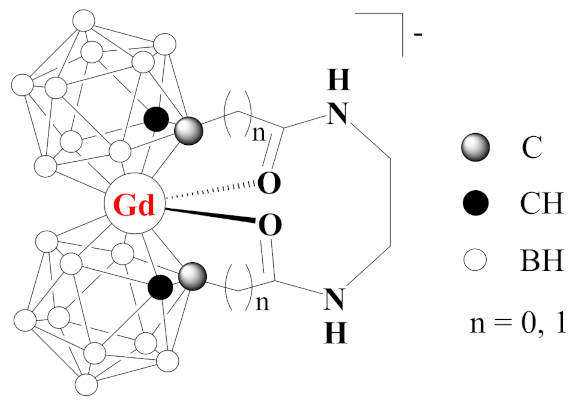
2. Results and Discussion
3. Materials and Methods
3.1. General Methods
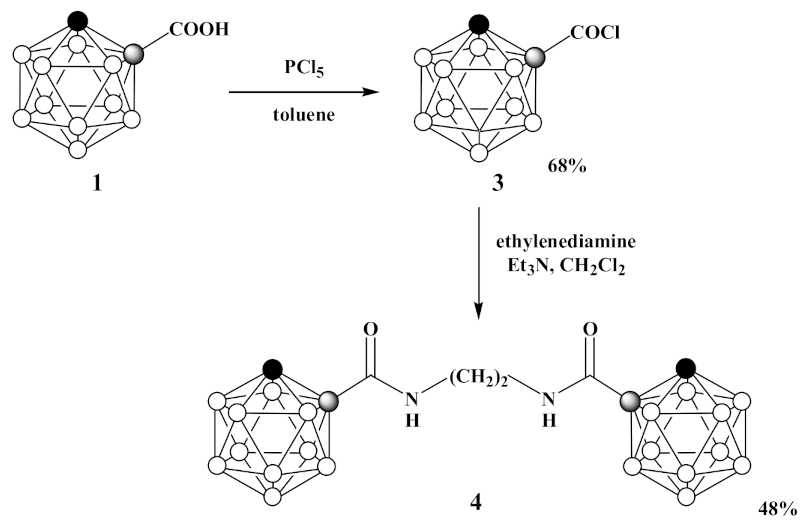
3.2. Synthesis of 1-Cl(O)C-1,2-C2B10H11 (3)
3.3. Synthesis of 1,1′-(CH2NH(O)C-1,2-C2B10H11)2 (4)
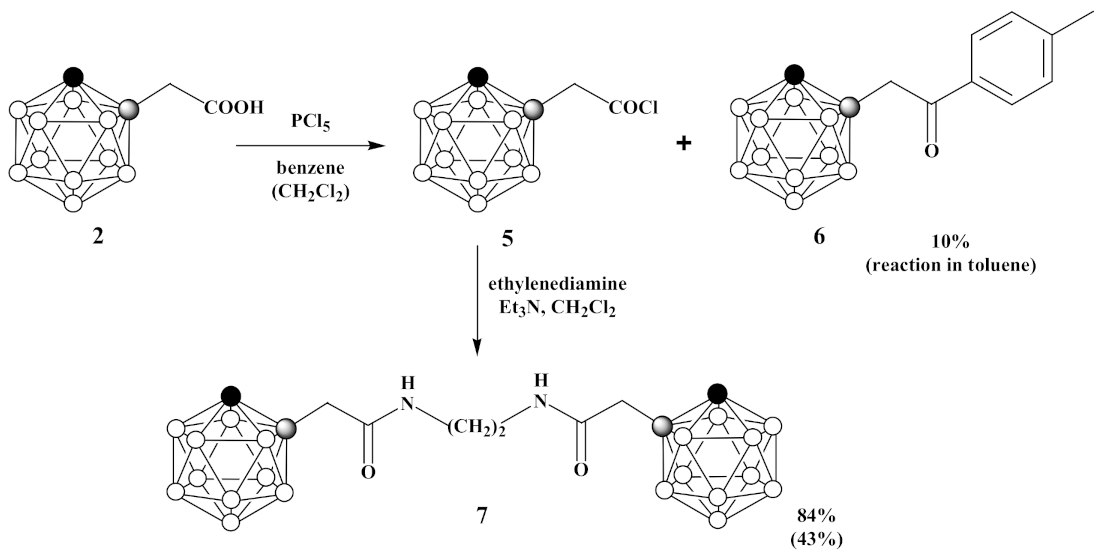
3.4. Synthesis of 1-Cl(O)CCH2-1,2-C2B10H11 (5), 1,1′-(CH2NH(O)CCH2-1,2-C2B10H11)2 (6) and 1-(p-CH3C6H4(O)CCH2)-1,2-C2B10H11 (7)
- (a)
- Under an argon atmosphere phosphorus pentachloride (1.69 g, 8.2 mmol) was slowly added to 1-HOOCCH2-1,2-C2B10H11 (2) (1.65 g, 8.2 mmol) in 30 mL of benzene and reaction mixture was stirred for 30 min. Then, the benzene and phosphoryl chloride were removed by distillation at 110 °C. The obtained 1-Cl(O)CCH2-1,2-C2B10H11 (5) was used in the next step without further purification. Under argon atmosphere mixture of ethylenediamine (0.27 mL, 4.1 mmol) and triethylamine (1.13 mL, 8.2 mmol) in 10 mL of dichloromethane was added dropwise to 1-Cl(O)CCH2-1,2-C2B10H11 (5) (1.80 g, 8.2 mmol) in 10 mL of dichloromethane and reaction mixture was stirred for 60 min. Thereafter, volatiles were removed under reduced pressure. The column chromatography on silica using ethyl acetate as eluent gave white solid of 1,1′-(CH2NH(O)CCH2-1,2-C2B10H11)2 (7) (1.47 g, yield 84%).
