
Conversion of Racemic Unnatural Amino Acids to Optically Pure Forms by a Coupled Enzymatic Reaction


Abstract
1. Introduction
2. Results and Discussion
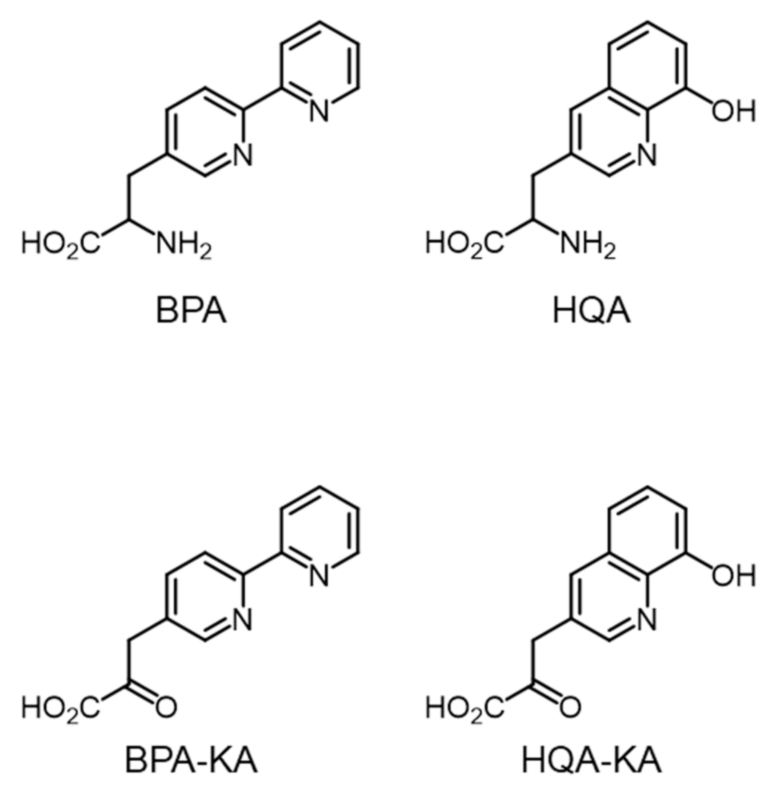
2.1. Metal-Chelating Amino Acids in GCE Technology
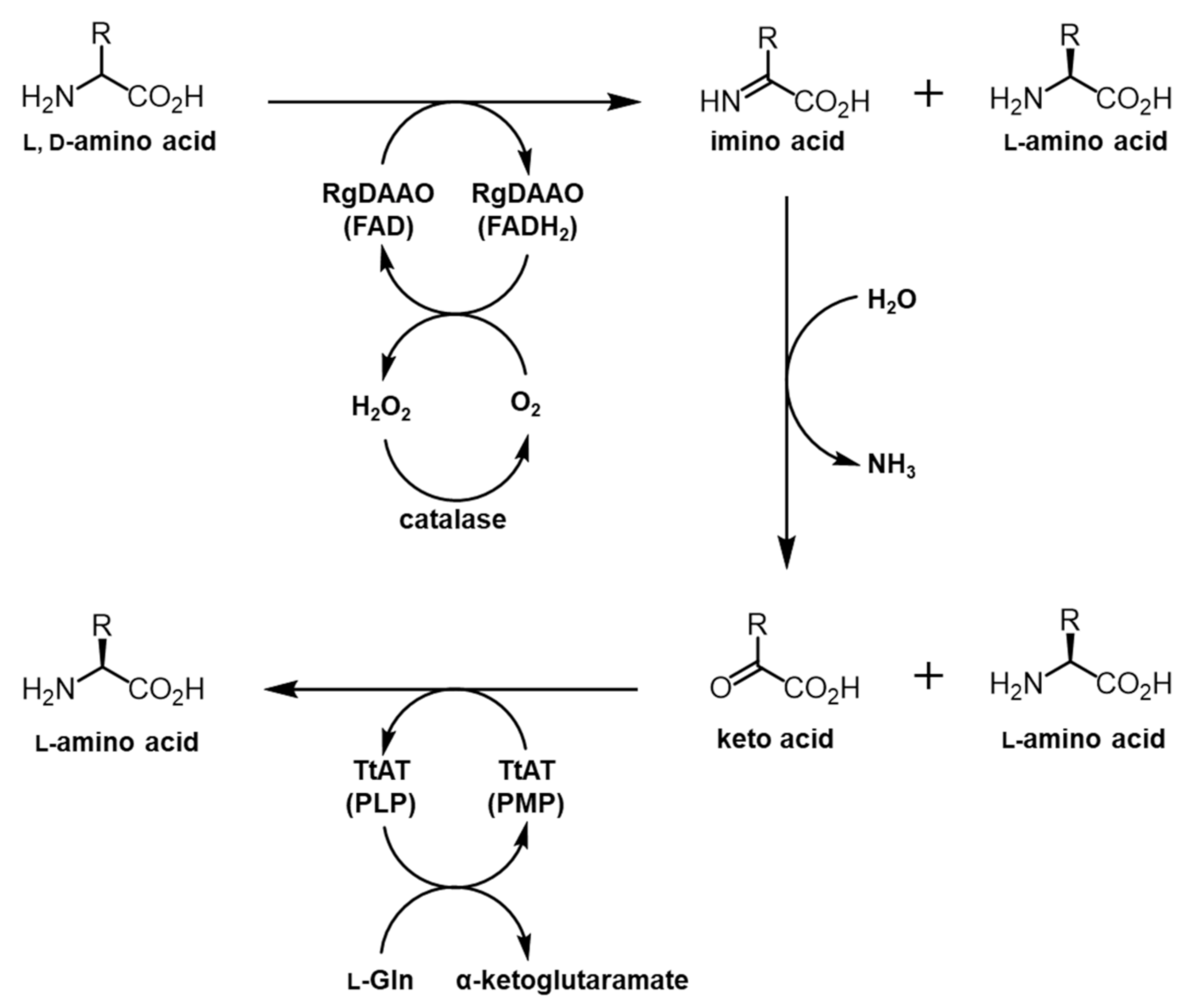
2.2. System Design for the Preparation of Optically Pure BPA and HQA
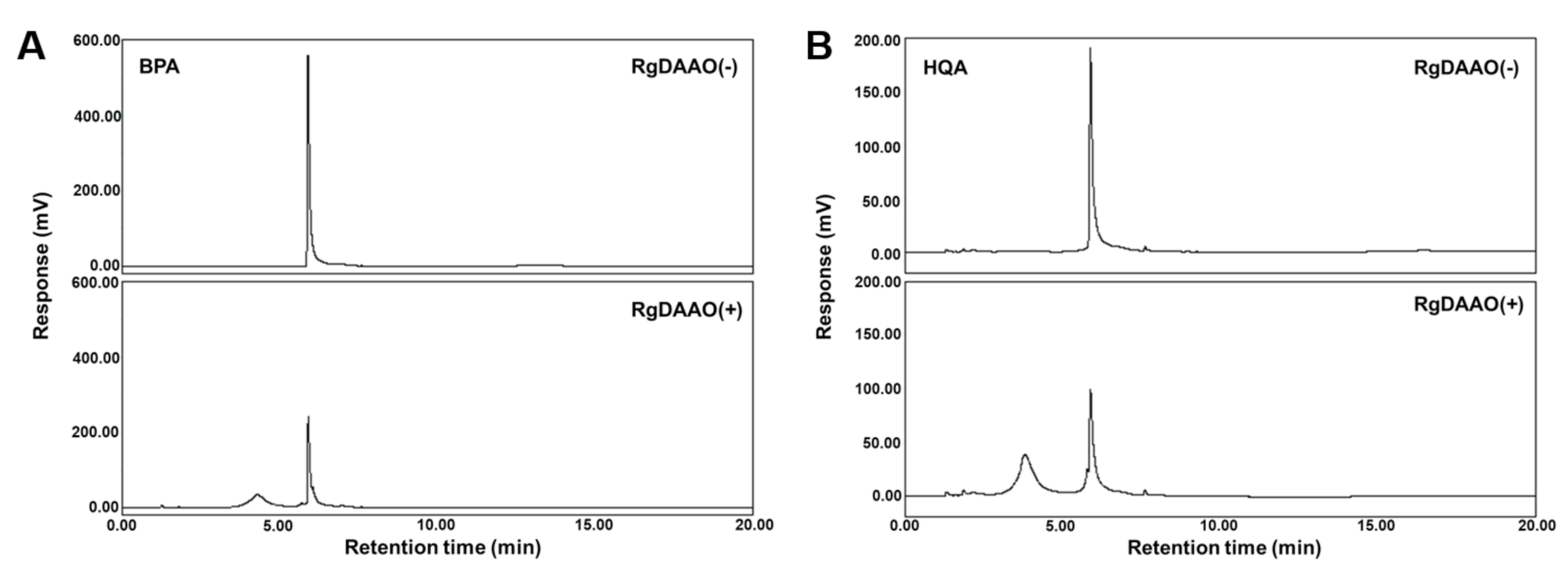
2.3. RgDAAO and TtAT Activity for BPA and HQA
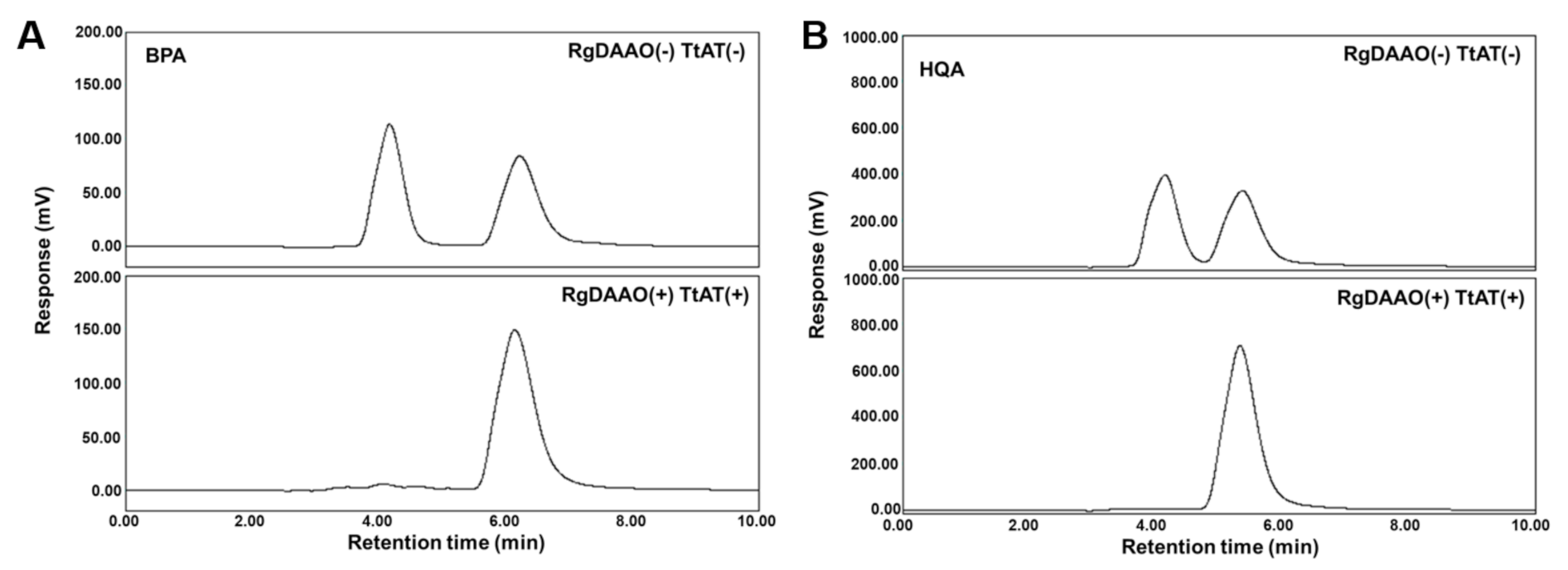
2.4. Enzymatic Preparation of Optically Pure BPA and HQA
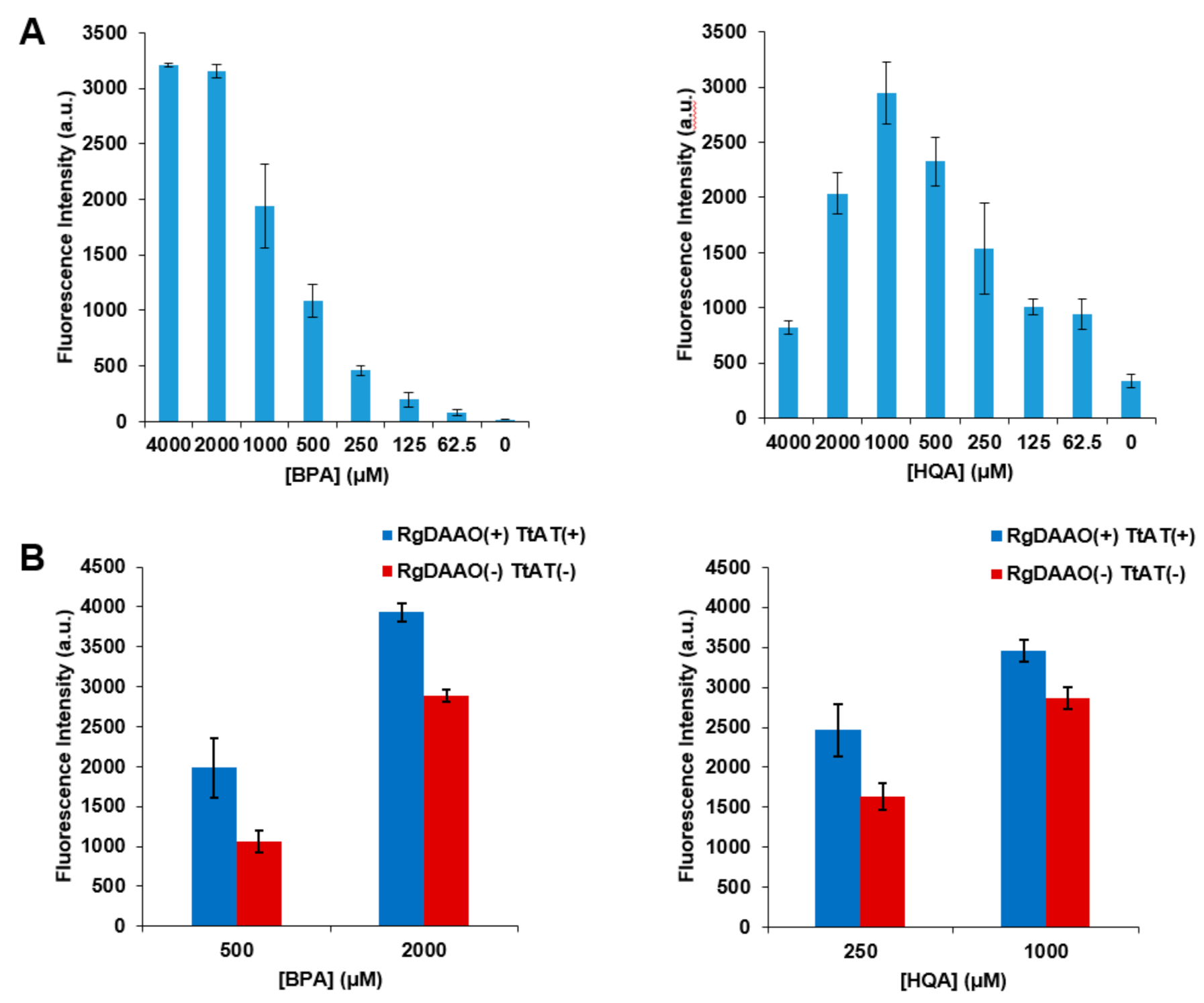
2.5. Application of Optically Pure BPA and HQA to GCE Experiments
3. Materials and Methods
3.1. Expression and Purification of RgDAAO
3.2. Expression and Purification of TtAT
3.3. RgDAAO Reaction
3.4. TtAT Reaction
3.5. Coupled Reaction of RgDAAO and TtAT
3.6. Application to the GCE Technique
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Cesari, M.; Rossi, G.P.; Sticchi, D.; Pessina, A.C. Is homocysteine important as risk factor for coronary heart disease? Nutr. Metab. Cardiovasc. Dis. 2005, 15, 140–147. [Google Scholar] [CrossRef]
- Wu, G. Amino acids: Metabolism, functions, and nutrition. Amino Acids 2009, 37, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Wolfson, R.L.; Sabatini, D.M. The dawn of the age of amino acid sensors for the mTORC1 pathway. Cell Metab. 2017, 26, 301–309. [Google Scholar] [CrossRef] [PubMed]