- (b)
- Under an argon atmosphere phosphorus pentachloride (0.21 g, 1.0 mmol) was slowly added to 1-HOOCCH2-1,2-C2B10H11 (2) (0.20 g, 1.0 mmol) in 5 mL of dichloromethane and reaction mixture was stirred for 10 min. Then, the reaction mixture was heated under reflux for 1 h. Thereafter, the dichloromethane and phosphoryl chloride were removed by distillation at 110 °C. The obtained 1-Cl(O)CCH2-1,2-C2B10H11 (5) was used in the next step without further purification. Under argon atmosphere mixture of ethylenediamine (0.03 mL, 0.5 mmol) and triethylamine (0.14 mL, 1.0 mmol) in 10 mL of dichloromethane was added dropwise to 1-Cl(O)CCH2-1,2-C2B10H11 (5) in 10 mL of dichloromethane and reaction mixture was stirred for 60 min. Thereafter, volatiles were removed under reduced pressure. The column chromatography on silica using a mixture of ethyl acetate and hexane (1:1, v/v) as eluent gave pure white solid of 1,1′-(CH2NH(O)CCH2-1,2-C2B10H11)2 (7) (0.18 g, yield 43%). 1,1′-(CH2NH(O)CCH2- 1,2-C2B10H11)2 (7): 1H-NMR (400 MHz, acetone-d6): δ 7.75 (2H, br s, NH), 4.98 (2H, br s, CHCarb), 3.32 (4H, t (1:1:1), J2N,H = 2.7 Hz, NCH2), 3.23 (4H, s, CH2), 3.1–1.3 (br m, BHCarb) ppm. 11B-NMR (128 MHz, acetone-d6): δ −2.8 (2B, d, J = 147 Hz), −5.6 (2B, d, J = 145 Hz), −9.7 (4B, d, J = 130 Hz), −10.6 (4B, d, J = 137 Hz), −11.7 (4B, d, J = 152 Hz), −13.0 (4B, d, J = 158 Hz) ppm. 13C-NMR (100 MHz, acetone-d6): δ 167.4 (CO), 71.6 (CCarbCH2), 61.0 (CCarbH), 43.3 (NCH2), 39.7 (CH2) ppm. HRMS (ESI): found m/z 429.4545 [М + H]+, C10H33B20N2O2, calculated for C10H33B20N2O2 [М + H]+ = 429.4549.
- (c)
- Under an argon atmosphere phosphorus pentachloride (0.42 g, 2.0 mmol) was slowly added to 1-HOOCCH2-1,2-C2B10H11 (2) (0.40 g, 2.0 mmol) in 10 mL of toluene and reaction mixture was stirred for 10 min. Then, the reaction mixture was heated under reflux for 30 min. Thereafter, the toluene and phosphoryl chloride were removed by distillation at 110 °C. A mixture of ethylenediamine (0.07 mL, 1.0 mmol) and triethylamine (0.28 mL, 2.0 mmol) in 10 mL of dichloromethane was added dropwise to solution of crude 1-Cl(O)CCH2-1,2-C2B10H11 (1.80 g, 8.2 mmol) in 10 mL of dichloromethane and reaction mixture was stirred for 60 min. Thereafter, volatiles were removed under reduced pressure. The crude material was purified by recrystallization from hexane to give pure colorless crystalline solid of 1-p-CH3C6H4(O)CCH2-1,2-C2B10H11 (6) (0.06 g, yield 10%). 1H-NMR (400 MHz, acetone-d6): δ 7.91 (2H, d, J = 8.2 Hz, CHAr), 7.37 (2H, d, J = 8.2 Hz, CHAr), 5.08 (1H, br s, CHCarb), 4.12 (2H, s, CH2), 2.41 (3H, s, CH3) ppm. 11B-NMR (128 MHz, acetone-d6): δ −2.9 (1B, d, J = 148 Hz), −5.3 (1B, d, J = 146 Hz), −9.7 (2B, d, J = 135 Hz), −10.6 (2B, d, J = 129 Hz), −11.8 (2B, d, J = 158 Hz), −12.9 (2B, d, J = 156 Hz) ppm. MS (ESI): found m/z 275.3 [М − H]−, C11H20B10O, calculated for C11H20B10O [М − H]− = 275.2.
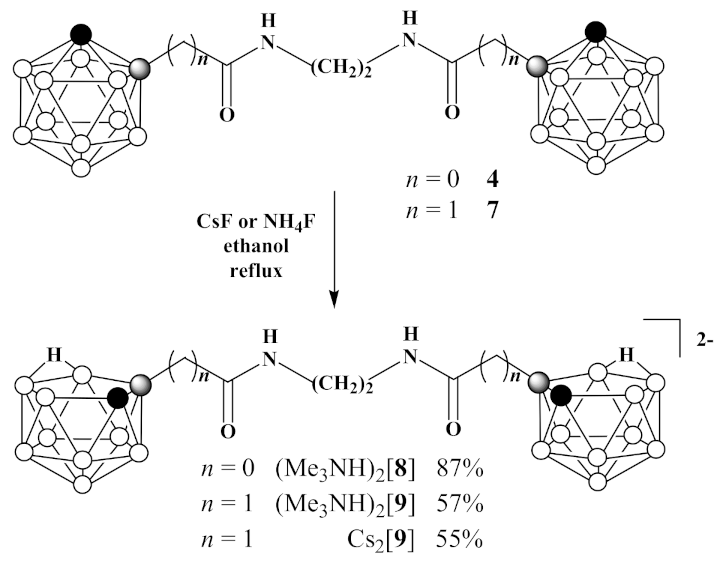
3.5. Synthesis of (Me3NH)2[7,7′(8′)-(CH2NH(O)C)-7,8-C2B9H11)2] ((Me3NH)2 [8])
3.6. Synthesis of Cs2[7,7′(8′)-(CH2NH(O)CCH2-7,8-C2B9H11)2] (Cs2 [9]) and (Me3NH)2[7,7′(8′)-(CH2NH(O)CCH2-7,8-C2B9H11)2] ((Me3NH)2[9])
- (a)
- Cesium fluoride (3.12 g, 20.5 mmol) was added to 1,1′-(CH2NH(O)CCH2-1,2-C2B10H11)2 (7) (1.47 g, 3.4 mmol) in 30 mL of ethanol and reaction mixture was heated under reflux for 48 h. Thereafter, the reaction mixture was filtered, volatiles were removed under reduced pressure. The column chromatography on silica using ethanol as eluent gave pure white solid of Cs2[7,7′(8′)-(CH2NH(O)CCH2-7,8-C2B9H11)2] (Cs2[9]) (1.24 g, yield 55%). 1H-NMR (400 MHz, acetone-d6): δ 7.40 (2H, br s, NH), 3.40 (4H, s, NCH2), 2.57 (2H, d, J = 14.9 Hz, CHH), 2.25 (2H, d, J = 14.9 Hz, CHH), 1.80 (2H, br s, CHCarb), −2.79 (2H, br q (1:1:1:1), J = 54 Hz, BHB) ppm. 11B-NMR (128 MHz, acetone-d6): δ −9.9 (2B, d, J = 137 Hz), −11.6 (2B, d, J = 151 Hz), −13.8 (4B, d, J = 133 Hz), −20.4 (6B, d, J = 130 Hz), −32.5 (2B, d, J = 118 Hz), −36.5 (2B, d, J = 138 Hz) ppm. HRMS (ESI): found m/z 203.7113 [М]2−, C10H32B18N2O2, calculated for C10H32B18N2O2 [М]2− = 203.7133.
- (b)
- Ammonium fluoride (0.11 g, 3.0 mmol) was added to 1,1′-(CH2NH(O)C-1,2-C2B10H11)2 (7) (0.09 g, 0.2 mmol) in 30 mL of ethanol and reaction mixture was heated under reflux until the disappearance of starting material on TLC. Thereafter, volatiles were removed under reduced pressure and to the residue water (5 mL) was added. The aquatic solution was filtered and added to trimethylamine hydrochloride (0.10 g, 1.0 mmol) in 5 mL of water to form precipitate. White solid was filtered and dried over P2O5 to give (Me3NH)2[7,7′(8′)-(CH2NH(O)C-7,8-C2B9H11)2] ((Me3NH)2[9]) (0.06 g, yield 57%). 1H-NMR (400 MHz, acetone-d6): δ 7.15 (br s, NH), 3.36 (4H, m, NCH2), 3.20 (18H, s, Me3NH+), 2.55 (2H, d, J = 15.0 Hz, CHH), 2.24 (2H, d, J = 15.0 Hz, CHH), 1.81 (2H, br s, CHCarb), −2.71 (2H, br q (1:1:1:1), J = 42 Hz, BHB) ppm. 11B-NMR (128 MHz, acetone-d6): δ −10.1 (2B, d, J = 133 Hz), −11.2 (2B, d, J = 138 Hz), −13.7 (2B, d, J = 170 Hz), −15.0 (2B, d, J = 135 Hz), −19.4 (4B, d, J = 130 Hz), −21.0 (2B, d, J = 156 Hz), −32.7 (2B, dd, J1 = 128 Hz, J2 = 42 Hz), −36.6 (2B, d, J = 135 Hz) ppm.
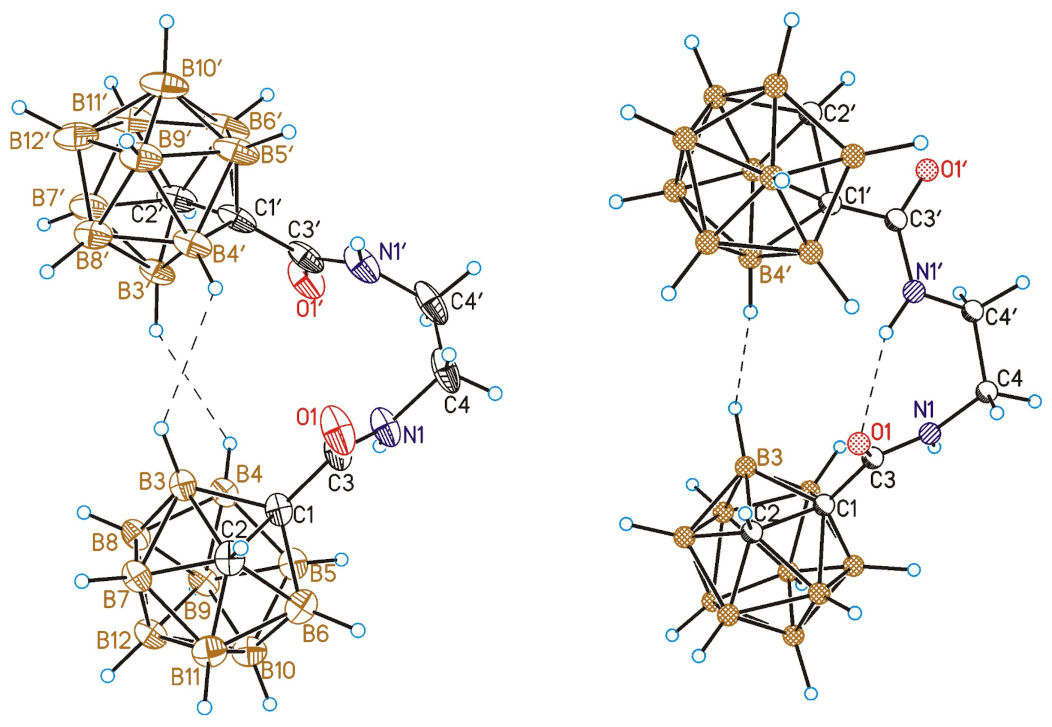
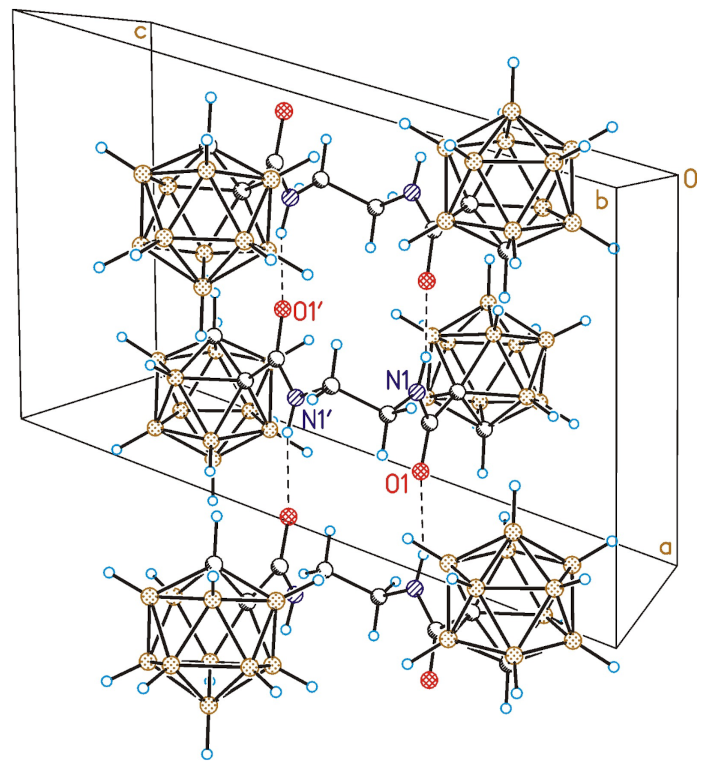
3.7. Single Crystal X-ray Diffraction Study
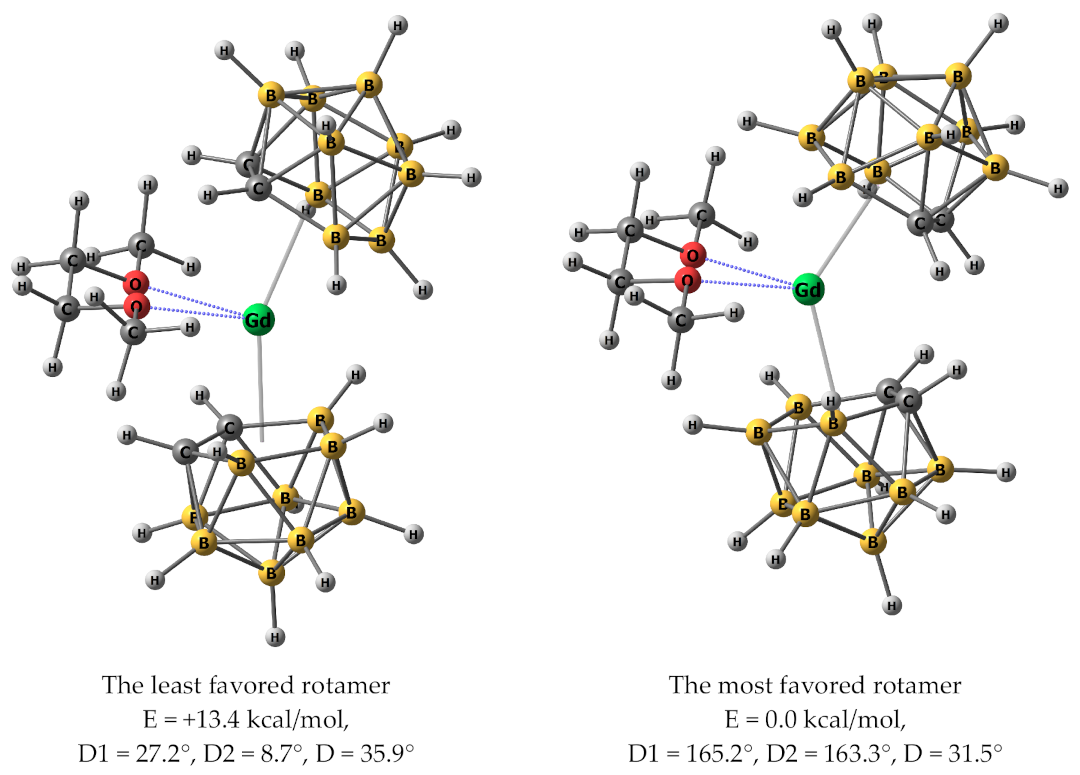
3.8. Quantum-Chemical Calculations
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Takagaki, M.; Kazuko, U.; Hosmane, N.S. An overview of clinical and biological aspects of current boron neutron capture therapy (BNCT) for cancer treatment. In Handbook of Boron Science: With Applications in Organometallics, Catalysis, Materials and Medicine. Volume 4: Boron in Medicine; Hosmane, N.S., Eagling, R., Eds.; World Scientific: London, UK, 2019; pp. 101–143. [Google Scholar] [CrossRef]
- Bregadze, V.I.; Sivaev, I.B. Polyhedral boron compounds for BNCT. In Boron Science: New Technologies and Applications; Hosmane, N.S., Ed.; CRC Press: Boca Raton, FL, USA, 2012; pp. 181–207. [Google Scholar] [CrossRef]
- Sibrian-Vazquez, M.; Vicente, M.G.H. Boron tumor delivery for BNCT: Recent developments and perspectives. In Boron Science: New Technologies and Applications; Hosmane, N.S., Ed.; CRC Press: Boca Raton, FL, USA, 2012; pp. 209–242. [Google Scholar] [CrossRef]
- Xuan, S.; Vicente, M.G.H. Recent advances in boron delivery agents for boron neutron capture therapy (BNCT). In Boron-Based Compounds: Potential and Emerging Applications in Biomedicine; Hey-Hawkins, E., Viñas, C., Eds.; JohnWiley & Sons Ltd.: Chichester, UK, 2018; pp. 298–342. [Google Scholar] [CrossRef]
- Ali, F.; Hosmane, N.S.; Zhu, Y. Boron chemistry for medical applications. Molecules 2020, 25, 828. [Google Scholar] [CrossRef]
- Hu, K.; Yang, Z.; Zhang, L.; Xie, L.; Wang, L.; Xu, H.; Josephson, L.; Liang, S.H.; Zhang, M.-R. Boron agents for neutron capture therapy. Coord. Chem. Rev. 2020, 405, 213139. [Google Scholar] [CrossRef]
- Wittig, A.; Michel, J.; Moss, R.L.; Stecher-Rasmussen, F.; Arlinghaus, H.F.; Bendel, P.; Mauri, P.L.; Altieri, S.; Hilger, R.; Salvadori, P.A.; et al. Boron analysis and boron imaging in biological materials for Boron Neutron Capture Therapy (BNCT). Critical Rev. Oncol./Hematol. 2008, 68, 66–90. [Google Scholar] [CrossRef] [PubMed]