- Paul, B.D.; Sbodio, J.I.; Xu, R.; Vandiver, M.S.; Cha, J.Y.; Snowman, A.M.; Snyder, S.H. Cystathionine γ-lyase deficiency mediates neurodegeneration in Huntington’s disease. Nature 2014, 508, 96–100. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.; Bazer, F.W.; Cudd, T.A.; Meininger, C.J.; Spencer, T.E. Recent advances in nutritional sciences argjnine nutrition and fetal development. Am. Soc. Nutr. Sci. 2004, 2626–2630. [Google Scholar]
- Xue, Y.P.; Cao, C.H.; Zheng, Y.G. Enzymatic asymmetric synthesis of chiral amino acids. Chem. Soc. Rev. 2018, 47, 1516–1561. [Google Scholar] [CrossRef]
- D’Este, M.; Alvarado-Morales, M.; Angelidaki, I. Amino acids production focusing on fermentation technologies – A review. Biotechnol. Adv. 2018, 36, 14–25. [Google Scholar] [CrossRef] [PubMed]
- Nájera, C.; Sansano, J.M. Catalytic asymmetric synthesis of α-amino acids. Chem. Rev. 2007, 107, 4584–4671. [Google Scholar] [CrossRef]
- Toda, F. Enantiomer Separations; Springer: Dordrecht, The Netherlands, 2003; ISBN 1402023367. [Google Scholar]
- Ahuja, S. Chiral separations, applications and technology. J. Liq. Chromatogr. Relat. Technol. 1998, 21, 1076–1078. [Google Scholar] [CrossRef]
- Subramanian, G. (Ed.) Chiral Separation Techniques: A Practical Approach; Wiley VCH: Weinheim, Germany, 2000; ISBN 3527298754. [Google Scholar]