- Protti, N.; Deagostino, A.; Boggio, P.; Alberti, D.; Geninatti Crich, S. New boronated compounds for an imaging-guided personalized neutron capture therapy. In Boron-Based Compounds: Potential and Emerging Applications in Biomedicine; Hey-Hawkins, E., Viñas, C., Eds.; John Wiley & Sons Ltd.: Chichester, UK, 2018; pp. 389–415. [Google Scholar] [CrossRef]
- Tolmachev, V.; Koziorowski, J.; Sivaev, I.; Lundqvist, H.; Carlsson, J.; Orlova, A.; Gedda, L.; Olsson, P.; Sjöberg, S.; Sundin, A. closo-Dodecaborate(2-) as a linker for iodination of macromolecules. Aspects on conjugation chemistry and biodistribution. Bioconjug. Chem. 1999, 10, 338–345. [Google Scholar] [CrossRef] [PubMed]
- Tolmachev, V.; Bruskin, A.; Sivaev, I.; Lundqvist, H.; Sjöberg, S. Radiobromination of closo-dodecaborate anion. Aspects of labelling chemistry in aqueous solution using Chloramine-T. Radiochim. Acta 2002, 90, 29–235. [Google Scholar] [CrossRef]
- Munck af Rosenschöld, P.M.; Verbakel, W.F.A.R.; Ceberg, C.P.; Stecher-Rasmussen, F.; Persson, B.R.R. Toward clinical application of prompt gamma spectroscopy for in vivo monitoring of boron uptake in boron neutron capture therapy. Med. Phys. 2001, 28, 787–795. [Google Scholar] [CrossRef]
- Caravan, P.; Ellison, J.J.; McMurry, T.J.; Lauffer, R.B. Gadolinium(III) chelates as MRI contrast agents: Structure, dynamics, and applications. Chem. Rev. 1999, 99, 2293–2352. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.-T.; Chuang, K.-H. Gd(III) chelates for MRI contrast agents: From high relaxivity to “smart”, from blood pool to blood–brain barrier permeable. Med. Chem. Commun. 2012, 3, 552–565. [Google Scholar] [CrossRef]
- Li, X.; Sun, Y.; Ma, L.; Liu, G.; Wang, Z. The renal clearable magnetic resonance imaging contrast agents: State of the art and recent advances. Molecules 2020, 25, 5072. [Google Scholar] [CrossRef] [PubMed]
- Hosmane, N.S.; Maguire, J.A.; Zhu, Y.; Takagaki, M. Boron and Gadolinium Neutron Capture Therapy for Cancer Treatment; World Scientific Publishing: Hackensack, NJ, USA, 2012; p. 272. [Google Scholar] [CrossRef]
- Deagostino, A.; Protti, N.; Alberti, D.; Boggio, P.; Bortolussi, S.; Altieri, S.; Geninatti Crich, S. Insights into the use of gadolinium and gadolinium/boron-based agents in imaging-guided neutron capture therapy applications. Future Med. Chem. 2016, 8, 899–917. [Google Scholar] [CrossRef] [PubMed]
- Nemoto, H.; Cai, J.; Nakamura, H.; Fujiwara, M.; Yamamoto, Y. The synthesis of a carborane gadolinium–DTPA complex for boron neutron capture therapy. J. Organomet. Chem. 1999, 581, 170–175. [Google Scholar] [CrossRef]
- Nakamura, H.; Fukuda, H.; Girald, F.; Kobayashi, T.; Hiratsuka, J.; Akaizawa, T.; Nemoto, H.; Cai, J.; Yoshida, K.; Yamamoto, Y. In vivo evaluation of carborane gadolinium-DTPA complex as an MR imaging boron carrier. Chem. Pharm. Bull. 2000, 48, 1034–1038. [Google Scholar] [CrossRef]
- Aime, S.; Barge, A.; Crivello, A.; Deagostino, A.; Gobetto, R.; Nervi, C.; Prandi, C.; Toppino, A.; Venturello, P. Synthesis of Gd(III)-C-palmitamidomethyl-C’-DOTAMA-C6-o-carborane: A new dual agent for innovative MRI/BNCT applications. Org. Biomol. Chem. 2008, 6, 4460–4466. [Google Scholar] [CrossRef] [PubMed]
- Geninatti-Crich, S.; Alberti, D.; Szabo, I.; Deagostino, A.; Toppino, A.; Barge, A.; Ballarini, F.; Bortolussi, S.; Bruschi, P.; Protti, N.; et al. MRI-guided neutron capture therapy by use of a dual gadolinium/boron agent targeted at tumour cells through upregulated low-density lipoprotein transporters. Chem. Eur. J. 2011, 17, 8479–8486. [Google Scholar] [CrossRef]
- Alberti, D.; Toppino, A.; Geninatti Crich, S.; Meraldi, C.; Prandi, C.; Protti, N.; Bortolussi, S.; Altieri, S.; Aime, S.; Deagostino, A. Synthesis of a carborane-containing cholesterol derivative and evaluation as a potential dual agent for MRI/BNCT applications. Org. Biomol. Chem. 2014, 12, 2457–2467. [Google Scholar] [CrossRef] [PubMed]
- Goswami, L.N.; Cai, Q.; Ma, L.; Jalisatgi, S.S.; Hawthorne, M.F. Synthesis, relaxation properties and in vivo assessment of a carborane-GdDOTA-monoamide conjugate as an MRI blood pool contrast agent. Org. Biomol. Chem. 2015, 13, 8912–8918. [Google Scholar] [CrossRef]
- Goswami, L.N.; Ma, L.; Kueffer, P.J.; Jalisatgi, S.S.; Hawthorne, M.F. Synthesis and relaxivity studies of a DOTA based nanomolecular chelator assembly supported by an icosahedral closo-B122− core for MRI: A click chemistry approach. Molecules 2013, 18, 9034–9048. [Google Scholar] [CrossRef] [PubMed]
- Goswami, L.N.; Ma, L.; Chakravarty, S.; Cai, Q.; Jalisatgi, S.S.; Hawthorne, M.F. Discrete nanomolecular polyhedral borane scaffold supporting multiple gadolinium(III) complexes as a high performance MRI contrast agent. Inorg. Chem. 2013, 52, 1694–1700. [Google Scholar] [CrossRef]
- Goswami, L.N.; Ma, L.; Cai, Q.; Sarma, S.J.; Jalisatgi, S.S.; Hawthorne, M.F. cRGD peptide-conjugated icosahedral closo-B122− core carrying multiple Gd3+-DOTA Chelates for αvβ3 integrin-targeted tumor imaging (MRI). Inorg. Chem. 2013, 52, 1701–1709. [Google Scholar] [CrossRef] [PubMed]
- Port, M.; Idée, J.-M.; Medina, C.; Robic, C.; Sabatou, M.; Corot, C. Efficiency, thermodynamic and kinetic stability of marketed gadolinium chelates and their possible clinical consequences: A critical review. BioMetals 2008, 21, 469–490. [Google Scholar] [CrossRef]
- McDonald, J.S.; McDonald, R.J. MR imaging safety considerations of gadolinium-based contrast agents: Gadolinium retention and nephrogenic systemic fibrosis. Magn. Reson. Imaging Clin. N. Am. 2020, 28, 497–507. [Google Scholar] [CrossRef]
- Unruh, C.; Van Bavel, N.; Anikovskiy, M.; Prenner, E.J. Benefits and detriments of gadolinium from medical advances to health and ecological risks. Molecules 2020, 25, 5762. [Google Scholar] [CrossRef]
- Grimes, R.N. Carboranes, 3rd ed.; Academic Press: London, UK, 2016; pp. 711–903. [Google Scholar] [CrossRef]
- Lebedev, V.N.; Shemyakin, N.F.; Solodovnikov, S.P.; Zakharkin, L.I. The synthesis of the bis(π-7,8-dicarbaboryl)gadolinium (III) anion. Organomet. Chem. USSR 1988, 1, 401–402. [Google Scholar]
- Anufriev, S.A.; Sivaev, I.B.; Nakamura, H. Two possible ways to combine boron and gadolinium for Gd-guided BNCT. A concept. Phosphorus Sulfur Silicon Relat. Elem. 2020, 195, 910–917. [Google Scholar] [CrossRef]