- Soloshonok, V.A.; Ellis, T.K.; Ueki, H.; Ono, T. Resolution/deracemization of chiral α-amino acids using resolving reagents with flexible stereogenic centers. J. Am. Chem. Soc. 2009, 131, 7208–7209. [Google Scholar] [CrossRef]
- Huh, J.W.; Yokoigawa, K.; Esaki, N.; Soda, K. Total conversion of racemic pipecolic acid into the L-enantiomer by a combination of enantiospecific oxidation with D-amino acid oxidase and reduction with sodium borohydride. Biosci. Biotechnol. Biochem. 1992, 56, 2081–2082. [Google Scholar] [CrossRef]
- Huh, J.W.; Yokoigawa, K.; Esaki, N.; Soda, K. Synthesis of l-proline from the racemate by coupling of enzymatic enantiospecific oxidation and chemical non-enantiospecific reducation. J. Ferment. Bioeng. 1992, 74, 189–190. [Google Scholar] [CrossRef]
- Turner, N.J. Enzyme catalysed deracemisation and dynamic kinetic resolution reactions. Curr. Opin. Chem. Biol. 2004, 8, 114–119. [Google Scholar] [CrossRef] [PubMed]
- Rosini, E.; Melis, R.; Molla, G.; Tessaro, D.; Pollegioni, L. Deracemization and stereoinversion of α-amino acids by L-amino acid deaminase. Adv. Synth. Catal. 2017, 359, 3773–3781. [Google Scholar] [CrossRef]
- Liu, C.C.; Schultz, P.G. Adding new chemistries to the genetic code. Annu. Rev. Biochem. 2010, 79, 413–444. [Google Scholar] [CrossRef]