- Teixidor, F.; Pedrajas, J.; Rojo, I.; Viñas, C.; Kivekäs, R.; Sillanpää, R.; Sivaev, I.; Bregadze, V.; Sjöberg, S. Chameleonic capacity of [3,3′-Co(1,2-C2B9H11)2]- in coordination. Generation of the highly uncommon S(thioether)-Na bond. Organometallics 2003, 22, 3414–3423. [Google Scholar] [CrossRef]
- Kazakov, G.S.; Stogniy, M.Y.; Sivaev, I.B.; Suponitsky, K.Y.; Godovikov, I.A.; Kirilin, A.D.; Bregadze, V.I. Synthesis of crown ethers with the incorporated cobalt bis(dicarbollide) fragment. J. Organomet. Chem. 2015, 798, 196–203. [Google Scholar] [CrossRef]
- Kasar, R.A.; Knudsen, G.M.; Kahl, S.B. Synthesis of 3-amino-1-carboxy-o-carborane and an improved, general method for the synthesis of all three C-amino-C-carboxycarboranes. Inorg. Chem. 1999, 38, 2936–2940. [Google Scholar] [CrossRef] [PubMed]
- Haushalter, R.C.; Butler, W.M.; Rudolph, R.W. The preparation and characterization of several meso- tetracarboranylporphyrins. J. Am. Chem. Soc. 1981, 103, 2620–2627. [Google Scholar] [CrossRef]
- Stanko, V.I.; Klimova, A.I.; Chapovskii, Y.A.; Klimova, T.P. Reactions of nucleophilic reagents with esters, ketones, and amides of the carborane series. J. Gen. Chem. USSR 1966, 36, 1779–1786. [Google Scholar]
- Kahl, S.B.; Joel, D.D.; Finkel, G.C.; Micca, P.L.; Nawrocky, M.M.; Coderre, J.A.; Slatkin, D.N. A carboranyl porphyrin for boron neutron capture therapy of brain tumors. In Clinical Aspects of Neutron Capture Therapy; Fairchild, R.G., Bond, V.P., Woodhead, A.D., Vivirito, K., Eds.; Springer: Boston, MA, USA, 1989; pp. 193–203. [Google Scholar] [CrossRef]
- Evstigneeva, R.P.; Luzgina, V.N.; Ol’shevskaya, V.A.; Zakharkin, L.I. Synthesis of o- and m-carborane-containing derivatives of 5,10,15,20-tetra-(p-aminophenyl)porphyrin. Dokl. Akad. Nauk 1997, 357, 637–639. [Google Scholar]
- Scholz, M.; Blobaum, A.L.; Marnett, L.J.; Hey-Hawkins, E. ortho-Carbaborane derivatives of indomethacin as cyclooxygenase (COX)-2 selective inhibitors. Bioorg. Med. Chem. 2012, 20, 4830–4837. [Google Scholar] [CrossRef] [PubMed]
- Nie, Y.; Wang, Y.; Miao, J.; Bian, D.; Zhang, Z.; Cui, Y.; Sun, G. Synthesis and structural characterization of o-carboranylamides with direct cage-amide bond. Dalton Trans. 2014, 43, 5083–5094. [Google Scholar] [CrossRef] [PubMed]
- Xu, B.; Wang, Y.-P.; Yao, Z.-J.; Jin, G.-X. Metal-induced B–H bond activation: Reactions between half-sandwich Ir and Rh complexes with carboranylthioamide. Dalton Trans. 2015, 44, 1530–1533. [Google Scholar] [CrossRef]
- Scholz, M.S.; Wingen, L.M. Synthesis of dicarba-closo-dodecaborane-1-carboxamides. Inorg. Chem. 2017, 56, 5510–5513. [Google Scholar] [CrossRef]
- Orlova, A.V.; Zinin, A.I.; Malysheva, N.N.; Kononov, L.O.; Sivaev, I.B.; Bregadze, V.I. Conjugates of polyhedral boron compounds with carbohydrates. 1. New approach to the design of selective agents for boron neutron capture therapy of cancer. Russ. Chem. Bull. 2003, 52, 2766–2768. [Google Scholar] [CrossRef]
- Kononov, L.O.; Orlova, A.V.; Zinin, A.I.; Kimel, B.G.; Sivaev, I.B.; Bregadze, V.I. Conjugates of polyhedral boron compounds with carbohydrates. 2. Unexpected easy closo- to nido-transformation of a carborane–carbohydrate conjugate in neutral aqueous solution. J. Organomet. Chem. 2005, 690, 2769–2774. [Google Scholar] [CrossRef]