- Lang, K.; Chin, J.W. Cellular incorporation of unnatural amino acids and bioorthogonal Labeling of Proteins. Chem. Rev. 2014, 114, 4764–4806. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Zheng, S.; Liu, H.; Chen, P.R. Illuminating biological processes through site-specific protein labeling. Chem. Soc. Rev. 2015, 44, 3405–3417. [Google Scholar] [CrossRef] [PubMed]
- Jung, J.E.; Lee, S.Y.; Park, H.; Cha, H.; Ko, W.; Sachin, K.; Kim, D.W.; Chi, D.Y.; Lee, H.S. Genetic incorporation of unnatural amino acids biosynthesized from α-keto acids by an aminotransferase. Chem. Sci. 2014, 5, 1881–1885. [Google Scholar] [CrossRef]
- Kim, S.; Sung, B.H.; Kim, S.C.; Lee, H.S. Genetic incorporation of l-dihydroxyphenylalanine (DOPA) biosynthesized by a tyrosine phenol-lyase. Chem. Commun. 2018, 54, 3002–3005. [Google Scholar] [CrossRef]
- He, J.; Li, S.; Deng, Y.; Fu, H.; Laforteza, B.N.; Spangler, J.E.; Homs, A.; Yu, J. Ligand-controlled C(sp3)–H arylation and olefination in synthesis of unnatural chiral α –amino acids. Science 2014, 343, 1216–1220. [Google Scholar] [CrossRef] [PubMed]
- Xiang, Z.; Wang, L. Enantiospecific synthesis of genetically encodable fluorescent unnatural amino acid l-3-(6-acetylnaphthalen-2-ylamino)-2-aminopropanoic acid. J. Org. Chem. 2011, 76, 6367–6371. [Google Scholar] [CrossRef]
- Ko, W.; Kumar, R.; Kim, S.; Lee, H.S. Construction of bacterial cells with an active transport system for unnatural amino acids. ACS Synth. Biol. 2019, 8, 1195–1203. [Google Scholar] [CrossRef]