- Orlova, A.M.; Kononov, L.O.; Kimel, B.G.; Sivaev, I.B.; Bregadze, V.I. Conjugates of polyhedral boron compounds with carbohydrates. 4. Hydrolytic stability of carborane–lactose conjugates depends on the structure of a spacer between the carborane cage and sugar moiety. Appl. Organomet. Chem. 2006, 20, 416–420. [Google Scholar] [CrossRef]
- Likhosherstov, L.M.; Novikova, O.S.; Kononov, L.O.; Orlova, A.V.; Sivaev, I.B.; Bregadze, V.I. Conjugates of polyhedral boron compounds with carbohydrates. 5. Synthesis of glycoconjugates of closo-ortho-carborane and N-acyl-β-lactosylamines with various spacers. Russ. Chem. Bull. 2007, 56, 2105–2108. [Google Scholar] [CrossRef]
- Likhosherstov, L.M.; Novikova, O.S.; Kononov, L.O.; Sivaev, I.B.; Bregadze, V.I. Conjugates of polyhedral boron compounds with carbohydrates. 6. Synthesis of glycoconjugates of closo-ortho-carborane with β-lactosylamine and β-D-galactopyranosyl- amine derivatives as galectin bi- and trivalent ligands. Russ. Chem. Bull. 2009, 58, 446–449. [Google Scholar] [CrossRef]
- Nie, Y.; Wang, Y.; Miao, J.; Hu, C.; Zhang, Z.; Cui, Y.; Li, Y. Facile deboronation of some o-carboranylamides. Eur. J. Inorg. Chem. 2017, 4559–4567. [Google Scholar] [CrossRef]
- Bühl, M.; Hnyk, D.; Machaček, J. Computational study of structures and properties of metallaboranes: Cobalt bis(dicarbollide). Chem. Eur. J. 2005, 11, 4109–4120. [Google Scholar] [CrossRef]
- Kazheva, O.N.; Alexandrov, G.G.; Kravchenko, A.V.; Kosenko, I.D.; Lobanova, I.A.; Sivaev, I.B.; Filippov, O.A.; Shubina, E.S.; Bregadze, V.I.; Starodub, V.A.; et al. Molecular conductors with a 8-hydroxy cobalt bis(dicarbollide) anion. Inorg. Chem. 2011, 50, 444–450. [Google Scholar] [CrossRef] [PubMed]
- Anufriev, S.A.; Erokhina, S.A.; Suponitsky, K.Y.; Godovikov, I.A.; Filippov, O.A.; Fabrizi de Biani, F.; Corsini, M.; Chizhov, A.O.; Sivaev, I.B. Methylsulfanyl-stabilized rotamers of cobalt bis(dicarbollide). Eur. J. Inorg. Chem. 2017, 4444–4451. [Google Scholar] [CrossRef]
- Anufriev, S.A.; Suponitsky, K.Y.; Filippov, O.A.; Sivaev, I.B. Synthesis and structure of methylsulfanyl derivatives of nickel bis(dicarbollide). Molecules 2019, 24, 4449. [Google Scholar] [CrossRef]
- Stogniy, M.Y.; Anufriev, S.A.; Shmal’ko, A.V.; Antropov, S.M.; Anisimov, A.A.; Suponitsky, K.Y.; Filippov, O.A.; Sivaev, I.B. The unexpected reactivity of 9-iodo-nido-carborane: From nucleophilic substitution reactions to the synthesis of tricobalt tris(dicarbollide) Na[4,4′,4”-(MeOCH2CH2O)3-3,3′,3”-Co3(μ3-O)(μ3-S)(1,2-C2B9H10)3]. Dalton Trans. 2021, 50, 2671–2688. [Google Scholar] [CrossRef] [PubMed]
- Stogniy, M.Y.; Kazheva, O.N.; Chudak, D.M.; Shilov, G.V.; Filippov, O.A.; Sivaev, I.B.; Kravchenko, A.V.; Starodub, V.A.; Buravov, L.I.; Bregadze, V.I.; et al. Synthesis and study of C-substituted methylthio derivatives of cobalt bis(dicarbollide). RSC Adv. 2020, 10, 2887–2896. [Google Scholar] [CrossRef]
- Armarego, W.L.F.; Chai, C.L.L. Purification of Laboratory Chemicals, 6th ed.; Butterworth-Heinemann: Burlington, MA, USA, 2009. [Google Scholar] [CrossRef]
- APEX2 and SAINT; Bruker AXS Inc.: Madison, WI, USA, 2014.