- Ko, W.; Kim, S.; Jo, K.; Lee, H.S. Genetic incorporation of recycled unnatural amino acids. Amino Acids 2016, 48, 357–363. [Google Scholar] [CrossRef]
- Xie, J.; Liu, W.; Schultz, P.G. A genetically encoded bidentate, metal-binding amino acid. Angew. Chem. Int. Ed. 2007, 46, 9239–9242. [Google Scholar] [CrossRef]
- Lee, H.S.; Schultz, P.G. Biosynthesis of a site-specific DNA cleaving protein. J. Am. Chem. Soc. 2008, 130, 13194–13195. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.S.; Spraggon, G.; Schultz, P.G.; Wang, F. Genetic incorporation of a metal-ion chelating amino acid into proteins as a biophysical probe. J. Am. Chem. Soc. 2009, 131, 2481–2483. [Google Scholar] [CrossRef] [PubMed]
- Park, N.; Ryu, J.; Jang, S.; Lee, H.S. Metal ion affinity purification of proteins by genetically incorporating metal-chelating amino acids. Tetrahedron 2012, 68, 4649–4654. [Google Scholar] [CrossRef]
- Curti, B.; Ronchi, S.; Simonetta, P.M. D- and L-amino acid oxidases. In Chemistry and Biochemistry of Flavoenzyme; CRC Press: Boca Raton, FL, USA, 1992; Volume III, pp. 69–94. [Google Scholar]
- Nakano, S.; Kozuka, K.; Minamino, Y.; Karasuda, H.; Hasebe, F.; Ito, S. Ancestral L-amino acid oxidases for deracemization and stereoinversion of amino acids. Commun. Chem. 2020, 3, 1–11. [Google Scholar] [CrossRef]
- Molla, G.; Melis, R.; Pollegioni, L. Breaking the mirror: L-amino acid deaminase, a novel stereoselective biocatalyst. Biotechnol. Adv. 2017, 35, 657–668. [Google Scholar] [CrossRef]
- Caligiuri, A.; D’Arrigo, P.; Rosini, E.; Tessaro, D.; Molla, G.; Servi, S.; Pollegioni, L. Enzymatic conversion of unnatural amino acids by yeast D-amino acid oxidase. Adv. Synth. Catal. 2006, 348, 2183–2190. [Google Scholar] [CrossRef]
- Hosonol, A.; Mizuguchi, H.; Hayashi, H.; Goto, M.; Miyahara, I.; Hirotsu, K.; Kagamiyama, H. Glutamine:phenylpyruvate aminotransferase from an extremely thermophilic bacterium, thermus thermophilus HB8. J. Biochem. 2003, 134, 843–851. [Google Scholar] [CrossRef] [PubMed]
- Rosini, E.; Caldinelli, L.; Piubelli, L. Assays of D-amino acid oxidase activity. Front. Mol. Biosci. 2018, 4. [Google Scholar] [CrossRef] [PubMed]
- Smith, R.M.; Martell, A.E.; Motekaitis, R.J. NIST Critically Selected Stability Constants of Metal Complexes Database; NIST Stand. Ref. Database 46; U.S. Department of Commerce Technology Administration, National Institute of Standards and Technology, Standard Reference Data Program: Gaithersburg, MD, USA, 2004.






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Substrate | kcat (min−1) | Km (mM) | kcat/Km (min−1mM−1) |
|---|---|---|---|
| NPA | 1710 ± 85 | 0.052 ± 0.0087 | 3.3 × 104 |
| BPA | 448 ± 20 | 0.087 ± 0.010 [b] | 0.52 × 104 |