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Cryst. C 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Neese, F. Software update: The ORCA program system, version 4.0. WIREs Comp. Mol. Sci. 2017, 8, e1327. [Google Scholar] [CrossRef]
- van Wüllen, C. Molecular density functional calculations in the regular relativistic approximation: Method, application to coinage metal diatomics, hydrides, fluorides and chlorides, and comparison with first-order relativistic calculations. J. Chem. Phys. 1998, 109, 392. [Google Scholar] [CrossRef]
- Adamo, C.; Barone, V. Toward reliable density functional methods without adjustable parameters: The PBE0 model. J. Chem. Phys. 1999, 110, 6158. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef]
- Medvedev, M.G.; Bushmarinov, I.S.; Sun, J.W.; Perdew, J.P.; Lyssenko, K.A. Density functional theory is straying from the path toward the exact functional. Science 2017, 355, 49–52. [Google Scholar] [CrossRef]
- Suponitsky, K.Y.; Burakov, N.I.; Kanibolotsky, A.L.; Mikhailov, V.A. Multiple noncovalent bonding in halogen complexes with oxygen organics. I. Tertiary amides. J. Phys. Chem. A 2016, 120, 4179–4190. [Google Scholar] [CrossRef] [PubMed]
- Suponitsky, K.Y.; Tafur, S.; Masunov, A.E. Applicability of hybrid density functional theory methods to calculation of molecular hyperpolarizability. J. Chem. Phys. 2008, 129, 044109. [Google Scholar] [CrossRef]
- Suponitsky, K.Y.; Masunov, A.E.; Antipin, M.Y. Conformational dependence of the first molecular hyperpolarizability in the computational design of nonlinear optical materials for optical switching. Mendeleev. Commun. 2008, 18, 265–267. [Google Scholar] [CrossRef]
- Pantazis, D.A.; Neese, F. All-electron scalar relativistic basis sets for the lanthanides. Chem. Theory Comput. 2009, 5, 2229–2238. [Google Scholar] [CrossRef]
- Jaoul, A.; Nocton, G.; Clavaguéra, C. Assessment of density functionals for computing thermodynamic properties of lanthanide complexes. Chem. Phys. Chem. 2017, 18, 2688–2696. [Google Scholar] [CrossRef] [PubMed]
- Bader, R.F.W. Atoms in Molecules. A Quantum Theory; Clarendon Press: Oxford, UK, 1990. [Google Scholar]
- Keith, T.A. AIMAll (Version 15.05.18); TK Gristmill Software: Overland Park, KS, USA, 2015. [Google Scholar]
- Espinosa, E.; Molins, E.; Lecomte, C. Hydrogen bond strengths revealed by topological analyses of experimentally observed electron densities. Chem. Phys. Lett. 1998, 285, 170–173. [Google Scholar] [CrossRef]
- Espinosa, E.; Alkorta, I.; Rozas, I.; Elguero, J.; Molins, E. About the evaluation of the local kinetic, potential and total energy densities in closed-shell interactions. Chem. Phys. Lett. 2001, 336, 457–461. [Google Scholar] [CrossRef]
- Dalinger, I.L.; Serushkina, O.V.; Muravyev, N.V.; Meerov, D.B.; Miroshnichenko, E.A.; Kon’kova, T.S.; Suponitsky, K.Y.; Vener, M.V.; Sheremetev, A.B. Azasydnone—novel “green” building block for designing high energetic compounds. J. Mater. Chem. A 2018, 6, 18669–18676. [Google Scholar] [CrossRef]
- Sheremetev, A.B.; Aleksandrova, N.S.; Semyakin, S.S.; Suponitsky, K.Y.; Lempert, D.B. Synthesis and characterization of 3-(5-(fluorodinitromethyl)-1H-1,2,4-triazol-3-yl)-4-nitrofurazan: A novel promising energetic component of boron-based fuels for rocket ramjet engines. Chem. Asian J. 2019, 14, 4255–4261. [Google Scholar] [CrossRef] [PubMed]
- Dmitrienko, A.O.; Karnoukhova, V.A.; Potemkin, A.A.; Struchkova, M.I.; Kryazhevskikh, I.A.; Suponitsky, K.Y. The influence of halogen type on structural features of compounds containing α-halo-α,α-dinitroethyl moieties. Chem. Heterocycl. Comp. 2017, 53, 532–539. [Google Scholar] [CrossRef]
- Anufriev, S.A.; Sivaev, I.B.; Suponitsky, K.Y.; Godovikov, I.A.; Bregadze, V.I. Synthesis of 10-methylsulfide and 10-alkylmethylsulfonium nido-carborane derivatives: B-H···π Interactions between the B-H-B hydrogen atom and alkyne group in 10-RC≡CCH2S(Me)-7,8-C2B9H11. Eur. J. Inorg. Chem. 2017, 4436–4443. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Asawa, Y.; Arsent’eva, A.V.; Anufriev, S.A.; Anisimov, A.A.; Suponitsky, K.Y.; Filippov, O.A.; Nakamura, H.; Sivaev, I.B. Synthesis of Bis(Carboranyl)amides 1,1′-μ-(CH2NH(O)C(CH2)n-1,2-C2B10H11)2 (n = 0, 1) and Attempt of Synthesis of Gadolinium Bis(Dicarbollide). Molecules 2021, 26, 1321. https://doi.org/10.3390/molecules26051321
Asawa Y, Arsent’eva AV, Anufriev SA, Anisimov AA, Suponitsky KY, Filippov OA, Nakamura H, Sivaev IB. Synthesis of Bis(Carboranyl)amides 1,1′-μ-(CH2NH(O)C(CH2)n-1,2-C2B10H11)2 (n = 0, 1) and Attempt of Synthesis of Gadolinium Bis(Dicarbollide). Molecules. 2021; 26(5):1321. https://doi.org/10.3390/molecules26051321
Chicago/Turabian StyleAsawa, Yasunobu, Aleksandra V. Arsent’eva, Sergey A. Anufriev, Alexei A. Anisimov, Kyrill Yu. Suponitsky, Oleg A. Filippov, Hiroyuki Nakamura, and Igor B. Sivaev. 2021. "Synthesis of Bis(Carboranyl)amides 1,1′-μ-(CH2NH(O)C(CH2)n-1,2-C2B10H11)2 (n = 0, 1) and Attempt of Synthesis of Gadolinium Bis(Dicarbollide)" Molecules 26, no. 5: 1321. https://doi.org/10.3390/molecules26051321
APA StyleAsawa, Y., Arsent’eva, A. V., Anufriev, S. A., Anisimov, A. A., Suponitsky, K. Y., Filippov, O. A., Nakamura, H., & Sivaev, I. B. (2021). Synthesis of Bis(Carboranyl)amides 1,1′-μ-(CH2NH(O)C(CH2)n-1,2-C2B10H11)2 (n = 0, 1) and Attempt of Synthesis of Gadolinium Bis(Dicarbollide). Molecules, 26(5), 1321. https://doi.org/10.3390/molecules26051321