| HQA | 124 ± 0.4 | 0.043 ± 0.0094 [b] | 0.29 × 104 |
| Substrate | Conversion (%) [a] | Enantiomeric Excess (%) | |
|---|---|---|---|
| RgDAAO Reaction | Coupled Reaction | ||
| BPA | 97 | 95 | 95 |
| HQA | 98 | 98 | 98 |
| Samples | GFP Intensity | |||
|---|---|---|---|---|
| BPA | HQA | |||
| 500 μM | 2000 μM | 250 μM | 1000 μM | |
| l-form [a] | 1983 | 3937 | 2456 | 3456 |
| d,l-form [b] | 1059 (1942) [c] | 2885 (3211) [c] | 1633 (2326) [c] | 2868 (2036) [c] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, H.; Kim, D.; Kim, S.; Lee, H.S. Conversion of Racemic Unnatural Amino Acids to Optically Pure Forms by a Coupled Enzymatic Reaction. Molecules 2021, 26, 1274. https://doi.org/10.3390/molecules26051274
Lee H, Kim D, Kim S, Lee HS. Conversion of Racemic Unnatural Amino Acids to Optically Pure Forms by a Coupled Enzymatic Reaction. Molecules. 2021; 26(5):1274. https://doi.org/10.3390/molecules26051274
Chicago/Turabian StyleLee, Hannae, Dongchan Kim, Sooin Kim, and Hyun Soo Lee. 2021. "Conversion of Racemic Unnatural Amino Acids to Optically Pure Forms by a Coupled Enzymatic Reaction" Molecules 26, no. 5: 1274. https://doi.org/10.3390/molecules26051274
APA StyleLee, H., Kim, D., Kim, S., & Lee, H. S. (2021). Conversion of Racemic Unnatural Amino Acids to Optically Pure Forms by a Coupled Enzymatic Reaction. Molecules, 26(5), 1274. https://doi.org/10.3390/molecules26051274